

Abstract
Platelets not only play an essential role in hemostasis after vascular injury but are also involved in the development of coronary artery disease (CAD) and cerebrovascular lesions. Patients with CAD and cerebral ischemia are recommended to undergo antiplatelet therapy, but they have an increased incidence of major bleeding complications. Both assessment of the platelet activation status and response to antiplatelet therapy in each patient are highly desired. β-Amyloid precursor protein (APP) 770 is expressed in vascular endothelial cells, and its extracellular region, a soluble form of APP770 (sAPP770, also called nexin-2), is proteolytically cleaved for shedding. Abundant sAPP770 is also released from activated platelets. In this study, we used peripheral blood samples from patients with CAD and control subjects and evaluated sAPP770 as a specific biomarker for platelet activation. First, the plasma levels of sAPP770 correlated well with those of the soluble form CD40 ligand (CD40L), an established biomarker for platelet activation. Additionally, flow cytometry analysis using peripheral blood cells showed that CD40L expression is up-regulated in activated T cells, whereas APP770 expression is negligible in all blood cell types except platelets. Following stimulation with collagen or ADP, aggregating platelets immediately released sAPP770. Finally, patients with dual antiplatelet therapy showed significantly lower levels of plasma sAPP770 than those with no therapy. Taken together, our data show that plasma sAPP770 could be a promising biomarker for platelet activation.
Keywords: amyloid precursor protein, CD40L, platelet, DAPT, angina pectoris, biomarker, cardiovascular disease, heart failure, ischemia, plasma, shedding
The primary function of platelets is to stop hemorrhage after vascular injury, but recent accumulating reports have revealed their important role in the development of atherosclerotic lesions (1, 2). Rupture of these lesions triggers the acute onset of arterial thrombosis, which could lead to acute coronary syndrome (ACS) and ischemic stroke.
Various antiplatelet drugs have been developed to treat and prevent atherothrombosis (3). Low-dose aspirin inhibits platelet cyclooxygenase-1, thereby reducing the synthesis of thromboxane A2, a platelet activator. Thienopyridine, an antagonist of P2Y12, which is a major ADP receptor on platelets, inhibits ADP-induced platelet activation. In the 1990s, dual antiplatelet therapy (DAPT; aspirin plus a P2Y12 inhibitor) was shown to reduce the incidence of stent thrombosis after percutaneous interventions (4). Conversely, antiplatelet therapy increases the incidence of major bleeding complications (mostly in the upper gastrointestinal tract) (3). How to set up the duration of DAPT in coronary artery disease (CAD) patients remains unclear. Although the ex vivo measurement of platelet responsiveness to various agonists can evaluate the function of platelets, such measurements show no correlation with platelet activation in vivo (1).
Activated platelets lead to the release of granular components and relocalization of membrane proteins to the cell surface. Upon platelet activation, surface P-selectin is expressed on the platelet membrane (5) and mediates platelet–endothelial interactions (2). P-selectin is considered the “gold standard” marker of platelet activation. Circulating monocyte-platelet aggregates can be detected by flow cytometry analysis and are sensitive laboratory markers for platelet activation (6). CD40 ligand (CD40L) is cryptic in unstimulated T cells and platelets but is rapidly presented to the cell surface after their activation (7, 8), leading to the induction of endothelial cells that produce various inflammatory mediators (2, 8, 9) and up-regulate functional membrane proteins, as well as to the induction of atherosclerosis (10, 11). Furthermore, a soluble form of CD40L (sCD40L) is released from activated T cells (12) and platelets. High plasma levels of sCD40L have been observed in patients with unstable angina (13) and active systemic lupus erythematosus (14) and are associated with an increased risk of vascular events in healthy women (15). However, no practical biomarker is available to evaluate platelet activation.
Amyloid precursor protein (APP) is a membrane protein that generates β-amyloid (Aβ) peptide, and the accumulation of Aβ and neurofibrillary tangles in the brain triggers the onset of Alzheimer's disease (16, 17). APP has three kinds of alternatively spliced mRNA isoforms: APP695, APP751, and APP770 (18, 19). APP695 is predominantly expressed in neurons (20), whereas APP770 is found in vascular endothelial cells (21) and platelets (22). The soluble form of APP770 (sAPP770) is released upon platelet activation, and plasma sAPP770 levels are significantly higher in ACS patients (23). In this study, by analyzing blood samples from control subjects and CAD patients, we found a significant correlation between the plasma levels of sAPP770 and sCD40L and that APP770 expression is limited to platelets in blood cells. Moreover, compared with those in control subjects and CAD patients with aspirin alone, the plasma sAPP770 levels were significantly lower in CAD patients with DAPT. Our finding raises the possibility that plasma sAPP770 can be a useful platelet activation marker.
Results
The sAPP770 levels correlate with the sCD40L levels in plasma
We first performed a validation study of APP770 ELISA using heparinized and citrated plasma and found that the dilution linearity, recovery rate, and coefficient validation were similar to those of EDTA plasma and serum, as reported in the manufacturer's data sheet (Fig. S1). Because sCD40L is an established laboratory marker for platelet activation, we then compared the performance of plasma sAPP770 with that of sCD40L. The plasma samples from healthy volunteers were diluted at 1:5 or 1:75 and used to determine sCD40L and sAPP770 levels using sandwich ELISA (Fig. 1A). The plasma sAPP770 level was high (27.3 ± 4.0 ng/ml), whereas the plasma sCD40L level was markedly lower (105 ± 34 pg/ml); 4 of 10 plasma samples showed undetectable sCD40L levels (zero absorbance; Fig. 1B). Because it is expected that CAD patients have higher levels of sCD40L (13) and sAPP770 (23), we next analyzed heparinized plasma from CAD patients and control subjects, such as hypertension, diabetes, and sleep apnea syndrome. Again, 31 of 67 samples from CAD patients and 12 of 53 samples from control subjects show undetectable sCD40L levels, and the remaining samples were then used to measure sAPP770 levels. We found a significant correlation between the sAPP770 and sCD40L levels (r = 0.4492; p < 0.0001) (Fig. 1C), raising the possibility of sAPP770 as a promising biomarker for platelet activation.
Figure 1.

sAPP770 levels correlate with sCD40L levels in plasma. A, heparinized plasma samples from healthy volunteers were used to determine the plasma volume to quantify the sCD40L and sAPP770 levels within a dynamic range. B, summary of the measurement of the plasma sCD40L and sAPP770 levels in healthy volunteers. C, heparinized plasma samples from CAD patients (n = 36) and control subjects, n = 40) were used to determine the plasma volume to quantify the sCD40L and sAPP770 levels. The scatter plot shows the plasma sCD40L and sAPP770 levels. Using Prism, we calculated Pearson's correlation coefficient.
APP770 expression is limited to platelets in peripheral blood
We next asked whether human peripheral blood cell types other than platelets express APP770 or another APP isoform. To this end, we used two antibodies, anti-APP770 and anti-APP 22C11, the latter of which recognizes all APP isoforms. In CD13+ cells, which represent myeloid cells, APP770 expression was not detectable (Fig. 2A), whereas the other APP isoform was weakly expressed. We next isolated lymphocytes from peripheral blood cells and found that none of the APP isoforms were detectable in both T and B cells (Fig. 2, B and C). We also employed a biochemical approach to confirm that APP770 expression is limited to platelets. We first isolated platelet-free blood cells by repeated centrifugation. Using magnetic beads, we then isolated leukocytes positive for CD45, which is expressed at high levels on all cells of hematopoietic origin except erythrocytes and platelets. To isolate the limited populations of leukocytes, we sequentially incubated platelet-free blood cells with magnetic beads conjugated with different antibodies: anti-CD3 (for T cells), anti-CD19 (for B cells), and anti-CD14 (for monocytes). Both APP and APP770 signals were undetectable in these cell types (Fig. 2D). From platelet-rich plasma (PRP), we also isolated platelets, which showed positive staining for platelet markers such as CD42b and CD40L, and found that the isolated platelets expressed APP770. T cells show weak CD40L signal. It is known that CD40L surface expression is markedly induced in activated T cells (7, 24). We next asked whether activated lymphocytes could induce APP770 expression. We found that T cells activated with phorbol 12-myristate 13-acetate and ionomycin showed strong cell surface CD40L expression, whereas activated lymphocytes did not express detectable levels of surface APP770 expression (Fig. 2E).
Figure 2.

AP770 is detectable only in platelets, not in other blood cell types. Human peripheral blood mononuclear cells, isolated from blood samples, were analyzed by flow cytometry. A–C, cell surface APP or APP770 expression was evaluated in CD13+ peripheral myeloid cells (A), CD3+ T-lymphocytes (B), and CD19+ B-lymphocytes (C) using anti-APP (22C11) and anti-hAPP770 antibodies. (D) Leukocytes, T cells, B cells, monocytes, and platelets from human blood samples were lysed in T-PER reagent containing protease inhibitor mixture and were used for Western blotting analysis with antibodies against CD45, CD3, CD19, CD14, CD42b, CD40L, APP770, APP (22C11), and β-actin. E, human peripheral lymphoid cells, isolated from blood samples, were incubated in the presence or absence of cell stimulation mixture for 5 h and then were subjected to flow cytometry analysis with anti-CD40L and APP770 antibodies. Cont, control.
APP770 is expressed on the platelet surface and is released upon platelet activation
The APP isoform with the Kunitz type II protease inhibitor domain, APP751/770, has been historically described as protease nexin-2, a membrane protein and α-granule protein in platelets (22, 25). Nevertheless, intracellular localization of membrane-bound APP remains unclear. Next, we assessed whether surface APP770 expression is found in platelets before and after activation. Resting platelets showed high APP770 expression (Fig. 3A). After thrombin-mediated platelet activation, as judged by staining with PAC-1, which recognizes activated glycoprotein IIa/IIIb, surface APP770 expression did not increase, but the number of APP770low platelets was decreased. The results indicate that membrane relocalization following platelet activation affects the surface APP770 levels. We next stimulated platelets with the strong agonist collagen or weak agonist ADP and then measured sAPP770 release. Both agonists caused the release of sAPP770 upon platelet aggregation (Fig. 3, B and C). Notably, low levels of ADP led to a platelet aggregation curve that was biphasic (an initial wave of aggregation (primary wave), followed by a secondary wave of aggregation), and the elevation of sAPP770 was delayed. The results indicate that the secondary wave of aggregation is mainly responsible for the sAPP770 release. Because markedly high levels of sAPP770 were released from platelets, we next analyzed the α- and β-site cleavage products of APP770, sAPP770α, sAPP770β, and Aβ40/42, following platelet activation with the strong agonist thrombin. Although both sAPPα and sAPPβ were found to be released from platelets, Aβ40/42 was undetectable (Fig. 3, D and E). The higher sAPP770 level than the total value of sAPPα plus sAPPβ could be explained by the different standard samples used in the different ELISA systems (APP770, sAPP-α, and sAPP-β), which made direct comparison of the measured APP770 values difficult. To analyze the proteolytic cleavage of platelet APP following stimulation in more detail, we performed Western blotting analysis using anti–N-terminal APP antibody 22C11 and anti–C-terminal APP antibody (which detects the C-terminal fragments (CTFs) of APP) (26). After platelet activation, a 22C11-positive signal corresponding to full-length APP was markedly reduced (Fig. 3F) in response to sAPP770 production. Although we observed a major signal corresponding to CTF-α/β (as demonstrated by an approximate molecular mass of 12 kDa), other CTFs were undetectable.
Figure 3.

APP770 is abundantly expressed in platelets and released upon platelet activation. A, human platelets were stimulated with thrombin (1 unit/ml) for 15 min and then were analyzed by flow cytometry with FITC-labeled PAC-1, or anti-APP770 and FITC-labeled anti-rabbit secondary antibodies. B, human platelets or PRP, stirred in the aggregometer, were stimulated with type I collagen (1, 3, and 5 µg/ml) or ADP (1, 3, and 5 µm). Typical platelet aggregation curves, stimulated with ADP, are shown. C, at 2, 4, and 6 min after stimulation, the platelet suspensions were centrifuged, and the supernatants were used to determine the sAPP770 levels, which are expressed as means ± S.D. (n = 3). D, schematic of APP770 and its major proteolytic cleavage products, as well as site-specific anti-APP antibodies. E and F, human platelets were stimulated with thrombin (1 unit/ml) for 5 min, and the platelet supernatants were used to measure sAPP770, sAPP-α, sAPP-β, and Aβ40/42 by sandwich ELISA (E); the data are expressed as means ± S.D. (n = 4). Before and after stimulation with thrombin, the platelet pellets were analyzed by Western blotting for APP, APP CTF, and α-tubulin (loading control) (F). Cont, control.
sAPP770 levels are elevated in heparinized blood samples
To determine whether blood sAPP770 is a useful biomarker for platelet activation, we first asked how different blood-collecting tubes would affect the blood sAPP770 levels. Analysis of blood samples from healthy human volunteers showed that the sAPP770 level was 10.4 ng/ml in citrated plasma, 36.0 ng/ml in heparinized plasma, and 416.0 ng/ml in serum (Fig. 4A). It is considered that proteolytic cleavage of endothelial APP770 results in the release of sAPP770 (23) and increased plasma sAPP770. The 10-fold higher serum sAPP770 levels than plasma sAPP770 are likely due to platelets becoming fully activated and degranulated in serum to release sAPP770. Similarly, we hypothesized that platelets are relatively unstable in heparinized blood samples, and partially activated platelets contribute to the increased sAPP770 levels. To confirm this hypothesis, we kept both citrated and heparinized blood samples at room temperature for up to 2 h and isolated plasma at the appropriate intervals. As expected, the sAPP770 levels were stable in citrated plasma, but gradual sAPP770 elevation was observed in heparinized plasma, with the highest value at 1 h (Fig. 4B). We next studied the fluctuation of the blood sAPP770 levels throughout the day. We found that serum sAPP770 levels did not vary throughout the day. However, citrated plasma sAPP770 levels were higher but not significant in the morning (Fig. 4C), which could be related to the previous report showing a marked circadian rhythm in the frequency of onset of ACS, with a peak from 6 a.m. to 12 p.m. (27).
Figure 4.

Blood-collecting methods affect blood sAPP770 levels. A, citrated plasma, heparinized plasma, and serum samples collected from healthy volunteers at 8:30–9:30 a.m. were analyzed to determine the sAPP770 levels (n = 10). Serum sAPP770 was measured at a 1:150 dilution ratio and is expressed as the mean ± S.D. **, p < 0.01; one-way ANOVA with Tukey's multiple comparison test. B, blood samples collected in citrated or heparin-containing tubes were kept at 25 °C for 0–2 h and then were centrifuged at 1,000 × g for 15 min to collect plasma and measure the sAPP770 levels (duplicate measurement). C, citrated plasma and serum samples, collected from healthy volunteers at 8:30–9:30 a.m., 12:30–1:30 p.m., and 5:00–6:00 p.m., were analyzed to determine the sAPP770 levels (n = 10). All data are expressed as means ± S.D. N.S., not significant; one-way ANOVA with Tukey's multiple comparison test.
Blood sAPP770 levels are significantly lower in CAD patients prescribed DAPT
We next anticipated that plasma sAPP770 levels would be reduced by antiplatelet therapy. Because the sAPP770 levels in heparinized plasma are variable by the time between the sample collection and the assay, we decided to measure sAPP770 levels in citrated plasma samples in control subjects with neither CAD nor antiplatelet therapy, CAD patients with aspirin only, and CAD patients with DAPT. We found that the levels of citrated plasma sAPP770 in CAD patients with DAPT were significantly lower than those of control subjects without therapy (Fig. 5). The results suggest that blood levels of sAPP770 are decreased by the effect of antiplatelet therapy, and we concluded that plasma sAPP770 could be a promising marker for platelet activity in CAD subjects.
Figure 5.
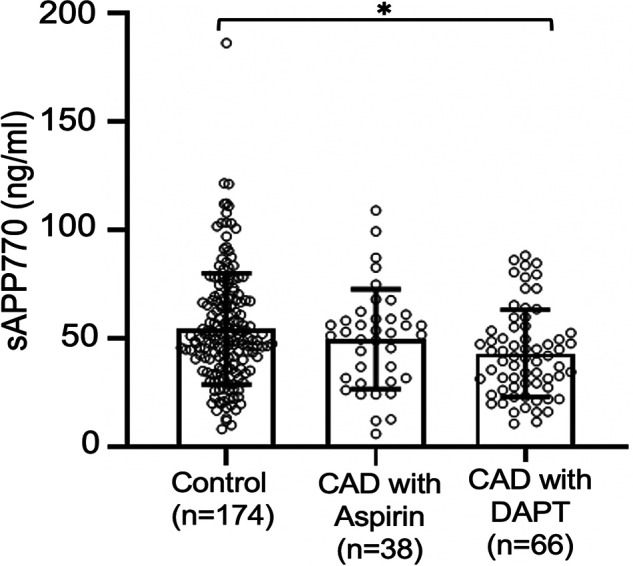
Plasma sAPP770 levels are significantly lower in patients with DAPT. Citrated plasma from CAD patients and control subjects were analyzed to quantify the sAPP770 levels at a 1:40 ratio. Plasma sAPP770 levels in patients without antiplatelet therapy (control subjects; n = 174), in CAD patients receiving aspirin only (n = 38), and in CAD patients receiving DAPT (n = 66). All the data are expressed as means ± S.D. and are shown as dots. *, p < 0.01; one-way ANOVA with Tukey's multiple comparison test.
Discussion
In this study, we show that APP770 is abundantly expressed in platelets but undetectable in other peripheral blood cells. Because platelets are abundant in peripheral blood, we repeated centrifugation several times to unambiguously eliminate platelets from other blood cell types; otherwise, APP770 is detected in different blood cell types. Flow cytometry analysis of platelets showed surface APP770 expression, whereas sandwich ELISA detected substantial levels of released sAPP770, sAPP770α, and sAPP770β following platelet activation. In our previous study using anti–C-terminal APP and APP770 antibodies, membrane-bound APP accounted for less than 10% of total APP in platelets (23). APP has mainly two processing pathways, α-site cleavage initiated by ADAM (“a disintegrin and metalloprotease”) and BACE1-mediated β-site cleavage, which produces pathogenic Aβ by subsequent γ-site cleavage (28, 29). Western blotting analysis using anti–C-terminal antibody showed that a significant amount of full-length APP disappears following platelet activation. Both ADAM and BACE1 proteases have been identified in platelets (30), indicating that sAPP770 could be produced from membrane-bound APP770 in platelets. A recent study using immunoprecipitation-coupled MS analysis showed that plasma Aβ is a marker for Alzheimer's disease (31). Contrary to our finding that abundant levels of sAPP770α and sAPP770β are released from platelets, Aβ40/42 was undetectable. It is reported that equivalent levels of sAPP770β and Aβ (∼0.03 pmol/ml each) are detected in the culture media of endothelial cells (21, 23). Thus, γ-secretase machinery may be lacking or significantly impaired in platelets.
Another interesting aspect is the functional role of platelet APP770. sAPP770 is a tight-binding inhibitor of coagulation factors IXa (32) and XIa (33). Anticoagulant properties of sAPP770 have been demonstrated in transgenic mouse models because sAPP770 overexpression in platelets both reduces in vivo cerebral thrombosis and increases intracerebral hemorrhage (34).
In our previous study, ACS patients exhibited higher levels of plasma sAPP770 than with normal control subjects (23). Although patients with antiplatelet therapy might have lower levels of plasma sAPP770 than those of control cases, our current study has several limitations because of the treatment policy. In the present study, control subjects were considered free from CAD or thrombosis, and they received no antiplatelet therapy. Because all CAD patients had already received antiplatelet therapy by their attending physicians when they first visited our hospital, plasma samples from CAD patients without antiplatelet therapy nor plasma samples before administration of antiplatelet therapy were unavailable. Nevertheless, we found that CAD patients with DAPT had lower levels of plasma sAPP770 than control subjects without therapy. Thus, plasma sAPP770 could be a promising marker for platelet activity. Regarding monitoring the platelet response to antiplatelet therapy, the VerifyNow system has been broadly used (35, 36). VerifyNow is a rapid system that measures platelet aggregation ex vivo; therefore, the function of platelets could be evaluated. Although plasma sAPP770 levels reflect the platelet activation status in vivo, such as local embolism and thrombosis, such states cannot be assessed by VerifyNow. Moreover, VerifyNow is affected by anemia, thrombocytopenia, and high triglyceride levels (37–39), and these conditions are co-morbidities of CAD patients. Whether plasma sAPP770 levels are affected by these factors warrants future investigation. A combination study using both systems would provide broader information about the platelet activation status and appropriate dose or duration of antiplatelet therapy in each patient.
Experimental procedures
Subjects
Human studies performed here were approved by the Ethical Committees of Fukushima Medical University (approval number 29378) and abided by the Declaration of Helsinki principles. Human blood samples were collected from healthy volunteers (n = 10), CAD patients, and control subjects at Fukushima Medical University. CAD patients were defined as those who had undergone percutaneous intervention and were prescribed aspirin or DAPT, whereas control subjects (hypertension, diabetes, and sleep apnea syndrome, etc.) had no history of CAD nor antiplatelet therapy.
Materials
The materials used in this study were sourced as follows: T-PER tissue extraction reagent from Thermo Fisher Scientific; complete protease inhibitor mixture from Roche; protein molecular weight standards from Bio-Rad; recombinant APP770 from BioLegend; cell stimulation mixture from Invitrogen; all other chemicals from Sigma or Wako Chemicals. The commercially available antibodies used were mouse monoclonal anti-APP (22C11, Merck), anti–α-tubulin (Sigma), anti–β-actin (Sigma), PE–anti-CD13 (BioLegend), PE–anti-CD3 (BioLegend), PE–anti-CD19 (BioLegend), FITC–anti-CD154 (BioLegend), and FITC–PAC-1 (BD Bioscience), and rabbit polyclonal anti-hAPP770 (IBL-Japan), anti-hAPP(C) (IBL-Japan), anti-CD40L (GeneTex), anti-CD42b (GeneTex), anti-CD45 (Proteintech), anti-CD3 (Abcam), anti-CD14 (Abcam), and anti-CD19 (Abcam). CD45, CD3, CD9, and CD14 microbeads were from Miltenyi Biotec. The Quantikine ELISA kit for human CD40L (R&D, DCDL40), APP770 ELISA system (IBL-Japan, 27736), sAPP α ELISA system (IBL-Japan, 27734), sAPP β−w ELISA system (IBL-Japan, 27732), β-amyloid(1–40) ELISA Kit (Wako, 298-64601), and β-amyloid(1–42) ELISA kit (Wako, 296-64401) were used according to the manufacturers' protocols.
Blood collection and cell preparation
To prepare human sera, human blood samples were collected using InsepackTM II (511462; SEKISUI), incubated at 25 °C for 15 min, and centrifuged at 1,800 × g for 10 min. To prepare heparinized or citrate plasma, blood samples were collected using InsepackTM II (500763; SEKISUI) or Venoject® II (VP-CA050K70; TERUMO), respectively, and were centrifuged at 1,000 × g for 15 min at 4 °C within 30 min of collection. The supernatant was additionally centrifuged at 10,000 × g for 10 min at 4 °C. Human peripheral lymphocytes isolated from human blood samples using Ficoll-PaqueTM Plus (GE Healthcare Life Sciences) according to the manufacturer's protocol were suspended in 10% fetal bovine serum in Dulbecco's modified Eagle's medium (high glucose) and were activated with the cell stimulation mixture (1:500 dilution) for 5 h. To isolate human granulocytes, 3 ml of Ficoll-PaqueTM Plus reagent (d = 1.077) was layered onto 3 ml of lymphocyte separation solution (d = 1.119, Nacalai) in a 15-ml tube. Blood samples diluted with PBS (5 ml, 1:1 dilution) were then layered and centrifuged at 700 × g for 30 min at 25 °C, after which the granulocyte-rich lower layer was collected.
Platelet preparation and aggregation
Blood samples (30 ml) were collected from healthy volunteers on the day of the experiment using Venoject tubes containing 3.2% sodium citrate. PRP (2.3 × 108–2.7 × 108 platelets/ml) was collected by centrifugation at 200 × g for 20 min, and platelets were collected from the PRP by centrifugation at 900 × g for 10 min in the presence of 4 µm citrate. The platelet pellet was resuspended in modified HEPES–Tyrode buffer (134 mm NaCl, 12 mm NaHCO3, 2.9 mm KCl, 0.34 mm NaH2PO4, 1 mm CaCl2, 5 mm HEPES, 5 mm glucose, pH 7.4) to a density of 2.5 × 108 platelets/ml. Aliquots (200 µl) of PRP or platelets were used for platelet aggregation assays, which were performed in siliconized glass cuvettes at 37 °C with constant stirring at 1,000 rpm in a TPA-4C aggregometer (Tokyo Photoelectric Co.). Platelet aggregation was initiated by adding type I collagen (final concentration, 1, 3, or 5 µg/ml) or ADP (final concentration, 1, 3, or 5 μm). At each time point, the PRP or platelet suspensions were centrifuged at 1,800 × g for 2 min, and the supernatants were used to measure the levels of sAPP770, sAPPα, sAPPβ, Aβ40, and Aβ42 by sandwich ELISA.
Flow cytometry analysis
The cells, as single-cell suspensions in FACS buffer (1% BSA and 0.1% NaN3 in PBS), were incubated with the fluorescent-labeled primary antibody for 30 min at 4 °C. If necessary, the cells were incubated with the primary antibody for 1 h at 4 °C, followed by incubation with the fluorescent-labeled secondary antibody for 30 min at 4 °C. The cells were next analyzed by flow cytometry using a FACSCalibur instrument (BD Biosciences). Detailed descriptions of the primary antibodies and secondary antibodies are provided in Table S1.
Western blotting
Human blood samples, collected using Venoject tubes, were centrifuged at 1,000 × g for 30 min at 25 °C to obtain PRP and blood cells. The latter was suspended in PBS and centrifuged twice to completely remove platelets, and the sample was divided into two. Lymphocyte separation solution (d = 1.119) was layered onto one of the blood cell suspensions, followed by centrifugation at 400 × g for 30 min at 25 °C. From the intermediate layers, leukocytes, but not erythrocytes and platelets, were isolated using CD45 microbeads. Ficoll-PaqueTM Plus reagent (d = 1.077) was layered onto another blood cell suspension and then was centrifuged at 700 × g for 30 min at 25 °C. The intermediate layers were sequentially reacted with CD14 microbeads, CD19 microbeads, and CD3 microbeads to isolate monocytes, B cells, and T cells, respectively. The cell lysates (10–80 µg of proteins) were separated using 5–20% SDS-PAGE and were transferred to nitrocellulose membranes. To detect the C-terminal fragment of APP, the platelet lysates before and after activation were separated using a 16.5% SDS-polyacrylamide gel (p-PAGEL, ATTO), and electrophoresis was performed in Tris–Tricine buffer (Ez-RunT, AE-1415, ATTO). After transfer, the nitrocellulose membranes were boiled in PBS for 5 min. After incubation with 5% nonfat dried milk in TBS containing 0.1% Tween 20, the membranes were incubated with the primary antibody, followed by incubation with the horseradish peroxidase–conjugated secondary antibody. Western Lightning ECL Pro (PerkinElmer) was used to detect the bound antibodies. The intensities of the protein signals were quantified using an ImageQuant LAS-4000 mini instrument (GE Healthcare). Detailed descriptions of the primary antibodies and secondary antibodies are provided in Table S1.
Statistical analysis
All analyses were performed using GraphPad Prism (Statcon).
Data availability
All data are contained within the article and supporting information.
Supplementary Material
Acknowledgments
We thank Dr. Tatsuro Segawa (IBL-Japan) and Dr. Yuji Tsuji (Toyobo) for technical assistance regarding the validation study of APP770 ELISA.
Author contributions—S. M., T. M., T. Y., and T. K. resources; S. M., M. T., K. O., and N. Y. data curation; S. M. and K. K. formal analysis; S. M. and S. K. funding acquisition; S. M. methodology; S. M., A. Y., and S. K. writing-original draft; S. M., A. Y., T. M., T. K., M. T., K. O., H. S., N. Y., K. K., Y. T., and S. K. writing-review and editing; A. Y. and S. K. conceptualization; A. Y., M. T., K. O., N. Y., and S. K. investigation; H. S., Y. T., and S. K. supervision.
Funding and additional information—This work was supported by AMED Grant JP18am0101036 (to S. K.) and KAKENHI Grants 16K08601 (to S. K.) and MO18K0747 (to S. M.).
Conflict of interest—This work was partially supported by Toyobo Co., Ltd.
- CAD
- coronary artery disease
- APP
- amyloid precursor protein
- ACS
- acute coronary syndrome
- DAPT
- dual antiplatelet therapy
- CD40L
- CD40 ligand
- s
- soluble form
- Abs
- absorbance
- Aβ
- β-amyloid
- PRP
- platelet-rich plasma
- CTF
- C-terminal fragment
- PE
- Phycoerythrin
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Davì G., and Patrono C. (2007) Platelet activation and atherothrombosis. N. Engl. J. Med. 357, 2482–2494 10.1056/NEJMra071014 [DOI] [PubMed] [Google Scholar]
- 2. Ruggeri Z. M. (2002) Platelets in atherothrombosis. Nat. Med. 8, 1227–1234 10.1038/nm1102-1227 [DOI] [PubMed] [Google Scholar]
- 3. Patrono C., Coller B., FitzGerald G. A., Hirsh J., and Roth G. (2004) Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 126, 234S–264S 10.1378/chest.126.3_suppl.234S [DOI] [PubMed] [Google Scholar]
- 4. Leon M. B., Baim D. S., Popma J. J., Gordon P. C., Cutlip D. E., Ho K. K., Giambartolomei A., Diver D. J., Lasorda D. M., Williams D. O., Pocock S. J., Kuntz R. E., and Stent Anticoagulation Restenosis Study Investigators (1998) A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. N. Engl. J. Med. 339, 1665–1671 10.1056/NEJM199812033392303 [DOI] [PubMed] [Google Scholar]
- 5. Larsen E., Celi A., Gilbert G. E., Furie B. C., Erban J. K., Bonfanti R., Wagner D. D., and Furie B. (1989) PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 59, 305–312 10.1016/0092-8674(89)90292-4 [DOI] [PubMed] [Google Scholar]
- 6. Furman M. I., Barnard M. R., Krueger L. A., Fox M. L., Shilale E. A., Lessard D. M., Marchese P., Frelinger A. L. 3rd, Goldberg R. J., and Michelson A. D. (2001) Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J. Am. Coll. Cardiol. 38, 1002–1006 10.1016/S0735-1097(01)01485-1 [DOI] [PubMed] [Google Scholar]
- 7. Armitage R. J., Fanslow W. C., Strockbine L., Sato T. A., Clifford K. N., Macduff B. M., Anderson D. M., Gimpel S. D., Davis-Smith T., and Maliszewski C. R. and (1992) Molecular and biological characterization of a murine ligand for CD40. Nature 357, 80–82 10.1038/357080a0 [DOI] [PubMed] [Google Scholar]
- 8. Henn V., Slupsky J. R., Gräfe M., Anagnostopoulos I., Förster R., Müller-Berghaus G., and Kroczek R. A. (1998) CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 391, 591–594 10.1038/35393 [DOI] [PubMed] [Google Scholar]
- 9. Urbich C., Dernbach E., Aicher A., Zeiher A. M., and Dimmeler S. (2002) CD40 ligand inhibits endothelial cell migration by increasing production of endothelial reactive oxygen species. Circulation 106, 981–986 10.1161/01.cir.0000027107.54614.1a [DOI] [PubMed] [Google Scholar]
- 10. Mach F., Schönbeck U., Sukhova G. K., Bourcier T., Bonnefoy J. Y., Pober J. S., and Libby P. (1997) Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 94, 1931–1936 10.1073/pnas.94.5.1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mach F., Schönbeck U., Sukhova G. K., Atkinson E., and Libby P. (1998) Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature 394, 200–203 10.1038/28204 [DOI] [PubMed] [Google Scholar]
- 12. Graf D., Müller S., Korthauer U., van Kooten C., Weise C., and Kroczek R. A. (1995) A soluble form of TRAP (CD40 ligand) is rapidly released after T cell activation. Eur. J. Immunol. 25, 1749–1754 10.1002/eji.1830250639 [DOI] [PubMed] [Google Scholar]
- 13. Aukrust P., Müller F., Ueland T., Berget T., Aaser E., Brunsvig A., Solum N. O., Forfang K., Frøland S. S., and Gullestad L. (1999) Enhanced levels of soluble and membrane-bound CD40 ligand in patients with unstable angina: possible reflection of T lymphocyte and platelet involvement in the pathogenesis of acute coronary syndromes. Circulation 100, 614–620 10.1161/01.cir.100.6.614 [DOI] [PubMed] [Google Scholar]
- 14. Kato K., Santana-Sahagún E., Rassenti L. Z., Weisman M. H., Tamura N., Kobayashi S., Hashimoto H., and Kipps T. J. (1999) The soluble CD40 ligand sCD154 in systemic lupus erythematosus. J. Clin. Invest. 104, 947–955 10.1172/JCI7014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schönbeck U., Varo N., Libby P., Buring J., and Ridker P. M. (2001) Soluble CD40L and cardiovascular risk in women. Circulation 104, 2266–2268 10.1161/hc4401.099447 [DOI] [PubMed] [Google Scholar]
- 16. Selkoe D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 10.1152/physrev.2001.81.2.741 [DOI] [PubMed] [Google Scholar]
- 17. De Strooper B. (2010) Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 90, 465–494 10.1152/physrev.00023.2009 [DOI] [PubMed] [Google Scholar]
- 18. Ponte P., Gonzalez-DeWhitt P., Schilling J., Miller J., Hsu D., Greenberg B., Davis K., Wallace W., Lieberburg I., and Fuller F. (1988) A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature 331, 525–527 10.1038/331525a0 [DOI] [PubMed] [Google Scholar]
- 19. Tanzi R. E., McClatchey A. I., Lamperti E. D., Villa-Komaroff L., Gusella J. F., and Neve R. L. (1988) Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer's disease. Nature 331, 528–530 10.1038/331528a0 [DOI] [PubMed] [Google Scholar]
- 20. Wertkin A. M., Turner R. S., Pleasure S. J., Golde T. E., Younkin S. G., Trojanowski J. Q., and Lee V. M. (1993) Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular β-amyloid or A4 peptides. Proc. Natl. Acad. Sci. U.S.A. 90, 9513–9517 10.1073/pnas.90.20.9513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kitazume S., Tachida Y., Kato M., Yamaguchi Y., Honda T., Hashimoto Y., Wada Y., Saito T., Iwata N., Saido T., and Taniguchi N. (2010) Brain endothelial cells produce amyloid β from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 285, 40097–40103 10.1074/jbc.M110.144626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bush A. I., Martins R. N., Rumble B., Moir R., Fuller S., Milward E., Currie J., Ames D., Weidemann A., Fischer P., et al. (1990) The amyloid precursor protein of Alzheimer's disease is released by human platelets. J. Biol. Chem. 265, 15977–15983 [PubMed] [Google Scholar]
- 23. Kitazume S., Yoshihisa A., Yamaki T., Oikawa M., Tachida Y., Ogawa K., Imamaki R., Hagiwara Y., Kinoshita N., Takeishi Y., Furukawa K., Tomita N., Arai H., Iwata N., Saido T., et al. (2012) Soluble amyloid precursor protein 770 is released from inflamed endothelial cells and activated platelets: a novel biomarker for acute coronary syndrome. J. Biol. Chem. 287, 40817–40825 10.1074/jbc.M112.398578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ludewig B., Henn V., Schröder J. M., Graf D., and Kroczek R. A. (1996) Induction, regulation, and function of soluble TRAP (CD40 ligand) during interaction of primary CD4+ CD45RA+ T cells with dendritic cells. Eur. J. Immunol. 26, 3137–3143 10.1002/eji.1830261246 [DOI] [PubMed] [Google Scholar]
- 25. Van Nostrand W. E., Schmaier A. H., Farrow J. S., and Cunningham D. D. (1990) Protease nexin-II (amyloid β-protein precursor): a platelet α-granule protein. Science 248, 745–748 10.1126/science.2110384 [DOI] [PubMed] [Google Scholar]
- 26. Willem M., Tahirovic S., Busche M. A., Ovsepian S. V., Chafai M., Kootar S., Hornburg D., Evans L. D., Moore S., Daria A., Hampel H., Müller V., Giudici C., Nuscher B., Wenninger-Weinzierl A., et al. (2015) η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526, 443–447 10.1038/nature14864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Müller J. E., Stone P. H., Turi Z. G., Rutherford J. D., Czeisler C. A., Parker C., Poole W. K., Passamani E., Roberts R., and Robertson T. and (1985) Circadian variation in the frequency of onset of acute myocardial infarction. N. Engl. J. Med. 313, 1315–1322 10.1056/NEJM198511213132103 [DOI] [PubMed] [Google Scholar]
- 28. Haass C., Kaether C., Thinakaran G., and Sisodia S. (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2, a006270 10.1101/cshperspect.a006270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thinakaran G., and Koo E. H. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 10.1074/jbc.R800019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang K., Hynan L. S., Baskin F., and Rosenberg R. N. (2006) Platelet amyloid precursor protein processing: a bio-marker for Alzheimer's disease. J. Neurol. Sci. 240, 53–58 10.1016/j.jns.2005.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakamura A., Kaneko N., Villemagne V. L., Kato T., Doecke J., Doré V., Fowler C., Li Q. X., Martins R., Rowe C., Tomita T., Matsuzaki K., Ishii K., Ishii K., Arahata Y., et al. (2018) High performance plasma amyloid-β biomarkers for Alzheimer's disease. Nature 554, 249–254 10.1038/nature25456 [DOI] [PubMed] [Google Scholar]
- 32. Schmaier A. H., Dahl L. D., Rozemuller A. J., Roos R. A., Wagner S. L., Chung R., and Van Nostrand W. E. (1993) Protease nexin-2/amyloid β protein precursor: a tight-binding inhibitor of coagulation factor IXa. J. Clin. Invest. 92, 2540–2545 10.1172/JCI116863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Nostrand W. E. (1995) Zinc (II) selectively enhances the inhibition of coagulation factor XIa by protease nexin-2/amyloid β-protein precursor. Thromb. Res. 78, 43–53 10.1016/0049-3848(95)00033-X [DOI] [PubMed] [Google Scholar]
- 34. Xu F., Previti M. L., and Van Nostrand W. E. (2007) Increased severity of hemorrhage in transgenic mice expressing cerebral protease nexin-2/amyloid β-protein precursor. Stroke 38, 2598–2601 10.1161/STROKEAHA.106.480103 [DOI] [PubMed] [Google Scholar]
- 35. Malinin A., Pokov A., Spergling M., Defranco A., Schwartz K., Schwartz D., Mahmud E., Atar D., and Serebruany V. (2007) Monitoring platelet inhibition after clopidogrel with the VerifyNow-P2Y12(R) rapid analyzer: the Verify thrombosis risk assessment (VERITAS) study. Thromb. Res. 119, 277–284 10.1016/j.thromres.2006.01.019 [DOI] [PubMed] [Google Scholar]
- 36. Lee P. Y., Chen W. H., Ng W., Cheng X., Kwok J. Y., Tse H. F., and Lau C. P. (2005) Low-dose aspirin increases aspirin resistance in patients with coronary artery disease. Am. J. Med. 118, 723–727 10.1016/j.amjmed.2005.03.041 [DOI] [PubMed] [Google Scholar]
- 37. Kim Y. G., Suh J. W., Park J. J., Oh I. Y., Yoon C. H., Cho Y. S., Youn T. J., Chae I. H., and Choi D. J. (2014) Different influences of hematocrit on the results of two point-of-care platelet function tests, the VerifyNow assay and multiple electrode platelet aggregometry. PLoS One 9, e114053 10.1371/journal.pone.0114053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Werkum J. W., Harmsze A. M., Elsenberg E. H., Bouman H. J., ten Berg J. M., and Hackeng C. M. (2008) The use of the VerifyNow system to monitor antiplatelet therapy: a review of the current evidence. Platelets 19, 479–488 10.1080/09537100802317918 [DOI] [PubMed] [Google Scholar]
- 39. Wang J. C., Aucoin-Barry D., Manuelian D., Monbouquette R., Reisman M., Gray W., Block P. C., Block E. H., Ladenheim M., and Simon D. I. (2003) Incidence of aspirin nonresponsiveness using the Ultegra rapid platelet function assay–ASA. Am. J. Cardiol. 92, 1492–1494 10.1016/j.amjcard.2003.08.072 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article and supporting information.