

The cytolethal distending toxin B subunit (CdtB) induces significant cytotoxicity and inflammation in many cell types that are involved in the pathogenesis of postinfectious irritable bowel syndrome (PI-IBS). However, the underlying mechanisms remain unclear. This study tested the potential role of Rab small GTPase 5a (Rab5a) in the process. We tested mRNA and protein expression of proinflammatory cytokines (interleukin-1β [IL-1β] and IL-6) in THP-1 macrophages by quantitative PCR (qPCR) and enzyme-linked immunosorbent assays (ELISAs), respectively.
KEYWORDS: postinfectious irritable bowel syndrome, cytolethal distending toxin B, Rab5a, cytotoxicity, inflammation
ABSTRACT
The cytolethal distending toxin B subunit (CdtB) induces significant cytotoxicity and inflammation in many cell types that are involved in the pathogenesis of postinfectious irritable bowel syndrome (PI-IBS). However, the underlying mechanisms remain unclear. This study tested the potential role of Rab small GTPase 5a (Rab5a) in the process. We tested mRNA and protein expression of proinflammatory cytokines (interleukin-1β [IL-1β] and IL-6) in THP-1 macrophages by quantitative PCR (qPCR) and enzyme-linked immunosorbent assays (ELISAs), respectively. In the primary colonic epithelial cells, Cdt treatment induced a CdtB-Rab5a-cellugyrin association. Rab5a silencing, by target small hairpin RNAs (shRNAs), largely inhibited CdtB-induced cytotoxicity and apoptosis in colon epithelial cells. CRISPR/Cas9-mediated Rab5a knockout also attenuated CdtB-induced colon epithelial cell death. Conversely, forced overexpression of Rab5a intensified CdtB-induced cytotoxicity. In THP-1 human macrophages, Rab5a shRNA or knockout significantly inhibited CdtB-induced mRNA expression and production of proinflammatory cytokines (IL-1β and IL-6). Rab5a depletion inhibited activation of nuclear factor-κB (NF-κB) and Jun N-terminal protein kinase (JNK) signaling in CdtB-treated THP-1 macrophages. Rab5a appears essential for CdtB-induced cytotoxicity in colonic epithelial cells and proinflammatory responses in THP-1 macrophages.
INTRODUCTION
Epidemiological studies have shown that a significant number of patients develop irritable bowel syndrome (IBS) after a bout of infectious gastroenteritis (IGE), a condition known as postinfectious IBS (PI-IBS) (1, 2). Months, or even years, after IGE, patients with PI-IBS may present with typical IBS symptoms, including persistent diarrhea, abdominal pain, cramping, abnormal laboratory values, weight loss, or particular endoscopic findings (1–4). Gut microbiota dysregulation and dysbiosis are involved in PI-IBS pathogenesis. A dysbiotic microbiota produces substances and antigenic products, which promote infiltration and activation of neutrophils and lymphocytes, causing an increased production of multiple proinflammatory cytokines, including interleukin-1β (IL-1β) and IL-6, among others (1, 3, 4). The detailed pathological mechanisms of PI-IBS are unclear (1, 3, 4).
A significant element in the dysbiotic microbiota involved in the PI-IBS pathogenesis is a cytolethal distending toxin (Cdt) (1, 5, 6). Cdt is produced by Campylobacter jejuni and other Gram-negative bacteria (7), causing genotoxic stress to the cell. This stress leads to growth arrest and eventual apoptotic cell death, perturbing the innate immune responses (8). Cdt is composed of three subunits, CdtA, CdtB, and CdtC (7). Patients with PI-IBS have increased levels of anti-CdtB antibodies (1, 5, 6). CdtA and CdtC bind to the cell surface, a mechanism responsible for the subsequent delivery of the active subunit CdtB to intracellular compartments (9, 10). Cdt membrane binding occurs in the context of cholesterol/sphingomyelin-rich membrane microdomains, also called lipid rafts (9, 10). In the CdtB endocytosis process, several targets, including fucose moieties and glycosphingolipids, will bind to CdtB, a process essential for mediating its cytotoxic and proinflammatory activity (9, 10). CdtB can induce significant cytotoxicity and inflammatory responses in several cell types (7, 10–13). However, the underlying mechanisms remain unknown.
Rab small GTPase (Rab) family proteins interact with various adaptor and effector proteins, regulating several different cellular behaviors, including cell growth, development, and membrane trafficking (14, 15). Of these behaviors, Rab5a is essential for endolysosomal system biogenesis (16, 17). Studies have shown that Rab5a silencing inhibits the formation of early endosomes, late endosomes, and lysosomes (16, 17). Furthermore, Rab5a is required for the progression of early and late endosomes (16, 17). Considering the critical role of Rab5a in membrane trafficking, we tested the potential involvement of Rab5a in CdtB-induced actions in primary human colonic epithelial cells and THP-1 human macrophages.
RESULTS
Cdt holotoxin treatment induces CdtB-Rab5a-cellugyrin association in human colonic epithelial cells.
Cdt binds to the cell surface, leading to the subsequent delivery of the active subunit CdtB into intracellular compartments (9, 10). The present study aimed to test the potential role of Rab5a in this process, using the coimmunoprecipitation (co-IP) assay. The primary cultured human colonic epithelial cells were treated with Cdt holotoxin. Since CdtB is the active subunit, we referred to this treatment as “CdtB” treatment (same for all in vitro studies). As shown, following treatment, CdtB immunoprecipitated with Rab5a (Fig. 1, IP), indicating a CdtB-Rab5a association. Cellugyrin (or synaptogyrin-2) plays an essential role in the internalization and traffic of CdtB to the intracellular target sites, mediating its functions (11). The co-IP assay results revealed that cellugyrin is in the CdtB-Rab5a complex (Fig. 1, IP), suggesting a potential role of Rab5a in CdtB internalization and traffic. CdtB treatment did not affect the expression of Rab5a and cellugyrin in colonic epithelial cells (Fig. 1, input). Thus, Cdt holotoxin treatment induces a CdtB-Rab5a-cellugyrin association in human colonic epithelial cells.
FIG 1.
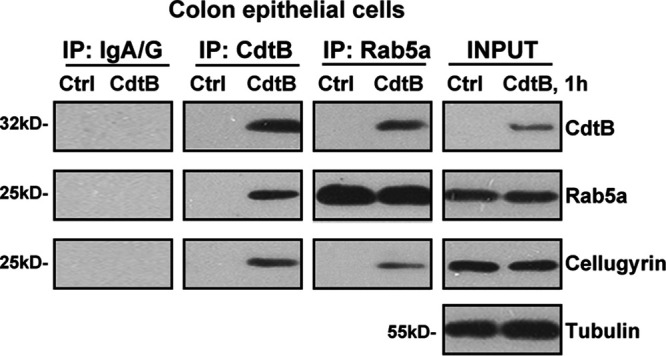
Cdt holotoxin treatment induces a CdtB-Rab5a-cellugyrin association in human colonic epithelial cells. Primary human colonic epithelial cells were treated with Cdt holotoxin (labeled as “CdtB,” 2 μg/ml, same for all figures) for 1 h, and CdtB-Rab5a-cellugyrin association (“IP”) and expression (“input”) were tested by the coimmunoprecipitation (co-IP) assay and the Western blotting assay, respectively. “IP: IgA/G” is the control for the co-IP assay. The experiments shown were repeated three times, and similar results were obtained.
Rab5a shRNA inhibits CdtB-induced cytotoxicity and apoptosis in human colonic epithelial cells.
We used the small hairpin RNA (shRNA) strategy to silence Rab5a so we could study the potential function of Rab5a in CdtB-induced cytotoxicity. As described, a set of two different lentiviral Rab5a shRNAs, with nonoverlapping sequences (“S1/S2”), were individually transfected into primary human colonic epithelial cells. Stable cells were established by puromycin selection. An analysis of Rab5a mRNA levels by qPCR assay showed that Rab5a mRNA was significantly downregulated in the Rab5a shRNA-expressing stable cells (Fig. 2A), with more than 90% Rab5a mRNA reduction compared with the control cells (Fig. 2A, scrambled shRNA [sc-shRNA]). Rab5a mRNA expression remained unaffected by CdtB treatment (Fig. 2A). Western blotting assay results further confirmed a more than 90% to 95% knockdown of the Rab5a protein in the stable cells with Rab5a shRNA (Fig. 2B). Meanwhile, cellugyrin expression was not affected by Rab5a shRNA (Fig. 2B). In control (sc-shRNA) epithelial cells, CdtB treatment induced significant cytotoxicity, as evidenced by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) optical density (OD) reduction (Fig. 2C) and medium lactate dehydrogenase (LDH) release (Fig. 2D). Importantly, CdtB-induced cytotoxicity was attenuated significantly in Rab5a shRNA-expressing stable cells (Fig. 2C and D). These results indicate that Rab5a is necessary for CdtB-induced cytotoxicity against the epithelial cells.
FIG 2.

Rab5a shRNA inhibits CdtB-induced cytotoxicity and apoptosis in human colonic epithelial cells. Stable primary human colonic epithelial cells, with Rab5a shRNA (“S1/S2,” two different sequences) or the scrambled shRNA (sc-shRNA), were either left untreated (“Ctrl,” same for all figures) or treated with Cdt holotoxin (2 μg/ml) for the applied time, and the expression of the listed genes was tested by qPCR (A) and Western blotting (B); cell viability and cell death were tested by an MTT assay (C) and an LDH medium release assay (D), respectively. Caspase-3/-9 activation (E and F) and cell apoptosis (G) were tested by the assays mentioned in the text. Rab5a mRNA was normalized to GAPDH mRNA (same for all figures). Rab5a and cellugyrin protein expression were normalized to the loading control GAPDH/tubulin (B and F). Cell apoptosis was tested by the annexin V-FACS assay (H and I). Bars represent means ± SD (same for all figures). *, P < 0.05 versus Ctrl treatment of sc-shRNA cells. #, P < 0.05 versus CdtB treatment of sc-shRNA cells. The experiments reflected in this figure were repeated three times, and similar results were obtained.
Our further studies of control colonic epithelial cells revealed significant CdtB-induced apoptosis activation, as evidenced by the caspase-3 activation (Fig. 2E), caspase-3/caspase-9 cleavages (Fig. 2F), single-stranded DNA (ssDNA) accumulation (Fig. 2G), and increased annexin V-positive cells (Fig. 2H and I). Notably, CdtB-induced apoptosis activation was attenuated significantly in Rab5a-silenced cells (Fig. 2E to I). Rab5a shRNA did not affect the functions of colonic epithelial cells (Fig. 2C to I). Together, these results show that Rab5a shRNA significantly inhibits CdtB-induced cytotoxicity and apoptosis in human colonic epithelial cells.
Rab5a knockout inhibits CdtB-induced cytotoxicity and apoptosis in human colonic epithelial cells.
To exclude the potential off-target effect of the Rab5a shRNAs, we used the CRISPR/Cas9 gene-editing method to knock out Rab5a. As described, the lenti-CRISPR/Cas9-GFP construct with Rab5a sgRNA (see the Methods section) was transfected into the colonic epithelial cells. Puromycin was added to select the stable (Rab 5a knockout [“Rab5a-KO”]) cells. Using qPCR and Western blotting, we confirmed that Rab5a mRNA (Fig. 3A) and protein (Fig. 3B) were depleted entirely in Rab5a-KO cells, with cellugyrin remaining unaffected (Fig. 3B). In line with the shRNA results, compared with the control cells, Rab5a-KO cells were resistant to CdtB, showing significantly decreased viability reduction (Fig. 3C), cell death (Fig. 3D), and apoptosis (Fig. 3E). Rab5a-KO alone was ineffective (Fig. 3C to E). This evidence further added support to the hypothesis that Rab5a is required for CdtB-induced cytotoxicity in human colonic epithelial cells.
FIG 3.

Rab5a knockout inhibits CdtB-induced cytotoxicity and apoptosis in human colonic epithelial cells. Stable primary human colonic epithelial cells, with the lenti-CRISPR/Cas9-GFP Rab5a-KO construct (“Rab5a-KO” cells) or the empty vector (“CRISPR-C”) were treated with Cdt holotoxin (2 μg/ml) for the applied time; the expression of the listed genes was tested by qPCR (A) and Western blotting (B); cell viability and cell death were tested by an MTT assay (C) and an LDH medium release assay (D), respectively; cell apoptosis was quantitatively analyzed by the annexin V-FACS assay (E). *, P < 0.05 versus Ctrl treatment of CRISPR-C cells. #, P < 0.05 versus CdtB treatment of CRISPR-C cells. The experiments reflected in this figure were repeated three times, and similar results were obtained.
Rab5a overexpression enhances CdtB-induced cytotoxicity in human colonic epithelial cells.
The above results show that Rab5a silencing (Fig. 2) or knock out (Fig. 3) potently inhibited CdtB-induced cytotoxicity in human colonic epithelial cells. Therefore, we hypothesized that forced Rab5a overexpression might further enhance CdtB-induced cytotoxicity. To test this hypothesis, we added the adenovirus-encoding Rab5a cDNA (“Ad-Rab5a”) to the human colonic epithelial cells. Two stable cell lines were established by puromycin selection, namely, “Rab5a-OE (L1/L2).” The qPCR assay results confirmed that Rab5a mRNA levels increased more than 3-fold in the Rab5a-OE cells (Fig. 4A), regardless of CdtB treatment (Fig. 4A). Rab5a protein expression, tested by a Western blotting assay, was also significantly increased (Fig. 4B). Importantly, compared with control cells (with empty vector), CdtB-induced viability (MTT OD) reduction (Fig. 4C), cell death (LDH medium release) (Fig. 4D), and apoptosis (annexin V assay) (Fig. 4E and F) were significantly enhanced in Rab5a-OE (L1/L2) cells. Rab5a overexpression by itself was ineffective in colonic epithelial cell functions (Fig. 4C to F). The results confirmed that forced Rab5a overexpression potentiated CdtB-induced cytotoxicity in human colonic epithelial cells, further verifying the essential role of Rab5a in mediating CdtB functions.
FIG 4.

Rab5a overexpression enhances CdtB-induced cytotoxicity in human colonic epithelial cells. Stable primary human colonic epithelial cells, with Rab5a cDNA adenovirus or “Rab5-OE (L1/L2),” as well as vector control cells (“Vec”), were treated with Cdt holotoxin (2 μg/ml) for the applied time, and the expression of the listed genes was tested by qPCR (A) and Western blotting (B); cell viability and cell death were tested by an MTT assay (C) and an LDH medium release assay (D), respectively; cell apoptosis was tested by the annexin V-FACS assay (E and F). *, P < 0.05 versus Ctrl treatment of Vec cells. #, P < 0.05 versus CdtB treatment of Vec cells. The experiments reflected in this figure were repeated four times, and similar results were obtained.
Rab5a is necessary for CdtB-induced proinflammatory responses in THP-1 human macrophages.
Macrophages exposed to CdtB can be activated to produce proinflammatory cytokines, including IL-1β and IL-6 (13). To test the possible role played by Rab5a in the process, genetic methods were again employed in THP-1 human macrophages. The lentiviral CdtB shRNA (Fig. 2, S2) or the lenti-CRISPR/Cas9-GFP Rab5a-KO construct (Fig. 3) were transfected into THP-1 macrophages, and stable cells were established via puromycin selection. As shown, Rab5 mRNA and protein levels were significantly downregulated in THP-1 macrophages with the Rab5a shRNA or Rab5a-KO construct (Fig. 5A and B). Unlike the human colonic epithelial cells, treatment with CdtB failed to induce significant cytotoxicity to THP-1 macrophages, as cell viability remained unchanged (Fig. 5C).
FIG 5.

Rab5a is required for CdtB-induced proinflammatory responses in THP-1 human macrophages. Stable THP-1 human macrophages, with Rab5a shRNA (“S2”) or the lenti-CRISPR/Cas9 Rab5a knockout construct (Rab5a-KO cells), as well as the parental control THP-1 macrophages (“C”), were treated with Cdt holotoxin (2 μg/ml) for the applied time; the expression of the listed genes was tested by qPCR (A, D, and E) and Western blotting (B and I); cell viability was tested by an MTT assay (C); the production of listed cytokines in the medium supernatants was tested by ELISA (F and G); p65 DNA-binding activity was tested as well (H). *, P < 0.05 versus Ctrl treatment of C cells. #, P < 0.05 versus CdtB treatment of C cells. The experiments reflected in this figure were repeated three times, and similar results were obtained.
In line with a previous study (13), CdtB treatment in THP-1 macrophages induced significant mRNA expression of IL-1β (Fig. 5D) and IL-6 (Fig. 5E). Furthermore, the ELISA results showed that CdtB induced the production of IL-1β (Fig. 5F) and IL-6 (Fig. 5G). Of note, in THP-1 macrophages, Rab5a shRNA or Rab5a-KO inhibited CdtB-induced mRNA expression (Fig. 5D and E) and secretion (Fig. 5F and G) of IL-1β and IL-6. Thus, Rab5a is also necessary for CdtB-induced proinflammatory actions. For the mechanism study, in THP-1 macrophages, CdtB-induced activation of NF-κB and Jun N-terminal protein kinase (JNK) signaling was significantly inhibited by Rab5a shRNA or Rab5a-KO (Fig. 5H and I). The increased NF-κB (p65) DNA-binding activity (Fig. 5H) and p65 phosphorylation (Fig. 5I) led to NF-κB activation. Collectively, Rab5a is also important for CdtB-induced NF-κB-JNK signaling activation and proinflammatory cytokine production in THP-1 human macrophages.
DISCUSSION
Cdt is produced by Gram-negative bacteria implicated in the pathogenesis of PI-IBS (18). CdtB can induce significant cytotoxicity and inflammatory responses in many cell types (7, 10–13). CdtB traffic is vital for its functions (18). The underlying mechanisms of cellular uptake and delivery of CdtB in host cells, as well as the translocation of CdtB to the cell organelles, are only partially understood (18). Rab5a is a key protein regulating membrane traffic and endocytosis (16, 17, 19). The results of the current study suggest that Rab5a could be a pivotal player in mediating CdtB-induced actions. Following Cdt stimulation, Rab5a associated with CdtB and its interaction protein, cellugyrin (11), in primary human colonic epithelial cells. Noticeably, Rab5 silencing, by targeted shRNAs, largely attenuated CdtB-induced cytotoxicity and apoptosis in the colonic epithelial cells. Moreover, CRISPR/Cas9-induced Rab5a knockout inhibited CdtB-induced colonic epithelial cell death and apoptosis. Conversely, using an adenovirus construct, Rab5a overexpression further exacerbated CdtB-induced cytotoxicity. Therefore, Rab5a might participate in CdtB cell uptake and traffic, mediating CdtB-induced cytotoxicity against human colonic epithelial cells.
It is known that CdtB, the active subunit of Cdt, can function as a lipid phosphatase to degrade the signaling lipid phosphatidylinositol-3, 4, 5-triphosphate (PIP3), thereby blocking the prosurvival phosphatidylinositol 3-kinase (PI3K)-AKT-mammalian target of rapamycin (mTOR) signaling to trigger cell death and apoptosis (9, 10, 13, 18). Existing studies have proposed that CdtB could function as a DNase to cause double-strand breaks (DSBs), resulting in cell apoptosis as well (9, 10, 13). Other mechanisms of mediating CdtB-induced cytotoxicity have also been proposed (9, 10, 13). Therefore, further studies are necessary to test the link between Rab5a and these proposed mechanisms by CdtB.
CdtB could also provoke significant inflammatory responses, serving as another important contributor of PI-IBS and other inflammatory diseases (9, 10, 18). Increased inflammation may lead to the overproduction of 5-hydroxytryptamine (5-HT), neuronal growth factors, and cytokines, eventually resulting in increased colonic motility and diarrhea (9, 10, 18). Adding CdtB to macrophages and other immune cells can provoke significant proinflammatory responses (13). The results of the present study suggest that Rab5a is important for mediating CdtB-induced proinflammation activity in cultured macrophages. In THP-1 cells, Rab5a depletion inhibited CdtB-induced mRNA expression and the production of proinflammatory cytokines (IL-1β and IL-6). At the molecular level, Rab5a shRNA or Rab5a-KO inhibited CdtB-induced activation of NF-κB-JNK cascades. Thus, Rab5a could mediate CdtB-induced NF-κB and JNK activation to induce proinflammatory cytokine production, causing a profound inflammation response. The underlying mechanisms may warrant further characterization.
Although CdtB induces NF-κB and JNK activation, it is not sufficient to induce IL-1β maturation and release by itself. Thus, we propose that a second signal may be required, potentially involving the activation of the inflammasome. Previous studies have shown that Cdt activates the NLRP3/caspase-1 inflammasome, leading to the upregulation of IL-1β (20, 21). A recent study has demonstrated that Cdt can also induce the release of IL-1β via the noncanonical (caspase-4) inflammasomes (22). Additionally, it has revealed that a whole range of cytokines appear to be regulated by Cdt-induced macrophages, beyond IL-1β and IL-6, such as tumor necrosis factor alpha (TNF-α), IL-18, IL-10, and IL-12 (21, 23). Therefore, the involvement of the inflammasome in cytokine regulation by CdtB requires further confirmation.
Heterogeneity in outcome measures, as well as a low strength of evidence, have precluded recommendations on the optimal treatment of PI-IBS by a single specific agent (24). Indeed, there are no management guidelines or expert recommendations for unified PI-IBS management thus far (24). The results here show that Rab5a is essential for CdtB-induced cytotoxicity and proinflammatory responses, suggesting that Rab5a could be a novel and important therapeutic target for CdtB-associated PI-IBS.
MATERIALS AND METHODS
Research ethics.
The study protocols adhered to the principles of the Declaration of Helsinki. The ethical review boards of the authors’ institutions approved the study protocol.
Chemicals and reagents.
Using a previously described protocol (13), Cdt holotoxin was synthesized by Genechem (Shanghai, China). The isolated toxin was tested by SDS-PAGE, and it contained all three Cdt subunits, namely, CdtA (18 kDa), CdtB (32 kDa), and CdtC (20 kDa). Puromycin and MTT were obtained from Sigma-Aldrich (St. Louis, MO). Cell culture reagents were purchased from HyClone Co. (Logan, UT). All antibodies were purchased from Abcam (Shanghai, China) and Cell Signaling Technology (Shanghai, China).
Cell culture.
The THP-1 human macrophages (25) were purchased from the Cell Bank of Shanghai Institute of Biological Science (Shanghai, China). The macrophages were maintained in RPMI 1640 medium with 10% fetal bovine serum (FBS), under standard conditions (13). The primary human colonic epithelial cells were provided by Lu and colleagues (26–28) and cultured, as described previously (26–28).
Cell death assay.
Lactate dehydrogenase (LDH) medium release was examined to quantitatively measure cell death intensity, using a two-step LDH assay kit (TaKaRa, Tokyo, Japan) (29).
Cell viability assay.
A routine MTT assay was used to examine cell viability. Cells (5 × 103 cells per well) were seeded in 96-well tissue culture plates. Following the applied treatments, MTT optical density (OD) values were recorded at a wavelength of 490 nm.
Western blotting and co-IP assays.
The detailed protocol for the Western blotting assay was previously described (30). The same set of lysates was run in parallel (“sister”) gels to test different proteins. The band intensity (total gray value) was quantified using the ImageJ software (NIH). For the co-IP assay, total cell lysates (1,000 μg per treatment) were precleared by adding protein A/G Sepharose beads (Biyuntian, Wuxi, China). The cleared lysate samples were incubated with anti-CdtB or anti-Rab5a antibody for 12 h. Afterward, the Sepharose beads were added back to the lysates for another 3 to 4 h. The CdtB-Rab5a-cellugyrin association was tested using the Western blotting assay.
Rab5 shRNA.
The shRNA sequences 5′-AAACTAGTACTTCTGGGAGAGTC-3′ (S1) and 5′-AATTTCAAGAGAGTACCATTGGG-3′ (S2) targeting human Rab5a were subcloned in a lentiviral GV248 construct (Genechem, Shanghai, China). The lentivirus was filtered, enriched, and added to cultured cells for 12 h. Next, a fresh renewed medium was added. We incubated the plates for another 12 h. Stable cells were selected by puromycin (3.0 μg/ml) for 4 more days. Control cells were infected with scrambled control shRNA lentiviral particles (sc-108080; Santa Cruz Biotech).
Rab5 knockout.
The Rab5a small guide RNA (sgRNA), with the targeted DNA sequence 5′-AAGACCCAACGGGCCAAATA-3′, was inserted into the lenti-CRISPR-GFP plasmid (31). The latter was then transfected into colonic epithelial cells. Next, the GFP-positive cells were sorted by fluorescence-activated cell sorting (FACS). Thereafter, a fresh renewed medium was added, and plates were incubated for another 12 h. Stable cells were selected by puromycin (3.0 μg/ml) for 4 more days, followed by Rab5a knockout screening for stable Rab5 knockout (“Rab5-KO”) cells.
Rab5a overexpression.
The Rab5a cDNA was cloned from the human colonic epithelial cells and inserted into the pDC315 construct (Genechem). HEK-293 cells were transfected with the construct along with the pBHG lox ΔE1,3 Cre plasmid (Genechem) (32) to generate the Rab5a expression adenovirus Ad-Rab5a. The virus was then filtered, enriched, and added to cultured cells for 12 h. Thereafter, a fresh renewed medium was added, and the plates were incubated for another 12 h. The stable cells were selected by puromycin (3.0 μg/ml) for 4 more days. Rab5a overexpression was confirmed by Western blotting and quantitative real-time PCR.
Caspase-3 activity assay.
Caspase-3 activity was tested using a previously described protocol (33). Briefly, for each treatment, 20-μg cytosolic proteins were incubated with the caspase-3 assay buffer (33) together with the caspase-3 substrate. After incubation, the caspase-3 activity was quantified and normalized to the control.
ssDNA ELISA.
Cells were seeded in 96-well tissue culture plates (3 × 103 cells per well). The cellular levels of ssDNA were examined by the ApoStrand ELISA apoptosis detection kit (Biomol, Plymouth Meeting, PA) (34). ssDNA ELISA OD values were recorded at a wavelength of 405 nm.
Annexin V assay.
In brief, cells were harvested, washed, and incubated with annexin V and propidium iodide (PI) (each at 5 μg/ml). Afterward, the cells were subjected to FACS analysis (BD, Shanghai, China). Annexin V ratios were recorded.
qPCR assay.
Following the indicated treatment, TRIzol (Sigma) was added to extract total cellular RNA. After RNA quantification, 500 ng RNA of each treatment was mixed with SYBR green PCR master mix (Applied Biosystems) along with 100 nM primers. The ABI Prism 7600H Fast real-time PCR system was employed for the qPCR, using the cycle threshold (ΔΔCT) method for the quantification of targeted mRNAs, and GAPDH mRNA was used as the internal control. All the mRNA primers were purchased from Genechem (Shanghai, China). Primer sequences used in this study are listed in Table 1.
TABLE 1.
Primer sequences utilized in the study
| Human gene | Forward sequence (5′–3′) | Reverse sequence (5′–3′) |
|---|---|---|
| Rab5a | ACTTCTGGGAGAGTCCGCTGTT | GTGTCATCAAGACATACAGTTTGG |
| IL-6 | AAGCCAGAGCTGTGCAGATGAGTA | CTTGGTCACCGACGTCCTGT |
| IL-1β | CCGACCACCACTACAGCAAG | GGGCAGGGAACCAGCATCTT |
| GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA |
NF-κB activity assay.
As described in previous studies (35–37), NF-κB (p65) DNA-binding activity was examined using the TransAM ELISA kit (Active Motif, Carlsbad, CA), analyzing 1.0 μg of the nuclear protein extracts per treatment. The OD value of each treatment group was always normalized to the control.
ELISA of cytokines.
THP-1 cells were seeded in 24-well tissue culture plates at a density of 2 × 104 cells per well. Cytokine concentrations in the medium supernatant were measured using the IL-1β and IL-6 BD OptEIA ELISA kits (BD Biosciences, San Jose, CA). In each instance, the amount of cytokine was determined by using a standard curve.
Statistical analysis.
Means ± standard deviations (SDs) were calculated for replicate experiments. Data were analyzed by one-way analysis of variance (ANOVA) followed by a Scheffe’s f-test using the SPSS 18.0 software (SPSS Inc., Chicago, IL). The two-tailed unpaired t test (Excel 2017) was used to identify statistically significant differences between the two groups. A probability (P) value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This work is supported by the National Natural Science Foundation of China Young Scientist Program (no. 81302957), Zhejiang Provincial Natural Science Foundation of China (no. LY17H270003), Zhejiang Province Institute of Science and Technology of Special Foundation (no. 2017F30044), Zhejiang Province Key Research Project of Traditional Chinese Medicine (no. 2018ZZ005), Zhejiang Province Project of Traditional Chinese Medicine (no. 2019ZA034), and Zhejiang Medical and Health Science and Technology Plan Project (no. 2018KY326).
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Downs IA, Aroniadis OC, Kelly L, Brandt LJ. 2017. Postinfection irritable bowel syndrome: the links between gastroenteritis, inflammation, the microbiome, and functional disease. J Clin Gastroenterol 51:869–877. doi: 10.1097/MCG.0000000000000924. [DOI] [PubMed] [Google Scholar]
- 2.Dai C, Jiang M. 2012. The incidence and risk factors of post-infectious irritable bowel syndrome: a meta-analysis. Hepatogastroenterology 59:67–72. doi: 10.5754/hge10796. [DOI] [PubMed] [Google Scholar]
- 3.Ghoshal UC, Gwee KA. 2017. Post-infectious IBS, tropical sprue and small intestinal bacterial overgrowth: the missing link. Nat Rev Gastroenterol Hepatol 14:435–441. doi: 10.1038/nrgastro.2017.37. [DOI] [PubMed] [Google Scholar]
- 4.Schwille-Kiuntke J, Frick JS, Zanger P, Enck P. 2011. Post-infectious irritable bowel syndrome—a review of the literature. Z Gastroenterol 49:997–1003. doi: 10.1055/s-0031-1281581. [DOI] [PubMed] [Google Scholar]
- 5.Pimentel M, Morales W, Rezaie A, Marsh E, Lembo A, Mirocha J, Leffler DA, Marsh Z, Weitsman S, Chua KS, Barlow GM, Bortey E, Forbes W, Yu A, Chang C. 2015. Development and validation of a biomarker for diarrhea-predominant irritable bowel syndrome in human subjects. PLoS One 10:e0126438. doi: 10.1371/journal.pone.0126438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poropatich KO, Walker CL, Black RE. 2010. Quantifying the association between Campylobacter infection and Guillain-Barre syndrome: a systematic review. J Health Popul Nutr 28:545–552. doi: 10.3329/jhpn.v28i6.6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taieb F, Petit C, Nougayrede JP, Oswald E. 2016. The enterobacterial genotoxins: cytolethal distending toxin and colibactin. EcoSal Plus 7:10.1128/ecosalplus.ESP-0008-2016. doi: 10.1128/ecosalplus.ESP-0008-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belibasakis GN, Bostanci N. 2014. Inflammatory and bone remodeling responses to the cytolethal distending toxins. Cells 3:236–246. doi: 10.3390/cells3020236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerding DN, Johnson S, Rupnik M, Aktories K. 2014. Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes 5:15–27. doi: 10.4161/gmic.26854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerra L, Cortes-Bratti X, Guidi R, Frisan T. 2011. The biology of the cytolethal distending toxins. Toxins (Basel) 3:172–190. doi: 10.3390/toxins3030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boesze-Battaglia K, Walker LP, Dhingra A, Kandror K, Tang HY, Shenker BJ. 2017. Internalization of the active subunit of the Aggregatibacter actinomycetemcomitans cytolethal distending toxin is dependent upon cellugyrin (synaptogyrin 2), a host cell non-neuronal paralog of the synaptic vesicle protein, synaptogyrin 1. Front Cell Infect Microbiol 7:469. doi: 10.3389/fcimb.2017.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boesze-Battaglia K, Alexander D, Dlakić M, Shenker BJ. 2016. A journey of cytolethal distending toxins through cell membranes. Front Cell Infect Microbiol 6:81. doi: 10.3389/fcimb.2016.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shenker BJ, Walker LP, Zekavat A, Dlakic M, Boesze-Battaglia K. 2014. Blockade of the PI-3K signalling pathway by the Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces macrophages to synthesize and secrete pro-inflammatory cytokines. Cell Microbiol 16:1391–1404. doi: 10.1111/cmi.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porther N, Barbieri MA. 2015. The role of endocytic Rab GTPases in regulation of growth factor signaling and the migration and invasion of tumor cells. Small GTPases 6:135–144. doi: 10.1080/21541248.2015.1050152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A. 2007. Rab GTPases at a glance. J Cell Sci 120:3905–3910. doi: 10.1242/jcs.015909. [DOI] [PubMed] [Google Scholar]
- 16.Göhre V, Vollmeister E, Bölker M, Feldbrügge M. 2012. Microtubule-dependent membrane dynamics in Ustilago maydis: trafficking and function of Rab5a-positive endosomes. Commun Integr Biol 5:485–490. doi: 10.4161/cib.21219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torres VA, Stupack DG. 2011. Rab5 in the regulation of cell motility and invasion. Curr Protein Pept Sci 12:43–51. doi: 10.2174/138920311795659461. [DOI] [PubMed] [Google Scholar]
- 18.Lai CK, Chen YA, Lin CJ, Lin HJ, Kao MC, Huang MZ, Lin YH, Chiang-Ni C, Chen CJ, Lo UG, Lin LC, Lin H, Hsieh JT, Lai CH. 2016. Molecular mechanisms and potential clinical applications of Campylobacter jejuni cytolethal distending toxin. Front Cell Infect Microbiol 6:9. doi: 10.3389/fcimb.2016.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woodman PG. 2000. Biogenesis of the sorting endosome: the role of Rab5. Traffic 1:695–701. doi: 10.1034/j.1600-0854.2000.010902.x. [DOI] [PubMed] [Google Scholar]
- 20.Belibasakis GN, Johansson A. 2012. Aggregatibacter actinomycetemcomitans targets NLRP3 and NLRP6 inflammasome expression in human mononuclear leukocytes. Cytokine 59:124–130. doi: 10.1016/j.cyto.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 21.Shenker BJ, Ojcius DM, Walker LP, Zekavat A, Scuron MD, Boesze-Battaglia K. 2015. Aggregatibacter actinomycetemcomitans cytolethal distending toxin activates the NLRP3 inflammasome in human macrophages, leading to the release of proinflammatory cytokines. Infect Immun 83:1487–1496. doi: 10.1128/IAI.03132-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shenker BJ, Walker LM, Zekavat Z, Ojcius DM, Huang P-R, Boesze-Battaglia K. 2020. Cytolethal distending toxin-induced release of interleukin-1β by human macrophages is dependent upon activation of glycogen synthase kinase 3β, spleen tyrosine kinase (Syk) and the noncanonical inflammasome. Cell Microbiol 22:e13194. doi: 10.1111/cmi.13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ando-Suguimoto ES, da Silva MP, Kawamoto D, Chen C, DiRienzo JM, Mayer MP. 2014. The cytolethal distending toxin of Aggregatibacter actinomycetemcomitans inhibits macrophage phagocytosis and subverts cytokine production. Cytokine 66:46–53. doi: 10.1016/j.cyto.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 24.Torbicki E, Oh J, Mishra S, Page AV, Boggild AK. 2015. Interventions for post-infectious irritable bowel syndrome: a systematic review of treatment efficacy. Trop Dis Travel Med Vaccines 1:1. doi: 10.1186/s40794-015-0002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang C, Wang X, Xie Y, Cai X, Yu N, Hu Y, Zheng Z. 2018. 4-Octyl itaconate activates Nrf2 signaling to inhibit pro-inflammatory cytokine production in peripheral blood mononuclear cells of systemic lupus erythematosus patients. Cell Physiol Biochem 51:979–990. doi: 10.1159/000495400. [DOI] [PubMed] [Google Scholar]
- 26.Chen MB, Zhang Y, Wei MX, Shen W, Wu XY, Yao C, Lu PH. 2013. Activation of AMP-activated protein kinase (AMPK) mediates plumbagin-induced apoptosis and growth inhibition in cultured human colon cancer cells. Cell Signal 25:1993–2002. doi: 10.1016/j.cellsig.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 27.Lu PH, Chen MB, Ji C, Li WT, Wei MX, Wu MH. 2016. Aqueous Oldenlandia diffusa extracts inhibits colorectal cancer cells via activating AMP-activated protein kinase signalings. Oncotarget 7:45889–45900. doi: 10.18632/oncotarget.9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, Cui J-F, Chen M-B, Liu C-Y, Liu F, Zhang Q-D, Zou J, Lu P-H. 2015. The preclinical evaluation of the dual mTORC1/2 inhibitor INK-128 as a potential anti-colorectal cancer agent. Cancer Biol Ther 16:34–42. doi: 10.4161/15384047.2014.972274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji F, Mao L, Liu Y, Cao X, Xie Y, Wang S, Fei H. 2015. K6PC-5, a novel sphingosine kinase 1 (SphK1) activator, alleviates dexamethasone-induced damages to osteoblasts through activating SphK1-Akt signaling. Biochem Biophys Res Commun 458:568–575. doi: 10.1016/j.bbrc.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS, Marshall J, Jiang M, Chu WM. 2009. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal 2:ra17. doi: 10.1126/scisignal.2000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai C, Zhang X, Xie D, Tang P, Li C, Zuo Y, Jiang B, Xue C. 2017. Targeting PP2A activates AMPK signaling to inhibit colorectal cancer cells. Oncotarget 8:95810–95823. doi: 10.18632/oncotarget.21336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall J, Zhou XZ, Chen G, Yang SQ, Li Y, Wang Y, Zhang ZQ, Jiang Q, Birnbaumer L, Cao C. 2018. Antidepression action of BDNF requires and is mimicked by Galphai1/3 expression in the hippocampus. Proc Natl Acad Sci U S A 115:E3549–E3558. doi: 10.1073/pnas.1722493115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao L-P, Ji C, Lu P-H, Li C, Xu B, Gao H. 2013. Oxygen glucose deprivation (OGD)/re-oxygenation-induced in vitro neuronal cell death involves mitochondrial cyclophilin-D/P53 signaling axis. Neurochem Res 38:705–713. doi: 10.1007/s11064-013-0968-5. [DOI] [PubMed] [Google Scholar]
- 34.Ye X, Xie J, Huang H, Deng Z. 2018. Knockdown of MAGEA6 activates AMP-activated protein kinase (AMPK) signaling to inhibit human renal cell carcinoma cells. Cell Physiol Biochem 45:1205–1218. doi: 10.1159/000487452. [DOI] [PubMed] [Google Scholar]
- 35.Li P, Wu Y, Li M, Qiu X, Bai X, Zhao X. 2015. AS-703026 inhibits LPS-iInduced TNFalpha production through MEK/ERK dependent and independent mechanisms. PLoS One 10:e0137107. doi: 10.1371/journal.pone.0137107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li P, Fan JB, Gao Y, Zhang M, Zhang L, Yang N, Zhao X. 2016. miR-135b-5p inhibits LPS-induced TNFalpha production via silencing AMPK phosphatase Ppm1e. Oncotarget 7:77978–77986. doi: 10.18632/oncotarget.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu YH, Li Q, Li P, Liu B. 2016. GSK621 activates AMPK signaling to inhibit LPS-induced TNFalpha production. Biochem Biophys Res Commun 480:289–295. doi: 10.1016/j.bbrc.2016.10.001. [DOI] [PubMed] [Google Scholar]