

Group A Streptococcus (GAS) is the etiologic agent of numerous high-morbidity and high-mortality diseases. Infections are typically highly proinflammatory. During the invasive infection necrotizing fasciitis, this is in part due to the GAS protease SpeB directly activating interleukin-1β (IL-1β) independent of the canonical inflammasome pathway. The upper respiratory tract is the primary site for GAS colonization, infection, and transmission, but the host-pathogen interactions at this site are still largely unknown.
KEYWORDS: Streptococcus pyogenes, group A streptococcus, inflammation, neutrophils, pathogenesis, proteases, respiratory pathogens, virulence factors
ABSTRACT
Group A Streptococcus (GAS) is the etiologic agent of numerous high-morbidity and high-mortality diseases. Infections are typically highly proinflammatory. During the invasive infection necrotizing fasciitis, this is in part due to the GAS protease SpeB directly activating interleukin-1β (IL-1β) independent of the canonical inflammasome pathway. The upper respiratory tract is the primary site for GAS colonization, infection, and transmission, but the host-pathogen interactions at this site are still largely unknown. We found that in the murine nasopharynx, SpeB enhanced IL-1β-mediated inflammation and the chemotaxis of neutrophils. However, neutrophilic inflammation did not restrict infection and instead promoted GAS replication and disease. Inhibiting IL-1β or depleting neutrophils, which both promote invasive infection, prevented GAS infection of the nasopharynx. Mice pretreated with penicillin became more susceptible to GAS challenge, and this reversed the attenuation from neutralization or depletion of IL-1β, neutrophils, or SpeB. Collectively, our results suggest that SpeB is essential to activate an IL-1β-driven neutrophil response. Unlike during invasive tissue infections, this is beneficial in the upper respiratory tract because it disrupts colonization resistance mediated by the microbiota. This provides experimental evidence that the notable inflammation of strep throat, which presents with significant swelling, pain, and neutrophil influx, is not an ineffectual immune response but rather is a GAS-directed remodeling of this niche for its pathogenic benefit.
INTRODUCTION
Group A Streptococcus (GAS; Streptococcus pyogenes) is a leading cause of infectious mortality and is responsible for over half a million deaths annually (1). Death is primarily due to invasive infection, including sepsis, necrotizing fasciitis, and toxic shock syndrome, or to autoimmune diseases, most prominently acute rheumatic fever and rheumatic heart disease. The nasopharyngeal mucosa and associated lymphoid tissues are the most common site for infection (strep throat pharyngitis) and the primary carriage site for dissemination of GAS between individuals and to other sites of the body (2). Humans are transiently colonized by GAS throughout childhood but can be culture positive at any point in their lives, often without overt symptoms of disease (3, 4).
Infection starts at the mucosal and epidermal surfaces, where GAS adheres to and invades skin keratinocytes and epithelial cells (5–8) to gain access to lymphoid tissues (9). This leads to inflammation of the mucosa, swelling of the tonsils, and formation of white patches of neutrophilic pus, characteristic of pharyngitis. Many molecular details of the host-pathogen interactions at this site remain unknown. Similar to how some enteric pathogens translocate across intestinal barriers, GAS can penetrate deeper into the mucosa through M cells and by disrupting cell junctions (2). It is unknown to what extent cell and tissue invasion is an essential feature of pharyngitis; however, the intracellular population of bacteria is protected from some antibiotics and may act as a reservoir for recurrent infection (4).
GAS inoculated intranasally into mice adheres to, colonizes, and invades the nasopharyngeal mucosa and nasopharynx-associated lymphoid tissues (NALT). The human NALT includes the palatine tonsils and adenoids, making it anatomically distinct from the murine NALT, which lacks defined lymphoid follicles (9). However, GAS is still trophic toward murine NALT, and the inflammation, neutrophil infiltration, and pathology in mice resemble human disease and constitute a useful model for examining host-pathogen interactions relevant for human pharyngitis (6, 9–17). Many virulence factors important at other body sites are also essential at this site, including capsule (18), superantigens (14), SpyCEP (19), ScpA (15), and the regulator CovRS (13). M protein, a multifunctional virulence factor anchored on the cell surface that is the target for serological typing, is dispensable (6).
Some GAS virulence factors act to induce inflammation. During nasopharyngeal infection, the superantigen SpeA forces T cell antigen receptors (TCR) to engage the major histocompatibility complex (MHC) class II molecules of antigen-presenting cells in an antigen-independent manner (14). This induces excessive T cell activation, which is highly proinflammatory and promotes nasopharyngeal infection (20). During invasive skin infections, the protease SpeB is also strongly proinflammatory and directly activates the proinflammatory cytokine interleukin 1β (IL-1β), which is inert until an inhibitory domain is proteolytically removed, bypassing its ordinary activation by host caspase-family proteases (21). IL-1β represses bacterial growth during invasive skin infections; neutrophil ablation or IL-1β neutralization enhances GAS growth in murine models of invasive infection and is a risk factor for invasive infections in humans (21). GAS can evade IL-1β-mediated restriction by inactivating SpeB through spontaneous mutation in the CovRS/CsrRS regulators (21, 22), a frequent observation made with isolates from invasive diseases but not pharyngitis (23–27).
Here, we used a murine model of disease to examine SpeB-mediated activation of IL-1β within the nasopharynx. In contrast to our observations in models of invasive skin and soft tissue infection, activation of IL-1β is not restrictive and instead promotes infection. IL-1 signaling is required for neutrophil recruitment, which is also required for infection, potentially to overcome microbial interference. Together, these results have allowed us to examine how inflammation can promote pathogenesis and helped decipher how the cost-benefit of this strategy for GAS changes according to host immune statue and infection site.
RESULTS
Anakinra antagonizes GAS colonization of the nasopharynx.
Anakinra inhibition of IL-1 signaling increases GAS burden during murine invasive infection (21). The more common site for GAS to reside in is the upper respiratory tract. Here, we used the established murine nasopharyngeal infection model (6, 9, 13, 20) to assess anakinra’s effect at this location. Mice given anakinra intravenously and then inoculated with GAS intranasally had significantly reduced GAS titers (Fig. 1A). Proinflammatory cytokines were quantified using multiplexed enzyme-linked immunosorbent assay (ELISA), and IL-1β, IL-6, tumor necrosis factor alpha (TNF-α), and IL-12 were all found to be significantly induced by infection (Fig. 1B). Anakinra treatment reduced IL-1β and IL-6 levels. These effects are likely due to both reduced bacterial burden and IL-1β-mediated induction of IL-6 and autoregulation (28), but other cytokines were not significantly impacted (Fig. 1B). IL-1β retaining the amino-terminal inhibitory domain is detected by ELISA even though it lacks proinflammatory activity, so we quantified how much proinflammatory signal was present using transgenic IL-1 receptor reporter cells that detect processed, active IL-1β but not the noninflammatory full-length protein (21, 29); IL-1 signaling was completely inhibited by anakinra (Fig. 1C). IL-1α, another agonist for the IL-1 receptor that is typically membrane anchored (28), was not present at detectable levels.
FIG 1.

Anakinra antagonizes GAS colonization of the nasopharynx. Induction of inflammatory cytokines and their contribution to GAS infection. C57BL/6 mice were treated with anakinra (50 μg/kg) or PBS control and inoculated intranasally with 108 CFU of GAS M1T1 5448. Mice were euthanized after 24 or 72 h, and the nasopharynxes were subjected to lavage to quantify GAS CFU by dilution plating (A), quantify cytokines with a Meso Scale Diagnostics multiplex ELISA (means are displayed; comparisons indicated reach significance only for IL-1β and IL-6) (B), and measure levels of IL-1 signaling using a transgenic IL-1 receptor reporter specific for active IL-1α and IL-1β (C). Data are means ± standard deviations (SD) (n = 10 each) and are representative of at least 3 experiments. ND, none detected; *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ns, not significant.
SpeB and caspase-1 contribute to IL-1β generation in the nasopharynx.
Similar to anakinra treatment, mice deficient in the IL-1 receptor (IL1R1−/−) rapidly clear intranasally inoculated GAS (Fig. 2A). Levels of IL-1β were also reduced, consistent with both its autoregulation (28, 30) and the reduced burden of GAS. We further examined the pathways involved using mice lacking caspase-1 and caspase-11 (casp-1/11−/−), the inflammasome proteases that are typically essential for IL-1β maturation (28). Intranasally inoculated Casp-1/11−/− mice had a more modestly, but significantly, reduced GAS burden and induced less total IL-1β (Fig. 2A). We recently showed that in place of caspase-1 and caspase-11, the GAS protease SpeB can directly mature IL-1β and induce IL-1 signaling (21). Intranasally inoculated ΔspeB GAS induced significantly less total IL-1β and less active IL-1 signal and was highly attenuated (Fig. 2B). During skin infection, GAS growth and invasion are increased by anakinra (21), but anakinra does not reverse the attenuation of ΔspeB GAS in the nasopharynx (Fig. 2B). Thus, inflammasome-associated caspases also contribute to GAS growth in the nasopharynx, and not its restriction. However, caspase-mediated IL-1 signaling is not essential for GAS, unlike SpeB-mediated IL-1 signaling. Together, these observations are consistent with a model where the proinflammatory activity of IL-1β activated by SpeB promotes upper respiratory tract infection.
FIG 2.

SpeB and caspase-1 contribute to IL-1β generation in the nasopharynx. Effects of IL-1 and inflammasome signaling on GAS survival. (A) C57BL/6, casp-1/11−/−, or IL1R1−/− mice were inoculated intranasally with 108 CFU of wild-type GAS M1T1 5448. Mice were euthanized after 24 h, nasopharyngeal lavage fluids were plated to enumerate CFU, and cytokine levels were quantified by ELISA and IL-1R reporter assay. (B and C) Role of SpeB and CovRS in GAS survival. C57BL/6 mice were given anakinra (50 μg/kg) or PBS and infected as described above with wild-type, ΔspeB, or AP covS-frameshift GAS, and CFU and cytokines were examined as described above. (D) Role of host pathways in selection for covS-frameshift SpeB− clones. Isolated colonies from the experiments for panels A, B, and C were screened for SpeB hydrolysis of azocasein, and the fraction where SpeB activity is lost is indicated. ND, none detected (for experiments where no GAS was recoverable from the mouse). Data are means ± SD (n = 5 each) and are representative of at least 3 experiments. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ns, not significant.
To further investigate how SpeB contributes to infection of the nasopharynx, we used an isogenic strain of M1T1 GAS (5448) with a mutation in CovS (covS). Mutations in CovS are frequently observed in isolates from human invasive infections (21, 25–27, 31), and this strain with a CovS mutation was isolated after animal passage (AP). CovS mutation represses SpeB and greatly induces other important virulence factors, including capsule, streptolysin O, and streptokinase, which during invasive infections may mitigate the attenuation due to loss of SpeB (21, 22). AP covS GAS induced less IL-1β, were attenuated in the nasopharynx, like ΔspeB GAS (Fig. 2C), and did not have the hypervirulent phenotype observed with covS GAS during invasive skin and soft tissue infections (21, 26). covS (SpeB−) GAS mutants spontaneously arise at high frequency during invasive infections, accounting for up to 10% of the recoverable bacteria at 24 h (21, 26, 32). No isolates of this genotype were detected in the nasopharynx, and recovery was unaffected by IL-1 (IL1R1−/− or anakinra-treated mice) or the inflammasome (casp-1/11−/− mice) (Fig. 2D). Together, these data show a requirement for SpeB and its essential regulator CovS in the nasopharynx that is not shared in the skin, and they also show that this correlates with the ability to induce IL-1β.
IL-1-dependent neutrophil recruitment promotes GAS nasopharyngeal infection.
Since inflammation and GAS CFU were both decreased by anakinra treatment, we examined which immune cells were altered by this treatment. After intravenous administration of anakinra and intranasal inoculation with GAS, cells present at the infection site were removed by lavage and examined by flow cytometry. The only population significantly changed in frequency was Ly6G+ CD11b+ neutrophils (Fig. 3A), consistent with their robust recruitment during human infections (2). All neutrophils during GAS infection expressed high levels of IL-1 receptor (IL-1R), which were lessened in anakinra-treated mice (Fig. 3B). While IL-1β may drive neutrophil activation via this mechanism, it is not a conventional chemokine, so it likely promotes chemotaxis through the induction of IL-8 (CXCL8, murine homologs KC/MIP-2), CCL2, gamma interferon (IFN-γ), complement factors, and other known chemokines and their receptors (Fig. 1B) (33, 34).
FIG 3.

IL-1-dependent neutrophil recruitment promotes GAS nasopharyngeal infection. IL-1R signaling effects on nasopharyngeal cell populations. (A) C57BL/6 mice were treated with anakinra (50 μg/kg) or PBS control. inoculated intranasally with 108 CFU of GAS M1T1 5448, and euthanized after 24 h (as described for Fig. 1A), and nasopharyngeal lavage cells were analyzed by cytometry with the markers for neutrophils (CD11bhi Ly6Ghi), dendritic cells (CD11chi), macrophages (CD11bhi Ly6Glow), T cells (CD3+), and B cells (CD45R+ B220+) and expressed as a percentage of total live cells. (B) Effects of IL-1R signaling inhibition on IL-1R expression. Neutrophils (CD11bhi Ly6Ghi) were further examined for expression of IL-1R1 under each condition. (C) Neutrophil effects on GAS survival in the nasopharynx. Mice treated 24 h earlier with anti-Ly6G (100 μg), isotype IgG (100 μg), or PBS were infected with 108 CFU of GAS M1T1 5448 as described above, and neutrophils (CD11bhi Ly6Ghi) and CFU were quantified after 24 h. (D) Human neutrophils were incubated with anakinra (blue) or PBS only (purple) and infected with GAS at a multiplicity of infection of 10. Aliquots were removed for CFU counts at the indicated intervals. Data are means ± SD (n = 5 each) and are representative of at least 3 experiments. ND, none detected; *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ns, not significant.
Since neutrophil influx is a major feature of our model and natural infections, we examined the contribution of neutrophils to disease by using an anti-Ly6G antibody to deplete their numbers. While neutrophils are essential for restricting growth in models of invasive skin and soft tissue infection (21, 26, 35), neutrophil ablation completely blocked GAS infection of the nasopharynx (Fig. 3C). Neutrophils effectively killed GAS in vitro, and this was not impacted by anakinra (Fig. 3D). Together, these results suggest that neutrophil recruitment initiated by IL-1 signaling may have compensatory benefits for GAS in the nasopharynx that do not occur in the skin and soft tissue.
IL-1 mediates microbial interference in the nasopharynx.
Nasopharyngeal shedding of Streptococcus pneumoniae is inhibited by the anti-inflammatory corticosteroid dexamethasone (36). Antagonizing IL-1-mediated inflammation may not be integral to S. pneumoniae infection, as we find it is for GAS (Fig. 1A), since IL-1β deficiency instead prolonged S. pneumoniae colonization (37). Nonetheless, this suggests more broadly that inflammation in some instances may promote nasopharyngeal infections. Mice were intranasally inoculated with the nonpathogenic Streptococcaceae species Lactococcus lactis, and after 24 h, this organism could be detected in the nasopharynx at levels similar to those of GAS (Fig. 4A). IL-1β was induced to significantly lower levels by L. lactis but was enhanced by L. lactis transformed to express SpeB (7), which led to clearance of the bacterium within 24 h (Fig. 4A). The attenuation of SpeB-expressing L. lactis was reversed by anakinra (Fig. 4B), demonstrating that activation of IL-1β by SpeB is the major mechanism driving attenuation of SpeB-expressing L. lactis in the nasopharynx. Thus, inflammation that is beneficial to GAS is harmful to L. lactis. Intranasal coinoculation of GAS and L. lactis decreased recovery of both species (Fig. 4C). During coculture in rich medium (Todd-Hewitt broth), growth was not antagonistic (Fig. 4D), indicating that host factors are involved in the mutual antagonization between these species.
FIG 4.
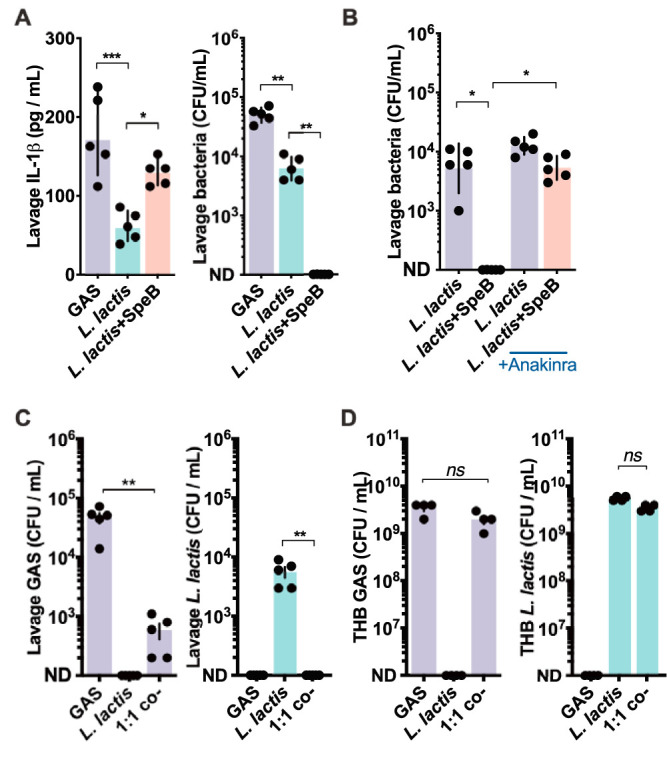
IL-1 mediates microbial interference in the nasopharynx. (A) SpeB-dependent effects on IL-1β and growth. C57BL/6 mice were inoculated intranasally with 108 CFU of GAS, L. lactis, or SpeB-expressing L. lactis. IL-1β was quantified by ELISA and CFU by dilution plating from nasopharyngeal lavage samples collected after 24 h infection. (B) IL-1β-dependent effects on L. lactis growth. C57BL/6 mice treated with anakinra (50 μg/kg) or PBS control were infected and CFU enumerated after 24 h as described above. (C) In vivo competition experiment with GAS and L. lactis. C57BL/6 mice were inoculated with 108 CFU GAS, L. lactis, or a mix of both (5 × 107 each; 1:1), and CFU were enumerated after 24 h as described above. (D) In vitro competition experiment with GAS and L. lactis. GAS (104 CFU), L. lactis (104 CFU), or a mix of both (5 × 103 CFU each; 1:1) were grown 18 h in 3 ml Todd-Hewitt broth at 37°C in 5% CO2; then, the CFU of each were enumerated by dilution and differential plating. Data are means ± SD (n = 5 each) and are representative of at least 3 experiments. ND, none detected; *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ns, not significant.
Antibiotic pretreatment promotes GAS growth in the nasopharynx.
The nasopharyngeal microbiota has the potential to interact with GAS and L. lactis in similar ways. Of note, early human prospective studies show that some microbiomes correlate with resistance to GAS (38) that is lost when their community structure is disrupted by penicillin treatment (39). Administration of penicillin as a single dose either 24 or 48 h before infection significantly increases GAS growth in the murine nasopharynx (Fig. 5A), in agreement with these human studies. When the interval between penicillin treatment and infection is short, GAS does not survive due to its susceptibility. In mice pretreated with penicillin, the inhibition of GAS growth previously observed with anakinra inhibition of IL-1 (Fig. 1A) or neutrophil ablation (Fig. 4C) is reversed (Fig. 5B). Furthermore, attenuation of ΔspeB GAS in the nasopharynx (Fig. 2B) is reversed in mice pretreated with penicillin (Fig. 5C). Thus, penicillin pretreatment removes the proinflammatory requirements for SpeB, IL-1, and neutrophils during GAS infection of the nasopharynx.
FIG 5.

Antibiotic pretreatment promotes GAS growth in the nasopharynx. Growth of GAS in the upper respiratory tract of antibiotic pretreated mice. (A) C57BL/6 mice were given 5,000 U penicillin intraperitoneally at the indicated intervals (−48, −24, −2, or +1 h relative to inoculation time), and 108 CFU of GAS 5448 was delivered intranasally. Mice were euthanized 24 h postinoculation, and GAS CFU were quantified from nasopharyngeal lavage. (B) C57BL/6 mice were treated as described above, with uniform administration of penicillin 24 h preinfection, anti-Ly6G 24 h preinfection, or anakinra 4 h preinfection. (C) C57BL/6 mice were given penicillin 24 h preinfection and infected with 108 CFU of wild-type or ΔspeB GAS 5448 as described above, and CFU were enumerated after 24 h. Data are means ± SD (n = 5 each) and are representative of at least 3 experiments. *, P < 0.05; **, P < 0.005; ns, not significant.
DISCUSSION
IL-1 signaling induces antimicrobial effector mechanisms that restrict most pathogens, including GAS causing invasive infections. Pharmacological inhibition of this critical proinflammatory pathway is a risk factor for severe GAS infection in humans (21). We show that in the nasopharynx, IL-1 signaling instead promotes GAS infection, highlighting a fundamental difference in inflammation and immunity between infection sites. Neutrophils are major immune cells recruited during infections of the nasopharynx and skin, and this recruitment is dependent on IL-1 signaling at both sites (21, 26, 28). Neutrophil influx in invasive skin and soft tissue infection is host protective (2, 19, 35). In contrast, this neutrophil infiltrate in the nasopharynx, which is a major component of strep throat pharyngitis, promotes infection.
Inflammation may be broadly beneficial for GAS in the nasopharynx. Other than SpeB, the superantigen SpeA also strongly induces inflammation by activating T cells; both the toxin and T cells are required for infection in this model (20, 40). Additional GAS proteins may similarly contribute to infection by not only acting as conventional virulence factors but also promoting inflammation as pathogen-associated molecular patterns (PAMPs). The strong resistance of GAS to immune effectors may function in part to help the organism survive the hyperinflammatory state induced by SpeB and superantigens, and this resistance would be dispensable in a less-inflamed host. covS frameshift mutations that repress speB and greatly induce most other virulence factors readily arise during invasive skin and soft tissue infections of human (23, 24) and mice (25). These mutations do not occur in human (31, 41, 42) or murine (21, 26, 27, 43, 44) upper respiratory tract infections, supporting our observation that CovS and SpeB are required in the nasopharynx. covS mutants have a reduced ability to colonize and to induce less IL-1β and less neutrophil infiltration and are more resistant to neutrophils and other immune cells (21, 26, 45–47). Accordingly, IL-1β and neutrophils are essential for selection of covS mutation during invasive skin infections (21, 45), though we report a reciprocal role in the nasopharynx. Since covS mutations are not fixed in the population, despite these seemingly beneficial activities that are advantageous at invasive sites, we infer that attenuation in the nasopharynx is a bottleneck exerting strong selection on the species.
Diverse other pathogens activate specific inflammatory pathways to antagonize competing microbes, disrupt membrane barrier function, promote dissemination, or acquire nutrients. Most notably, S. pneumoniae shedding from the nasopharynx is inhibited by the broadly immunosuppressant corticosteroid dexamethasone (36) but not IL-1 signaling (48), which instead promotes S. pneumoniae clearance from the nasopharynx (49). IL-1 signaling still promotes neutrophil infiltration during S. pneumoniae infection, suggesting that the underlying roles for inflammation in disease differ between these species. Other microbes of the upper respiratory tract, including the pathogens Haemophilus influenzae, Staphylococcus aureus, Moraxella catarrhalis, and Neisseria meningitidis and the microbiota, primarily other Streptococcus, Haemophilus, and Neisseria species (50), may similarly be impacted by inflammation and immunomodulation.
Consistent with our observation that penicillin pretreatment 24 h prior to infection restores GAS colonization, previous studies examining treatment failure with penicillin (51), recurrent infection (38), and infection secondary to antibiotics for other indications (39) support a role of antibiotics in human susceptibility. Our data support the hypothesis that GAS colonization may be mediated by the nasopharyngeal microbiota. Resident species are able to directly kill GAS in vitro (52), and their presence correlates with resistance to GAS infection (38). Resistance to GAS infection is lost when their community structure is disrupted by penicillin treatment (39). Recapitulating this observation in our animal model, we found that penicillin pretreatment increased susceptibility to GAS in the nasopharynx and removed the replication defects observed upon targeted ablation of inflammatory responses. These results suggest that the distinctive inflammatory pathology of strep throat is key for pathogenesis. Investigation into microbiome changes due to penicillin pretreatment of the murine nasopharynx may provide insight into specific species that antagonize GAS. If these or functionally similar species are present in the human oropharynx, they may naturally antagonize human colonization and infection.
Our study demonstrates that in the absence of IL-1 signaling in the nasopharynx, there is decreased colonization of GAS, the opposite of what is observed during invasive skin and soft tissue infection. The decrease in GAS colonization stems from a reduction in inflammation and neutrophil infiltrate. Pretreatment with antibiotics increases host permissiveness for GAS in the nasopharynx, implicating a role for the microbiota in colonization resistance. These observations establish a connection between inflammation, antibiotics, and host permissiveness that has immediate clinical implications for this challenging pathogen.
MATERIALS AND METHODS
Bacterial strains.
GAS M1T1 5448, its isogenic ΔspeB mutant, Lactococcus lactis, and the speB complementation vector pSpeB were previously described (7, 21, 53). GAS strains were statically grown at 37°C and 5% CO2 in Todd-Hewitt broth (Difco), washed twice with phosphate-buffered saline (PBS; pH 7.4), and diluted to a multiplicity of infection (MOI) of 10 or 100 for in vitro infections.
Experimental infection.
Seven- to 8-week-old wild-type C57BL/6 mice of both sexes (Jackson Laboratory) were used for experiments; casp-1/11−/− and IL1R1−/− C57BL/6 mice were described previously (21). Experimental treatments of these mice included intravenous delivery of 50 mg/kg anakinra (Kineret; inhibits both human and murine IL-1R1 [21]) 4 h preinfection, intraperitoneal delivery of 5,000 U penicillin G (Pfizer) 24 h preinfection or as indicated, neutrophil depletion with 100 μg anti-Ly6G monoclonal antibody (MAb) (1A8) or isotype IgG (both from BioXCell) 24 h preinfection. Mouse groups were routinely inoculated intranasally with 108 CFU GAS slowly administered via micropipette in 10 μl PBS divided between nostrils and were allowed to aspirate the inoculum via the normal breathing process. This volume resulted in no respiration into the lung, which can cause a necrotizing pneumonia and rapid death. At various intervals, the mice were euthanized, and the nasopharynxes were subjected to lavage by insertion of a catheter through a midline incision in the trachea through which 100 μl PBS was flushed and collected from the nose. Samples were prepared from this fluid for cytometry or for the quantification of cytokines and/or GAS CFU.
Cytometry and cytokine measurements.
Cytokine levels in cell-free tissue homogenate were quantified by multiple ELISAs with the mouse proinflammatory panel 1, following the manufacturer’s instructions (K15048G; Meso Scale Diagnostics). Cells were treated with GolgiStop (BD), fixed with 2% paraformaldehyde with Fc block (BD Biosciences) before and after, and incubated with anti-CD3–BV605, anti-CD11c–BUV, anti-CD11b–PECF594, anti-Gr-1/Ly6–APCH7, anti-CD40–BV650, anti-CD80–BUV737, and anti-CD86–APCR700 (all from BioLegend) to assess T cells, monocytes, macrophages, dendritic cells, and neutrophils. Depletion was confirmed by analyzing populations in blood obtained by cardiac puncture, with Ly6G, CD11b, and CD11c, as described above. Flow cytometry was performed on a BD LSRII or LSRFortessa X-20 system and analyzed using FlowJo 20 (TreeStar).
Transgenic IL-1R reporter cells were used to measure matured IL-1β essentially as previously described (21). These reporter cells were modified (29) to use a luciferase-based instead of an alkaline phosphatase-based readout, to eliminate signal interference from bacterial and murine alkaline phosphatases. Luciferase activity was measured with Steady-Luc luciferin (Biotium) on a multimode plate reader (PerkinElmer).
SpeB activity.
Total SpeB activity was measured in individual isolates by hydrolysis of azocasein (Sigma) by previously described methods (21, 54). At least 12 isolates from each biological replicate were examined, and phenotypic conversion to the covS (SpeB−) phenotype is expressed as the percentage of isolates with a heritable loss of casein proteolysis.
Neutrophil isolation and infection.
Blood was collected from healthy male and female human volunteers, who provided informed consent, with approval by the Emory University Institutional Review Board. Neutrophils were isolated using PolyMorphPrep (Axis-Shield) as previously described (55). Neutrophil viability and concentration were assessed microscopically using 0.04% trypan blue; then, neutrophils were diluted to 105 cells in PBS and infected with 106 CFU of GAS. Aliquots were removed at various intervals, incubated for 2 min with 0.02% Triton X-100, and plated for CFU enumeration.
Statistical analysis.
Statistical analyses were performed using Prism 8 (GraphPad). Values are expressed as means and standard errors unless otherwise specified. Differences were determined using the Mann-Whitney U test (paired) or the Kruskal-Wallis test with Dunn’s postanalysis (multiple groups) unless otherwise specified.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health NIH/NIAID K22 AI130223 (C.N.L.) and startup funds from Emory University.
We thank Victor Nizet for bacterial strains, the Emory Multiplexed Immunoassay Core, the Children's Healthcare of Atlanta and Emory University's Children’s Clinical and Translational Discovery Core, and the Jacob Kohlmeier lab for technical support, and Jacqueline Kimmey and all members of the LaRock lab for critical manuscript review.
REFERENCES
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Walker MJ, Barnett TC, McArthur JD, Cole J, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin Microbiol Rev 27:264–301. doi: 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmes MC, Williams R. 1954. The distribution of carriers of Streptococcus pyogenes among 2,413 healthy children. J Hyg (Lond) 52:165–179. doi: 10.1017/s0022172400027376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan EL, Chhatwal GS, Rohde M. 2006. Reduced ability of penicillin to eradicate ingested group A streptococci from epithelial cells: clinical and pathogenetic implications. Clin Infect Dis 43:1398–1406. doi: 10.1086/508773. [DOI] [PubMed] [Google Scholar]
- 5.Okada N, Liszewski MK, Atkinson JP, Caparon M. 1995. Membrane cofactor protein (CD46) is a keratinocyte receptor for the M protein of the group A streptococcus. Proc Natl Acad Sci U S A 92:2489–2493. doi: 10.1073/pnas.92.7.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson EL, Cole JN, Olson J, Ryba B, Ghosh P, Nizet V. 2014. The fibrinogen-binding M1 protein reduces pharyngeal cell adherence and colonization phenotypes of M1T1 group A Streptococcus. J Biol Chem 289:3539–3546. doi: 10.1074/jbc.M113.529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnett TC, Liebl D, Seymour LM, Gillen CM, Lim JY, LaRock CN, Davies MR, Schulz BL, Nizet V, Teasdale RD, Walker MJ. 2013. The globally disseminated M1T1 clone of group A streptococcus evades autophagy for intracellular replication. Cell Host Microbe 14:675–682. doi: 10.1016/j.chom.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soderholm AT, Barnett TC, Korn O, Rivera-Hernandez T, Seymour LM, Schulz BL, Nizet V, Wells CA, Sweet MJ, Walker MJ. 2018. Group A Streptococcus M1T1 intracellular infection of primary tonsil epithelial cells dampens levels of secreted IL-8 through the action of SpyCEP. Front Cell Infect Microbiol 8:160. doi: 10.3389/fcimb.2018.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park H-S, Francis KP, Yu J, Cleary PP. 2003. Membranous cells in nasal-associated lymphoid tissue: a portal of entry for the respiratory mucosal pathogen group A streptococcus. J Immunol 171:2532–2537. doi: 10.4049/jimmunol.171.5.2532. [DOI] [PubMed] [Google Scholar]
- 10.Bessen D, Fischetti VA. 1988. Passive acquired mucosal immunity to group A streptococci by secretory immunoglobulin A. J Exp Med 167:1945–1950. doi: 10.1084/jem.167.6.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dale JB, Baird RW, Courtney HS, Hasty DL, Bronze MS. 1994. Passive protection of mice against group A streptococcal pharyngeal infection by lipoteichoic acid. J Infect Dis 169:319–323. doi: 10.1093/infdis/169.2.319. [DOI] [PubMed] [Google Scholar]
- 12.Wessels MR, Bronze MS. 1994. Critical role of the group A streptococcal capsule in pharyngeal colonization and infection in mice. Proc Natl Acad Sci U S A 91:12238–12242. doi: 10.1073/pnas.91.25.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alam FM, Turner CE, Smith K, Wiles S, Sriskandan S. 2013. Inactivation of the CovR/S virulence regulator impairs infection in an improved murine model of Streptococcus pyogenes naso-pharyngeal infection. PLoS One 8:e61655. doi: 10.1371/journal.pone.0061655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasper KJ, Zeppa JJ, Wakabayashi AT, Xu SX, Mazzuca DM, Welch I, Baroja ML, Kotb M, Cairns E, Cleary PP, Haeryfar SMM, McCormick JK. 2014. Bacterial superantigens promote acute nasopharyngeal infection by Streptococcus pyogenes in a human MHC class II-dependent manner. PLoS Pathog 10:e1004155. doi: 10.1371/journal.ppat.1004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park H-S, Cleary PP. 2005. Active and passive intranasal immunizations with streptococcal surface protein C5a peptidase prevent infection of murine nasal mucosa-associated lymphoid tissue, a functional homologue of human tonsils. Infect Immun 73:7878–7886. doi: 10.1128/IAI.73.12.7878-7886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dileepan T, Linehan JL, Moon JJ, Pepper M, Jenkins MK, Cleary PP. 2011. Robust antigen specific Th17 T cell response to group A Streptococcus is dependent on IL-6 and intranasal route of infection. PLoS Pathog 7:e1002252. doi: 10.1371/journal.ppat.1002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Dileepan T, Briscoe S, Hyland KA, Kang J, Khoruts A, Cleary PP. 2010. Induction of TGF-beta1 and TGF-beta1-dependent predominant Th17 differentiation by group A streptococcal infection. Proc Natl Acad Sci U S A 107:5937–5942. doi: 10.1073/pnas.0904831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vega LA, Sanson MA, Shah BJ, Flores AR. 2020. Strain-dependent effect of capsule on transmission and persistence in an infant mouse model of group A Streptococcus infection. Infect Immun 88:e00709-19. doi: 10.1128/IAI.00709-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zinkernagel AS, Timmer AM, Pence MA, Locke JB, Buchanan JT, Turner CE, Mishalian I, Sriskandan S, Hanski E, Nizet V. 2008. The IL-8 protease SpyCEP/ScpC of group A Streptococcus promotes resistance to neutrophil killing. Cell Host Microbe 4:170–178. doi: 10.1016/j.chom.2008.07.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeppa JJ, Kasper KJ, Mohorovic I, Mazzuca DM, Haeryfar SMM, McCormick JK. 2017. Nasopharyngeal infection by Streptococcus pyogenes requires superantigen-responsive Vβ-specific T cells. Proc Natl Acad Sci U S A 114:10226–10231. doi: 10.1073/pnas.1700858114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LaRock CN, Todd J, LaRock DL, Olson J, O’Donoghue AJ, Robertson AAB, Cooper MA, Hoffman HM, Nizet V. 2016. IL-1β is an innate immune sensor of microbial proteolysis. Sci Immunol 1:eaah3539. doi: 10.1126/sciimmunol.aah3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aziz RK, Pabst MJ, Jeng A, Kansal RG, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134. doi: 10.1046/j.1365-2958.2003.03797.x. [DOI] [PubMed] [Google Scholar]
- 23.Chatellier S, Ihendyane N, Kansal RG, Khambaty F, Basma H, Norrby-Teglund A, Low DE, McGeer A, Kotb M. 2000. Genetic relatedness and superantigen expression in group A Streptococcus serotype M1 isolates from patients with severe and nonsevere invasive diseases. Infect Immun 68:3523–3534. doi: 10.1128/iai.68.6.3523-3534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. 2000. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun 68:6362–6369. doi: 10.1128/iai.68.11.6362-6369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. 2001. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis 183:1043–1054. doi: 10.1086/319291. [DOI] [PubMed] [Google Scholar]
- 26.Walker MJ, Hollands A, Sanderson-Smith ML, Cole J, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 27.Cole J, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, Nizet V. 2010. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio 1:e00191-10. doi: 10.1128/mBio.00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LaRock CN, Nizet V. 2015. Inflammasome/IL-1β responses to streptococcal pathogens. Front Immunol 6:518. doi: 10.3389/fimmu.2015.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun J, LaRock DL, Skowronski EA, Kimmey JM, Olson J, Jiang Z, O’Donoghue AJ, Nizet V, LaRock CN. 2020. Role of inflammasome-independent activation of IL-1β by the Pseudomonas aeruginosa protease LasB. bioRxiv 2020.05.18.101303. doi: 10.1101/2020.05.18.101303. [DOI] [PMC free article] [PubMed]
- 30.Toda Y, Tsukada J, Misago M, Kominato Y, Auron PE, Tanaka Y. 2002. Autocrine induction of the human pro-IL-1 beta gene promoter by IL-1 beta in monocytes. J Immunol 168:1984–1991. doi: 10.4049/jimmunol.168.4.1984. [DOI] [PubMed] [Google Scholar]
- 31.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maamary PG, Zakour NLB, Cole J, Hollands A, Aziz RK, Barnett TC, Cork AJ, Henningham A, Sanderson-Smith ML, McArthur JD, Venturini C, Gillen CM, Kirk JK, Johnson DR, Taylor WL, Kaplan EL, Kotb M, Nizet V, Beatson SA, Walker MJ. 2012. Tracing the evolutionary history of the pandemic group A streptococcal M1T1 clone. FASEB J 26:4675–4684. doi: 10.1096/fj.12-212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LaRock CN, Cookson BT. 2013. Burning down the house: cellular actions during pyroptosis. PLoS Pathog 9:e1003793. doi: 10.1371/journal.ppat.1003793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinarello CA, Simon AK, van der Meer JWM. 2012. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hidalgo-Grass C, Mishalian I, Dan-Goor M, Belotserkovsky I, Eran Y, Nizet V, Peled A, Hanski E. 2006. A streptococcal protease that degrades CXC chemokines and impairs bacterial clearance from infected tissues. EMBO J 25:4628–4637. doi: 10.1038/sj.emboj.7601327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zafar MA, Wang Y, Hamaguchi S, Weiser JN. 2017. Host-to-host transmission of Streptococcus pneumoniae is driven by its inflammatory toxin, pneumolysin. Cell Host Microbe 21:73–83. doi: 10.1016/j.chom.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lemon JK, Weiser JN. 2015. Degradation products of the extracellular pathogen Streptococcus pneumoniae access the cytosol via its pore-forming toxin. mBio 6:e02110-14. doi: 10.1128/mBio.02110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crowe CC, Sanders WE. 1973. Bacterial interference. II. Role of the normal throat flora in prevention of colonization by group A Streptococcus. J Infect Dis 128:527–532. doi: 10.1093/infdis/128.4.527. [DOI] [PubMed] [Google Scholar]
- 39.Sanders CC, Sanders WE, Harrowe DJ. 1976. Bacterial interference: effects of oral antibiotics on the normal throat flora and its ability to interfere with group A streptococci. Infect Immun 13:808–812. doi: 10.1128/IAI.13.3.808-812.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dan JM, Havenar-Daughton C, Kendric K, Al-Kolla R, Kaushik K, Rosales SL, Anderson EL, LaRock CN, Vijayanand P, Seumois G, Layfield D, Cutress RI, Ottensmeier CH, Arlehamn CSL, Sette A, Nizet V, Bothwell M, Brigger M, Crotty S. 2019. Recurrent group A Streptococcus tonsillitis is an immunosusceptibility disease involving antibody deficiency and aberrant TFH cells. Sci Transl Med 11:eaau3776. doi: 10.1126/scitranslmed.aau3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog 6:e1000832. doi: 10.1371/journal.ppat.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shea PR, Beres SB, Flores AR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Ayers SD, Webb P, Willey BM, Low DE, Musser JM. 2011. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc Natl Acad Sci U S A 108:5039–5044. doi: 10.1073/pnas.1016282108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiebig A, Loof TG, Babbar A, Itzek A, Koehorst JJ, Schaap PJ, Nitsche-Schmitz DP. 2015. Comparative genomics of Streptococcus pyogenes M1 isolates differing in virulence and propensity to cause systemic infection in mice. Int J Med Microbiol 305:532–543. doi: 10.1016/j.ijmm.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 44.Horstmann N, Tran CN, Brumlow C, DebRoy S, Yao H, Nogueras Gonzalez G, Makthal N, Kumaraswami M, Shelburne SA. 2018. Phosphatase activity of the control of virulence sensor kinase CovS is critical for the pathogenesis of group A streptococcus. PLoS Pathog 14:e1007354. doi: 10.1371/journal.ppat.1007354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Liu G, Feng W, Zhou Y, Liu M, Wiley JA, Lei B. 2014. Neutrophils select hypervirulent CovRS mutants of M1T1 group A Streptococcus during subcutaneous infection of mice. Infect Immun 82:1579–1590. doi: 10.1128/IAI.01458-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LaRock CN, Nizet V. 2015. Cationic antimicrobial peptide resistance mechanisms of streptococcal pathogens. Biochim Biophys Acta 1848:3047–3054. doi: 10.1016/jbbamem.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hollands A, Pence MA, Timmer AM, Osvath SR, Turnbull L, Whitchurch CB, Walker MJ, Nizet V. 2010. Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group A streptococcus M1T1 clone. J Infect Dis 202:11–19. doi: 10.1086/653124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lysenko ES, Ratner AJ, Nelson AL, Weiser JN. 2005. The Role of innate immune responses in the outcome of interspecies competition for colonization of mucosal surfaces. PLoS Pathog 1:e1. doi: 10.1371/journal.ppat.0010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuipers K, Lokken KL, Zangari T, Boyer MA, Shin S, Weiser JN. 2018. Age-related differences in IL-1 signaling and capsule serotype affect persistence of Streptococcus pneumoniae colonization. PLoS Pathog 14:e1007396. doi: 10.1371/journal.ppat.1007396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, Kolter R. 2010. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. mBio 1:e00129-10. doi: 10.1128/mBio.00129-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eagle H. 1952. Experimental approach to the problem of treatment failure with penicillin: I. Group A streptococcal infection in mice. Am J Med 13:389–399. doi: 10.1016/0002-9343(52)90293-3. [DOI] [PubMed] [Google Scholar]
- 52.Sanders E. 1969. Bacterial interference. I. Its occurrence among the respiratory tract flora and characterization of inhibition of group A streptococci by viridans streptococci. J Infect Dis 120:698–707. doi: 10.1093/infdis/120.6.698. [DOI] [PubMed] [Google Scholar]
- 53.LaRock CN, Döhrmann S, Todd J, Corriden R, Olson J, Johannssen T, Lepenies B, Gallo RL, Ghosh P, Nizet V. 2015. Group A streptococcal M1 protein sequesters cathelicidin to evade innate immune killing. Cell Host Microbe 18:471–477. doi: 10.1016/j.chom.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwinn LA, Khosravi A, Aziz RK, Timmer AM, Doran KS, Kotb M, Nizet V. 2007. Genetic characterization and virulence role of the RALP3/LSA locus upstream of the streptolysin s operon in invasive M1T1 group A Streptococcus. J Bacteriol 189:1322–1329. doi: 10.1128/JB.01256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dohrmann S, LaRock CN, Anderson EL, Cole JN, Ryali B, Stewart C, Nonejuie P, Pogliano J, Corriden R, Ghosh P, Nizet V. 2017. Group A streptococcal M1 protein provides resistance against the antimicrobial activity of histones. Sci Rep 7:43039. doi: 10.1038/srep43039. [DOI] [PMC free article] [PubMed] [Google Scholar]