

Abstract
Marine natural products are an important source of lead compounds against many pathogenic targets. Herein, we report the discovery of lobosamides A-C from a marine actinobacterium Micromonospora sp., representing three new members of a small but growing family of bacterially produced polyene macrolactams. The lobosamides display growth inhibitory activity against the protozoan parasite Trypanosoma brucei (Lobosamide A IC50 = 0.8 μM), the causative agent of human African trypanosomiasis (HAT). The biosynthetic gene cluster of the lobosamides was sequenced and assembled and suggests a conserved cluster organization amongst the 26-membered macrolactams. While determination of the relative and absolute configurations of many members of this family is lacking, the absolute configurations of the lobosamides were deduced using a combination of chemical modification, detailed spectroscopic analysis, and bioinformatics. We implemented a “molecules-to-genes-to-molecules” approach to determine the prevalence of similar clusters in other bacteria, which led to the discovery of two additional macrolactams, mirilactams A and B from Actinosynnema mirum. These additional analogs have allowed us to identify specific structure activity relationships that contribute to the antitrypanosomal activity of this class. This approach illustrates the power of combining chemical analysis and genomics in the discovery and characterization of natural products as new lead compounds for neglected disease targets.
INTRODUCTION
Neglected tropical diseases (NTDs) continue to be a significant global health burden, with over 500 million people at risk in endemic areas. Despite this, less than 5% of worldwide funding for NTDs has been allocated for the most neglected diseases.1 Human African trypanosomiasis (HAT) remains a serious problem, with 70 million people at risk in Africa and an estimated 30,000 cases annually.2 HAT is caused by two different subspecies of the protozoan parasite Trypanosoma brucei and can result in death if left untreated. The development of new HAT treatments has been extremely slow, with a majority of the four clinically used drugs having been discovered over 40 years ago.3 In addition, current HAT therapeutics have drawbacks in administration, toxicity, and cost. For example, melarsoprol, which remains the first-line treatment in many regions,4 has a 5% drug-induced death rate.5 New compounds in clinical trials, such as SCYX-71586 and fexinidazole,7 as well as new formulations, such as nifurtimox-eflornithine combination therapy (NECT), hold promise for improving HAT treatment worldwide.8 However, the relatively few available drugs and validated drug targets for HAT makes continued research for parasite-specific antitrypanosomal lead compounds an important priority.9
Natural products have historically been an important source of clinically relevant compounds, and the emergence of the marine environment as a rich source of new chemical entities has heralded numerous recent successes, with many marine natural products currently in preclinical or clinical trials.10 Marine bacteria have been particularly prolific producers of important compounds, such as salinosporamide A, which is currently in late-phase clinical trials for multiple myeloma.11 In addition to many other biological activities, marine natural products are an important source of lead compounds for global health targets. Representatives of multiple classes of marine natural products have shown activity against T. brucei parasites including polyketides,12 alkaloids,13,14 diketopiperazines,15 peptides,16 and others.8
As part of our ongoing program to discover new lead compounds for NTDs, we have screened our bacterially-derived marine natural products library against T. b. brucei. Herein, we report a family of structurally novel polyene macrolactams, lobosamides A-C (1 – 3, Figure 1), which are produced by a marine sediment-derived Micromonospora sp., with lobosamide A exhibiting sub-micromolar antitrypanosomal activity. To date, these molecules represent the first polyene macrolactams with reported activity against T. brucei parasites. In order to confirm the structures and deduce the full absolute configurations for these new molecules we sequenced the genome of the producing Micromonospora sp. and identified the biosynthetic gene cluster (BGC) responsible for their production. Using a combination of detailed spectroscopic and genome sequence analyses we assigned the full absolute configurations of the lobosamides, providing a rare example of the complete absolute configurational assignment for a member of this compound class.
Figure 1.
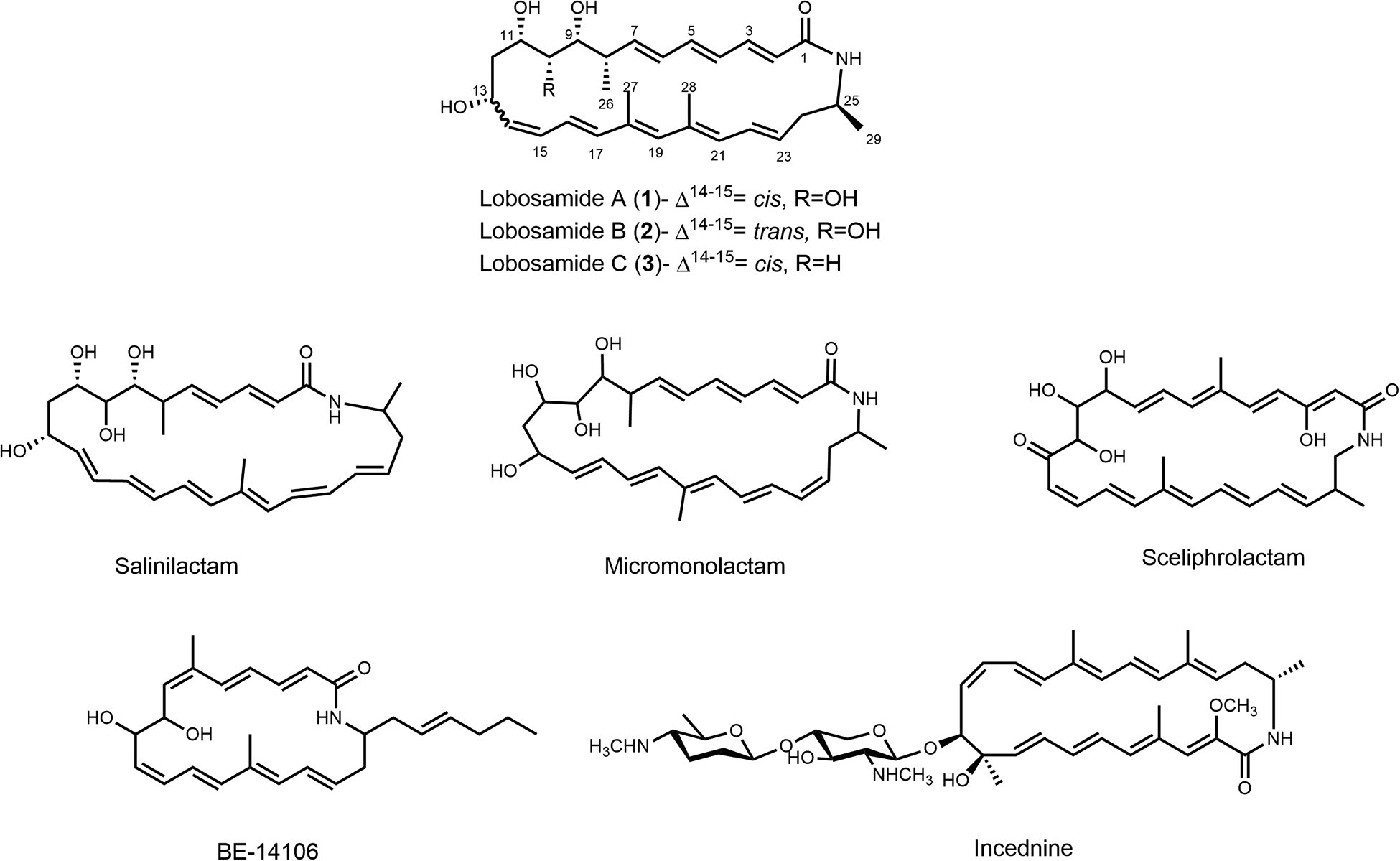
Structures of the lobosamides and related polyene macrolactams.
The increased availability of bacterial genomes and BGCs has the potential to change the way natural products discovery is conducted.17 In this study we have implemented a “molecules-to-genes-to-molecules” approach by using the newly elucidated lobosamide structure and assembled BGC as a query sequence to identify similar BGCs in other organisms that have not yet been chemically annotated. This genome mining strategy, which uses the genetic information found in biosynthetic gene clusters to predict molecular frameworks, is gaining importance in natural product discovery platforms.18,19 Using this approach, we identified a homologous BGC in the full genome sequence of a distantly related marine actinobacterium, Actinosynnema mirum (ATCC 29888), which we predicted to produce a related macrocyclic lactam.20 Using the predicted structure to guide the isolation, we identified two additional 26-membered macrolactams, which provided structure activity relationship (SAR) information about the features necessary for antitrypanosomal activity. The success of this strategy provides further support for the parallel implementation of molecular biology and analytical chemistry techniques for the discovery of natural products with unique chemical and biological features.
RESULTS AND DISCUSSION
Identification and structure elucidation of lobosamides A-C
Initial screening of our marine bacterially-derived natural product library against axenic T. b. brucei parasites revealed a prefraction that exhibited significant trypanocidal activity and no measurable cytoxicity towards mammalian cells in our phenotypic assay.21 This prefraction was produced by Micromonospora sp. RL09–050-HVF-A isolated from a marine sediment sample collected from Point Lobos in the Monterey Bay, CA. Secondary screening of a one-compound, one-well ‘peak library’ (Supplementary Information) identified two related compounds in the trace as responsible for the biological activity. Isolation by reversed-phase (RP)-HPLC led to the discovery of lobosamides A and B (Figure 1). Analysis of the (+)-HRESIFTMS (obsd. [M+Na]+ at m/z 506.2882) and NMR data for lobosamide A (1) gave the molecular formula C29H41O5N. Interpretation of the one- and two-dimensional NMR spectra (gCOSY, gTOCSY, gHSQC) showed the presence of two conjugated polyene systems, as well as four oxymethine protons, two vinyl methyl groups, two aliphatic methyl groups, two aliphatic methines, and two sets of diastereotopic methylene protons. The 1H spectrum of 1 also exhibited an exchangeable signal at δ 7.58 (Table S1). Analysis of the HMBC spectrum connected the two large spin systems through a 1,3-dimethyl moiety, and the 26-membered macrocycle was closed around a secondary amide, thus completing the planar structure of 1 (Figure 2A).
Figure 2.
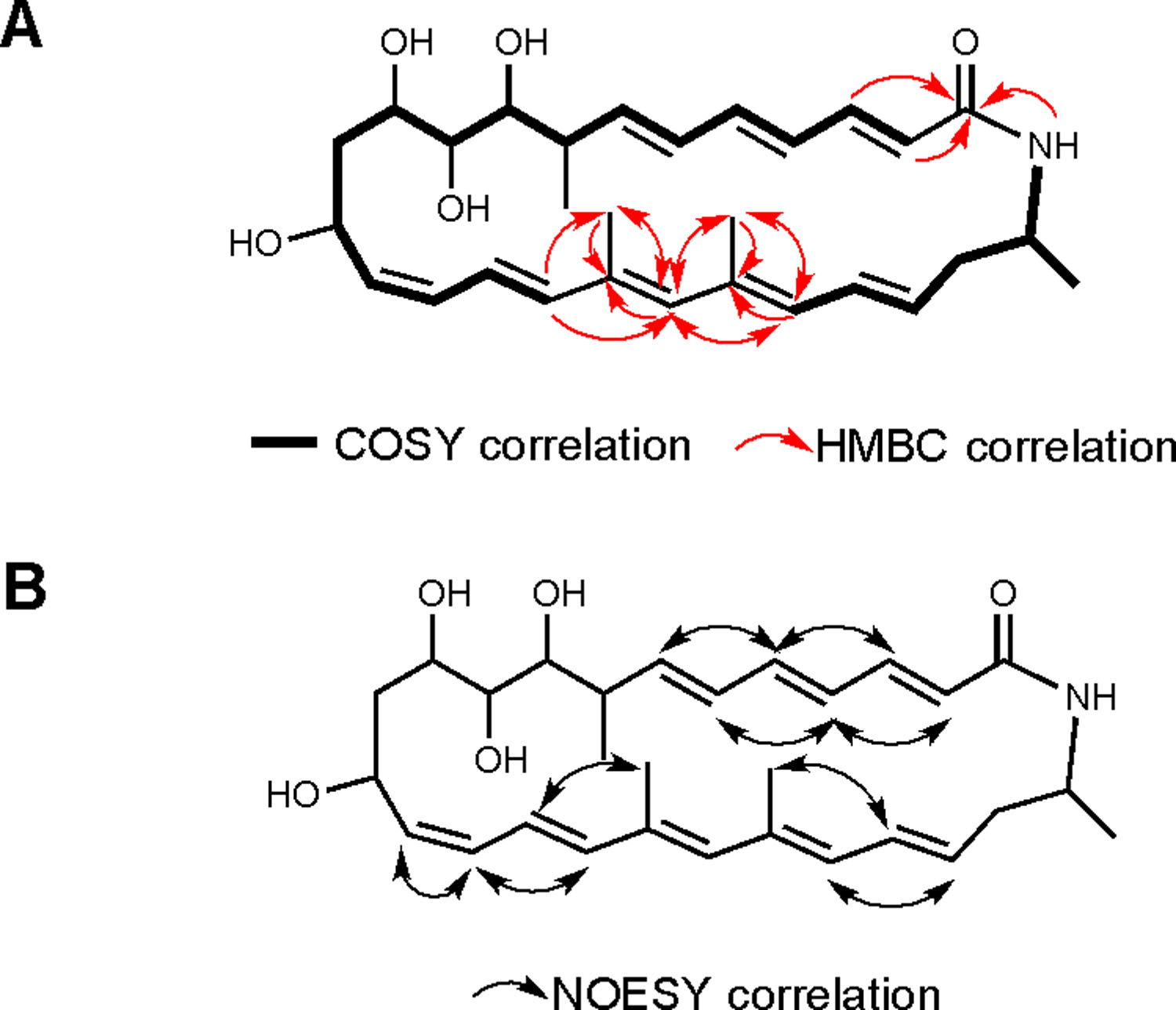
Structure elucidation of the lobosamides. (A) Two large spin systems were identified by COSY correlations (bold bonds) and connected through a 1,3-dimethyl moiety in the polyene system using HMBC correlations (red arrows). The 26-membered macrocycle was closed through an amide linkage. (B) The configurations of the 8 alkenes were determined using NOESY correlations (black arrows) and coupling constant analysis.
The geometries of the eight double bonds in lobosamide A were determined by analysis of coupling constant information and NOESY correlations (Figure 2B, and Table S1). A similar analysis for lobosamide B (2) identified it as a geometrical isomer of 1, with the C-14-C-15 olefin having a trans geometry based on its characteristically large coupling constant (3JHH = 15.0 Hz) and ROESY analysis (Table S2 and Figure S1). Further analysis of the biosynthetic products from this Micromonospora sp. revealed a third compound, lobosamide C (3), which had a similar UV profile to compounds 1 and 2. Analysis of the (+)-HRESITOFMS (obsd. [M+H]+ at m/z 467.3048) and NMR data revealed that lobosamide C shares an identical planar structure with lobosamide A, with the exception of the absence of a hydroxyl group at C-10. While compound 3 exhibited significant overlap in the alkene region of the 1H NMR spectrum, DQF-COSY and gROESY experiments showed that compound 3 has the same double bond configuration as lobosamide A (Table S4 and Figure S1).
The lobosamides are part of a relatively small, but growing family of bacterially biosynthesized macrolactams, which includes salinilactam,22 incednine,23 and micromonlactam,24 amongst others25–28 (Figure 1). The 26-membered macrocyclic core of the lobosamides is shared by salinilactam and micromonolactam, but the structures of the lobosamides differ from these compounds by the hydroxylation and methylation patterns, as well as the length and configuration of the polyene systems. While many macrolactams display antibacterial and anticancer activities, the lobosamides are the first reported compounds in this family with activity against T. brucei.
Relative configurations of lobosamides A-C
Configurational assignments for this class of molecules are challenging, due to the number of chiral centers and the instability of the polyene system. Most of the reported polyene macrolactam structures do not have the full absolute configurations assigned.24–27,29–32 These challenges also apply to the lobosamides, which are produced in low titer (~250 μg/L) and are sensitive to light, heat, and acidic conditions. However, using a combination of spectroscopic, synthetic and genomic approaches, we have deduced the complete absolute configurations for all chiral centers in these new metabolites.
Initially, lobosamide C (3) was reacted with 2,2-dimethoxypropane, resulting in acetonide formation exclusively between the hydroxyl groups at C-9 and C-11. Chemical shift analysis of the geminal methyl groups revealed that the newly formed 6-membered ring adopted a chair confirmation and these two hydroxyl groups were syn (Figure 3A).33 Key ROESY correlations from H-13 to the axial proton at H-11 and the equatorial proton H-12b revealed that these three atoms were on the same face of the molecule and that the hydroxyl group at C-13 was syn to the hydroxyls at C-9 and C-11 (Figure 3A). Strong ROESY correlations from the axial proton H-9 to both H-8 and Me-26 indicated these atoms adopted a gauche configuration (3JH-8, H-9 = 3.5 Hz). A key ROESY correlation between Me-26 and the equatorial proton H-10b indicated an anti relationship between the hydroxyl group at C-9 and Me-26 (Figure 3A). Furthermore, the orientations of both polyene chains were determined using ROESY analysis, highlighted by correlations between Me-26 and H-6, H-7 and H-8, and H-13 and H-16 (Figure 3A).
Figure 3.
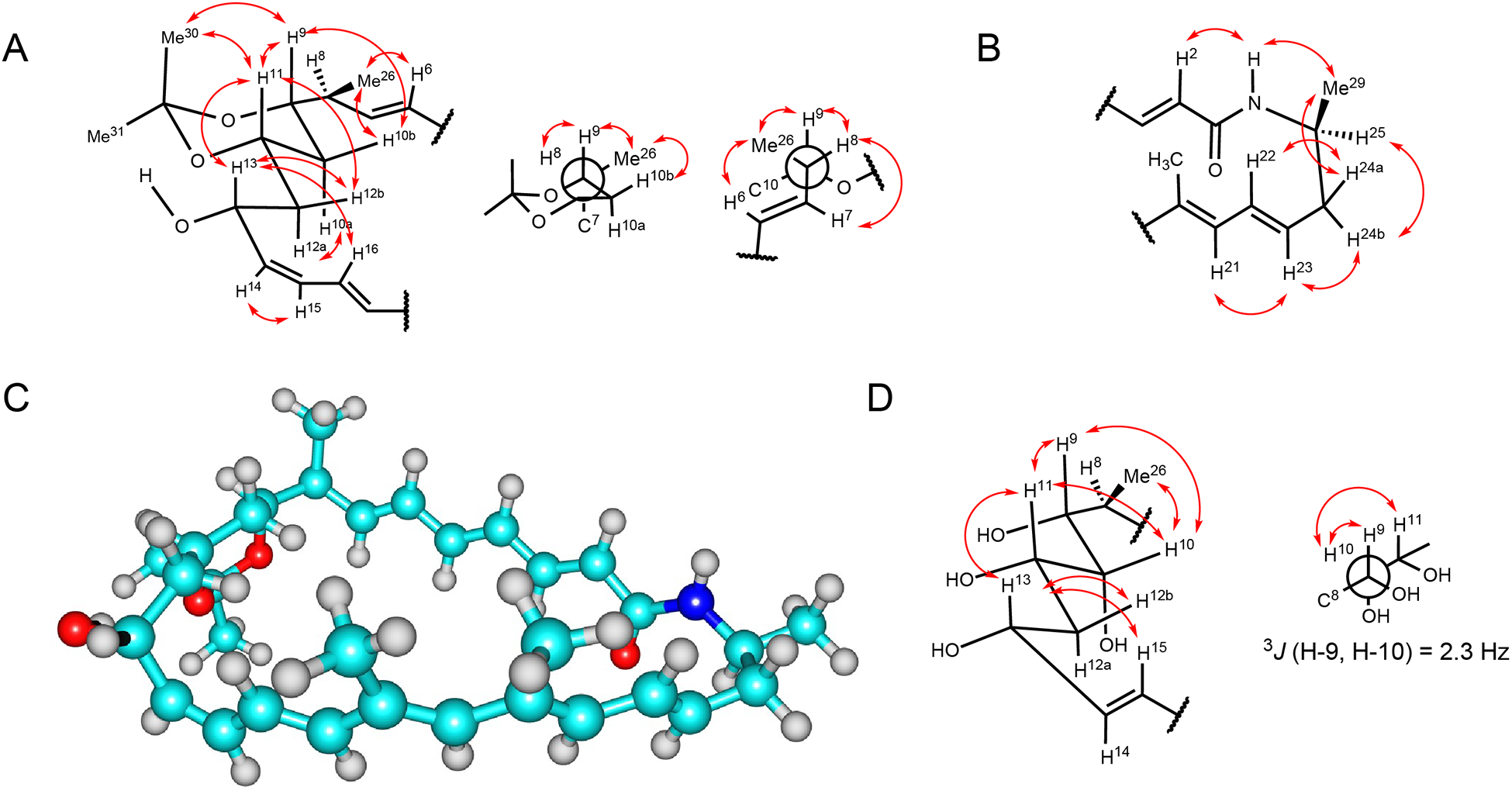
Key ROESY correlations used in determining the relative configuration of the lobosamides. (A) Analysis of lobosamide C acetonide allowed the assignment of the hydroxyl group at C-13, the methyl group at C-8, and the relative configuration of both polyene systems. (B) The relative orientation of the distal methyl group at C-25 was determined by key ROESY correlations to both polyene chains. (C) Three dimensional structure of lobosamide C acetonide determined by DFT molecular modeling. (D) Coupling constant and ROESY analysis of lobosamide B provided the relative configuration of the hydroxyl at C-10.
The rigidity of the two conjugated alkene systems and the determination of their relative orientations to the stereocenters in the western portion of the molecule allowed us to utilize the strong ROESY correlations between each alkene chain and the eastern portion of the molecule to also determine the relative configuration of the distal methyl group at C-25 (Figure 3B, for detailed explanation see Supplementary Information). The two alkene chains are arranged with the pi-systems facing one another across the ring, making it possible to identify the sets of alkene protons on the top and bottom faces of the molecule (Figure 3A). Because of the rigidity of these pi-systems, key ROESY correlations from the top face in the western portion can be extended through the alkene chain to the top face of the eastern portion of the molecule, where further ROESY correlations define the atoms on this top face (Figure 3B). Using complementary analyses for protons on the bottom face, it was therefore possible to deduce the relative configuration at C-25 based on ROESY correlations from H-25 and Me-29 to atoms on the bottom and top faces, respectively (Figure 3B).
To provide additional evidence for this assignment, minimized energy structures for both diastereomers at C-25 were modeled using DFT calculations, allowing conformational flexibility around this stereocenter by only imposing nOe constraints in the acetonide ring at the western portion of the molecule. Only the diastereomer predicted from our relative configurational assignment (Figure 3C) was consistent with the ROESY correlations observed in the eastern portion of the molecule, while the opposite configuration violated multiple observed correlations (Supplementary Information). Lobosamides A and B have very similar NMR correlations, but differ from lobosamide C by the inclusion of an additional hydroxyl group at C-10. ROESY correlations from H-10 to H-9, H-11, and Me-26 and coupling constant analysis (3JH-9, H-10 = 2.3 Hz) revealed that the hydroxyl at C-10 was on the same face as the hydroxyls at C-9 and C-11 (Figure 3D), thus completing the relative configurational assignments for all three natural products.
Identification and characterization of the lobosamide biosynthetic gene cluster
The lobosamides represent new members in the small family of 26-membered macrolactams, which includes salinilactam22 and micromonolactam.24 While structurally similar, we sought to determine how the chemical diversity in this family is encoded on a genetic level, as well as how their BGC organization compares to related macrocyclic lactams. We used Single Molecule Real-Time Sequencing technology (SMRT, Pacific Biosciences®) to obtain a complete genome sequence of Micromonospora sp. RL09–050-HVF-A (Supplementary Information). This technology proved valuable in overcoming the challenges of assembling BGCs from actinobacteria, particularly their high GC content (>70%) and repetitive nature (especially in modular polyketide synthase and nonribosomal peptide synthetase BGCs). We used AntiSmash to identify and annotate the polyketide synthase (PKS) clusters in this organism, which led to the localization of the putative cluster responsible for lobosamide biosynthesis (lob) (Genbank accession number: KT209587).34 Two lines of evidence provide support that lob is indeed the BGC responsible for lobosamides biosynthesis: a) lob is the only PKS BGC in the complete genome of Micromonospora sp. RL09–050-HVF-A that is in agreement with the structure of lobosamides, and b) lob is syntenic, shares high percent identity and conserved overall PKS domains architecture, with BGCs involved in the biosynthesis of closely related macrolactams (for salinilactam and micromonolactam BGCs, see Figure 5 and Table S7). Among the notable features in this BGC are a set of genes involved in the biosynthesis of the 3-aminobutyrate starter unit. The mechanism of this enzymatic sequence was recently characterized for incednine, and involves the formation of β-glutamate and subsequent stereospecific decarboxylation to form (S)-3-aminobutyrate (Figure 4B).35 This mechanism was proposed for micromonolactam and is likely also present in salinilactam, which contain methyl branches adjacent to the nitrogen atom in the macrocycle.22,24 Both LobP and LobO, which are proposed to catalyze these reactions (Figure 4B), share high amino acid sequence identity to the homologous gene products in the BGCs of salinilactam (94% and 89%, respectively) and incednine (70% and 71%, respectively). The cluster also contains genes responsible for a natural amino acid protecting group strategy to prevent premature macrocyclization, which was characterized in vicenistatin biosynthesis and hypothesized to be found in other macrolactams36 (Figure 4B).
Figure 5.
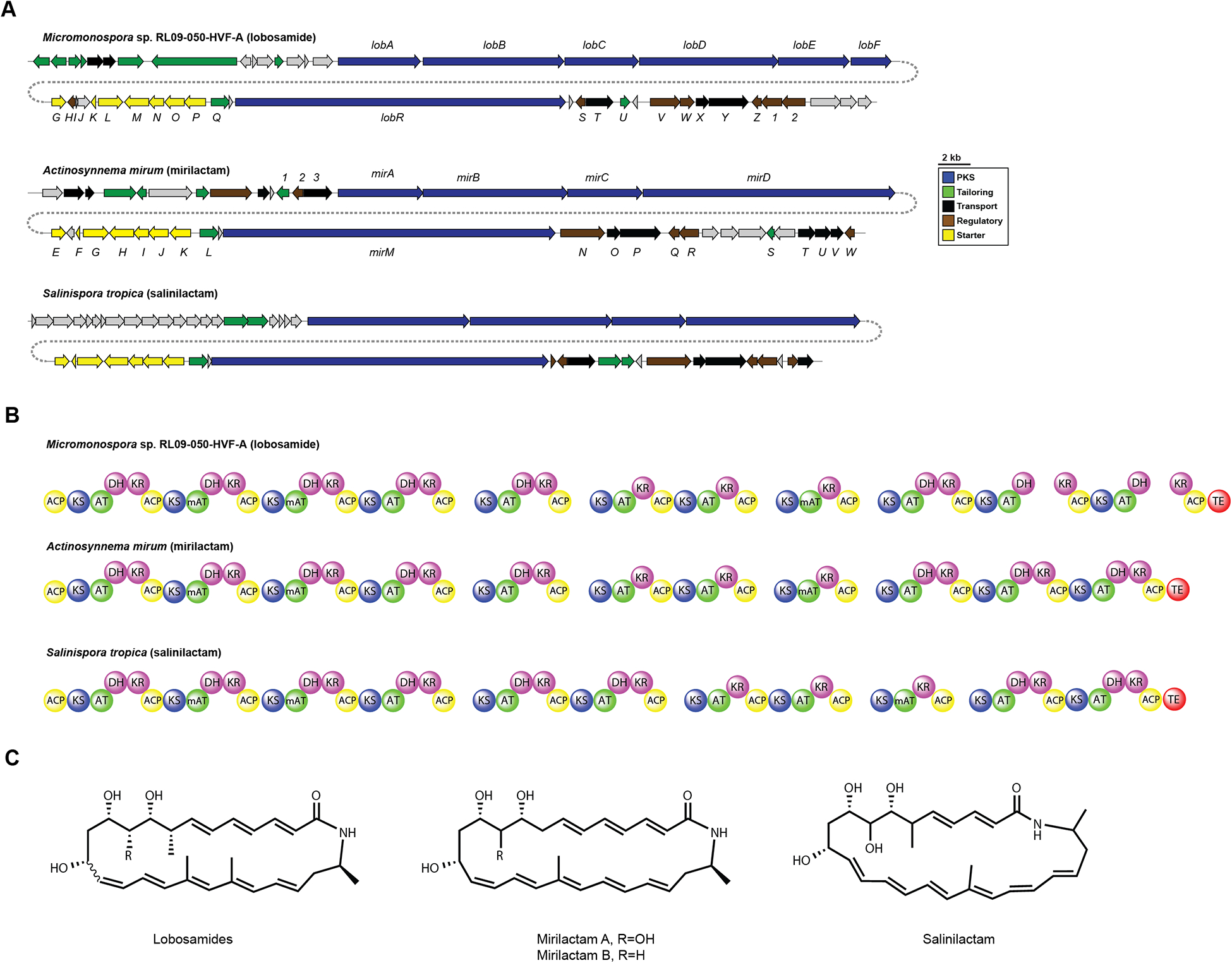
Comparative analysis of related polyene BGCs. (A) Comparison of the lobosamide, mirilactam, and salinilactam BGC organization. (B) Domain organization of the polyketide synthase modules. (C) Structures of the lobosamides, mirilactams, and salinilactam.
Figure 4.

Proposed biosynthesis of the lobosamides. (A) Organization of the lobosamide biosynthetic gene cluster (lob). (B) Biosynthesis of the 3-aminobutyrate starter unit from glutamate. (C) Module organization and proposed biosynthesis of the lobosamides.
Despite our assignment of the relative configurations of the lobosamides, multiple attempts to determine the absolute configuration of these molecules using chemical derivatization were unsuccessful due to isomerization, degradation, and scarcity of the natural product. However, genetic analyses of ketoreductase (KR) domains have shown that the absolute configurations of the resultant hydroxyl groups can be accurately predicted based on key conserved residues in the amino acid sequence.37–39 This technique has also been used to assign the configurations of a number of recently reported natural products, many of which have subsequently been verified by total synthesis or chemical derivatization.40–44 In the lobosamide gene cluster, all three KR domains were classified as A-type (Figure S2), which agreed with our analysis of the spectroscopic data that indicated the three hydroxyls were syn, and allowed us to assign their stereochemistry as 9R, 11S, 13R. Based on these data and the relative configurational assignments obtained from spectroscopic analyses and molecular modeling, we therefore assigned the full absolute configuration of lobosamides A and B as 8S, 9R, 10R, 11S, 13R, 25S. Lobosamide C shares the same arrangement of functional groups at each chiral center, but the change in priority due to the absence of the oxidation at C-10 results in an absolute configuration of 8S, 9S, 11S, 13R, 25S. Notably, these assignments also agree with bioinformatic predictions of the methyl group at C-8 and the recent report that the enzymes that catalyze starter unit biosynthesis exclusively produce (S)-3-aminobutyrate.38,45
Genome-guided discovery of the mirilactams
As part of our analysis of the lobosamide gene cluster, we sought to assess the diversity of this class of molecules by determining whether similar clusters existed in other bacteria. Using the lobosamide gene cluster as a search query, we identified a highly similar gene cluster in the genome of another actinomycete, Actinosynnema mirum (ATCC 29888).20 Although this strain is commercially available and the complete genome has been published, only three natural products have been reported from A. mirum: the nocardicidins,46 a mycosporine-like amino acid,47 and the novel siderophore mirubactin,48 from which the gene cluster was identified using bioinformatics. A comparative analysis of the gene clusters from the lobosamides, salinilactam, and A. mirum, revealed a highly conserved organization of genes and percent identity in the encoded proteins (Figure 5A and Table S7). A bioinformatic analysis of the polyketide modules in the A. mirum cluster revealed that the chemical scaffold of the biosynthetic product from this cluster should be structurally identical to the lobosamides (Figure 5B). We obtained the A. mirum strain and used UPLC-ESITOFMS to perform a metabolomic analysis of culture extracts. Despite the absence of the lobosamides from A. mirum extracts, we identified a metabolite with a UV profile indicative of a conjugated polyene moiety, which differed in mass from the lobosamides by approximately 28 amu. Purification of the target molecules from A. mirum extracts resulted in the discovery of two additional polyene macrolactams, designated mirilactams A and B (4 and 5, respectively, Figure 5C). The mirilactams differ from the lobosamides by the absence of the methyl groups at C-8 and C-20 (Figure 5C). The acyltransferase domains at these positions are predicted to load a methylmalonyl extender unit akin to the lobosamides, suggesting a relaxed substrate specificity in the mirilactam BGC. The discovery of the mirilactams exemplifies the utility of integrating genomics in natural products discovery but highlights the importance of careful chemical analysis of gene products from bioinformatic predictions.
Biological activities against T. b. brucei
With five related structures in hand, we assessed the activity of these molecules against T. brucei parasites to determine the specific structural elements required for activity. The biological activities of all five analogs are summarized in Table 1. Although all five compounds share the same 26-membered polyene macrocylic core, only lobosamides A and B showed significant activity (IC50 < 10 μM) against T. brucei. Lobosamide B exhibits attenuated activity compared to lobosamide A, suggesting that the isomerization of the double bond at C-14-C-15 changes the conformation of the molecule unfavorably. Lobosamide C, which lacks the hydroxyl group at C-10, was inactive at the highest concentration tested (10 μM), suggesting that oxidation at this position is important for the biological activity of these compounds. Neither mirilactam displayed antitrypanosomal activity, despite their high structural similarity to the lobosamides. While data from the lobosamides predicted that mirilactam B, which does not have the hydroxyl at C-10, would be inactive, the lack of activity exhibited by mirilactam A suggests that the methylation pattern of these molecules is also important. The mirilactams lack both the aliphatic methyl at C-8 and the vinyl methyl at C-20. These structural deviations from the lobosamides have abolished their antiparasitic activity, indicating that these positions play an important role. As part of our lead discovery program for global health targets, we assessed the cytotoxicity of our extracts in mammalian cells in order to uncover therapeutically relevant compounds. None of the macrolactams discovered in this work showed cytotoxicity to mammalian cells at the highest concentrations tested (Table 1), suggesting that the observed antitrypanosomal activity likely occurs via a parasite-specific mechanism.
Table 1.
Summary of the biological activities of the lobosamides and mirilactams.
| Compound | IC50 T. b. brucei | IC50 mammalian cells |
| Lobosamide A | 0.8 μM | > 66 μM (T98G cells) |
| Lobosamide B | 6.1 μM | > 66 μM (T98G cells) |
| Lobosamide C | > 10 μM | > 33 μM (HeLa cells) |
| Mirilactam A | > 10 μM | Not determined |
| Mirilactam B | > 10 μM | > 33 μM (HeLa cells) |
Polyene macrolides, such as amphotericin, are known antibiotics that exert their activity by interaction and pore formation in membranes.49 These interactions are generally driven by the sterol composition of membranes,50 which allows for selectivity amongst the many known macrolide structures. Recently, the polyene macrolactam heronamide C was shown to target membranes in yeast.51 Heronamide C contains a post-polyketide assembly hydroxyl group akin to the lobosamides, which displayed more potent membrane affinity and biological activity compared to 8-deoxyheronamide C, which lacks the post-assembly oxidation. This is similar to the differential activity observed between lobosamide A (IC50 = 0.8 μM) and lobosamide C (IC50 > 10 μM). Many of the reported polyene macrolactam structures contain post-assembly hydroxyl groups, raising the possibility that these oxidations are an evolutionarily favorable contribution to the biological activities of this class. While the instability of the polyene systems in the lobosamides likely prevents their continued development as lead compounds for HAT, the parasite-specific activity displayed by these compounds makes them intriguing candidates for target identification studies. Little is known about the mechanism of action of many HAT drugs, and the number of validated targets in T. brucei is small. Future studies on the mode of action of the lobosamides will prove valuable to find novel targets in the parasite for further development.
CONCLUSION
The lobosamides are new members of a small, but growing family of polyene macrolactam natural products. They represent the first members of this family with significant activity against T. b. brucei. Based on the successes of polyene macrolides as antibiotics, further investigation into the antitrypanosomal activity of macrolactams is warranted. The identification and assembly of the biosynthetic gene cluster of the lobosamides has presented a conserved organization of the biosynthetic gene clusters for 26-membered macrolactams and was instrumental in the assignment of the absolute configuration of lobosamides. The “molecules-to-genes-to-molecules’ approach used in the discovery of the mirilactams has further expanded the members of this class and highlights the utility of using a bioinformatic approach to expand the chemical diversity of lead compounds. Additionally, the mirilactams have provided valuable SAR information for the biological activity of the lobosamides. Further investigations into the mode of action and biological activity of this class against T. brucei may provide important lead compounds and therapeutic targets for further development against HAT.
EXPERIMENTAL
General experimental procedures.
Solvents used for HPLC and LCMS chromatography were HPLC grade and were used without further purification. NMR spectroscopy and high-resolution mass spectrometry data were obtained for all compounds. NMR spectra were acquired on a 600 MHz spectrometer equipped with a 5 mm HCN triple resonance cryoprobe and referenced to residual solvent proton and carbon signals. HRMS data were acquired using University of California, Berkeley QB3/Chemistry Mass Spectrometry Facility’s multimode electrospray ionization (ESI) Fourier transfer mass spectrometer (FTMS) or an electrospray ionization (ESI) time-of-flight (TOF) mass spectrometer.
Isolation, Cultivation, and Extraction of Micromonospora sp. RL09–050-HVF-A
Micromonospora sp. RL09–050-HVF-A was isolated from a marine sediment sample collected from Point Lobos in the Monterey Bay, CA (Carmel, CA) at a depth of 15 m. This collection was made under permit number MBNMS-2009–022 from the Monterey Bay National Marine Sanctuary. The strain was isolated on HVF medium (10.0 g starch, 1.7 g KCl, 0.01 g FeSO4·7H2O, 0.5 mg thiamine, 0.5 mg riboflavin, 0.5 mg nicotonic acid, 0.5 mg pyridoxine HCl, 0.5 mg p-aminobenzoic acid, 0.5 mg myoinositol, 0.25 mg biotin, 3.0 g KNO3, 20.0 mg CaCO3, 0.5 g MgSO4·7H2O and 0.5 g Na2HPO4, 18 g agar, 1 L Milli-Q water) supplemented with 50 mg/L cycloheximide and 50 mg/L nalidixic acid. Further isolation was performed on MB medium (37.4 g Difco™ Marine Broth, 18 g agar, 1 L Milli-Q water). The pure culture was cultivated in 2.8 L Fernbach flasks containing 500 mL fermentation medium GNZ (10.0 g glucose, 5.0 g NZ-amine, 1.0 g CaCO3, 20.0 g starch, 5.0 g yeast extract), a stainless steel spring, and 20.0 g Amberlite XAD-16 adsorbent resin. The culture was shaken at 200 rpm for 4 days. At the end of the fermentation period, the cells and resin were removed by vacuum filtration using Whatman glass microfiber filters and washed with deionized water. The resin and cells from each culture flask were extracted with 250 mL of 1:1 methanol/dichloromethane. The organic extract was removed from the cells and resin by vacuum filtration and concentrated in vacuo. The crude organic extract, given extract code RLUS1353, was subjected to solid phase extraction using a Supelco-Discovery C18 cartridge (5 g) and eluted using a MeOH/H2O step gradient (40 mL; 10% MeOH, 20% MeOH, 40% MeOH, 60% MeOH, 80% MeOH, 100% MeOH, 100% EtOAc) to afford seven fractions. The 10% MeOH fraction was discarded and the remaining six (fractions A – F) concentrated to dryness in vacuo. Analysis by LCMS revealed that lobosamides A and B were present in the D (80% MeOH) fraction, while lobosamide C was present in the E (100% MeOH) fraction. Mirilactam A was present in the D fraction, while mirilactam B was present in the E (100% MeOH) fraction from the A. mirum extract.
Purification of the lobosamides and mirilactams
Lobosamides A and B were purified from prefraction RLUS1353D by RP-HPLC (Agilent Eclipse XDB-C18 150 × 4.6 mm, 5 μm) using an isocratic separation (50% MeOH, 50% H2O + 0.02% formic acid, 1 mL min−1, lobosamide A tR = 31.7 min., ~250 μg/L lobosamide B tR = 45.9 min., ~250 μg/L). Lobosamide C was purified from prefraction RLUS1353E by RP-HPLC (Phenomenex Synergi Fusion-RP, 250 × 4.6 mm, 10 μm) using an isocratic separation (38% MeCN, 62% H2O + 0.02% formic acid, 2 mL min−1, tR = 14.6 min., ~300 μg/L).
Mirilactam A was purified from the D fraction of the A. mirum extract by RP-HPLC (Phenomenex Synergi Fusion-RP, 250 × 4.6 mm, 10 μm) by a gradient separation (45% MeOH, 55% H2O + 0.02% formic acid to 57% MeOH, 43% H2O + 0.02% formic acid over 17 min, 2 mL min−1, tR = 14.1 min.). Mirilactam B was purified from the E fraction of the A. mirum extract by RP-HPLC (Phenomenex Synergi Fusion-RP, 250 × 4.6 mm, 10 μm) by a gradient separation (45% MeOH, 55% H2O + 0.02% formic acid to 57% MeOH, 43% H2O + 0.02% formic acid over 17 min, 2 mL min−1, tR = 16.5 min.).
Chemical characterization of compounds
Lobosamide A (1): [α]D25= +67.5 (c 0.08, MeOH); UV (MeOH) λmax (log ε) = 245 (4.02), 253 (4.07), 295 (4.23) nm; See supporting information for NMR tables; IR (MeOH, cm−1): 3377.9, 2946.0, 2833.8, 2524.9, 2228.2, 2044.4, 1646.9, 1604.7, 14450.6, 1405.5, 1115.4, 1028.6; (+)-FTICRMS m/z 506.2882 [M+Na]+ (calcd. for C29H41O5NNa, 506.2877).
Lobosamide B (2): [α]D25= −18.1 (c 0.34, MeOH); UV (MeOH) λmax (log ε) = 241 (3.80), 249 (3.82), 294 (4.08) nm; See supporting information for NMR tables; IR (MeOH, cm−1): 3367.0, 2945.2, 2833.0, 2595.4, 2523.1, 2228.2, 2045.1, 1647.4, 1595.0, 1449.6, 1419.6, 1115.9, 1030.5; (+)-FTICRMS m/z 506.2884 [M+Na]+ (calcd. for C29H41O5NNa, 506.2877).
Lobosamide C (3): [α]D25= −203.9 (c 0.09, C5H5N); UV (MeOH) λmax (log ε) = 231 (3.75), 282 (3.76) nm; See supporting information for NMR tables; IR (MeOH, cm−1): 3350.2, 1945.2, 2832.8, 2595.8, 2523.3, 2229.1, 2045.2, 1646.9, 1595.5, 1450.4, 1417.3, 1115.5, 1031.6. (+) HRESITOFMS m/z 468.3112 [M+H]+ (calcd. for C29H42O4N, 468.3108).
Mirilactam A (4): See supporting information for NMR tables; (+) HRESITOFMS m/z 456.2746 [M+H]+ (calcd. for C29H42O4N, 456.2744).
Mirilactam B (5): [α]D25= −76.7 (c 0.15, MeOH) UV (MeOH) λmax (log ε) = 242 (3.62), 289 (3.81), 338 (3.32) sh, 356 (3.22) sh nm; See supporting information for NMR tables; IR (MeOH, cm−1): 3350.3, 2945.1, 2832.8, 2595.7, 2523.2, 2230.5, 2045.4, 1645.3, 1600.1, 1450.3, 1417.4, 1115.7, 1030.6. (+) HRESITOFMS m/z 440.2794 [M+H]+ (calcd. for C27H38O4N, 440.2795).
Synthesis of lobosamide C acetonide
Lobosamide C (2.6 mg) was dissolved in 2,2-dimethoxypropane (2 mL) and anhydrous MeOH (1 mL), and 3 mg pyridinium paratoluenesulfonate was added. The reaction was stirred for 18 h at room temperature under argon and then quenched with sat. NaHCO3. The aqueous phase was extracted 3 × with EtOAc and 2 × with CH2Cl2. The organic phase was dried under inert gas and purified by RP-HPLC (Agilent Eclipse XDB-C18 150 × 4.6 mm, 5 μm) using an isocratic separation (47% ACN 53% H2O + 0.02% formic acid over 17 min, 1.3 mL min−1, tR = 12.2 min.) to afford 770 μg of lobosamide C acetonide. (+)-HRESITOFMS m/z 508.3419 [M+H]+ (calcd. for C32H46NO4, 508.3421); 1H NMR (DMSO-d6, 600 MHz): δ 0.83 (1H, m, H-10a), 1.05 (3H, d, J = 7.0 Hz, H-26), 1.10 (3H, d, J = 6.5 Hz, H-29), 1.12 (1H, m, H-12a), 1.28 (3H, s, acetonide equatorial, H-31), 1.41 (3H, s, acetonide axial, H-30), 1.64 (3H, s, H-27), 1.71 (1H, m, H-12b)a, 1.72 (1H, m, H-10b)a, 1.80 (3H, s, H-28), 1.92 (1H, ddd, J = 3.0, 10.0, 10.0 Hz, H-24b), 2.42 (1H, ddd, J = 4.0, 4.0, 12.5 Hz, H-24a), 2.51 (1H, m, H-8), 3.90 (1H, ddd, J = 3.0, 3.5, 11.5 Hz, H-9), 4.03 (1H, m, H-25), 4.06 (1H, dddd, J = 2.5, 2.5, 10.5, 13.5 Hz, H-11), 4.50 (1H, m, H-13), 5.43 (1H, dd, J = 8.0, 10.5, H-14), 5.47 (1H, ddd, J = 5.0, 9.5, 15.0, H-23), 5.75 (1H, d, J = 11.0, H-21), 5.90 (1H, m, H-15)a, 5.91 (1H, d, J = 15.0, H-2), 6.01 (1H, s, H-19), 6.11 (1H, dd, J = 15.5, 3.5, H-7), 6.16 (1H, m, H-6)a, 6.19 (1H, m, H-16)a, 6.23 (1H, m, H-17)a, 6.24 (1H, m, H-22)a, 6.30 (1H, dd, J =14.5, 11.5, H-4), 6.58 (1H, dd, J = 14.5, 10.0, H-5), 7.50 (1H, d, J = 10.0, NH) aunresolved multiplicities due to signal overlap. 13C NMR (DMSO-d6, 600MHz)b: δ 13.4 (C-27), 14.4 (C- 26), 17.5 (C-28), 19.5 (C-30, acetonide axial), 21.2 (C-29), 29.6 (C-31, acetonide equatorial), 29.7 (C-10), 38.6 (C-8), 40.5 (C-24), 43.7 (C-25), 44.7 (C-12), 62.8 (C-13), 65.6 (C-11), 71.3 (C-9), 97.7 (ketal, acetonide), 124.2 (C-2), 126.3 (C-15), 128.3 (C-4), 128.6 (C-6), 129.0 (C-16), 129.3 (C-22), 130.0 (C-21), 130.8 (C-23), 132.0 (C-20), 133.7 (C-18), 136.3 (C-19), 136.4 (C-14), 138.3 (C-17), 139.1 (C-3), 139.2 (C-5), 139.7 (C-7), 165.2 (C-1), bchemical shifts determined from HSQC and HMBC NMR data.
Supplementary Material
ACKNOWLEDGMENT
We thank W.-R.Wong and K. Kurita for 16S analysis and DNA extraction, and R. S. Lokey and W. Bray for assistance with HeLa cell cytotoxicity measurements.
Funding Sources
This work was supported by the Sandler Center for Parasitic Disease Research (RGL and JHM), NIH DP2 OD007290 (MAF) and the UCSC Committee on Research Special Research Grant program (REG and RGL).
Footnotes
Supporting Information
NMR chemical shift tables and NMR spectra for compounds 1 – 5, list of genes for lob biosynthetic gene cluster, alignment of KR sequences for lob cluster, Cartesian coordinates for molecular modeling, graphical representations of minimum energy conformers for calculated lobosamide C stereoisomers. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- (1).Moran M, Guzman J, Ropars A-L, McDonald A, Jameson N, Omune B, Ryan S, and Wu L (2009) Neglected disease research and development: how much are we really spending? PLoS Med. 6, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).WHO. (2013) Human African Trypanosomiasis. World Heal. Organ [Google Scholar]
- (3).Stich A, Ponte-Sucre A, and Holzgrabe U (2013) Do we need new drugs against human African trypanosomiasis? Lancet Infect. Dis 13, 733–734. [DOI] [PubMed] [Google Scholar]
- (4).Simarro PP, Diarra A, Ruiz Postigo JA, Franco JR, and Jannin JG (2011) The human African trypanosomiasis control and surveillance programme of the World Health Organization 2000–2009: the way forward. PLoS Negl. Trop. Dis 5, e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Simarro PP, Franco J, Diarra A, Ruiz Postigo JA, and Jannin J (2012) Update on field use of the available drugs for the chemotherapy of human African trypanosomiasis. Parasitology 139, 842–846. [DOI] [PubMed] [Google Scholar]
- (6).Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, Jenks MX, Noe RA, Bowling TS, Mercer LT, Rewerts C, Gaukel E, Owens J, Parham R, Randolph R, Beaudet B, Bacchi CJ, Yarlett N, Plattner JJ, Freund Y, Ding C, Akama T, Zhang YK, Brun R, Kaiser M, Scandale I, and Don R (2011) Scyx-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis 5, e1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kaiser M, Bray MA, Cal M, Bourdin Trunz B, Torreele E, and Brun R (2011) Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother 55, 5602–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jones AJ, Grkovic T, Sykes ML, and Avery VM (2013) Trypanocidal activity of marine natural products. Mar. Drugs 11, 4058–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chatelain E, and Ioset JR (2011) Drug discovery and development for neglected diseases: The DNDi model. Drug Des. Devel. Ther 5, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gerwick WH, and Moore BS (2012) Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol 19, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B, Chandra J, Chauhan D, Cusack JC, Fenical W, Ghobrial IM, Groll M, Jensen PR, Lam KS, Lloyd GK, McBride W, McConkey DJ, Miller CP, Neuteboom ST, Oki Y, Ovaa H, Pajonk F, Richardson PG, Roccaro AM, Sloss CM, Spear MA, Valashi E, Younes A, and Palladino MA (2011) Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 11, 254–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Feng Y, Davis RA, Sykes M, Avery VM, Camp D, and Quinn RJ (2010) Antitrypanosomal cyclic polyketide peroxides from the Australian marine sponge Plakortis sp. J. Nat. Prod 73, 716–719. [DOI] [PubMed] [Google Scholar]
- (13).Rodenko B, Al-Salabi MI, Teka IA, Ho W, El-Sabbagh N, Ali JAM, Ibrahim HMS, Wanner MJ, Koomen GJ, and de Koning HP (2011) Synthesis of marine-derived 3-alkylpyridinium alkaloids with potent antiprotozoal activity. ACS Med. Chem. Lett 2, 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wright AD, Goclik E, König GM, and Kaminsky R (2002) Lepadins D-F: Antiplasmodial and antitrypanosomal decahydroquinoline derivatives from the tropical marine tunicate Didemnum sp. J. Med. Chem 45, 3067–3072. [DOI] [PubMed] [Google Scholar]
- (15).Watts KR, Ratnam J, Ang KH, Tenney K, Compton JE, McKerrow J, and Crews P (2010) Assessing the trypanocidal potential of natural and semi-synthetic diketopiperazines from two deep water marine-derived fungi. Bioorganic Med. Chem 18, 2566–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sanchez LM, Lopez D, Vesely BA, Della Togna G, Gerwick WH, Kyle DE, and Linington RG (2010) Almiramides A-C: Discovery and development of a new class of leishmaniasis lead compounds. J. Med. Chem 53, 4187–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Walsh CT, and Fischbach MA (2010) Natural products version 2.0: Connecting genes to molecules. J. Am. Chem. Soc 132, 2469–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Winter JM, Behnken S, and Hertweck C (2011) Genomics-inspired discovery of natural products. Curr. Opin. Chem. Biol 15, 22–31. [DOI] [PubMed] [Google Scholar]
- (19).Zerikly M, and Challis GL (2009) Strategies for the discovery of new natural products by genome mining. ChemBioChem 10, 625–633. [DOI] [PubMed] [Google Scholar]
- (20).Land M, Lapidus A, Mayilraj S, Chen F, Copeland A, Del Rio TG, Nolan M, Lucas S, Tice H, Cheng J-F, Chertkov O, Bruce D, Goodwin L, Pitluck S, Rohde M, Göker M, Pati A, Ivanova N, Mavromatis K, Chen A, Palaniappan K, Hauser L, Chang Y-J, Jeffries CC, Brettin T, Detter JC, Han C, Chain P, Tindall BJ, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, and Klenk H-P (2009) Complete genome sequence of Actinosynnema mirum type strain (101). Stand. Genomic Sci 1, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schulze CJ, Bray WM, Woerhmann MH, Stuart J, Lokey RS, and Linington RG (2013) “Function-first” lead discovery: mode of action profiling of natural product libraries using image-based screening. Chem. Biol 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, and Moore BS (2007) Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc. Natl. Acad. Sci. U. S. A 104, 10376–10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Futamura Y, Sawa R, Umezawa Y, Igarashi M, Nakamura H, Hasegawa K, Yamasaki M, Tashiro E, Takahashi Y, Akamatsu Y, and Imoto M (2008) Discovery of incednine as a potent modulator of the anti-apoptotic function of Bcl-xL from microbial origin. J. Am. Chem. Soc 130, 1822–1823. [DOI] [PubMed] [Google Scholar]
- (24).Skellam EJ, Stewart AK, Strangman WK, and Wright JLC (2013) Identification of micromonolactam, a new polyene macrocyclic lactam from two marine Micromonospora strains using chemical and molecular methods: clarification of the biosynthetic pathway from a glutamate starter unit. J. Antibiot 66, 431–441. [DOI] [PubMed] [Google Scholar]
- (25).Kojiri K, Nakajima S, Suzuki H, Kondo H, and Suda H (1992) A new macrocyclic lactam antibiotic, BE-14106. I. Taxonomy, isolation, biological activity and structural elucidation. J. Antibiot 45, 868–874. [DOI] [PubMed] [Google Scholar]
- (26).Oh DC, Poulsen M, Currie CR, and Clardy J (2011) Sceliphrolactam, a polyene macrocyclic lactam from a wasp-associated Streptomyces sp. Org. Lett 13, 752–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Thawai C, Kittakoop P, Tanasupawat S, Suwanborirux K, Sriklung K, and Thebtaranonth Y (2004) Micromonosporin A, a novel 24-membered polyene lactam macrolide from Micromonospora sp. isolated from peat swamp forest. Chem. Biodivers 1, 640–645. [DOI] [PubMed] [Google Scholar]
- (28).Shindo K, Kamishohara M, Odagawa A, Matsuoka M, and Kawai H (1993) Vicenistatin, a novel 20-membered macrocyclic lactam antitumor antibiotic. J. Antibiot 46, 1076–1081. [DOI] [PubMed] [Google Scholar]
- (29).Jørgensen H, Degnes KF, Dikiy A, Fjaervik E, Klinkenberg G, and Zotchev SB (2010) Insights into the evolution of macrolactam biosynthesis through cloning and comparative analysis of the biosynthetic gene cluster for a novel macrocyclic lactam, ML-449. Appl. Environ. Microbiol 76, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Schulz D, Nachtigall J, Geisen U, Kalthoff H, Imhoff JF, Fiedler H-P, and Süssmuth RD (2012) Silvalactam, a 24-membered macrolactam antibiotic produced by Streptomyces sp. Tü 6392*. J. Antibiot 65, 369–372. [DOI] [PubMed] [Google Scholar]
- (31).Schulz D, Nachtigall J, Riedlinger J, Schneider K, Poralla K, Imhoff JF, Beil W, Nicholson G, Fiedler H-P, and Süssmuth RD (2009) Piceamycin and its N-acetylcysteine adduct is produced by Streptomyces sp. GB 4–2. J. Antibiot 62, 513–518. [DOI] [PubMed] [Google Scholar]
- (32).Mitchell SS, Nicholson B, Teisan S, Lam KS, and Potts BCM (2004) Aureoverticillactam, a novel 22-atom macrocyclic lactam from the marine actinomycete Streptomyces aureoverticillatus. J. Nat. Prod 67, 1400–1402. [DOI] [PubMed] [Google Scholar]
- (33).Rychnovsky SD, Rogers BN, and Richardson TI (1998) Configurational assignment of polyene macrolide antibiotics using the [13C]acetonide analysis. Acc. Chem. Res 31, 9–17. [Google Scholar]
- (34).Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, and Weber T (2013) antiSMASH 2.0--a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, W204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Takaishi M, Kudo F, and Eguchi T (2012) A unique pathway for the 3-aminobutyrate starter unit from L-glutamate through β-glutamate during biosynthesis of the 24-membered macrolactam antibiotic, incednine. Org. Lett 14, 4591–4593. [DOI] [PubMed] [Google Scholar]
- (36).Shinohara Y, Kudo F, and Eguchi T (2011) A natural protecting group strategy to carry an amino acid starter unit in the biosynthesis of macrolactam polyketide antibiotics. J. Am. Chem. Soc 133, 18134–18137. [DOI] [PubMed] [Google Scholar]
- (37).Caffrey P (2003) Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. ChemBioChem 4, 654–657. [DOI] [PubMed] [Google Scholar]
- (38).Keatinge-Clay AT (2007) A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem. Biol 14, 898–908. [DOI] [PubMed] [Google Scholar]
- (39).Siskos AP, Baerga-Ortiz A, Bali S, Stein V, Mamdani H, Spiteller D, Popovic B, Spencer JB, Staunton J, Weissman KJ, and Leadlay PF (2005) Molecular basis of Celmer’s rules: Stereochemistry of catalysis by isolated ketoreductase domains from modular polyketide synthases. Chem. Biol 12, 1145–1153. [DOI] [PubMed] [Google Scholar]
- (40).Essig S, Bretzke S, Müller R, and Menche D (2012) Full stereochemical determination of ajudazols A and B by bioinformatics gene cluster analysis and total synthesis of Aaudazol B by an asymmetric ortholithiation strategy. J. Am. Chem. Soc 134, 19362–19365. [DOI] [PubMed] [Google Scholar]
- (41).Ishida K, Lincke T, and Hertweck C (2012) Assembly and Absolute Configuration of Short-Lived Polyketides from Burkholderia thailandensis. Angew. Chem. Int. Ed 51, 5470–5474. [DOI] [PubMed] [Google Scholar]
- (42).Jahns C, Hoffmann T, Müller S, Gerth K, Washausen P, Höfle G, Reichenbach H, Kalesse M, and Müller R (2012) Pellasoren: structure elucidation, biosynthesis, and total synthesis of a cytotoxic secondary metabolite from Sorangium cellulosum. Angew. Chem. Int. Ed 51, 5239–5243. [DOI] [PubMed] [Google Scholar]
- (43).Janssen D, Albert D, Jansen R, Müller R, and Kalesse M (2007) Chivosazole A—alucidation of the absolute and relative configuration. Angew. Chem. Int. Ed 46, 4898–4901. [DOI] [PubMed] [Google Scholar]
- (44).Laureti L, Song L, Huang S, Corre C, Leblond P, Challis GL, and Aigle B (2011) Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc. Natl. Acad. Sci. U. S. A 108, 6258–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Takaishi M, Kudo F, and Eguchi T (2013) Identification of the incednine biosynthetic gene cluster: characterization of novel β-glutamate-β-decarboxylase IdnL3. J. Antibiot 66, 691–699. [DOI] [PubMed] [Google Scholar]
- (46).Watanabe K, Okuda T, Yokose K, Furumai T, and Maruyama HB (1983) Actinosynnema mirum, a new producer of nocardicin antibiotics. J. Antibiot 36, 321–324. [DOI] [PubMed] [Google Scholar]
- (47).Miyamoto KT, Komatsu M, and Ikeda H (2014) Discovery of gene cluster for mycosporine-like amino acid biosynthesis from Actinomycetales microorganisms and production of a novel mycosporine-like amino acid by heterologous expression. Appl. Environ. Microbiol 80, 5028–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Giessen TW, Franke KB, Knappe TA, Kraas FI, Bosello M, Xie X, Linne U, and Marahiel MA (2012) Isolation, structure elucidation, and biosynthesis of an unusual hydroxamic acid ester-containing siderophore from Actinosynnema mirum. J. Nat. Prod 75, 905–914. [DOI] [PubMed] [Google Scholar]
- (49).Bolard J, Legrand P, Heitz F, and Cybulska B (1991) One-sided action of amphotericin B on cholesterol-containing membranes is determined by its self-association in the medium. Biochemistry 30, 5707–5715. [DOI] [PubMed] [Google Scholar]
- (50).Koukalová A, Pokorná Š, Fišer R, Kopecký V, Humpolíčková J, Černý J, and Hof M (2015) Membrane activity of the pentaene macrolide didehydroroflamycoin in model lipid bilayers. Biochim. Biophys. Acta 1848, 444–452. [DOI] [PubMed] [Google Scholar]
- (51).Sugiyama R, Nishimura S, Matsumori N, Tsunematsu Y, Hattori A, and Kakeya H (2014) Structure and biological activity of 8-deoxyheronamide C from a marine-derived Streptomyces sp.: Heronamides target saturated hydrocarbon chains in lipid membranes. J. Am. Chem. Soc 136, 5209–5212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.