

Abstract
Phenotype-guided natural products discovery is emerging as a useful new discovery tool that addresses challenges in early, unbiased natural product biological annotation. These high-content approaches yield screening results that report directly on the impact of test compounds on cellular processes in target organisms and can be used to predict the modes of action (MOAs) of bioactive constituents from primary screening data. In this study we explored the use of our recently implemented cytological profiling (CP) platform for the isolation of compounds with a specific, predefined mode of action, namely induction of mitotic arrest. Screening of a microbially-derived extract library revealed six extracts whose cytological profiles clustered closely with those of known antimitotic agents from the pure compound training set. Subsequent examination of one of these extracts revealed the presence of two separate bioactive constituents, each of which possessed a unique cytological profile. The first, diketopiperazine XR334 (3), recapitulated the observed antimitotic phenotype of the original extract, demonstrating that cytological profiling can be used for the targeted isolation of compounds with specific modes of action. The second, nocapyrone L (6), possessed a cytological profile that clustered with known calcium channel modulators, in line with previous published activities for this compound class, indicating that cytological profiling is a flexible and powerful platform for the de novo characterization of compound modes of action.
Although natural products have proven value in the area of cancer chemotherapeutic development, the process from the original collection to the biological annotation of natural products is labor intensive and time consuming; for example, the microtubule stabilizing activity of the natural product dictyostatin 1 was not established until 10 years after it was originally isolated.1 In addition, collection of the plant material containing paclitaxel occurred in the 1960s but its microtubule stabilizing activity was not published until 1979.2 Traditionally, targeted anticancer discovery for natural products is performed by screening crude extracts in high-throughput screens using reporter assays such as enzyme-linked immunosorbent assays (ELISAs) or reporter gene assays (RGAs). While these assays are high-throughput, they typically only report on a single molecular target or pathway, meaning that discovery is limited to a specific set of biological targets.3 These population measurements miss variations in individual cell dynamics and morphology and hits in these screens are therefore unable to account for off target affects, and are prone to false positives when broadly cytotoxic compounds are encountered. For example, screening campaigns for a selected target such as a specific enzyme can yield hits that are indistinguishable from compounds that are also active against a wider set of targets and whose development potential is therefore typically low.
Tremendous progress has been made in recent years to develop high content imaging systems capable of quantifying multiparametric cellular responses of drug treated cells.4,5 These data can be viewed as phenotypic “fingerprints” which have been successfully used to differentiate classes of drugs with discrete MOAs.6 This screening approach takes a global view of biological attributes of bioactive compounds by determining the overall effect of small molecules on cell morphology, rather than examining specific molecular targets or pathways as is common in target-based screening. While high content screening (HCS) is an extremely powerful tool, it is predominately used for pure compound analysis; therefore the time between sample collections and MOA annotation still exists as a bottleneck in drug discovery.7
Development of untargeted HCS of natural product extracts is providing a mechanism to circumvent this bottleneck by obtaining detailed mechanistic information for large natural product libraries in a high-throughput fashion. Elimination of lengthy purification and work up protocols for non-bioactive or broadly cytotoxic compounds rapidly prioritizes compounds with desired bioactivities. Since the emergence of automated image analysis, the development of algorithms that process and categorize images to predict MOAs has provided new opportunities for the analysis of compounds without published MOAs. In our effort to expedite hit-to-lead development from natural product libraries we have recently reported the development of a high content cytological profiling platform that uses image-based screening to directly visualize the phenotypic effects of natural product extracts on HeLa cell development.8,9 These reports analyzed the major compound clusters found from screening a pilot library of 312 extracts in order to validate this screening tool and identified both known and novel natural products. In this study we applied this technology to our entire 5304-member extract library and used these data to test the hypothesis that CP could be used to discover compounds with specific and predefined modes of action.
A number of approaches have been employed to derive multiparametric annotation for natural product extracts. Predominately applied to pure compound libraries, untargeted profiling utilizes bioactivity profile matching where compounds with similar phenotypic responses are inferred to have similar mechanisms of action. Among these, gene expression profiling and yeast gene deletion libraries have been used successfully to annotate natural product MOAs in an untargeted manner.10,11 Zebrafish imaging has also been utilized to characterize whole animal response of natural products and is one of the few whole organism approaches to untargeted natural products screening.12 A number of computational tools exist to aid in the interpretation of image-based screening data; however, to date there have been few examples of the application of these methods to unbiased natural products discovery.
To test the hypothesis that HCS screening can be used to discover bioactive compounds with pre-specified modes of action we explored our library for extracts displaying an antimitotic phenotype, and have demonstrated that this bioactivity profiling approach can be used to map a single specific constituent within this extract to this predefined biological activity. This proof of concept provides evidence that untargeted image-based screening can be used effectively for the discovery of compounds with specific mechanisms of action and opens the door for the use of this technology to discover compounds that target other high priority pathways and molecular targets from crude natural products in a high-throughput manner.
Induction of mitotic arrest is a validated anticancer mechanism, with tubulin being one of the most studied cancer targets, and the target of a number of chemotherapeutic agents.13 However, many mitosis-targeting treatments currently struggle with incomplete neoplasm eradication, resistance, and toxicity issues.14 Current antimitotic drug discovery has focused on analyzing specific aspects of mitosis, often accomplished by screening compounds in tubulin binding assays or in whole cell assays where the amount of mitotic arrest with respect to vehicle treated cells is analyzed. As an example, a high throughput ELISA assay has been used to discover new paclitaxel analogues with similar activity to the parent compound.15 In addition, biphenabulin, an antimitotic agent with nanomolar activity, was recently discovered utilizing whole cell imaging of cells stained with phosphorylated histone H3 (pHH3), a mitotic marker, to screen libraries of synthetic compounds created by diversity orientated synthesis of macrocycles with natural-product-like geometry.16 Our new study focused on determining whether the unbiased CP screen could be utilized to identify compounds that affect tubulin dynamics from complex natural product extracts, while still providing detailed yet untargeted biological annotations for all bioactive constituents.
RESULTS AND DISCUSSION
In the original report describing the development of cytological profiling for natural products discovery we evaluated a pilot library of 312 extracts and characterized the bioactive constituents from a subset of these bioactive fractions.8 We have now extended this analysis to our full natural products library, consisting of 5304 prefractionated extracts from marine-derived Actinobacteria. Cytological profiling of this entire library, followed by hierarchical clustering with the training set of 480 compounds from the Harvard Institute of Chemistry and Cell Biology collection (ICCB) possessing known MOAs revealed 41 discrete clusters, including 27 clusters which contained both bioactive extracts and reference compounds from the training set. Among these there are a number of expanded versions of clusters originally identified and annotated in our initial cytological profiling paper, including DNA synthesis inhibitors, vacuolar ATPase inhibitors, potassium channel inhibitors, and kinase inhibitors. In addition, this expanded cytological profiling set contains multiple clusters not observed in the original training set, many of which contain training set compounds with related MOAs. Finally, there are numerous smaller clusters that do not cluster well with compounds from the training set, suggesting that these extracts either contain compounds with MOAs not represented by members of the training set, or that these extracts contain multiple bioactive constituents that cause “mixed-mode” phenotypes that are not representative of any of the individual components, an inherent limitation of cytological profiling with complex mixtures.
Among these active clusters, one small subset clustered tightly with the known microtubule poisons nocodazole and paclitaxel, suggesting that these extracts contain compounds that disrupted tubulin dynamics. In general, CP fingerprints of known microtubule stabilizers and destabilizers cluster closely together but can be differentiated by comparing tubulin staining from DMSO controls. Tubulin stabilizers such as paclitaxel show a positive deviation while destabilizers such as colchicine and nocodazole exhibit a negative deviation in tubulin staining (Figure 1A). Analysis of the heatmap suggested that the clustering was predominately driven by an increase in the mitotic index. Inspection of the well images for extract RLUS1665D (Figure 1A) showed a strong mitotic stall and diminished tubulin staining consistent with the phenotypes observed for neighboring reference compounds that inhibit tubulin polymerization (Figure 1B). The precise order of extracts and reference compounds in the hierarchical clustering network is contingent upon both the number and concentration of compounds in the original input set. Clustering these active extracts with several different dilution series for the training set of known bioactives identified extract RLUS1665D as having the most reliable clustering with known antimitotic agents. We therefore prioritized RLUS1665D for further chemical evaluation.
Figure 1.
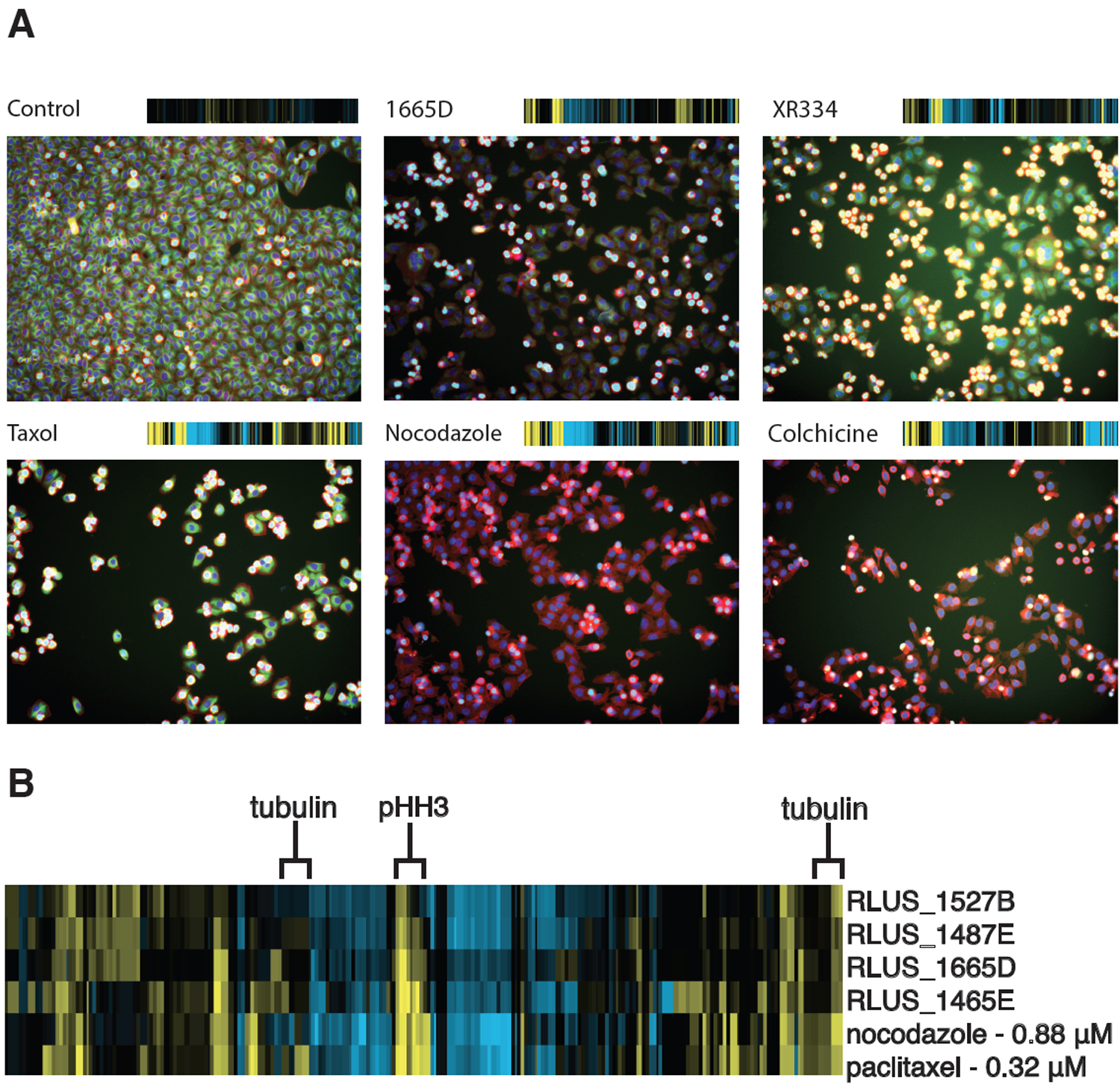
(A) Fluorescence images of drug-treated wells all showing deviations in tubulin (green) and mitotic cells (cyan) from the control. The corresponding phenotypic fingerprint generated from the image analysis is shown above each well image.(27) (B) Cytological profiling fingerprint of original natural products extracts and microtubule poisons. Positive deviations from DMSO-treated wells are displayed in yellow; negative deviations are displayed in blue. Features highlighted for tubulin and mitotic cells display an increase in mitotic cells and variable tubulin staining.
Our standard library preparation protocol involves organic extraction of microbial culture broths, followed by initial prefractionation on C18 SPE cartridges using a MeOH/H2O step gradient to generate seven prefractions for screening.8 An aliquot of prefraction RLUS1665D was first examined using our standard peak library separation protocol.8 In brief, extracts are analyzed by RP-HPLC using an analytical fraction collector to separate the eluent into deep well 96-well plates in one minute intervals (Phenomenex Synergi Fusion-RP 10 micron, 80 Å, 250 × 4.6 mm, 25:75% MeCN/H2O + 0.02% formic acid to 90:10% MeCN/H2O + 0.02% formic acid; 2 mL min−1 flow rate). A portion of this eluent (~5%) is diverted from the fraction collector to an ESI-single quadrupole mass spectrometer performing fast polarity switching to simultaneously acquire mass spectra in both positive and negative ionization modes. Collectively these analyses provide retention time, UV absorbance, and MS data for all constituents from each extract. When combined with screening data from the eluent plate these data can be used to connect individual constituents with observed bioactivities from primary screening results.
In the case of RLUS1665D the peak library revealed the presence of two major families of compounds, both of which showed weak phenotypes in the cytological profiling screen of the peak library. One limitation of peak library screening is that if an extract contains families of compounds with similar bioactivities but different retention times, separation into individual wells can decrease the titer in each well sufficiently to reduce the strength of the observed phenotype. In this instance, refermentation of the producing organism followed by a combination of normal phase silica gel and reversed phase HPLC steps provided fractions containing these two compound families, each of which displayed strong independent cytological profiles when reevaluated in the cytological profiling screen.
Examination of the screening results from the second round of fractionation indicated that one of these compound families recapitulated the original antimitotic phenotype, while the other family was strongly cytotoxic at the tested concentration, affording test wells with few cells (Figure S9, Supporting Information). This result highlights the second challenge with phenotypic screening, whereby testing at too high a concentration leads to a situation where few cells remain for image analysis. This ‘death phenotype’ affords cytological profiles where the majority of size and shape features are scored as strongly negative (due to the low cell count), which in turn precludes accurate MOA predictions. This limitation is resolved by screening test materials as fine-scale dilution series, typically 16 two-fold dilutions with concentration ranges 100 μM – 3 nM. Appropriate concentrations for clustering are selected by identifying the first dilution with a cell count within three standard deviations of the mean cell count for untreated control wells. In this instance, final purification by sequential stages of RP-HPLC led to the isolation of representatives from both of these compound classes. Screening of these pure compounds as dilutions series in the CP assay (Figure S10, Supporting Information) confirmed that one of these compounds displayed an antimitotic cytological profile, while the other clustered tightly with the known calcium channel modulators benzamil, BAY-K-8644, and A-23187 (Figure 3C). This result highlights how cytological profiling can be used to both focus on the discovery of compounds within a specific mechanistic class (antimitotic agents) while simultaneously annotating compounds with unknown MOAs (calcium channel modulators).
Figure 3.
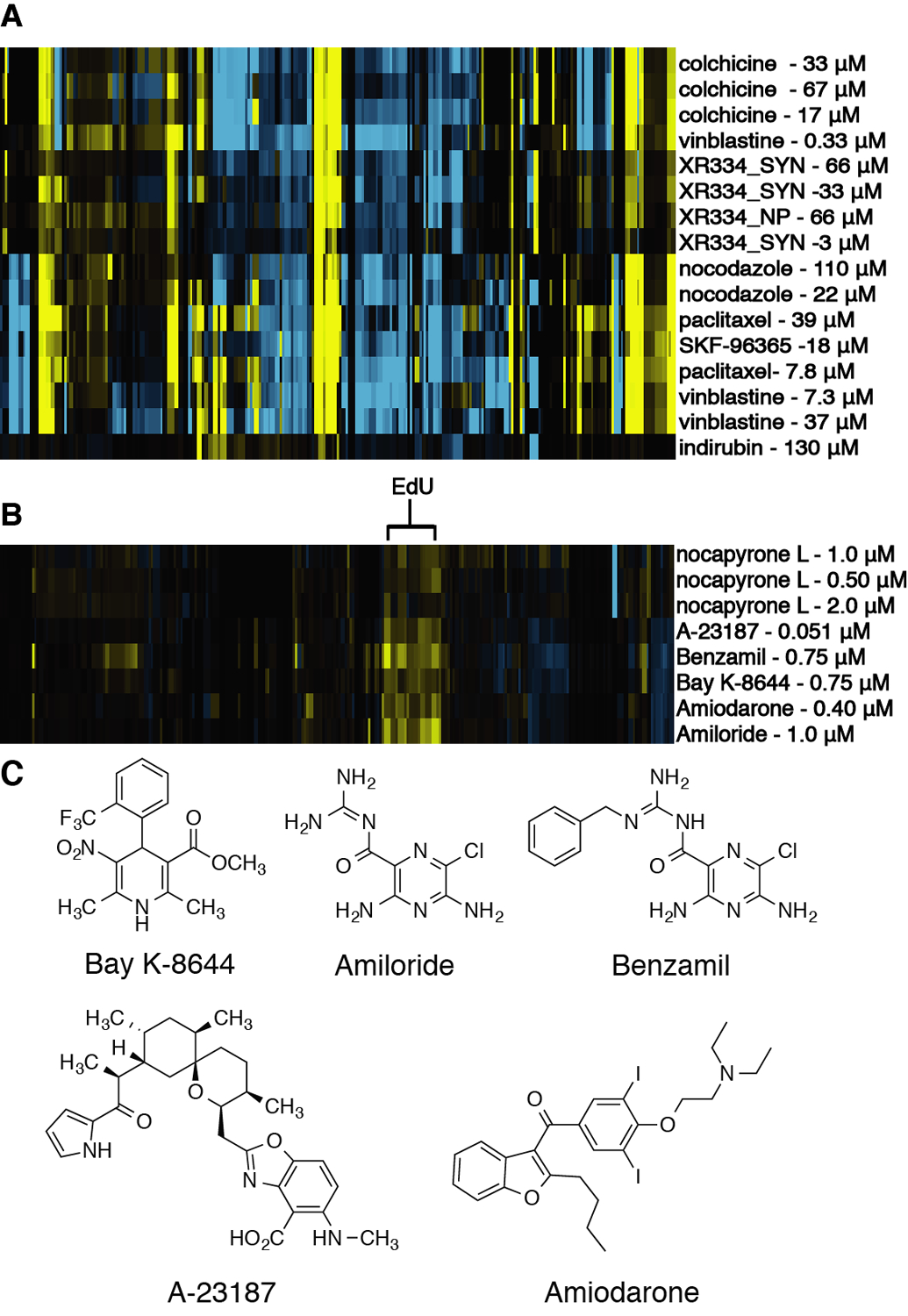
(A) Cytological profiling analysis of isolated XR334 (XR334_NP) and synthesized XR334 (XR334_SYN). (B) Cytological profiling analysis of nocapyrone L. (C) Chemical structures of compounds that clustered with nocapyrone L.
The identity of the antimitotic agent was determined through a combination of 1D and 2D NMR analyses, mass spectrometry, and total synthesis.17 Initial ESI-single-quadrupole LCMS analysis identified two mass spectrometric features consistent with the [M+H]+ and [M-H]− adducts with m/z values of 321.2 and 319.0, respectively. Subsequent HRESITOFMS analysis revealed an [M+Na]+ adduct ion with m/z 343.1875, consistent with the molecular formula C19H16N2O3. Examination of the UV-absorbance spectrum for this compound revealed strong absorbances at 250 and 352 nm, suggestive of a conjugated aromatic compound. A query of the Antimarin database revealed one compound, diketopiperazine XR334, which possessed a matching molecular formula, as well as identical NMR and UV spectroscopic features.18,19 Because this scaffold is synthetically accessible through a two-step condensation reaction between diacetyl-2,5-piperazinedione, benzaldehyde, and p-anisaldehyde, the structure of the isolated antimitotic agent was confirmed by total synthesis (Scheme S1, Supporting Information), and validated by NMR and HPLC-MS comparison with the original reported data (Figure S2, Supporting Information).19–21
In addition to the structural verification, the biological activity for the isolated natural product was confirmed through parallel CP screening for dilution series of both the natural and synthetic materials (Figure 3A). These results demonstrated that the synthetic and naturally occurring material possessed the same cytological profiles, and confirmed that the observed activity was caused by XR334, rather than any potent but minor constituents present as contaminants in the natural sample. Clustering of the CP fingerprints from the synthetic and natural samples of XR334 with the training set library revealed that XR334 clustered closely with other microtubule poisons, including nocodazole and vinblastine (Figure 3B).
XR334 is structurally similar to plinabulin (2), a synthetic analogue of the natural product phenylahistin (1). Plinabulin is both a vasculature disrupting and an antimicrotubule agent that maintains efficacy in vivo and works synergistically with docetaxel, a synthetic derivative of paclitaxel.17 Examination of the high-content images generated in the CP screen showed that XR334 displays moderate tubulin depolymerization compared to DMSO controls or strong destabilizers such as nocodazole. Therefore, while this clustering is predominately driven by the increased mitotic index, closer inspection of its effect on the microtubule cytoskeleton morphology indicates that it is possible to further subdivide this large cluster of microtubule poisons into several classes, based on their effect on tubulin polymerization. This is consistent with MOA studies performed on synthetic analogues of plinabulin.22–24
While screening approaches focused on antimitotic compounds would have successfully identified XR334 as the bioactive constituent, upon peak library generation they would be unable to annotate compounds within the extract with alternative MOAs. In addition to XR334, a second class of bioactive compounds was also identified from this extract. This second class of compounds shared similar UV profiles, and differed by sequential mass differences of 14 Da, consistent with variation in the number of methylene units in a set of structurally related analogues. Initial isolation of one of these family members afforded a white solid that gave an HRESITOFMS [M+H]+ ion at m/z 267.1957 consistent with the molecular formula C16H26O3. 1H and 13C NMR analyses revealed the presence of one carbonyl, one methoxy, and four methyl groups. Further NMR analysis identified nocapyrone L (6) (Figure 2) as a candidate match, which was subsequently confirmed by comparison of NMR and MS literature data.25 The remaining pyrone analogues from this series were identified as nocapyrones B (4) and H (5) through comparison of methylation patterns of 1H and COSY NMR spectra versus nocapyrone L and comparison with reported literature data.25–27
Figure 2.
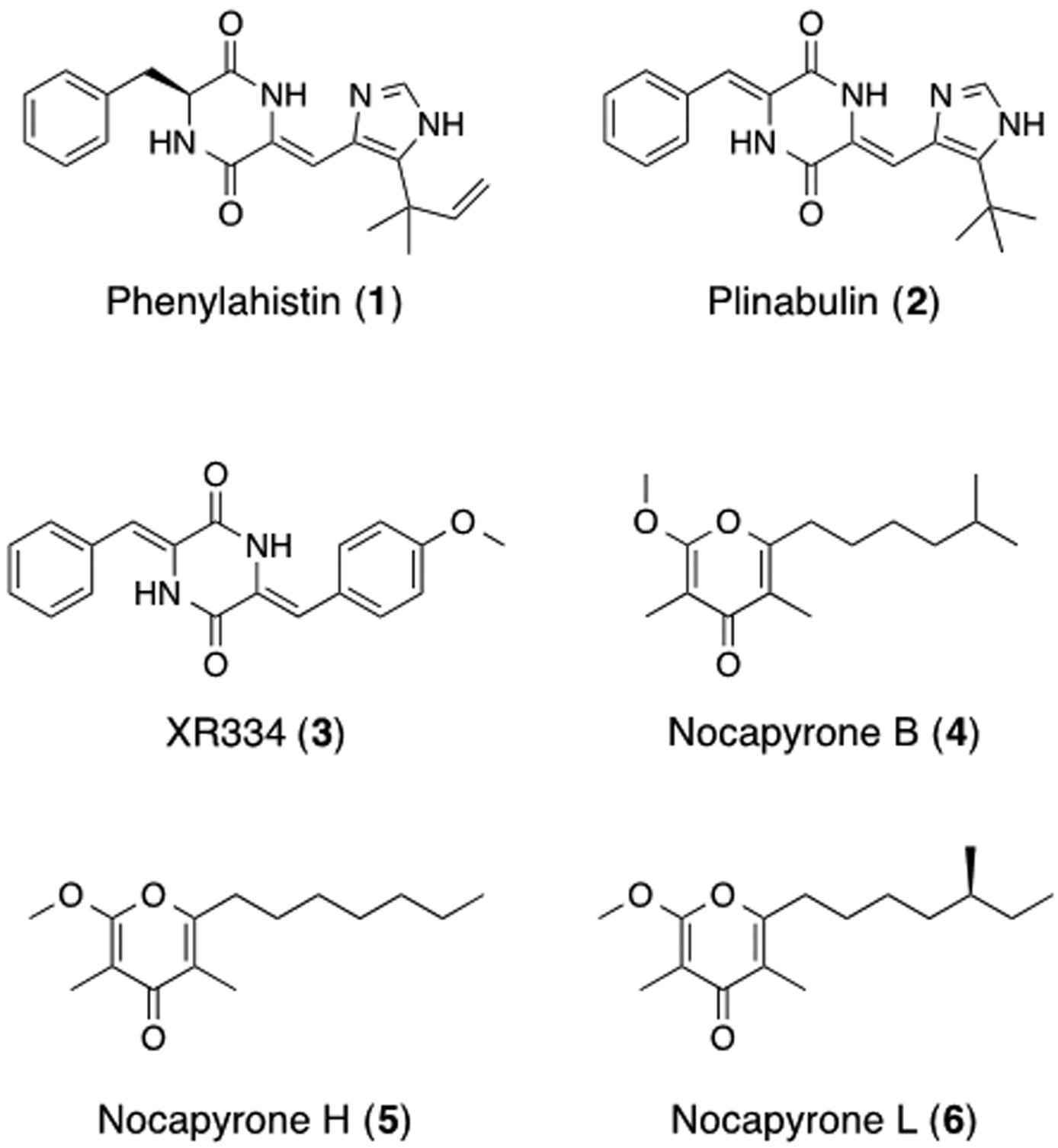
Structures of known antimitotic agents and compounds isolated from RLUS1665.
γ-Pyrones have been identified in a number of screening campaigns,26,27 and have previously been reported to impact intracellular calcium ion concentrations.25 In order to predict the MOA of these compounds against HeLa cells, a dilution series of the most active compound, nocapyrone L (6), was evaluated in the CP assay. Nocapyrone L was previously isolated from a venom duct of a sea snail, Conus rolani, and was found to be active in a calcium ion imaging assay of dissociated dorsal root ganglion neurons from mice and reported to modulate calcium ion levels.25 In line with previous reports, sub-lethal concentrations of compound 6 clustered closely with compounds that affect calcium ion channels from the ICCB library training set including Bay K-8644 and benzamil (Figure 3B). These results indicate that disruption of calcium ion channel function is likely the predominant MOA for the observed cytotoxicity of the nocapyrones in our assay system; this result demonstrates that the CP platform can differentiate between a wide array of biological mechanisms, even if these mechanisms are not directly reported on by the structural and cell cycle stains used in CP analysis.
In order to determine which cytological phenotypes underlie the clustering of the nocapyrones with the calcium ion channel modulators from the reference library, we used a modification of gene sets enrichment analysis (GSEA) using software publically available from the Broad Institute.10 GSEA was originally developed to identify gene sets (i.e. sets of genes grouped by function, biological process, co-regulation, etc.) that are enriched in genes whose up- or down- regulation are correlated with a particular phenotype (e.g. metastatic vs. non-metastatic) among a set of samples (e.g. tissue samples from different patients).28,29 For example, initial gene analysis of type 2 diabetes mellitus (DM2) revealed that no single gene could be correlated to biopsy samples taken from patients with DM2. While the expression of individual genes can have large variations within sample sets, if these genes are placed in gene sets based on their connection to specific biological pathways it is then possible to identify consistent correlations between gene set expression and specific phenotypes. Analysis of DM2 tissue samples using GSEA revealed the correlation between the expression of OXPHOS-CR genes and DM2.29
To apply this approach to the chemical genetic signatures derived from the cytological profiling screen, we grouped the CP features into 23 classes (analogous to the gene sets discussed above) based on the broad phenotypic signatures on which they report (e.g., nuclear shape, pHH3 intensity in large areas, cell count, mitotic index, etc.). Applying GSEA to our dataset, we replaced genes with CP features, gene sets with feature classes, phenotypes with annotated MOA classes, and samples with compound-dose instances. This allowed us to ask, for the calcium ion channel modulators and nocapyrones that cluster with them, which CP feature classes correlate significantly with the distinction between these compounds and the rest of the library. The advantage of this analysis is that it provides a quantifiable method for identifying key biological features that drive associations between test compounds in the cytological profiling clusters, even if these features are not readily observable by eye. Knowing which features are drivers of clustering for a specific group of compounds provides an additional tool for evaluating the biological significance of a given cluster by highlighting specific size and shape cell deviations that are positively correlated with the clustering of any specific group of compounds.
This “GSEA-CP” analysis identified enrichment in nuclear stain 5-ethynyl-2′-deoxyuridine (EdU) intensity features among those that correlated with the cluster containing the nocapyrones and calcium ion channel modulators (Figure 4). The EdU stain is a modified nucleoside analogue that reports on cells actively undergoing DNA synthesis. For each of the feature classes an enrichment score was generated. The enrichment score reflects the significance of a feature class in terms of how well its individual members are correlated with a designated phenotype. Expansion of these features enriched in this class for nocapyrone L (6) identified that six out of the seven enriched features from the GSEA analysis show significant positive deviation from the DMSO control (Figure S8, Supporting Information). Visual inspection of the images did not reveal this subtle phenotypic difference, highlighting the utility of computational image processing in classifying compounds by phenotype. Therefore, CP is a powerful tool capable of deciphering predetermined mechanisms from complex data sets, yet flexible enough to annotate MOAs of compounds with orthogonal bioactivities within the same extract.
Figure 4.
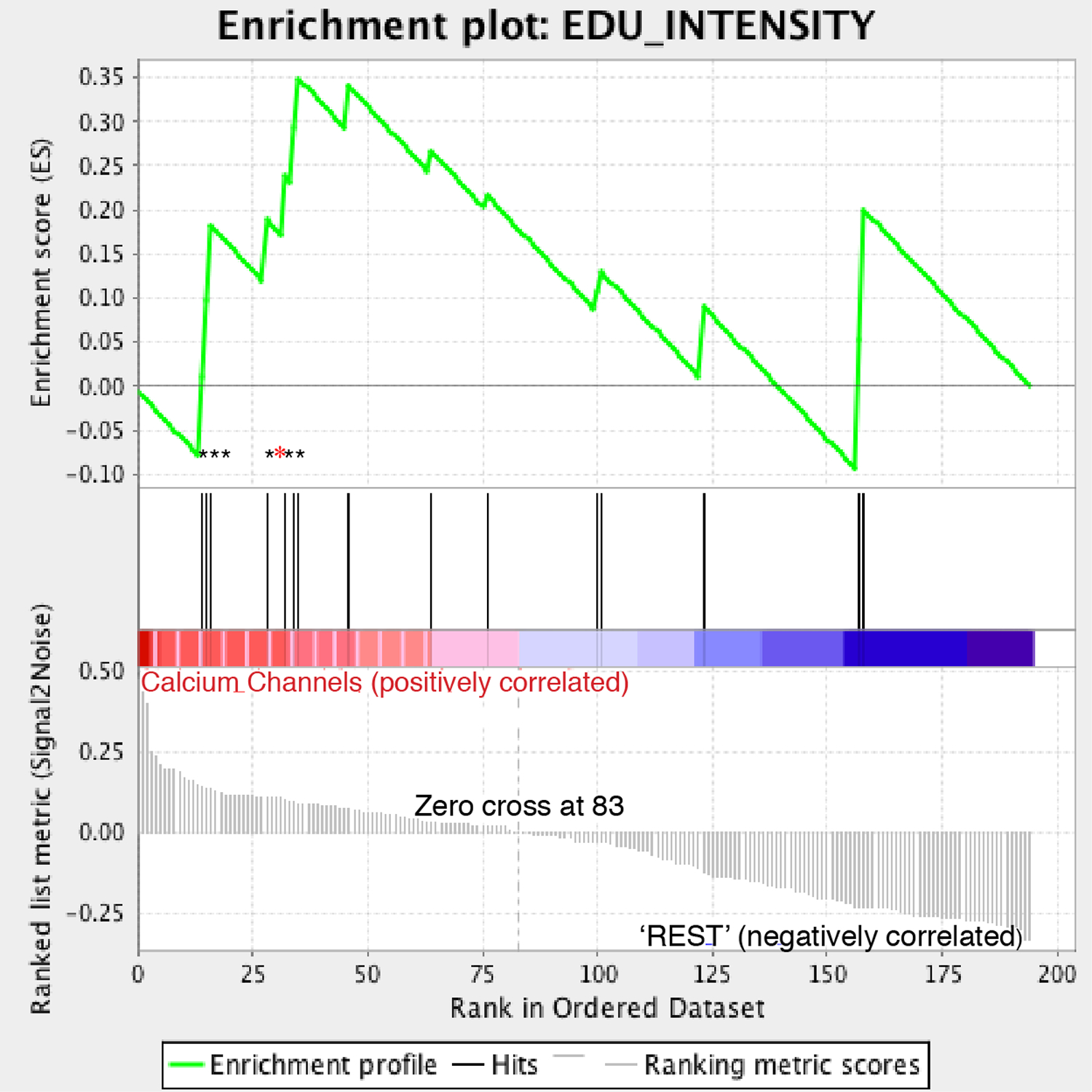
Gene sets enrichment analysis (GSEA) analysis of EdU features for calcium channels and nocapyrone L. Seven EdU features were identified to drive the clustering of this class as denoted by asterisks; six of these enriched features showed significant positive deviations for the cells treated with nocapyrone L as denoted by black asterisks.
In conclusion, this study demonstrates the utility of untargeted morphology-based screening for the broad classification of natural product MOAs. By quantifying specific morphological features of HeLa cell development under sub-lethal drug pressure, the CP platform is capable of distinguishing a large number of discrete phenotypes that in turn report on the pathways and mechanisms disrupted by drug treatment. We have shown that targeted phenotype matching from untargeted cytological profiling data can be used to identify compounds with specific predefined MOAs. In this case the antimitotic XR334 was directly targeted and identified from our complex natural product library through a combination of CP screening and analytical chemistry.
The parallel identification of nocapyrones B (4), H (5), and L (6) as predicted calcium ion channel modulators demonstrates that the CP platform can be used to discover and characterize bioactive constituents whether these bioactive compounds directly target processes reported on by the staining set (e.g. XR334, which disrupts tubulin dynamics) or not (e.g. nocapyrone L, which disrupts calcium channel function). These results therefore suggest that unbiased phenotypic profiling may be a valuable tool for the broad characterization of bioactive constituents from natural product libraries, and that this approach can be employed to identify compounds with specific biological MOAs by targeting relevant clusters in the CP profiles. In this study we have demonstrated the practicability of this approach by mining our natural product libraries for compounds with specific mechanisms, and validating their predicted MOAs through a combination of NP isolation, synthesis, and phenotypic image-based screening.
EXPERIMENTAL SECTION
General Experimental Procedures
Solvents for all chromatography were HPLC grade and used without further purification. Optical rotations were measured on a Jasco P-2000 polarimeter using a 10 mm or 100 mm path length cell at 589 nm. UV spectra were recorded on a Shimadzu UV-Visible spectrophotometer (UV-1800) with a path length of 1 cm. NMR spectra were acquired on a Varian Inova 600 MHz spectrometer equipped with a 5 mm H/C/N triple resonance cryoprobe, and referenced to residual solvent proton and carbon signals (δH 7.26, δC 77.1 for CDCl3 and δH 3.31, δC 49.0 for methanol-d4). HRMS data were acquired using an Agilent 6230 electrospray ionization (ESI) accurate-mass time-of-flight (TOF) liquid chromatograph-mass spectrometer.
Fermentation and Isolation
The producing organism, RL10–282-NTS-A, was isolated from a marine sediment sample collected by SCUBA near American Samoa. The strain was originally isolated on NTS medium (20.0 g of agar, 50.0 mg of nalidixic acid, 50.0 mg of cycloheximide, 20.0 g of starch, 0.5g of NaCl, 0.01 g of FeSO4•7H2O, 0.5 g MgSO4 •7 H2O, 0.5 g of K2PO4, 1.0 g of KNO3, 750 mL of 0.2 μm filtered seawater, 250 mL of Milli-Q H2O). Frozen stocks of environmental isolates were streaked onto fresh Marine Broth plates (37.4 g of Difco Marine Broth, 18 g of agar, 1 L of Milli-Q water) and incubated at 25°C until discrete colonies became visible. Selected colonies were inoculated into 10 mL of modified saline SYP (mSYP) media (10 g starch, 4 g peptone, 2 g yeast extract and 31.2 g instant ocean in 1 L of distilled H2O). The cultures were stepped up in stages at 7 day intervals by first inoculating 1.5 mL of the 10 mL cell cultures into 50 mL of mSYP (medium-scale), followed by inoculation of 40 mL of these medium-scale cell cultures into 1 L of the same broth also containing 20.0 g of Amberlite XAD-16 adsorbent resin in 2.8 L Fernbach flasks for 7 days. All cultures were incubated at 25°C containing glass beads for 10 mL cultures and stainless steal springs for 50 mL and 1 L cultures and shaken at 200 rpm.
The cells and resin were removed by vacuum filtration using Whatman glass microfiber filters and washed with deionized water. This cell/resin slurry was extracted with 250 mL of 1:1 MeOH/CH2Cl2, and the organic extract was removed by vacuum filtration and concentrated to dryness in vacuo. The crude organic extract, given extract code RLUS1665, was subjected to solid-phase extraction (SPE) using a Supelco-Discovery C18 cartridge (10 g) and eluted using a step gradient of 80 mL of MeOH/H2O solvent mixtures (10% MeOH, 20% MeOH (A), 40% MeOH (B), 60% MeOH (C), 80% MeOH (D), 100% MeOH (E) and finally with ethyl acetate (F) to afford seven fractions designated as prefractions A-F. The 10% MeOH fraction was discarded, and the remaining six were dried in vacuo. The 80% MeOH prefraction, RLUS1665D, was subjected to RP-HPLC (Phenomenex Synergi Fusion-RP 10 micron, 80 Å, 250 × 4.6 mm, 65:35% MeOH/H2O + 0.02% formic acid isocratic run over 25 min., 2 mLmin−1 flow rate) to afford compounds 3 – 6 which eluted at 5.75, 14.10, 15.70, and 21.95 minutes respectively.
Cytological Profiling Screening
Cytological profiling was performed at the UC Santa Cruz Chemical Screening Center using our standard screening protocol.8 In general, prefractions were tested at two concentrations (1:5 and 1:25 dilutions from master stock solutions). For stock solution preparation protocol see reference 8. Pure compounds were screened as two-fold dilution series (16 dilutions, 100 μM – 3 nM final testing concentrations). DMSO solutions (150 nL) of test compounds or extracts were added to two separate black-walled clear-bottomed 384 well plates seeded with HeLa cells and incubated at 37°C for 19 hr. Each plate was fixed and stained with the appropriate stain set (plate 1: Hoechst dye (DNA), anti-phosphohistone H3 antibody (mitotic marker), and EdU (S-phase) plate 2: Hoechst dye (DNA), TMR-phalloidin (actin), anti-tubulin Ab (tubulin); for staining protocol see reference 8) then imaged at four sites per well using an ImageXpress Micro epifluorescent microscope (Molecular Devices) with a 10x Nikon objective lens. Images were subsequently journaled to extract cell-by-cell features using Molecular Devices MetaXpress software, and these cell-by-cell values converted to final cytological profiles using our in-house data analysis pipeline.8 Finally, cytological profiles were clustered using Cluster 3.0,30 and visualized using Java Treeview.31
Synthesis of XR344
XR344 was synthesized from diacetyl-2,5-piperazinedione, benzaldehyde and p-anisaldehyde following established literature procedures (Scheme S1, Supporting Information).19 The structures of all synthetic products were confirmed by NMR and MS data, and comparison with literature values. Synthetic and naturally occurring XR334 were compared by NMR spectroscopy and LCMS co-injection (Supporting Information).
Supplementary Material
REFERENCES
- (1).Saito S-Y Prog. Mol. Subcell. Biol 2009, 46, 187–219. [DOI] [PubMed] [Google Scholar]
- (2).Altmann K-H; Gertsch JR Nat. Prod. Rep 2007, 24, 327–357. [DOI] [PubMed] [Google Scholar]
- (3).Feng Y; Mitchison TJ; Bender A; Young DW; Tallarico JA Nat. Rev. Drug Discovery 2009, 8, 567–578. [DOI] [PubMed] [Google Scholar]
- (4).Mitchison TJ ChemBioChem 2005, 6, 33–39. [DOI] [PubMed] [Google Scholar]
- (5).Sutherland JJ; Low J; Blosser W; Dowless M; Engler TA; Stancato LF Mol. Cancer Ther 2011, 10, 242–254. [DOI] [PubMed] [Google Scholar]
- (6).Towne DL; Nicholl EE; Comess KM; Galasinski SC; Hajduk PJ; Abraham VC J. Biomol. Screening 2012, 17, 1005–1017. [DOI] [PubMed] [Google Scholar]
- (7).Carpenter AE; Jones TR; Lamprecht MR; Clarke C; Kang IH; Friman O; Guertin DA; Chang JH; Lindquist RA; Moffat J; Golland P; Sabatini DM Genome Biol. 2006, 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Schulze CJ; Bray WM; Woerhmann MH; Stuart J; Lokey RS; Linington RG Chem. Biol 2013, 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Woehrmann MH; Bray WM; Durbin JK; Nisam SC; Michael AK; Glassey E; Stuart JM; Lokey RS Mol. BioSyst 2013, 9, 2604. [DOI] [PubMed] [Google Scholar]
- (10).Potts MB; Kim HS; Fisher KW; Hu Y; Carrasco YP; Bulut GB; Ou Y-H; Herrera-Herrera ML; Cubillos F; Mendiratta S; Xiao G; Hofree M; Ideker T; Xie Y; Huang LJ-S; Lewis RE; MacMillan JB; White MA Sci. Signaling 2013, 6, ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lopez A; Parsons AB; Nislow C; Giaever G; Boone C Prog. Drug. Res 2008, 66, 239–271. [DOI] [PubMed] [Google Scholar]
- (12).Crawford AD; Liekens S; Kamuhabwa AR; Maes J; Munck S; Busson R; Rozenski J; Esguerra CV; de Witte PAM PLoS ONE 2011, 6, e14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kaestner P; Bastians HJ Cell. Biochem 2010, 111, 258–265. [DOI] [PubMed] [Google Scholar]
- (14).Chan KS; Koh C-G; Li H-Y Cell Death Dis. 2012, 3, e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Roberge M; Cinel B; Anderson HJ; Lim L; Jiang X; Xu L; Bigg CM; Kelly MT; Andersen RJ Cancer Res. 2000, 60, 5052–5058. [PubMed] [Google Scholar]
- (16).Laraia L; Stokes J; Emery A; McKenzie GJ; Venkitaraman AR; Spring DR ACS Med. Chem. Lett 2014, 5, 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hayashi Y; Takeno H; Chinen T; Muguruma K; Okuyama K; Taguchi A; Takayama K; Yakushiji F; Miura M; Usui T; Hayashi Y ACS Med. Chem. Lett 2014, 5, 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).AntiMarin Database; Blunt JW; Munro MHG; Laatsch H, Eds.; University of Canterbury: Christchurch, New Zealand, and University of Göttingen; Göttingen, Germany, 2006. [Google Scholar]
- (19).Bryans J; Charlton P; Chicarelli-Robinson I; Collins M; Faint R; Latham C; Shaw I; Trew SJ Antibiot. 1996, 49, 1014–1021. [DOI] [PubMed] [Google Scholar]
- (20).Hayashi Y; Yamazaki-Nakamura Y; Yakushiji F Chem. Pharm. Bull 2013, 61, 889–901. [DOI] [PubMed] [Google Scholar]
- (21).Loughlin WA; Marshall RL; Carreiro A; Elson KE Bioorg. Med. Chem. Lett 2000, 10, 91–94. [DOI] [PubMed] [Google Scholar]
- (22).Yamazaki Y; Tanaka K; Nicholson B; Deyanat-Yazdi G; Potts B; Yoshida T; Oda A; Kitagawa T; Orikasa S; Kiso Y; Yasui H; Akamatsu M; Chinen T; Usui T; Shinozaki Y; Yakushiji F; Miller BR; Neuteboom S; Palladino M; Kanoh K; Lloyd GK; Hayashi YJ Med. Chem 2012, 55, 1056–1071. [DOI] [PubMed] [Google Scholar]
- (23).Mita MM; Spear MA; Yee LK; Mita AC; Heath EI; Papadopoulos KP; Federico KC; Reich SD; Romero O; Malburg L; Pilat M; Lloyd GK; Neuteboom STC; Cropp G; Ashton E; LoRusso PM Clin. Cancer Res 2010, 16, 5892–5899. [DOI] [PubMed] [Google Scholar]
- (24).Millward M; Mainwaring P; Mita A; Federico K; Lloyd GK; Reddinger N; Nawrocki S; Mita M; Spear MA Invest. New Drugs 2012, 30, 1065–1073. [DOI] [PubMed] [Google Scholar]
- (25).Lin Z; Torres JP; Ammon MA; Marett L; Teichert RW; Reilly CA; Kwan JC; Hughen RW; Flores M; Tianero MD; Peraud O; Cox JE; Light AR; Villaraza AJL; Haygood MG; Concepcion GP; Olivera BM; Schmidt EW Chem. Biol 2013, 20, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schneemann I; Ohlendorf B; Zinecker H; Nagel K; Wiese J; Imhoff JF J. Nat. Prod 2010, 73, 1444–1447. [DOI] [PubMed] [Google Scholar]
- (27).Wang F; Tian X; Huang C; Li Q; Zhang SJ Antibiot. 2011, 64, 189–192. [DOI] [PubMed] [Google Scholar]
- (28).Subramanian A; Tamayo P; Mootha VK; Mukherjee S; Ebert BL; Gillette MA; Paulovich A; Pomeroy SL; Golub TR; Lander ES; Mesirov JP Proc. Natl. Acad. Sci. U.S.A 2005, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mootha VK; Lindgren CM; Eriksson K-F; Subramanian A; Sihag S; Lehar J; Puigserver P; Carlsson E; Ridderstråle M; Laurila E; Houstis N; Daly MJ; Patterson N; Mesirov JP; Golub TR; Tamayo P; Spiegelman B; Lander ES; Hirschhorn JN; Altshuler D; Groop LC Nat. Genet 2003, 34, 267–273. [DOI] [PubMed] [Google Scholar]
- (30).de Hoon MJL; Imoto S; Nolan J; Miyano S Bioinformatics 2004, 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- (31).Saldanha AJ Bioinformatics 2004, 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.