

Supplemental Digital Content is available in the text.
Keywords: carbon monoxide, glycolysis, oxidative phosphorylation, platelet aggregation, tandem-mass spectrometry
Abstract
Objectives:
Carbon monoxide (CO) produced by haem oxygenases or released by CO-releasing molecules (CORM) affords antiplatelet effects, but the mechanism involved has not been defined. Here, we tested the hypothesis that CO–induced inhibition of human platelet aggregation is mediated by modulation of platelet bioenergetics.
Approach and Results:
To analyze the effects of CORM-A1 on human platelet aggregation and bioenergetics, a light transmission aggregometry, Seahorse XFe technique and liquid chromatography tandem-mass spectrometry–based metabolomics were used. CORM-A1–induced inhibition of platelet aggregation was accompanied by the inhibition of mitochondrial respiration and glycolysis. Interestingly, specific inhibitors of these processes applied individually, in contrast to combined treatment, did not inhibit platelet aggregation considerably. A CORM-A1–induced delay of tricarboxylic acid cycle was associated with oxidized nicotinamide adenine dinucleotide (NAD+) depletion, compatible with the inhibition of oxidative phosphorylation. CORM-A1 provoked an increase in concentrations of proximal (before GAPDH [glyceraldehyde 3-phosphate dehydrogenase]), but not distal glycolysis metabolites, suggesting that CO delayed glycolysis at the level of NAD+–dependent GAPDH; however, GAPDH activity was directly not inhibited. In the presence of exogenous pyruvate, CORM-A1–induced inhibition of platelet aggregation and glycolysis were lost, but were restored by the inhibition of lactate dehydrogenase, involved in cytosolic NAD+ regeneration, pointing out to the key role of NAD+ depletion in the inhibition of platelet bioenergetics by CORM-A1.
Conclusions:
The antiplatelet effect of CO is mediated by inhibition of mitochondrial respiration—attributed to the inhibition of cytochrome c oxidase, and inhibition of glycolysis—ascribed to cytosolic NAD+ depletion.
Highlights.
Carbon monoxide was identified to inhibit platelet aggregation, but the mechanism involved has not been defined.
We observed that antiplatelet effect of carbon monoxide is accompanied by inhibition of mitochondrial respiration and inhibition of glycolysis at the level of GAPDH (glyceraldehyde 3-phosphate dehydrogenase).
In the presence of exogenous pyruvate inhibitory effects of carbon monoxide on platelet aggregation and glycolysis were lost, but restored after inhibition of cytosolic NAD+ regeneration, suggesting a key role of NAD+ depletion in the observed effects.
Antiplatelet effect of carbon monoxide is mediated by inhibition of both ATP-generation processes, mitochondrial respiration at the level of cytochrome c oxidase, and glycolysis, attributed to cytosolic NAD+ depletion
See accompanying editorial on page 2344
Carbon monoxide (CO) delivered to an organism through inhalation is lethally toxic. Hence, for a long time, CO has been referred to as a silent killer. However, CO produced endogenously during haem degradation by haem oxygenase enzymes is an important endogenous signalling molecule implied in cellular responses to various stimuli.1 In turn, CO supplied in relatively low quantities by CO-releasing molecules (CORMs) affords cytoprotective and anti-inflammatory effects.1,2
Numerous reports provide evidence that CO, due to its antiplatelet and antithrombotic action, plays an important role in maintaining vascular homeostasis in vivo. For example, it was demonstrated that HO-1 (haem oxygenase 1)-derived CO protects against hepatic ischemia-reperfusion injury through the inhibition of platelet adhesion to the sinusoids.3 Moreover, HO-1-deficiency induces an acceleration of arterial thrombosis, which occurs through various mechanisms (including platelet activation), while CO inhalation4 or CO delivery by CORM-25 can rescue from prothrombotic phenotype of HO-1 deficiency. These reports confirm the importance of HO-1-derived CO in the regulation of vascular thromboresistance and suggest that CORMs may represent a novel class of antiplatelet agents. Indeed, CORM-A1, a prototypic CORM slowly releasing CO,6 was shown to afford antiplatelet and antithrombotic activities in vivo without any hypotensive effect, while CORM-3, which releases CO instantly, displayed both antithrombotic and hypotensive effects.7 Recently, various novel CORMs with tuneable CO-releasing properties have been synthetized, all of which were shown to display antiplatelet activity similar or even slightly more pronounced than CORM-A1,8 confirming the significant antiplatelet effects of CORMs.
Despite the firm evidence on the antiplatelet effects of CO in in vivo and in vitro studies, the mechanisms involved have not been elucidated so far. Even though high concentrations of gaseous CO act in platelets via sGC (soluble guanyl cyclase),9,10 the antiplatelet effect of CORM-3, in contrast to NO-donors, was not mediated by the activation of sGC.11,12 Recently, the involvement of glycoprotein-mediated HS1 phosphorylation was suggested to be involved in CORM-2-induced suppression of platelet activation by LPS (lipopolysaccharide).13 However, this mechanism does not explain the antiplatelet effect of CO reported for platelet aggregation induced by classical platelet agonists such as collagen or thrombin,11,12 which entail a mechanistically distinct intraplatelet pathways as compared with LPS-induced platelet activation.14
Platelet aggregation is an energy-demanding process.15,16 In resting or activated states, platelets draw energy from oxidative phosphorylation and glycolysis.17–19 However, besides a couple of works describing altered bioenergetics of platelets derived from patients with sickle cell disease,20 asthma,21 sepsis,22 Parkinson disease23 or diabetes mellitus,24 still little is known about the role of metabolism in the regulation of platelets’ function. A number of reports, including ours, have demonstrated that CO modulates cellular bioenergetics in various cells types,25–30 including endothelial cells.31–33
Given the fact that platelet aggregation is an energy-demanding process, we hypothesized that CO may modulate platelet activity through the modulation of platelet bioenergetics involving oxidative phosphorylation and glycolysis. In the present work, we have characterized the bioenergetics of platelets in resting and activated states, and analyzed the effects of CORM-A1, on mitochondrial respiration, glycolysis, and metabolome in platelets. We have chosen CORM-A1 because of its well documented antiplatelet action in in vitro and in vivo models.7,12 Here, we propose that the mechanism of antiplatelet effect of CO involves the inhibition of 2 major ATP-generating pathways in platelets—mitochondrial respiration and glycolysis, by the inhibition of cytochrome c oxidase and cytosolic nicotinamide adenine dinucleotide (NAD+) depletion, respectively.
Methods
The authors declare that all supporting data are available within the article in the Data Supplement.
Isolation of Human Platelets
Venous blood was obtained from male volunteers at the University Hospital Blood Bank Centre. Volunteer donors had not taken any medicines for the preceding 2 weeks. Informed consent was given by a volunteer before the blood withdrawal and study conformed to the principles outlined in the World Medical Association Declaration of Helsinki. Blood obtained from at least 3 donors per one independent experiment was collected into vials containing sodium citrate (3.2%, 9:1v/v) as an anticoagulant agent. Blood was centrifuged (260g, 15 minutes) followed by a centrifugation/washing cycle using prostacyclin-containing PBS (PBS containing albumin [1 g/L] and glucose [1 g/L]), according to the previously described method.34 Washed platelets (WP) were finally suspended in assay medium (Seahorse XF Base Medium Minimal DMEM supplemented with glucose [1 g/L] and glutamine [2 mmol/L], pH 7.4) at a density of 2×105 platelets/µL, unless otherwise stated. Contamination of neutrophils in WP was <1/106 platelets.
Platelet Aggregation Assay in Humans
Aggregation of blood platelets was assessed in WP using a dual channel aggregometer (CHRONO-LOG) according to the method previously described by Born.35 WP (500 µL) were equilibrated for 2 minutes at 37°C with a continuous stirring at 800 rpm and then stimulated with collagen or thrombin to cause aggregation. At the beginning of each experiment, concentrations of thrombin (in the range of 0.1–0.5 U/mL) that induced sub-maximum aggregation response were determined. CORM-A1, metabolic inhibitors, and other tested compounds were added 2 minutes before stimulation of platelets with collagen or thrombin. Transmittance was read within 6 minutes after stimulation of platelets with an agonist. The concentrations of CO released from CORM-A1 were measured with myoglobin assay (Figure I in the Data Supplement), as described previously36 with minor changes (Materials in the Data Supplement).
Analysis of Cellular Bioenergetics Using Extracellular Flux Technology
To measure mitochondrial function and glycolysis in isolated human platelets, a Seahorse XFe96 Analyzer was used (the details are in Materials in the Data Supplement). Briefly, freshly isolated platelets suspended in assay medium were introduced into the Seahorse XFe96-well plates (10×106 of platelets per well) followed by centrifugation (5 minutes, 700g) and incubation with bicarbonate-free low buffered assay medium (1 hour, 37°C) in air without CO2 before the beginning of the assay. Changes in oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were assessed over time by sequential injections of reagents in ports A, B, C, and D. Concentrations of oligomycin, FCCP (carbonyl cyanide 4-[trifluoromethoxy]phenylhydrazone), rotenone, and antimycin A were optimized (data not shown). Mitochondrial stress test enabled determination of the following key parameters of mitochondrial function: acute response, proton leak, maximal respiration, spare respiratory capacity, ATP production, and nonmitochondrial oxygen consumption (calculated as described in Materials in the Data Supplement). Additionally, we calculated acute response and oligomycin-induced changes in ECAR. We analyzed the effects of CORM-A1 on thrombin-induced changes in OCR (Δ [Thr-A1]mt. respiration) and in ECAR (Δ [Thr-A1]glycolysis), calculated based on the differences in OCR and ECAR before and after thrombin injection. Based on the experiments with oligomycin, we analyzed the ability of platelets pretreated with CORM-A1 and thrombin to further maximize their glycolysis in response to oligomycin, reflecting a spare glycolytic capacity (Δ [O-Thr]), while based on the experiments with FCCP/pyruvate, we analyzed the ability of platelets pretreated with CORM-A1 and thrombin to further maximize their mitochondrial respiration in response to FCCP, reflecting a spare respiratory capacity (Δ [FCCP-Thr]).
Lactate Measurements
Measurements of lactate were performed by the enzymatic photometric methods using an automatic Pentra 400 (Horiba, Japan) biochemical analyzer according to the manufacturer’s instructions. Samples were suspended in assay buffer at a density of 3×104 platelets/μL and treated with tested compounds for 8 minutes at 37°C or preincubated with tested compounds for 2 minutes and then activated with thrombin for 6 minutes at 37°C, followed by centrifugation (1000g, 10 minutes) and measurement of the concentration of lactate in supernatant.
Analysis of Intraplatelet Metabolites
Detection of intraplatelet metabolites was performed according to the protocol described previously,33,37 with minor changes. Briefly, WP (suspended in PBS containing 1 g/L glucose and 2 mmol/L glutamine, in a number of 500×106 per 0.5 mL per sample) were untreated or preincubated for 2 minutes with 300 μmol/L CORM-A1, followed by the addition of thrombin (0.1 U/mL) and further incubation for 6 minutes. The metabolism was then quenched by the addition of 0.5 mL of extraction solution (acetonitrile: methanol: water, 5:2:3, v/v/v) cooled to −80°C. Metabolites were extracted by sonication for 15 minutes on ice, centrifuged (15 000g, 15 minutes, 4°C) and lyophilized. Before analysis, the samples were reconstituted in water, injected into a liquid chromatography column, and analyzed on a QTRAP 5500 (Sciex, Framingham, MA) coupled with UFLC (ultra-fast liquid chromatography) Nexera (Shimadzu, Kyoto, Japan). Chromatography separation was achieved on an Acquity UPLC BEH C18 1.7 μm 3.0×100 mm analytical column (Waters, Milford, MA). Samples were analyzed twice in positive and negative ionization MRM (multiple reaction monitoring) mode. For the analysis in positive ionization using acetonitrile, 100 mmol/L ammonium formate (pH 5.0) 95:5 v/v and 5 mmol/L ammonium formate (pH 5.0) were used as a mobile phase using gradient elution. The total run time was 8 and 10 minutes for negative and positive ionization modes, respectively.
Analysis of GAPDH and PFK-1 Activities
GAPDH (glyceraldehyde 3-phosphate dehydrogenase) activity was analyzed in samples of WP (suspended in PBS containing 1 g/L glucose and 2 mmol/L glutamine, in a number of 300×106 per 0.5 mL per sample) untreated or incubated for 8 minutes with CORM-A1, processed as described by Schmidt and Dringen38 and measured according to the protocol described by Bisswanger.39 PFK-1 (phosphofructokinase 1) activity was analyzed followed by incubation of WP (500×106 per 0.5 mL per sample) with CORM-A1 for 8 minutes or 30 minutes, according to the protocol described previously.40,41 Details are described in Materials in the Data Supplement.
Analysis of Metabolic ATP Concentration
Measurements were performed in samples treated for 8 minutes with tested compounds, in the presence of apyrase (1 U/mL) using ATPlite 1step Luminescence Assay System (PerkinElmer).
Statistical Analysis
Statistical analysis was performed using ORIGINPRO 9.1 software (OriginLab Corporation, Northampton, MA) or Graphpad Prism software (GraphPad Software, La Jolla, CA). Results were expressed as means±SD. For statistical analysis, the data have been analyzed for normality, and 1-way ANOVA test was performed with Benferroni test; with P values being provided in the legends (*P<0.05, #P<0.01, $P<0.001, and &P<0.0001).
Results
Effects of CORM-A1 on Mitochondrial Respiration and Glycolysis in Resting Platelets
In resting human WP, CORM-A1 induced a concentration-dependent decrease in OCR and biphasic changes in ECAR as analyzed by the Seahorse XFe technique (Figure 1). A slight inhibitory effect of CORM-A1 on mitochondrial respiration (Figure 1A) was already visible at the concentrations of 10 and 30 μmol/L, with stronger effects reaching ≈50% to 60% of basal OCR inhibition at concentrations of 100 to 300 μmol/L. The mitochondrial stress test (Figure 1C) revealed that CORM-A1 induced a decrease in ATP-linked respiration, maximal respiration, and spare respiratory capacity, which all may have resulted from the inhibition of cytochrome c oxidase by CO. CORM-A1 induced a slight increase in proton leak, but did not affect nonmitochondrial oxygen consumption. CORM-A1 decreased also intraplatelet ATP concentration as assessed by biochemical assay (Figure 1D).
Figure 1.
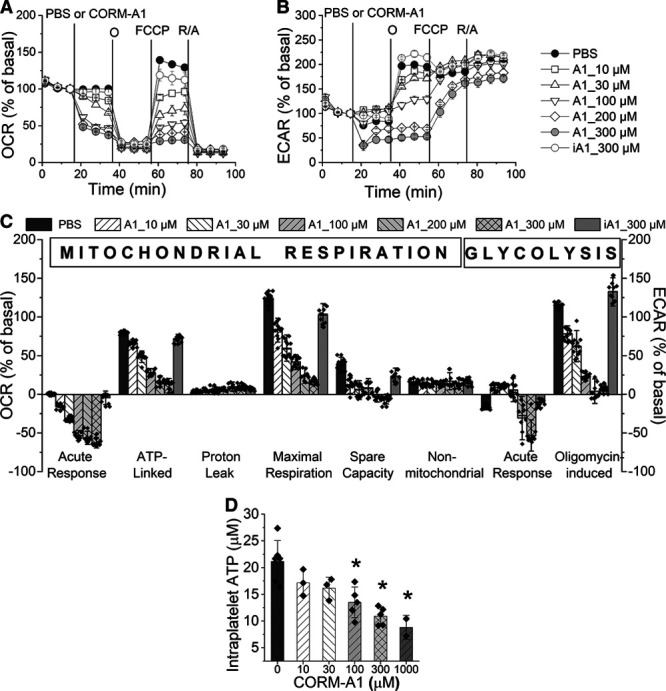
Effects of CORM-A1 on mitochondrial respiration and glycolysis in resting human platelets as monitored by Seahorse XFe96 Analyzer. Oxygen consumption rate (OCR; A) and extracellular acidification rate (ECAR; B) measurements of human washed platelets (WP) treated with PBS (control), CORM-A1 (A1; 10–300 μmol/L) or inactive CORM-A1 (iA1; 300 μmol/L) followed by sequential addition of oligomycin (1 μg/mL), FCCP (carbonyl cyanide 4-[trifluoromethoxy]phenylhydrazone)/pyruvate (0.3 μmol/L/1 mmol/L) and rotenone/antimycin A (0.5/0.5 μmol/L). Bioenergetic parameters of mitochondrial respiration and glycolysis (C) were calculated as described in Materials in the Data Supplement. Data represent the means±SD of 2 independent experiments; n=4–6 replicates in each experiment. D, Intracellular ATP concentration measured in platelets treated with PBS or CORM-A1 together with apyrase (1 U/mL) to remove released extracellular ATP. Data represent the means±SD of 3–5 independent experiments; *P<0.05 as compared with PBS group.
Biphasic effects of CORM-A1 on glycolysis comprised a slight increase in ECAR for the lower concentrations (10–100 μmol/L) and a decrease in ECAR for the higher concentrations (200 and 300 μmol/L) of CORM-A1 (Figure 1B). To confirm that all the described effects of CORM-A1 were induced by CO, inactive CORM-A1 was used, which neither induced changes in mitochondrial respiration nor in glycolysis. The levels of LDH (lactate dehydrogenase) released into the assay medium after CORM-A1 or lysis buffer treatment confirmed that CORM-A1 in the concentration range of 10 to 300 μmol/L did not affect viability of platelets (Figure II in the Data Supplement).
Effects of CORM-A1 on Mitochondrial Respiration and Glycolysis in Activated Platelets
To characterize the effects of CO on bioenergetics of activated platelets, mitochondrial stress test was performed in WP, which were pretreated with CORM-A1 and then activated with thrombin (0.1 U/mL). Thrombin induced a slight increase (by 11±2.2%) in OCR, which was further increased by FCCP (Figure 2A and 2E). In contrast to the mild activation of OCR, thrombin induced a substantial increase (by 116±0.7%) in ECAR, which was further increased by oligomycin (Figure 2D and 2E). In the presence of CORM-A1, the thrombin-induced increase in OCR was diminished, in particular for 100 or 300 μmol/L CORM-A1 as compared with control platelets (Δ [Thr-A1] in Figure 2E). When mitochondrial respiration was further activated with FCCP, CORM-A1 in a concentration-dependent manner in a wide range of concentrations (10–300 μmol/L) diminished the increase in OCR (Δ [FCCP-A1]). The effects of CORM-A1 on thrombin-activated glycolysis were more remarkable than those on thrombin-induced increase in OCR, due to stronger activation of glycolysis by thrombin. In the presence of 100 to 300 μmol/L CORM-A1, thrombin-activated glycolysis was substantially diminished. Only a slight increase in ECAR after thrombin (by around 30%) was observed for 100 μmol/L CORM-A1 (Δ [Thr-A1] in Figure 2), whereas 300 μmol/L CORM-A1 inhibited glycolysis so strongly that thrombin-induced increase in ECAR was abrogated (Δ [Thr-A1] in Figure 2E). CORM-A1 diminished also oligomycin-induced increase in glycolysis (Δ [O-A1]) in a concentration-dependent manner (10–300 μmol/L). To confirm that the observed changes in ECAR resulted from the changes in glycolytic flux, lactate extrusion by platelets was measured after thrombin stimulation with or without CORM-A1 (Figure 2F). Thrombin (0.1 U/mL) increased lactate release, whereas CORM-A1 in a concentration-dependent manner (100–1000 μmol/L) decreased lactate concentration in supernatant of resting or thrombin-stimulated platelets (Figure 2F).
Figure 2.
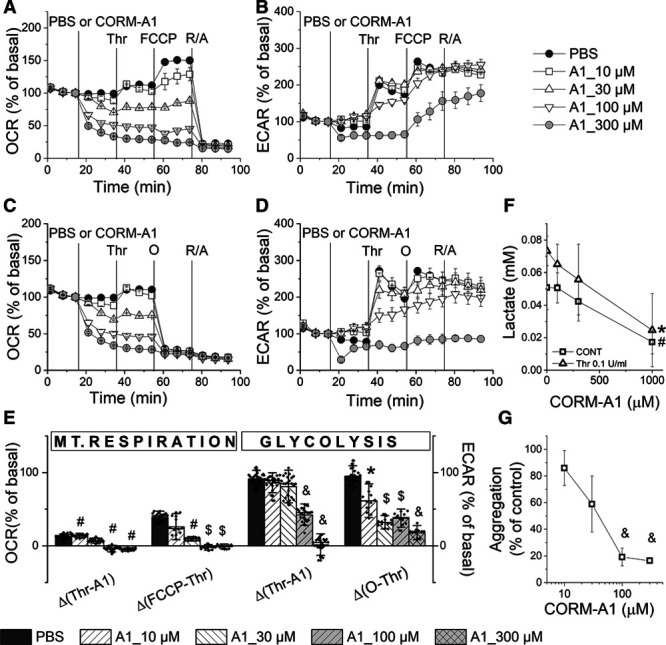
Effects of CORM-A1 on mitochondrial respiration and glycolysis in parallel to aggregation in activated platelets. Oxygen consumption rate (OCR; A and C) and extracellular acidification rate (ECAR; B and D) measurements of human washed platelets (WP) treated with PBS (control) or CORM-A1 (A1; 10–300 μmol/L) followed by the addition of thrombin (0.1 U/mL) and further sequential addition of FCCP (carbonyl cyanide 4-[trifluoromethoxy]phenylhydrazone)/pyruvate (0.3 μmol/L/1 mmol/L) and rotenone/antymycin A (R/A; 0.5/0.5 μmol/L; A and B) or oligomycin (1 μg/mL) and R/A (C and D). Bioenergetic parameters of mitochondrial respiration and glycolysis (E) were calculated as described in the Data Supplement. Data represent the means±SD of 3 independent experiments; n=4–8 replicates in each experiment. F, Concentration of lactate extruded from platelets (WP) treated with CORM-A1 (0, 100, 300, 1000 µmol/L; CONT) or CORM-A1 and thrombin (0.1 U/mL; Thr); data represent the means±SD of 4 independent experiments; n=2 replicates in each experiment. G, Aggregation of platelets (WP) treated with CORM-A1 (10, 30, 100, 300 µmol/L) and activated with thrombin (0.5 U/mL) as compared with control; data represent the means±SD of 4 independent experiments; n=2 replicates in each experiment. *P<0.05, #P<0.01, $P<0.001, &P<0.0001 as compared with control group.
The comparison of CORM-A1 effects on platelet bioenergetics and aggregation revealed that concentrations of CORM-A1, which afforded nearly complete inhibition of platelet aggregation (100–300 μmol/L, Figure 2G), also led to simultaneous and complete inhibition of glycolysis and mitochondrial respiration (100–300 μmol/L, Figure 2A through 2E). Bioenergetics of platelets treated with 10 or 30 μmol/L CORM-A1 was impaired to a lesser degree, what was associated with a weaker antiplatelet effect. Inactive CORM-A1 (300 μmol/L) did not affect platelet aggregation (99.8±2.61% of control, n=5) confirming the involvement of CO in CORM-A1–induced effects. Altogether, the concentration-dependent antiaggregatory effects of CORM-A1 were mirrored by the concentration-dependent inhibition of platelet bioenergetics.
Effects of Inhibitors of Mitochondrial Respiration and Glycolysis on Platelet Bioenergetics and Platelet Aggregation-Metabolic Plasticity of Platelets
To characterize the reliance of platelets on mitochondrial respiration versus glycolysis, the effects of oligomycin (inhibitor of ATP synthase), rotenone (inhibitor of complex I), antimycin A (inhibitor of complex III), UK-5099 (inhibitor of mitochondrial pyruvate carrier), 2-deoxy-D-glucose (2DG; competitive glycolytic inhibitor), and 3PO (inhibitor of PFKFB3 [6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3]) on OCR, ECAR, and platelet aggregation were investigated.
2DG profoundly inhibited glycolysis, whereas it barely affected mitochondrial respiration (Figure 3A and 3B). Only at the highest concentration of 100 mmol/L, 2DG significantly reduced OCR to 95±0.6% of basal (P<0.05). 3PO, another inhibitor of glycolysis, slightly reduced ECAR; however, even though the effect was statistically significant, it was not concentration dependent and was associated with concomitant reduction in OCR. In turn, oligomycin at 0.5 to 1 μg/mL reduced almost completely OCR, which was accompanied by a substantial increase in ECAR. Antimycin A and rotenone almost completely reduced OCR and highly increased ECAR, whereas UK-5099 slightly reduced OCR and increased ECAR. Notably, the complete inhibition of oxidative phosphorylation by oligomycin, antimycin A, or rotenone was compensated by almost a 3-fold increase in ECAR.
Figure 3.
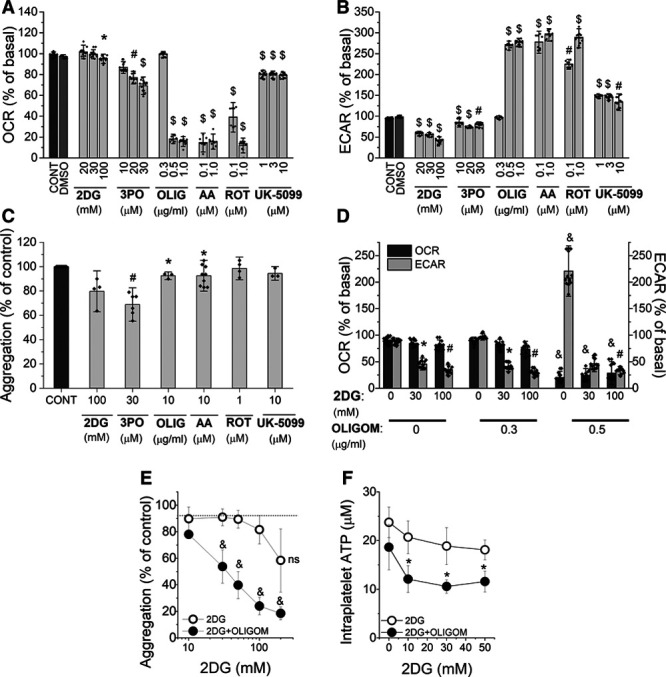
Effects of single or combined inhibition of mitochondrial respiration and glycolysis by metabolic inhibitors on platelet aggregation. Oxygen consumption rate (OCR; A) and extracellular acidification rate (ECAR; B) measurements of human washed platelets (WP) untreated (CONT) or treated with DMSO (as a vehicle), 2-deoxy-D-glucose (2DG), 3PO (3-[3-pyridinyl]-1-[4-pyridinyl]-2E-propen-1-one), oligomycin (OLIG), antimycin A (AA), rotenone (ROT) or UK-5099, presented as % of basal 20 minutes after addition of the reagents. Data represent the means±SD of 3 independent experiments; n=4–8 replicates in each experiment. C, Aggregation of platelets (WP) untreated (CONT) or treated with 2DG, 3PO, oligomycin, antimycin A, rotenone or UK-5099, and activated with thrombin (0.5 U/mL), presented as % of control; data represent the means±SD of 4 independent experiments, n=1–3 replicates in each experiment. D, OCR and ECAR measurements of WP treated with combined inhibitors: 2DG and oligomycin presented as % of basal 20 min after addition of the reagents. E, Aggregation of WP treated with combined inhibitors: 2DG and oligomycin (10 μg/mL) activated with thrombin (0.5 U/mL); presented as % of control. Data represent the means±SD of 3 independent experiments. F, Intraplatelet ATP concentration in WP treated with combined inhibitors: 2DG and oligomycin, measured in the presence of apyrase (1 U/mL). Data represent the means±SD of 3 independent experiments. *P<0.05, #P<0.01, $P<0.001, &P<0.0001 as compared with control group.
As shown in Figure 3C, those inhibitors which reduced exclusively mitochondrial respiration (oligomycin, antimycin A, rotenone, UK-5099) did not substantially inhibit platelet aggregation (less than by 10%). 2DG reduced platelet aggregation to 80% of control; however, at this concentration, 2DG also slightly reduced OCR. Among metabolic inhibitors, the most effective was 3PO, which at 30 μmol/L concentration reduced platelet aggregation to 68% of control (Figure 3C). However, as demonstrated in Figure 3A and 3B, 3PO reduced not only ECAR, but also OCR.
Effects of Simultaneous Inhibition of Mitochondrial Respiration and Glycolysis on Platelet Aggregation
Figure 3D demonstrates the bioenergetics of WP after addition of oligomycin and 2DG individually or in combination. Oligomycin at a concentration of 0.5 μg/mL, which in the absence of 2DG even slightly increased metabolic activity (OCR decreased to 20±4.5% of basal, but ECAR increased up to 221±20.8% of basal), in combination with 2DG, caused a profound inhibition of platelet bioenergetics that resulted in significant inhibition of platelet aggregation (Figure 3E). Indeed, oligomycin alone (even at a concentration of 10 µg/mL) inhibited platelet aggregation by only 8% (Figure 3E), 2DG alone (up to 100 mmol/L) by 21 %. Each of them alone only slightly decreased intraplatelet ATP concentration. However, combination of 2DG and oligomycin (10 µg/mL) induced a substantial fall in intraplatelet ATP concentration (Figure 3F) explaining a profound inhibition of platelet aggregation under combined 2DG and oligomycin treatment (Figure 3E).
Effect of CORM-A1 on Metabolic Pathways in Platelets—Metabolomic Analysis
To explore the effects of CORM-A1 on platelet bioenergetics, we quantified tricarboxylic acid cycle (TCA) and glycolysis metabolites by targeted metabolomic analysis in platelets pretreated with CORM-A1 and stimulated with thrombin. As presented in Figure 4, the concentrations of TCA cycle metabolites in platelets were not substantially changed at sixth minute after thrombin activation, whereas in the presence of CORM-A1, they were reduced (fumarate by 40% and malate by over 50%). Moreover, the CORM-A1–induced changes in TCA metabolites levels were accompanied by a reduction in NAD+ and an increase in NADH concentrations that is in agreement with inhibitory effects of CO on mitochondrial respiration in platelets. Interestingly, a sum of reduced and oxidized NAD (NADH together with NAD+) was significantly decreased, whereas a ratio of NADH/NAD+ was only slightly (nonsignificantly) increased after CORM-A1. In turn, glycolytic metabolites measured at sixth minute after thrombin stimulation were also not changed significantly, whereas CORM-A1 induced a massive increase in concentrations of proximal glycolytic metabolites. The concentration of hexose-6-P was increased 2-fold, while fructose-1,6-bis-phosphate and triose-phosphate (dihydroxyacetone phosphate [DHAP] and glyceraldehyde 3-phosphate [GA3P]) levels were >10-fold higher comparing to control platelets not treated with CORM-A1. Surprisingly, CORM-A1 did not induce changes in the concentrations of distal glycolytic metabolites (3-phospho-glicerate, phosphoenolpyruvate, and pyruvate; Figure 5). Noteworthy, the altered balance between proximal and distal glycolytic metabolites was associated with an increased diversion of glucose into pentose phosphate pathway (PPP). Indeed, CORM-A1 induced an increase in concentrations of PPP metabolites (ribose-5-phosphate, sedoheptulose-7-phosphate, and erythrose-4-phosphate) that was not associated with an increase in NADPH or NADPH/NADP+ ratio (Figure 5).
Figure 4.
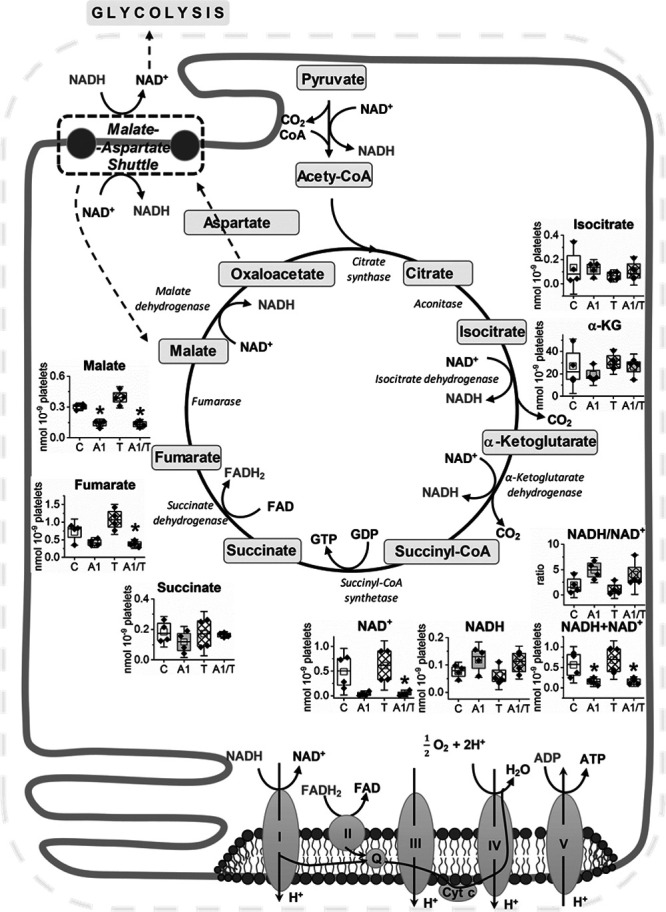
Effects of CORM-A1 on tricarboxylic acid cycle (TCA) metabolites and nicotinamide adenine dinucleotide (NAD+) content in platelets. Human washed platelets (WP) suspended in PBS containing glucose (1 g/L) and glutamine (2 mmol/L) were untreated (8 min; C), treated with CORM-A1 (300 μmol/L; 8 min; A1), treated with thrombin (0.1 U/mL; T) or treated with CORM-A1 and thrombin (2+6 min; A1/T). Data show amounts of metabolic by-products in TCA cycle. Data are presented as means±SD (nmoL/109 platelets) of 4 independent experiments; *P<0.05 vs control.
Figure 5.
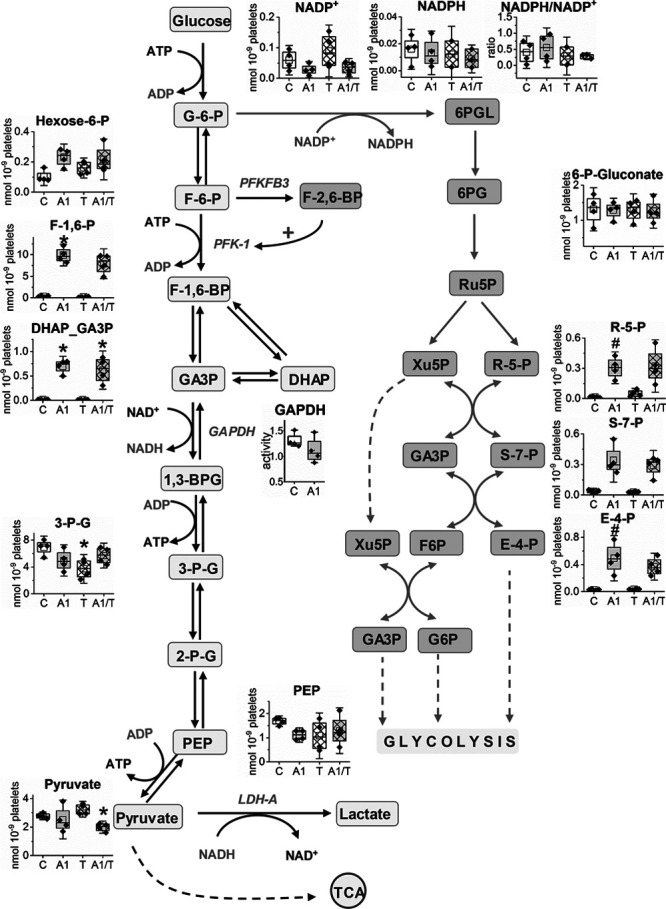
Effects of CORM-A1 on glycolysis and pentose phosphate pathway (PPP) metabolites in platelets. Human washed platelets (WP) suspended in PBS containing glucose (1 g/L) and glutamine (2 mmol/L) were untreated (8 min; C), treated with CORM-A1 (300 μmol/L; 8 min; A1), treated with thrombin (0.1 U/mL; T) or treated with CORM-A1 and thrombin (2+6 min; A1/T). Data show amounts of metabolic by-products in glycolysis and PPP. Data are presented as means±SD (nmoL/109 platelets) of 4 independent experiments. DHAP indicates dihydroxyacetone phosphate; F-1,6-BP, fructose-1,6-bis-phosphate; GA3P, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvate; and TCA, tricarboxylic acid cycle. *P<0.05 vs control.
These results, showing an activation of PPP and accumulation of proximal glycolytic metabolites, strongly suggest that the inhibition of glycolysis by CORM-A1 was targeted at the level of GAPDH. However, an activity of GAPDH in platelet was not inhibited directly by CORM-A1 (Figure 5). Similarly, another rate-limiting step of proximal glycolysis—the reaction catalyzed by PFK-1—was not modulated by CORM-A1, as evidence by the lack of effects of 8-minute long incubation with CORM-A1 on PFK-1 activity in platelets (data not shown). Only prolonged 30-minute incubation allowed to observe an inhibition of this enzyme (Figure IA), that was, however, not relevant to the inhibition of glycolysis by short-term exposure of CORM-A1. Another possible explanation for the inhibition of glycolysis at the level of GAPDH by CORM-A1 might have been linked directly to NAD+ depletion (Figure 4).
Reversal of Effects of Pyruvate on CORM-A1–Induced Inhibition of Platelet Aggregation and Glycolysis; Role of LDH and NAD+ Regeneration
As LDH is a major enzymatic source for NAD+ regeneration, we tested whether pyruvate could reverse antiplatelet effects of CORM-A1. As shown in Figure 6A, in the presence of pyruvate (1 mmol/L) the inhibitory effect of CORM-A1 on platelet aggregation was lost (Figure 6A) with concomitant increase in lactate release (Figure 6B) and ECAR (Figure 6C), in contrast to the absence of pyruvate (Figure 1). These data confirmed a high activity of LDH, catalyzing the conversion of exogenous pyruvate into lactate, accompanied by the regeneration of cytosolic NAD+ from NADH. Importantly, in the presence of 1 mmol/L pyruvate, the accumulation of proximal glycolysis intermediates (fructose-1,6-bis-phosphate and DHAP/GA3P; Figure 6D through 6F) was abrogated, and glycolysis flux was efficient again, as also evidenced by the lack of accumulation of PPP intermediates (ribose-5-phosphate, sedoheptulose-7-phosphate, erythrose-4-phosphate; Figure 6H through 6K). These results suggest that pyruvate–induced LDH–dependent regeneration of NAD+ reversed glycolysis inhibition at the level of GAPDH. Furthermore, these results demonstrate that antiaggregatory effect of CORM-A1 was dependent on NAD+ availability, which was modulated by LDH activity. To substantiate the finding on the role of LDH and pyruvate-dependent regeneration of NAD+ in the reversal of the CORM-A1–induced effects, we demonstrated that in the presence of GSK2837808A (LDH inhibitor; 5 μmol/L) the effect of pyruvate (100 μmol/L) was lost, and 300 μmol/L CORM-A1 inhibited platelet aggregation again (Figure 6P). Noteworthy, GSK2837808A in the absence of pyruvate, when added alone, only slightly reduced platelet aggregation, but in combination with CORM-A1 displayed significant antiplatelet effect (Figure 6P). Furthermore, combination of GSK2837808A with antimycin A resulted in a clear-cut antiaggregatory effect, even though neither of them given alone affected platelet aggregation (Figure 6Q). To test a putative functional role of NAD+ derived from malate-aspartate shuttle in platelets, we combined CORM-A1 or antimycin A with aminooxyacetic acid (an inhibitor of malate-aspartate shuttle), but in contrast to the combination of CORM-A1 with GSK2837808A, we did not observe enhancement of antiaggregatory effects of CORM-A1 (data not shown). Dimethyl malonate, which intracellularly is converted to malonate (an inhibitor of SDH [succinate dehydrogenase]), did not show significant antiaggregatory effects alone or in various combinations, excluding a possible role of SDH and malate-aspartate shuttle to regenerate NAD+ in CORM-A1–treated platelets.
Figure 6.
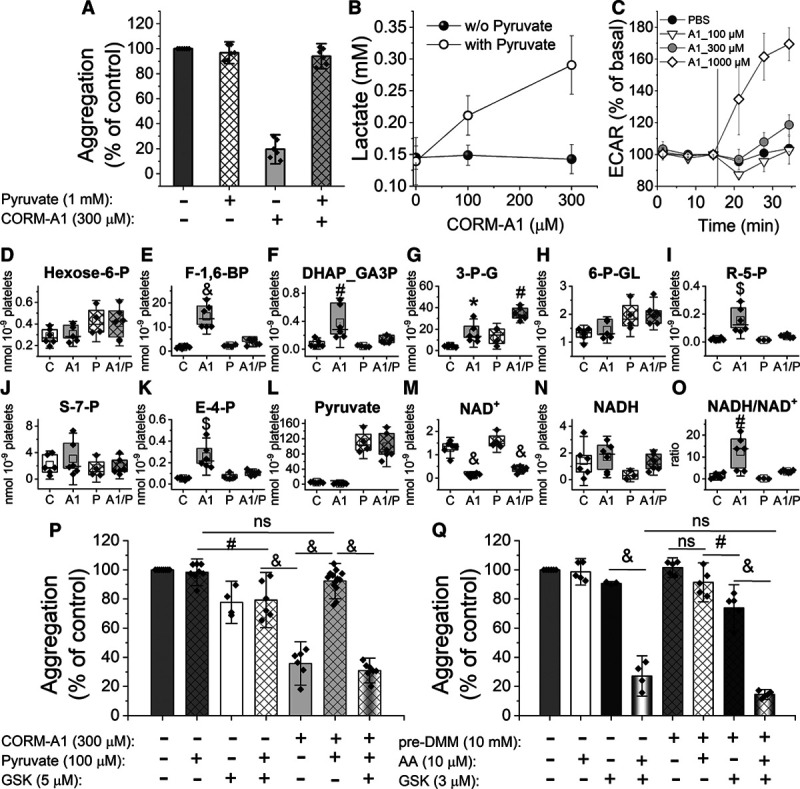
Effects of pyruvate on antiplatelet activity of CORM-A1 in platelets. Human washed platelets (WP) were suspended in assay medium (DMEM) supplemented with glucose (1 g/L) and glutamine (2 mmol/L) with or without pyruvate (1 mmol/L). A, Aggregation of WP treated with CORM-A1 (300 µmol/L) and activated with thrombin (0.1 U/mL) as compared with control; data represent the means±SD of 4 independent experiments; n=1–2 replicates in each experiment. B, Concentration of lactate extruded from platelets (WP) treated with CORM-A1 (0, 100, 300, 1000 µmol/L); data represent the means±SD of 3 independent experiments. C, Extracellular acidification rate (ECAR) measurements of WP treated with PBS (control) or CORM-A1 (A1; 100, 300, 1000 μmol/L) in DMEM supplemented with glucose (1 g/L), glutamine (2 mmol/L), and pyruvate (1 mmol/L); Seahorse XFe96 Analyzer. Data represent means±SD from a representative experiment, n=3–6 technical replicates. D–O, Human WP suspended in PBS containing glucose (1 g/L) and glutamine (2 mmol/L) without (C, A1) or with pyruvate (1 mmol/L; P, A1/P) were untreated or treated with CORM-A1 (300 µmol/L), followed by activation with thrombin (0.1 U/mL; 6 min); data demonstrate concentrations of selected metabolic by-products of glycolysis, PPP or mitochondrial respiration are presented as means±SD (nmol/109 platelets) of 3 independent experiments, n=2 replicates in each experiment. P, Aggregation of WP treated with pyruvate (100 µmol/L), CORM-A1 (300 µmol/L) and GSK2837808A (GSK; 5 µmol/L). Q, Aggregation of WP control or preincubated with dimethyl malonate (DMM; 15 min) followed by treatment with antimycin A (AA; 10 µmol/L) and GSK2837808A (GSK; 3 µmol/L). Data represent the means±SD of 4 independent experiments; n=1–3 replicates in each experiment. DHAP indicates dihydroxyacetone phosphate; F-1,6-BP, fructose-1,6-bis-phosphate; and NAD+, nicotinamide adenine dinucleotide. *P<0.05, #P<0.01, $P<0.001, &P<0.0001.
To further confirm, the key role of cytosolic NAD+ depletion in the antiplatelet effects of CORM-A1, we examined the effects of CORM-A1 on aggregation of platelets pretreated with 78c (0.3–20 μmol/L), an inhibitor of CD38—one of the major NAD+ consuming enzymes. The inhibition of CD38 by 1 µmol/L 78c blunted antiaggregatory effect of CORM-A1 by 13%, further supporting the key role of NAD+ depletion in antiplatelet effects of CORM-A1.
Discussion
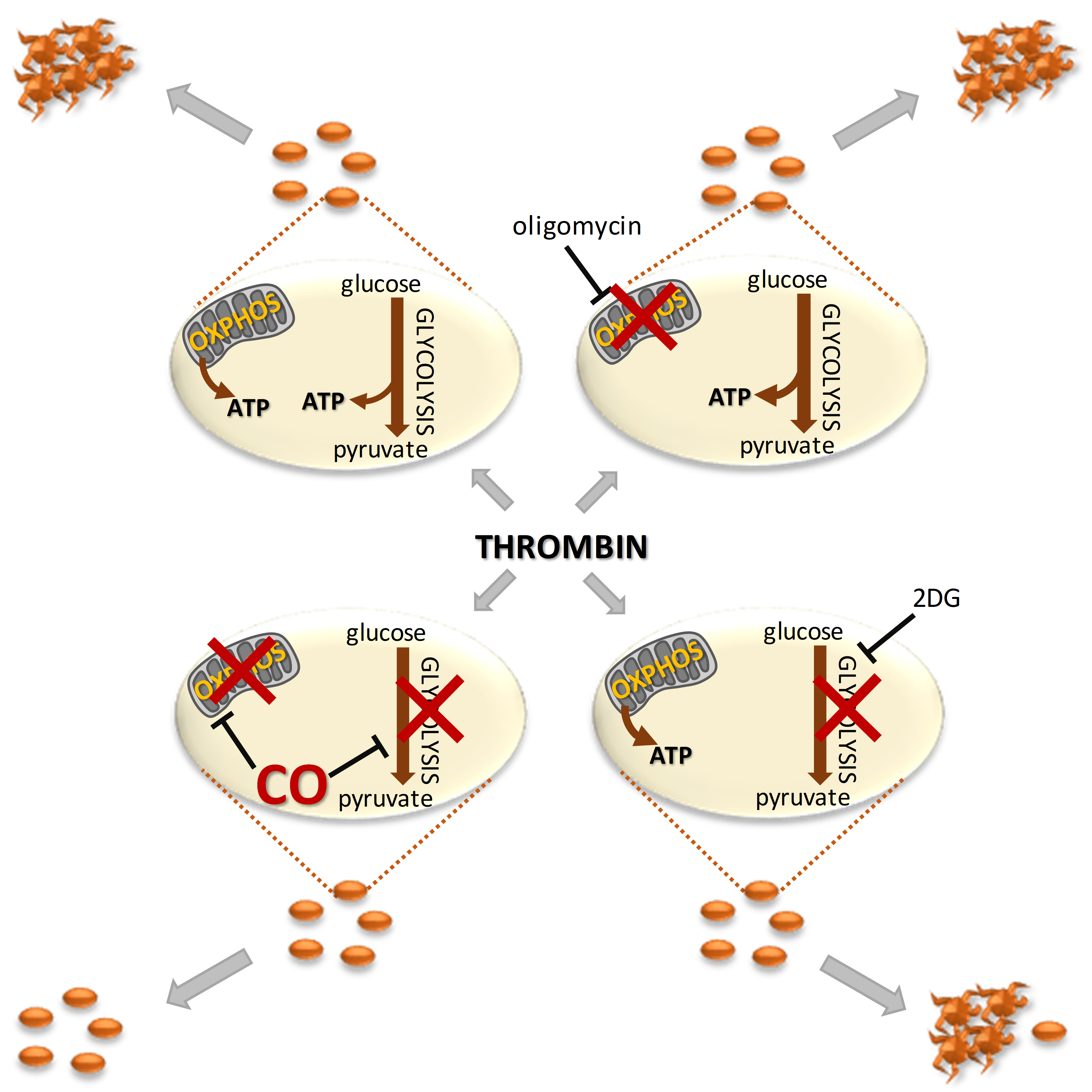
Platelet aggregation is an energy-demanding process,15,16 and recent evidence suggest a high substrate plasticity of metabolism in platelets.17,19,42,43 In the present work, to our knowledge for the first time, we provide evidence that CO affords antiplatelet effects by simultaneous and efficient inhibition of 2 major ATP–generating pathways: mitochondrial respiration, attributed to the inhibition of cytochrome c oxidase, and glycolysis, ascribed to cytosolic NAD+ depletion. Interestingly, an inhibition of glycolysis or mitochondrial respiration individually, using classical metabolic inhibitors, did not inhibit platelet aggregation because these 2 pathways compensate each other, as also suggested previously.19 Combined inhibition of glycolysis and mitochondrial respiration with oligomycin and 2DG, respectively, afforded pronounced platelet inhibition. In this context, our results showing that CORM-A1–induced a simultaneous inhibition of glycolysis and mitochondrial respiration underscores the ability of CO released from CORM-A1 to overcome metabolic plasticity of platelets. CORM-A1–derived CO effectively inhibits platelet aggregation by compromising at the same time 2 major pathways of platelet bioenergetics by distinct mechanisms culminating in NAD+ and ATP depletion. Given the fact that the sGC is a target for CO gas in platelets,9 but not for CORM-A111,12 we suspect that bioenergetic effects of CORM-A1 may not necessarily be shared by gaseous CO.
The effects of CO on mitochondrial respiration and glycolysis, although clearly observed for resting platelets, were more pronounced in platelets metabolically activated by thrombin, oligomycin (activating platelet glycolysis; Figure 3A and 3B), or FCCP (activating maximal mitochondrial respiration; Figure 1C). Platelets in the settings of prothrombotic activation increase energetic demands15,16,19 and, apparently in such settings, platelets also became more susceptible to the inhibitory effects of CO. Altogether, the concentration-dependent inhibition of platelet aggregation by CORM-A1 was accompanied by the concentration-dependent inhibition of platelet bioenergetics; both ensued in the same range of CORM-A1 concentrations and pertained to 2 major pathways of ATP generation, overcoming substrate plasticity of platelets.
Indeed, metabolic plasticity enables platelet aggregation even after inhibition of a single metabolic pathway,16,19 because platelets use diverse metabolic fuels—not only glucose, free fatty acids and glutamine, but also glycogen, citrate (added to the blood during collection), albumin, and acetate.17,19,42,44 Recently, Ravi et al19 demonstrated that the activation of platelets with thrombin results in metabolic reprogramming, leading to increased aerobic glycolysis, fatty acid oxidation and glutaminolysis, which all compensate each other. We confirmed platelet metabolic plasticity in our experimental set-up (Figure 3), demonstrating that selective inhibition of mitochondrial respiration (by oligomycin, antimycin A, rotenone) did not or only slightly (less than by 10%) inhibited platelet aggregation. Single inhibitors of mitochondrial respiration were almost ineffective in inhibition of platelet aggregation (Figure 3C), but inhibition of mitochondrial respiration was compensated by an almost 3-fold increase in glycolysis (Figure 3A and 3B), which probably met platelet aggregation energy requirements. In turn, glycolysis inhibition by 100 mmol/L 2DG slightly (by 21%) inhibited platelet aggregation, implying that glycolysis might be more important for platelet activity. It was suggested that in resting platelets ATP is produced mainly (65%) in glycolysis and only 35% in oxidative phosphorylation.18 Clearly, upon platelet activation, the contributions of these metabolic pathways may be different, depending on the substrates’ availability, underscoring platelets’ plasticity. Interestingly, 3PO is known as an inhibitor of glycolysis,45 but recently it was demonstrated to target and inhibit also TCA cycle and mitochondrial respiratory chain.46 In our model, 3PO appeared to inhibit not only glycolysis, but also oxygen consumption (Figure 3A and 3B), resulting in the inhibition of platelet aggregation in contrast to other metabolic inhibitors given individually. Combined inhibition of both glycolysis and oxidative phosphorylation with 2DG and oligomycin (Figure 3D and 3E) resulted in almost complete inhibition of metabolism and concomitant substantial reduction of platelet aggregation. Given extraordinary platelet metabolic plasticity, high efficacy of CO released from CORM-A1 in the inhibition of platelet aggregation rely on the inhibition of both oxidative phosphorylation and glycolysis and subsequent efficient blocking of ATP production necessary for platelet aggregation.
As regards inhibition of oxidative phosphorylation, we demonstrated that in resting platelets, CO released from CORM-A1 inhibited mitochondrial respiration in a concentration-dependent manner. A CORM-A1–induced fall in concentrations of fumarate and malate (Figure 4) confirms reduced TCA turnover in the presence of CO, while the imbalance in NAD+ and NADH concentrations, seen as increased NADH/NAD+ ratio, confirms the inhibition of oxidative phosphorylation by CO. Altogether, metabolomic results, together with the profile of CORM-A1–induced effects on OCR, provide evidence that CORM-A1 inhibited mitochondrial respiration in platelets, most probably by binding to cytochrome c oxidase, which is a well-known target for CO action.47 Moreover, CORM-A1–induced falls in fumarate and malate (Figure 4) may also suggest succinate dehydrogenase as another possible target for CO in mitochondria. However it seems to be of less significance for platelets’ mitochondrial respiration, as evidenced by the lack of antiplatelet effects of dimethyl malonate (Figure 6Q), a cell permeable precursor of succinate dehydrogenase inhibitor.48 Surprisingly, mitochondria in platelets were more susceptible to the inhibitory effects of CO, as compared with microglia30 or endothelial cells,31 where only higher concentrations of CO inhibited mitochondrial respiration. These results suggest an important regulatory role of mitochondrial ATP turnover in platelets.
Interestingly, CO modulated platelet glycolysis in a biphasic way—activated by lower and inhibited by higher concentrations of CORM-A1 (Figure 1). These results suggest that the observed activation of glycolysis at lower concentrations of CORM-A1 was a compensatory response to the inhibition of mitochondrial respiration. However, CORM-A1 at higher concentrations induced inhibition of mitochondrial respiration and glycolysis, the latter possibly, by separate cytosolic target for CO. In the presence of CORM-A1, we noted a strong increase in proximal metabolites of glycolysis (hexose-6-phosphate, fructose-1,6-bis-phosphate, DHAP and GA3P) and PPP, contrary to the metabolites of distal glycolysis (3-phospho-glicerate, phosphoenolpyruvate and pyruvate), which were unchanged (Figure 5). These results suggest that glycolysis inhibition by CO might be targeted at GAPDH, which contains a haem moiety, and CO–haem interaction may inhibit GAPDH.49 However, the activity of GAPDH in platelets was not inhibited by CORM-A1 (Figure 5). Another reason for the inhibition of GAPDH activity and subsequent inhibition of glycolysis (Figure 5) could be a substantial drop in NAD+ (Figure 4), primarily linked to the inhibition mitochondrial respiration by CO, and a subsequent increase in NADH/NAD+ ratio. However, due to NAD+ compartmentalization (mitochondria contain up to 70% of total NAD content50), it is necessary to consider a separate cytosolic NAD+ pool involved in glycolysis, which may be regenerated from the metabolism of pyruvate by LDH, or through the action of glycerol-3-phosphate shuttle or malate-aspartate shuttle.51 In the present work, we exclude the importance of malate-aspartate shuttle in NAD+ regeneration in platelets (lack of the effects of aminooxyacetic acid added alone or in combination with other compounds on platelet aggregation; data not shown), but we demonstrate that the addition of pyruvate: (1) induced LDH–dependent regeneration of NAD+ accompanying lactate extrusion, (2) reversed glycolysis inhibition at the level of GAPDH, and most importantly, (3) abrogated antiaggregatory effects of CORM-A1 (Figure 6). Indeed, in the presence of pyruvate, NADH/NAD+ ratio was normalized, and accumulation of proximal glycolysis and PPP intermediates were reversed (Figure 6D through 6F and 6H through 6K). Furthermore, inhibition of LDH–dependent regeneration of NAD+ by GSK2837808A resulted in the elimination of the effect of pyruvate (100 µmol/L) on CORM-A1–inhibited platelet aggregation. Inhibition of LDH with simultaneous inhibition of oxidative phosphorylation (Figure 6Q) reduced platelet aggregation. These results confirmed the key role of LDH–dependent NAD+ regeneration for glycolytic flux, the inhibition of which together with the inhibition of mitochondrial respiration resulted in the inhibition of platelet aggregation. Accordingly, NAD+ depletion and subsequent inhibition of glycolysis together with mitochondrial respiration are 2 major targets for CO–induced downregulation of platelet bioenergetics. In the present work, we indicate the importance of NAD+ in platelet glycolysis and aggregation, and suggest that CO, apart from inhibiting cytochrome c oxidase in mitochondria, has another cytosolic target(s) decreasing availability of NAD+, that was, however, not ultimately identified here.
Generally, among the enzymes involved in NAD+ turnover, there are NAD+–regenerating enzymes (as LDH, malate-aspartate shuttle, glycerol-3-phosphate shuttle), NAD+–salvage enzymes (as NAD+ synthase), and NAD+–consuming enzymes (sirtuins, poly [ADP-ribose] polymerases, cADP-ribose synthases).51 Little is known about the NAD+ turnover in platelets, however, it was shown that platelets contain an enzymatic machinery to metabolize nicotinamide riboside into NAD.52 We directed our attention towards CD38 (cADP-ribose synthase), which generates one molecule of cADP-ribose for every 100 molecules of NAD+ hydrolyzed, and plays a role of major regulator of cellular NAD+ availability.53 In fact, we found that 1 μmol/L 78c (inhibitor of CD38) induced a slight, but significant reduction of CORM-A1–induced inhibition of platelet aggregation, supporting the view that modulation of availability of NAD+ by CD38 or other enzymes of NAD+ turnover may influence antiplatelet effects of CORM-A1. However, a direct target for CO responsible for NAD+–deficiency still needs to be identified. It was demonstrated in pancreatic islets that CO activates CD38 through the activation of sGC.54 Nevertheless, in platelets CO released from CORMs does not act through sGC (see Refs11,12 and Figure IV in the Data Supplement) making CO/cGMP/CD38 pathway unlikely to operate in platelets.
Previously, the effects of CO on glycolysis were demonstrated among others in endothelial cells, whereby CO inhibited glycolysis and activated PPP.31–33 Some authors claim that CO-inhibition of glycolysis is attributed to the inhibition of CBS (cystathionine-β-synthase), which through the reduction of methylation of PFK-1/fructose bisphosphatase type-3 (PFKFB3; an activator of glycolytic flux) inhibited glycolysis on the level of PFK-1 and redirected glucose towards PPP.55 As shown in Figure 5, the concentration of fructose-1,6-phosphate as well as the combined concentration of DHAP and GA3P were increased in response to CORM-A1, whereas the concentrations of 3-P-glycerate, phosphoenolpyruvate and pyruvate were unchanged (1,3-bisphosphoglycerate was not analyzed). At first glance, the data could exclude the involvement of reduced activation of PFKFB3 in platelets in response to CO because increased, but not decreased concentration of fructose-1,6-phosphate was measured. Prolonged 30-minute incubation of platelets with CORM-A1 allowed to observe an inhibition of PFK-1 activity, however, not strong enough to avoid accumulation of fructose-1,6-bis-phosphate, DHAP and GA3P (Figure III in the Data Supplement). These data suggest that CBS inhibition, and a consequent PFKFB3 demethylation leading to PFK-1 inhibition, was not responsible for inhibition of glycolysis in platelets treated with CORM-A1, that was in turn ascribed here to NAD+ depletion.
Further studies are needed for a detailed explanation of biochemical mechanisms of CO action in platelets on NAD+ metabolome and elucidation whether haem-dependent or independent mechanisms are involved. Hemoproteins, such as voltage-gated K+ channels56 and epithelial Na+ channels,57 seem unlikely as targets for CO in platelets, as their link with NAD+– consuming processes is not obvious. In turn, large-conductance calcium-regulated K+ channel, known to display a relatively high affinity to CO, is not abundant in platelets.56
In summary, the results of the present work, to our knowledge for the first time, provide evidence that CO affords antiaggregatory effects by inhibition of mitochondrial respiration and glycolysis, attributed to the inhibition of cytochrome c oxidase and depletion of cytosolic NAD+, respectively. The results of our study not only uncover platelet-specific action of CO on bioenergetics, but also point out that metabolism-targeted antiplatelet therapeutic strategies may prove useful for inhibiting excessive platelet activation in various diseases.
Sources of Funding
This work was supported by The National Centre for Research and Development [STRATEGMED1/233226/11/NCBR/2015 to S. Chlopicki], Polish Ministry of Science and Higher Education (213965/E-338/S/2018), and The National Science Centre [2016/21/N/NZ7/02524 (PRELUDIUM) to B. Sitek]. The open-access publication of this article was funded by the Priority Research Area BioS under the program Excellence Initiative–Research University at the Jagiellonian University in Krakow.
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- 2DG
- 2-deoxy-D-glucose
- CBS
- cystathionine-β-synthase
- CO
- carbon monoxide
- CORM
- carbon monoxide-releasing molecule
- DHAP
- dihydroxyacetone phosphate
- ECAR
- extracellular acidification rate
- GA3P
- glyceraldehyde 3-phosphate
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- LDH
- lactate dehydrogenase
- LPS
- lipopolysaccharide
- NAD+
- nicotinamide adenine dinucleotide
- OCR
- oxygen consumption rate
- PFK-1
- phosphofructokinase 1
- PPP
- pentose phosphate pathway
- SDH
- succinate dehydrogenase
- sGC
- soluble guanyl cyclase
- TCA
- tricarboxylic acid cycle
- WP
- washed platelets
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.314284.
References
- 1.Ryter SW, Otterbein LE, Morse D, Choi AM. Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem. 2002;234-235:249–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motterlini R, Foresti R. Heme oxygenase-1 as a target for drug discovery. Antioxid Redox Signal. 2014;20:1810–1826. doi: 10.1089/ars.2013.5658 [DOI] [PubMed] [Google Scholar]
- 3.Tamura T, Kondo T, Ogawa K, Fukunaga K, Ohkohchi N. Protective effect of heme oxygenase-1 on hepatic ischemia-reperfusion injury through inhibition of platelet adhesion to the sinusoids. J Gastroenterol Hepatol. 2013;28:700–706. doi: 10.1111/jgh.12075 [DOI] [PubMed] [Google Scholar]
- 4.True AL, Olive M, Boehm M, San H, Westrick RJ, Raghavachari N, Xu X, Lynn EG, Sack MN, Munson PJ, et al. Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to the endothelium, which is rescued by inhaled carbon monoxide. Circ Res. 2007;101:893–901. doi: 10.1161/CIRCRESAHA.107.158998 [DOI] [PubMed] [Google Scholar]
- 5.Chen B, Guo L, Fan C, Bolisetty S, Joseph R, Wright MM, Agarwal A, George JF. Carbon monoxide rescues heme oxygenase-1-deficient mice from arterial thrombosis in allogeneic aortic transplantation. Am J Pathol. 2009;175:422–429. doi: 10.2353/ajpath.2009.081033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R, Green CJ. CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005;19:284–286. doi: 10.1096/fj.04-2169fje [DOI] [PubMed] [Google Scholar]
- 7.Kramkowski K, Leszczynska A, Mogielnicki A, Chlopicki S, Fedorowicz A, Grochal E, Mann B, Brzoska T, Urano T, Motterlini R, et al. Antithrombotic properties of water-soluble carbon monoxide-releasing molecules. Arterioscler Thromb Vasc Biol. 2012;32:2149–2157. doi: 10.1161/ATVBAHA.112.253989 [DOI] [PubMed] [Google Scholar]
- 8.Prieto L, Rossier J, Derszniak K, Dybas J, Oetterli RM, Kottelat E, Chlopicki S, Zelder F, Zobi F. Modified biovectors for the tuneable activation of anti-platelet carbon monoxide release. Chem Commun (Camb). 2017;53:6840–6843. doi: 10.1039/c7cc03642f [DOI] [PubMed] [Google Scholar]
- 9.Brüne B, Ullrich V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol. 1987;32:497–504 [PubMed] [Google Scholar]
- 10.Brüne B, Schmidt KU, Ullrich V. Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. Eur J Biochem. 1990;192:683–688. doi: 10.1111/j.1432-1033.1990.tb19276.x [DOI] [PubMed] [Google Scholar]
- 11.Chlopicki S, Olszanecki R, Marcinkiewicz E, Lomnicka M, Motterlini R. Carbon monoxide released by CORM-3 inhibits human platelets by a mechanism independent of soluble guanylate cyclase. Cardiovasc Res. 2006;71:393–401. doi: 10.1016/j.cardiores.2006.03.011 [DOI] [PubMed] [Google Scholar]
- 12.Chlopicki S, Lomnicka M, Fedorowicz A, Grochal E, Kramkowski K, Mogielnicki A, Buczko W, Motterlini R. Inhibition of platelet aggregation by carbon monoxide-releasing molecules (CO-RMs): comparison with NO donors. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:641–650. doi: 10.1007/s00210-012-0732-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu D, Liang F, Wang X, Cao J, Qin W, Sun B. Suppressive effect of CORM-2 on LPS-induced platelet activation by glycoprotein mediated HS1 phosphorylation interference. PLoS One. 2013;8:e83112 doi: 10.1371/journal.pone.0083112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hally KE, La Flamme AC, Harding SA, Larsen PD. The effects of aspirin and ticagrelor on Toll-like receptor (TLR)-mediated platelet activation: results of a randomized, cross-over trial. Platelets. 2019;30:599–607. doi: 10.1080/09537104.2018.1479520 [DOI] [PubMed] [Google Scholar]
- 15.Holmsen H, Kaplan KL, Dangelmaier CA. Differential energy requirements for platelet responses. A simultaneous study of aggregation, three secretory processes, arachidonate liberation, phosphatidylinositol breakdown and phosphatidate production. Biochem J. 1982;208:9–18. doi: 10.1042/bj2080009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corona de la Peña N, Gutiérrez-Aguilar M, Hernández-Reséndiz I, Marín-Hernández Á, Rodríguez-Enríquez S. Schulz C. Glycoprotein Ib activation by thrombin stimulates the energy metabolism in human platelets. PLoS One. 2017;12:e0182374 doi: 10.1371/journal.pone.0182374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karpatkin S. Studies on human platelet glycolysis. Effect of glucose, cyanide, insulin, citrate, and agglutination and contraction on platelet glycolysis. J Clin Invest. 1967;46:409–417. doi: 10.1172/JCI105542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akkerman JW, Holmsen H. Interrelationships among platelet responses: studies on the burst in proton liberation, lactate production, and oxygen uptake during platelet aggregation and Ca2+ secretion. Blood. 1981;57:956–966 [PubMed] [Google Scholar]
- 19.Ravi S, Chacko B, Sawada H, Kramer PA, Johnson MS, Benavides GA, O’Donnell V, Marques MB, Darley-Usmar VM. Metabolic plasticity in resting and thrombin activated platelets. PLoS One. 2015;10:e0123597 doi: 10.1371/journal.pone.0123597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardenes N, Corey C, Geary L, Jain S, Zharikov S, Barge S, Novelli EM, Shiva S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood. 2014;123:2864–2872. doi: 10.1182/blood-2013-09-529420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu W, Cardenes N, Corey C, Erzurum SC, Shiva S. Platelets from asthmatic individuals show less reliance on glycolysis. PLoS One. 2015;10:e0132007 doi: 10.1371/journal.pone.0132007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjövall F, Morota S, Hansson MJ, Friberg H, Gnaiger E, Elmér E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit Care. 2010;14:R214 doi: 10.1186/cc9337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schapira AH, Gu M, Taanman JW, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM. Mitochondria in the etiology and pathogenesis of Parkinson’s disease. Ann Neurol. 1998;443 Suppl 1S89–S98. doi: 10.1002/ana.410440714 [DOI] [PubMed] [Google Scholar]
- 24.Ran J, Guo X, Li Q, Mei G, Lao G. Platelets of type 2 diabetic patients are characterized by high ATP content and low mitochondrial membrane potential. Platelets. 2009;20:588–593. doi: 10.3109/09537100903288422 [DOI] [PubMed] [Google Scholar]
- 25.Lo Iacono L, Boczkowski J, Zini R, Salouage I, Berdeaux A, Motterlini R, Morin D. A carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radic Biol Med. 2011;50:1556–1564. doi: 10.1016/j.freeradbiomed.2011.02.033 [DOI] [PubMed] [Google Scholar]
- 26.Long R, Salouage I, Berdeaux A, Motterlini R, Morin D. CORM-3, a water soluble CO-releasing molecule, uncouples mitochondrial respiration via interaction with the phosphate carrier. Biochim Biophys Acta. 2014;1837:201–209. doi: 10.1016/j.bbabio.2013.10.002 [DOI] [PubMed] [Google Scholar]
- 27.Queiroga CS, Almeida AS, Alves PM, Brenner C, Vieira HL. Carbon monoxide prevents hepatic mitochondrial membrane permeabilization. BMC Cell Biol. 2011;12:10 doi: 10.1186/1471-2121-12-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reiter CE, Alayash AI. Effects of carbon monoxide (CO) delivery by a CO donor or hemoglobin on vascular hypoxia inducible factor 1α and mitochondrial respiration. FEBS Open Bio. 2012;2:113–118. doi: 10.1016/j.fob.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wegiel B, Gallo D, Csizmadia E, Harris C, Belcher J, Vercellotti GM, Penacho N, Seth P, Sukhatme V, Ahmed A, et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 2013;73:7009–7021. doi: 10.1158/0008-5472.CAN-13-1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson JL, Bouillaud F, Almeida AS, Vieira HL, Ouidja MO, Dubois-Randé JL, Foresti R, Motterlini R. Carbon monoxide reverses the metabolic adaptation of microglia cells to an inflammatory stimulus. Free Radic Biol Med. 2017;104:311–323. doi: 10.1016/j.freeradbiomed.2017.01.022 [DOI] [PubMed] [Google Scholar]
- 31.Kaczara P, Motterlini R, Rosen GM, Augustynek B, Bednarczyk P, Szewczyk A, Foresti R, Chlopicki S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: a role for mitoBKCa channels. Biochim Biophys Acta. 2015;1847:1297–1309. doi: 10.1016/j.bbabio.2015.07.004 [DOI] [PubMed] [Google Scholar]
- 32.Kaczara P, Motterlini R, Kus K, Zakrzewska A, Abramov AY, Chlopicki S. Carbon monoxide shifts energetic metabolism from glycolysis to oxidative phosphorylation in endothelial cells. FEBS Lett. 2016;590:3469–3480. doi: 10.1002/1873-3468.12434 [DOI] [PubMed] [Google Scholar]
- 33.Kaczara P, Proniewski B, Lovejoy C, Kus K, Motterlini R, Abramov AY, Chlopicki S. CORM-401 induces calcium signalling, NO increase and activation of pentose phosphate pathway in endothelial cells. FEBS J. 2018;285:1346–1358. doi: 10.1111/febs.14411 [DOI] [PubMed] [Google Scholar]
- 34.Radomski MW, Palmer RM, Read NG, Moncada S. Isolation and washing of human platelets with nitric oxide. Thromb Res. 1988;50:537–546. doi: 10.1016/0049-3848(88)90202-2 [DOI] [PubMed] [Google Scholar]
- 35.Born GV. Possible mechanisms of platelet aggregation by ADP and of its inhibition. Thromb Diath Haemorrh Suppl. 1967;26:173–174 [PubMed] [Google Scholar]
- 36.Fayad-Kobeissi S, Ratovonantenaina J, Dabiré H, Wilson JL, Rodriguez AM, Berdeaux A, Dubois-Randé JL, Mann BE, Motterlini R, Foresti R. Vascular and angiogenic activities of CORM-401, an oxidant-sensitive CO-releasing molecule. Biochem Pharmacol. 2016;102:64–77. doi: 10.1016/j.bcp.2015.12.014 [DOI] [PubMed] [Google Scholar]
- 37.Kus K, Kij A, Zakrzewska A, Jasztal A, Stojak M, Walczak M, Chlopicki S. Alterations in arginine and energy metabolism, structural and signalling lipids in metastatic breast cancer in mice detected in plasma by targeted metabolomics and lipidomics. Breast Cancer Res. 2018;20:148 doi: 10.1186/s13058-018-1075-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt MM, Dringen R. Differential effects of iodoacetamide and iodoacetate on glycolysis and glutathione metabolism of cultured astrocytes. Front Neuroenergetics. 2009;1:1 doi: 10.3389/neuro.14.001.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bisswanger H. Second. Practical Enzymology. 2011. Weinheim: Wiley-Blackwell; 117–119 [Google Scholar]
- 40.Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080 [DOI] [PubMed] [Google Scholar]
- 41.Ishikawa E, Ogushi S, Ishikawa T, Uyeda K. Activation of mammalian phosphofructokinases by ribose 1,5-bisphosphate. J Biol Chem. 1990;265:18875–18878 [PubMed] [Google Scholar]
- 42.Doery JC, Hirsh J, Cooper I. Energy metabolism in human platelets: interrelationship between glycolysis and oxidative metabolism. Blood. 1970;36:159–168 [PubMed] [Google Scholar]
- 43.Fink BD, Herlein JA, O’Malley Y, Sivitz WI. Endothelial cell and platelet bioenergetics: effect of glucose and nutrient composition. PLoS One. 2012;7:e39430 doi: 10.1371/journal.pone.0039430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guppy M, Abas L, Neylon C, Whisson ME, Whitham S, Pethick DW, Niu X. Fuel choices by human platelets in human plasma. Eur J Biochem. 1997;244:161–167. doi: 10.1111/j.1432-1033.1997.00161.x [DOI] [PubMed] [Google Scholar]
- 45.Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW, Quaegebeur A, Goveia J, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 46.Nukala SB, Baron G, Aldini G, Carini M, D’Amato A. Mass spectrometry-based label-free quantitative proteomics to study the effect of 3PO drug at cellular level. ACS Med Chem Lett. 2019;10:577–583. doi: 10.1021/acsmedchemlett.8b00593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6 [DOI] [PubMed] [Google Scholar]
- 48.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–470.e13. doi: 10.1016/j.cell.2016.08.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hannibal L, Collins D, Brassard J, Chakravarti R, Vempati R, Dorlet P, Santolini J, Dawson JH, Stuehr DJ. Heme binding properties of glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 2012;51:8514–8529. doi: 10.1021/bi300863a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. 2015;50:284–297. doi: 10.3109/10409238.2015.1028612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delabie W, Maes W, Devloo R, Van den Hauwe MR, Vanhoorelbeke K, Compernolle V, Feys HB. The senotherapeutic nicotinamide riboside raises platelet nicotinamide adenine dinucleotide levels but cannot prevent storage lesion. Transfusion. 2020;60:165–174. doi: 10.1111/trf.15556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chini EN. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des. 2009;15:57–63. doi: 10.2174/138161209787185788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rahman FU, Park DR, Joe Y, Jang KY, Chung HT, Kim UH. Critical roles of carbon monoxide and nitric oxide in Ca 2+ signaling for insulin secretion in pancreatic islets. Antioxidants Redox Signal. 2019;30:560–576. doi: 10.1089/ars.2017.7380 [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto T, Takano N, Ishiwata K, Ohmura M, Nagahata Y, Matsuura T, Kamata A, Sakamoto K, Nakanishi T, Kubo A, et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun. 2014;5:3480 doi: 10.1038/ncomms4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Silva HA, Carver JG, Aronson JK. Pharmacological evidence of calcium-activated and voltage-gated potassium channels in human platelets. Clin Sci (Lond). 1997;93:249–255. doi: 10.1042/cs0930249 [DOI] [PubMed] [Google Scholar]
- 57.Cerecedo D, Martínez-Vieyra I, Alonso-Rangel L, Benítez-Cardoza C, Ortega A. Epithelial sodium channel modulates platelet collagen activation. Eur J Cell Biol. 2014;93:127–136. doi: 10.1016/j.ejcb.2014.02.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.