

Supplemental Digital Content is available in the text.
Keywords: apolipoproteins E; atherosclerosis; cardiovascular diseases; coronary artery disease; RNA, untranslated
Abstract
Objective:
microRNAs are master regulators of gene expression with essential roles in virtually all biological processes. miR-217 has been associated with aging and cellular senescence, but its role in vascular disease is not understood.
Approach and Results:
We have used an inducible endothelium-specific knock-in mouse model to address the role of miR-217 in vascular function and atherosclerosis. miR-217 reduced NO production and promoted endothelial dysfunction, increased blood pressure, and exacerbated atherosclerosis in proatherogenic apoE−/− mice. Moreover, increased endothelial miR-217 expression led to the development of coronary artery disease and altered left ventricular heart function, inducing diastolic and systolic dysfunction. Conversely, inhibition of endogenous vascular miR-217 in apoE−/− mice improved vascular contractility and diminished atherosclerosis. Transcriptome analysis revealed that miR-217 regulates an endothelial signaling hub and downregulates a network of eNOS (endothelial NO synthase) activators, including VEGF (vascular endothelial growth factor) and apelin receptor pathways, resulting in diminished eNOS expression. Further analysis revealed that human plasma miR-217 is a biomarker of vascular aging and cardiovascular risk.
Conclusions:
Our results highlight the therapeutic potential of miR-217 inhibitors in aging-related cardiovascular disease.
Highlights.
miR-217 promotes endothelial dysfunction, aggravates atherosclerosis, and promotes cardiovascular dysfunction.
miR-217 regulates a hub of endothelial functions and triggers a coordinated dampening of eNOS (endothelial NO synthase) signaling pathways.
miR-217 is a biomarker of cardiovascular aging in humans.
Cardiovascular disease (CVD) is the leading cause of death in the Western world (http://www.who.int/cardiovascular_diseases/resources/atlas/en/), and the major risk factor for CVD is aging. CVD affects around 40% of people aged between 40 and 60 years and >80% of those aged over 85 years,1 with the most prevalent conditions being heart failure, hypertension, and coronary artery disease. A key determinant of the development of these CVD syndromes is the aging of the vasculature.2 The molecular pathways underlying vascular aging are not fully understood; however, it is known that one of the major contributors to atherosclerosis and CVD development is endothelial dysfunction.3
MicroRNAs (miRNAs) are small noncoding RNA molecules that fine-tune cellular homeostasis through the posttranscriptional control of gene networks. miRNAs play crucial roles in cardiovascular tissue homeostasis and disease, and there is growing interest in the development of miRNA-targeted therapies for CVD.4,5 Reduction or loss of the pre-miRNA processing enzyme Dicer in vivo disrupts various aspects of endothelial biology, including NO production and endothelial growth, demonstrating the relevance of miRNAs to endothelial function.6–8 Several studies have characterized the function of specific miRNAs that modulate endothelial cell physiology.9 miR-217 was identified as the most highly induced miRNA during in vitro human endothelial cell aging, resulting in the downregulation of the expression of Sirt1 (silent information regulator 1).10 Sirt1 is an NAD+-dependent deacetylase that regulates gene expression and plays an important role in longevity through the regulation of metabolic adaptation and cardiovascular protection.11 In endothelial cells, Sirt1 activates eNOS (endothelial NO synthase).12 eNOS-derived NO plays crucial roles in the regulation of vascular tone, cell proliferation, leukocyte adhesion, and platelet aggregation.13,14 However, knowledge remains limited about the pathophysiological impact of miR-217 expression on endothelial cells in vivo and the effect of miR-217 on the endothelial transcriptome.
In the present study, we used gain- and loss-of-function strategies to modify miR-217 expression in mice and thus decipher the impact of miR-217 on cardiovascular physiology and pathology. Transcriptome analysis showed that endothelial miR-217 expression is a master regulator of vascular signaling and decreases various eNOS stimulatory pathways. Accordingly, we found that miR-217 reduces NO production, promotes atherosclerosis, and impairs cardiovascular function. Moreover, we found increased miR-217 in human plasma from aged individuals and CVD patients. Our results thus reveal miR-217 as a promising therapeutic target and a novel biomarker of cardiovascular aging.
Experimental Procedures
Mice
Endothelial-specific inducible miR-217 C57BL/6J knock-in mice were generated by crossing Rosa26 miR-217 knock-in mice15 with the endothelium-specific tamoxifen-inducible VE-Cad (VE-cadherin) CreERT2 line.16 miR-217KI/+ VE-Cad CreERT2TG/+ mice (miR-217 mice) were crossed to the apoE−/− C57BL/6J proatherogenic background and fed a high-fat diet (HFD) containing 10.7% total fat and 0.75% cholesterol (Ssniff Spezialdiäten) for 7 to 12 weeks when indicated. In both models, Cre expression was induced by intraperitoneal administration of tamoxifen (1 mg tamoxifen/mouse for 5 consecutive days). When indicated, resveratrol was administrated during 1 week in drinking water at 1 mg/mL (400 mg/kg per day). In adherence to Arteriosclerosis, Thrombosis, and Vascular Biology Council guidelines, endothelium-dependent NO production, vessel contractility, and atherosclerosis studies were performed with groups of animals of the same sex, given that sex difference is a biological variable in these studies. All other studies were performed with equally distributed sex-matched experimental groups. Animal procedures were approved by the Centro Nacional de Investigaciones Cardiovasculares Ethics Committee and the Madrid regional authorities (PROEX 377/15) and conformed to EU Directive 2010/63EU and Recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes, enforced in Spanish law under Real Decreto 1201/2005.
Human Samples
Plasma from 41 disease-free nonagenarian individuals and from 42 healthy women aged <50 years and not diagnosed with any relevant disease and representative of the Spanish population was obtained from the Banco Nacional de ADN Carlos III-Salamanca University Biobank (http://www.bancoadn.org/en). Plasma samples from 32 patients diagnosed with coronary atherosclerosis and 34 age- and sex-matched healthy control individuals were obtained with informed consent at the Hospital Universitario 12 de Octubre (Madrid). The use of human samples in this project was approved by the Instituto de Salud Carlos III Ethics Committee (CEI PI 23_2017) and by the Institutional Review Board at the Instituto de Investigación Hospital 12 de Octubre (imas12; CEI 16/044).
Primary Mouse Lung Endothelial Cell Isolation and Culture
Lung endothelial cells were extracted from wild-type C57BL/6J mice. Lungs were disaggregated with 0.1% collagenase (Roche). Lung cell suspensions were seeded on a gelatin-collagen coated matrix (Sigma) and cultured for 24 hours in DMEM/F-12 supplemented with 20% fetal calf serum and bovine brain endothelial cell growth factor. Cells were then harvested, depleted of macrophages by negative magnetic selection with rat anti-mouse CD16/CD32 (cluster of differentiation 16/cluster of differentiation 32) antibody (BD Pharmingen) and sheep anti-rat IgG dynabeads (Invitrogen), and cultured for 48 hours. Mouse lung endothelial cells (MLECs) were isolated by positive magnetic selection using rat anti-mouse ICAM-2 (intercellular adhesion molecule 2) antibody (BD Pharmingen) and sheep anti-rat IgG dynabeads.
Cell Culture of Human Vein Endothelial Cells
Primary human vein endothelial cells were seeded on a gelatin-coated matrix (Sigma) and cultured for 3 days in DMEM/F-12 supplemented with 20% fetal calf serum, bovine brain endothelial cell growth factor, and different doses (from 0 to 100 µM) of resveratrol (Invivochem).
RNA Isolation and Real-Time Polymerase Chain Reaction
RNA was extracted with Trizol (Invitrogen) from cells or ultra-turrax disrupted aorta. RNA was extracted from plasma or serum with the miRCURY RNA biofluids isolation kit (Exiqon). For quantitative real-time polymerase chain reaction, total RNA was converted to cDNA using random primers (Roche), reverse transcribed with SuperScript II (Invitrogen), and analyzed by SYBR Green assay (Applied Biosystems) with mouse-specific primers: Sirt1 (forward) 5′-tgtgaagttactgcaggagtgtaaa-3′, (reverse) 5′-gcatagataccgtctcttgatctgaa-3′; TNFα (tumor necrosis factor-alpha; forward) 5′-agcccacgtcgtagcaaacca-3′, (reverse) 5′-acaacccatcggctggcacc-3′; IL (interleukin)-6 (forward) 5′-gaaaatctgctctggtcttctgg-3′, (reverse) 5′-ttttctgaccacagtgaggaatg-3′; IL-1Rα (forward) 5′-tagacatggtgcctattgacct-3′, (reverse) 5′-tcgtgactataaggggctcttc-3′; Icam (forward) 5′-cgctgtgctttgagaactgtg-3′, (reverse) 5′-atacacggtgatggtagcgga-3′; Aplnr (forward) 5′-ccttaccacctggtgaagac-3′, (reverse) 5′-aggtgcagtacggaaagac-3′; VEGF (vascular endothelial growth factor) R1 (forward) 5′-acagaaggagagcagaagtc-3′, (reverse) 5′-cttcatcgttacagcagcc-3′; VEGFA (forward) 5′-acagaaggagagcagaagtc-3′, (reverse) 5′-cttcatcgttacagcagcc3′; Lpar (lysophosphatidic acid receptor)-1 (forward) 5′-gaaagcaagcatgtggtgtg-3′, (reverse) 5′-cactctttgggtgacaagctc-3′; Adcy2 (adenylate cyclase 2; forward) 5′-ctttggagggaccgtgtctg-3′, (reverse) 5′-ccctcatgttgaagggaagc-3′; Nos3 (forward) 5′-atcacctacgacaccctcag-3′, (reverse) 5′-cggctctgtaacttccttgg-3′. Gapdh was used as a normalization control: (forward) 5′-tgaagcaggcatctgaggg-3′, (reverse) 5′-cgaaggtggaagagtgggag-3′. Human specific primers: SIRT1 (forward) 5′-cttcgcaactatacccagaac-3′, (reverse) 5′-cctcgtacagcttcacagtc-3′; NOS (NO synthase) 3 (forward) 5′-ctcatgggcacggtgatg-3′, (reverse) 5′-accacgtcatactcatccatacac-3′; GAPDH (forward) 5′-gaaggtgaaggtcggagtc-3′, (reverse) 5′-gaagatggtgatgggatttc-3′. For mature miR-217 quantification, RNA was converted to cDNA with the miRCURY LNA Universal RT kit (Exiqon). miR-217 was quantified with miR-217 miRCURY LNA primers (Exiqon). The presence of miR-217 was considered positive in samples in which all technical triplicates amplified before cycle 40. The normalization control for samples extracted from cells or tissue was U6. The normalization controls for serum and plasma samples were miR-16 and UniSp6 spike-in (Exiqon).
Immunohistochemistry
Mouse aortas were perfused with saline solution, cleaned under a microscope, excised, and fixed in formalin for 2 days before being embedded in paraffin blocks. Aortic cross sections were stained with antibodies against GFP (green fluorescent protein; Acris Antibodies) and CD31 (Abcam). GFP immunohistochemistry was quantified in the aorta intima using Tissuemorf software (Viopharm).
Isometric Tension Studies
Mice were euthanized by CO2 inhalation. The aorta and first-order mesenteric artery branches were removed and placed in cold (4°C) Krebs-Henseleit solution (KHS; 115 mmol/L NaCl, 25 mmol/L NaHCO3, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4.7H2O, 2.5 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 11.1 mmol/L glucose, and 0.01 mmol/L Na2EDTA) bubbled with a 95% O2-5% CO2 mixture (pH=7.4). Ring segments of mouse aorta and mesenteric arteries, ≈2 mm in length, were mounted in a small vessel chamber myograph (Danish Myotechnology) for the measurement of isometric tension. Two tungsten wires (40-mm diameter) were introduced through the lumen of the segments. After a 30-minute equilibration period in oxygenated KHS, arterial segments were stretched to their optimal lumen diameter for active tension development. Segment contractility was then tested with an initial exposure to high-K+ solution (K+-KHS: KHS containing 120 mmol/L KCl). The presence of endothelium was determined by the ability of 10 µmol/L acetylcholine to relax arteries precontracted with phenylephrine at ≈50% K+-KHS contraction. Afterward, a single concentration-response curve was recorded in response to acetylcholine (1 nmol/L to 30 µmol/L), phenylephrine (1 nmol/L to 0.1 mmol/L), and diethylamine NONOate (1 nmol/L to 30 µmol/L). In some experiments, L-NAME (Nω-nitro-L-arginine methyl ester; 100 µmol/L) was added 30 minutes before recording the phenylephrine concentration-response curves.
All drugs were dissolved in distilled water. Phenylephrine hydrochloride (catalog No. P6126), acetylcholine chloride (catalog No. A9101), diethylamine NONOate (catalog No. D5431), and L-NAME (catalog No. N5751) were purchased from Sigma-Aldrich.
Vasoconstrictor responses are expressed as a percentage of the tone generated by K+-KHS. Vasodilator responses are expressed as the percentage of the previous tone induced by phenylephrine in each case.
Quantification of NO Production
After equilibration for 60 minutes in HEPES buffer at 37°C, aortic segments were incubated for 45 minutes with the fluorescent probe 4,5-diaminofluorescein (2 µmol/L; Sigma-Aldrich; catalog No. D225). The medium was collected to measure baseline NO release, and the segments were incubated with phenylephrine (1 µmol/L, 5 minutes) and acetylcholine (10 µmol/L, 10 minutes). Thereafter, the medium was collected to measure agonist-induced NO release. Medium fluorescence was measured at room temperature using a spectrofluorimeter at an excitation wavelength of 492 nm and emission wavelength of 515 nm. To control for day-to-day fluorescence fluctuations, all experimental parameters were measured every day. NO release was calculated by subtracting baseline NO release from that evoked by acetylcholine. Blank samples were collected in the same way from segment-free medium to correct for background emission. The amount of NO released was expressed in arbitrary units/min·µg−1 protein. Variations in NO release were calculated as a percentage relative to controls.
Blood Pressure Measurements in Mice
Arterial blood pressure (BP) was measured in mouse tails using the automated BP-2000 Blood Pressure Analysis System (Visitech Systems). BP measurements were recorded with the tail-cuff method in restrained mice kept on a warmed 37°C surface. Before recording BP, mice were trained on the equipment over 5 consecutive days. After the training period, BP was measured in 3 independent aged-matched cohorts of HFD-fed mice. Twenty consecutive systolic and diastolic BP measurements were performed per mouse, and the last 10 readings were recorded and averaged.
Atherosclerosis Development Quantification
Atherosclerosis development was quantified in oil red O–stained aortic root heart cross sections from HFD-fed control and miR-217 gain- and loss-of-function apoE−/− mice. Saline-perfused hearts were fixed for 2 hours in 4% paraformaldehyde, embedded in optimal cutting temperature tissue embedding compound, and frozen at −80°C. Atherosclerotic plaque area and volumes were quantified by ImageJ analysis of oil red O–stained serial 80-µm spaced cryostat cross sections of aortic root, starting from the aortic sinus. Atherosclerosis plaque volume was calculated as the area under the curve in plots that quantify atherosclerosis lesion area through the aortic sinus and ascending aorta for each condition, as explained in detail in.17 The necrotic core within atherosclerotic lesions was quantified from Masson trichrome–stained cryostat cross sections as the area lacking extracellular matrix (total loss of collagen). Atherosclerotic plaques in the left coronary artery were quantified from oil red O–stained aortic root cryosections of HFD-fed control and miR-217 apoE−/− mice. Methods adhered to the American Heart Association guidelines for experimental atherosclerosis studies, which include recommendations for randomization, blinding, and data handling.
Immunofluorescence
For tissue immunofluorescence, hearts were fixed with 4% paraformaldehyde, incubated with 30% sucrose, embedded in optimal cutting temperature compound (Olympus), and frozen on dry ice. Ten-micrometer sections were permeabilized with 0.5% Triton X-100 (Sigma) and blocked with 2% BSA (Sigma) and 5% normal serum (Sigma) and stained with the following antibodies: anti-SMA-1 (smooth muscle actin-1) biotin (ThermoFisher Scientific), anti-MAC-2 (galectin 3; TebaBio), anti-CD68 (Serotec MCA1957), anti-VCAM-1 (vascular cell adhesion molecule 1; ab134047), biotinylated anti-ICAM-1 (553251 BD Pharmingen), anti-CD31 (Abcam ab28364 and MAB1398Z), and anti-active caspase-3 (R&D AF835) anti-mouse antibodies; goat anti-rabbit Alexa Fluor 568 and Alexa Fluor 488; goat anti-hamster IgG Alexa Fluor 647; goat anti-rabbit IgG Alexa Fluor 568; and chicken anti-rat IgG Alexa Fluor 488 and streptavidin Alexa Fluor 488 and Cy3. Slides were mounted with Prolong Gold (Life Technologies) after DAPI (4′,6-diamidino-2-phenylindole) staining, and images were captured with a Leica SPE or SP5 confocal microscope. Automated quantification of Mac2, CD68, SMCa (smooth muscle cell actin), and active caspase-3 immunofluorescence within atheroma plaques was performed by automatic threshold analysis of immunofluorescence images in Image J/Fiji.
Lipid Profile Biochemistry
Lipid profiline was performed in serum extracted from cohorts of 24-hour-fasted HFD-fed control and miR-217 apoE−/− mice with a Dimension RxL Max analyzer.
Echocardiography and Vascular Ultrasound
In vivo ultrasound studies were performed in 2 independent aged-matched cohorts of HFD-fed control and miR-217 apoE−/− mice. Mice anesthetized with 2% isoflurane mice were examined by high-frequency ultrasound at 30-μm resolution with a VEVO 2100 echography device (VisualSonics, Toronto, Canada). Images were analyzed with VEVO 2100 software, version 1.5.0. All recordings were made by a cardiac imaging technician blinded to animal genotype. Aortic and carotid artery maximal diameters were monitored in systole and diastole. For the assessment of left ventricular systolic function, parasternal standard 2-dimensional long- and short-axis views were acquired. Left ventricular ejection fraction and left ventricular end-diastolic volume were obtained from the long-axis view. For the study of diastolic function, mitral valve flow was evaluated using pulsed-wave Doppler echography in the 4-chamber apical view. Assessed parameters included early and late diastolic velocity peak waves (E and A, respectively), E/A ratio, and pulmonary venous diastolic and systolic profile flow from a modified angled 2-dimensional long-axis view.
miR-217 Inhibition by Lentiviral Infection
The miR-217 sponge construct (miR-217SPG) vector, including GFP and tandem sequences complementary to miR-217,15 was cloned into the pHRSIN lentiviral vector.18 Pseudotyped lentiviruses were produced by transient calcium phosphate transfection of HEK-293T (human embrionic kidney, ATCC CRL-1573 cell line) cells. Supernatants containing the lentiviral particles were collected 48 hours after removal of the calcium phosphate precipitate and ultracentrifuged for 2 hours at 26 000 rpm (Optima L-100 XP Ultracentrifuge; Beckman). Viruses were suspended in sterile PBS, stored at −80°C, and titrated by transduction into Jurkat cells. For in vivo transduction experiments, 4-month-old male apoE−/− mice were anesthetized (ketamine and xylazine), and a small incision was made to expose the right jugular vein. Virus solution (100 μL, 109 particles/mL in PBS) was inoculated directly into the right jugular vein 3 days before initiating the HFD regimen.
Transcriptome Studies in Primary Endothelial Cells
Primary microvascular endothelial cells were isolated from lungs of control and miR-217 apoE−/− mice by ICAM-2 selection, which provides a consistent method for obtaining large amounts of purified primary vascular endothelial cells. Cells were cultured as described above for 7 days in the presence of tamoxifen and were selected twice with ICAM-2 magnetic beads. Three biological replicates of each genotype were analyzed, each obtained from a pool of 3 mice. RNA was purified from MLEC pellets and treated with DNAse to avoid DNA contamination (Qiagen RNAeasy MiniKit). High RNA purity and integrity was confirmed by 2100 Bioanalyzer assessment. Sequencing libraries were prepared by the Centro Nacional de Investigaciones Cardiovasculares Genomics Unit according to the manufacturer’s protocol (NEB NEXT Ultra RNAseq Library Prep Kit; New England Biolabs) from 100-ng RNA per replicate and sequenced in an Illumina HiSeq 2500 platform. Sequencing read quality was assessed with FastQC. Illumina adapters were trimmed with Cutadapt 1.7.1, which also discarded reads shorter than 30 bp. The resulting reads were mapped against the mouse transcriptome (GRCm38 91, release 91) and quantified using RSEM v1.3.1. The RSEM strandedness reverse parameter was included to calculate expression from directional RNA sequencing data. Data were then processed with a deferential expression analysis pipeline that used the Bioconductor package LIMMA for normalization and deferential expression testing. Principal component analysis, performed by comparing the expression level of the set of 500 genes with the largest SD among samples, was conducted to remove samples with the highest variability within each genotype. The gene networks altered by miR-217 expression in endothelial cells were analyzed in 4 samples using Ingenuity Pathway Analysis software.
Statistical Analysis
Unless stated otherwise, data are expressed as mean±SEM. Data were analyzed for normality and equal variance. Statistical analysis was performed in GraphPad Prism 7.3, using the 2-tailed Student t test, the Mann-Whitney nonparametric test, or 2-way ANOVA followed by the Bonferroni post hoc test for continuous data or the Fisher test for categorical data. For statistical analysis in human nonagenarian and control cohorts, the association between the presence of miR-217 and traditional risk factors for which information was available plus lifestyle variables was tested by univariate logistic regression analysis. Significant variables (P<0.1) were further assessed in a multivariate model. The Fuster-Bewat score19,20 predicts individual CVD risk based on just 5 variables: BP, physical activity, body mass index (BMI), fruit and vegetable consumption, and smoking. We used a pseudo-Bewat score excluding the BP parameter because this information was not available in the cohort studied.
Data Disclosure
In line with Arteriosclerosis, Thrombosis, and Vascular Biology Transparency and Openness Promotion Guidelines, the data supporting the findings of this study are available upon reasonable request. Requests for dataset access should be sent to V.G. de Yébenes at vgarciay@ucm.es or to A.R. Ramiro at aramiro@cnic.es.
Results
Generation of a Mouse Model to Study the Function of miR-217 in Endothelial Aging
To assess the association of miR-217 expression with physiological endothelial cell aging, we first analyzed the expression of miR-217 in primary MLECs aged in vitro. miR-217 expression increased gradually as MLECs aged and became senescent (Figure IA in the Data Supplement). Increased miR-217 expression in aged MLECs was mirrored by downregulation of Sirt1 expression. These results are consistent with previous findings in human umbilical vein endothelial cells10 and indicate that miR-217 regulation of Sirt1 expression is conserved between mouse and human endothelial cells. We next analyzed whether vascular miR-217 expression was increased during aging in vivo. Aortic miR-217 expression was higher in old mice (≥100 weeks old) than in young mice (6–9 weeks old; Figure 1A). The expression of miR-217 thus increases during physiological endothelial cell aging.
Figure 1.
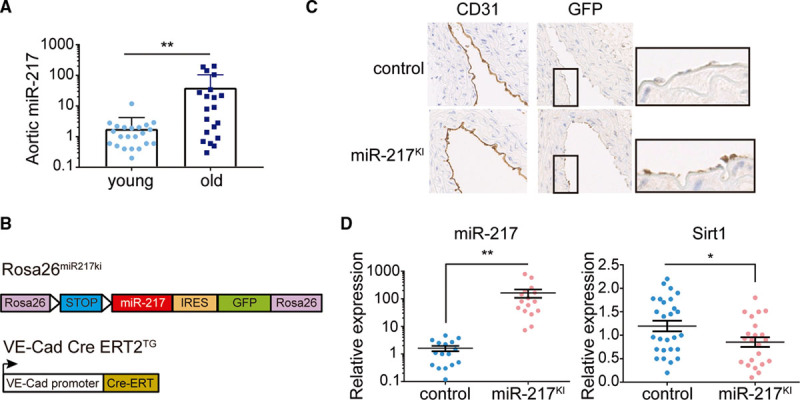
Generation of an endothelium-specific mouse model to study the function of miR-217 in vivo. A, miR-217 expression is associated with endothelial aging. qPCR analysis of miR-217 expression in aortas of young (6–9 wk old) and aged (2 y old) C57/Bl6 mice. B, Constructs used to generate the endothelium-specific miR-217 mouse model. In ROSA26miR217KI mice, the miR-217 precursor construct is inserted within the Rosa26 locus, where it is preceded by a LoxP-flanked transcriptional stop site. ROSA26miR217KI mice were crossed with endothelium-specific and tamoxifen-inducible VE-Cad (VE-cadherin) Cre ERT2TG mice to generate miR-217KI mice. C, Immunohistochemical detection of GFP expression in CD31+ aortic endothelial cells of VE-Cad CreERT2 control mice (miR-217+/+ VE-Cad CreERT2TG/+) and miR-217–overexpressing miR-217KI mice (miR-217KI/+ VE-Cad CreERT2TG/+) after tamoxifen (TMX) injection. D, qPCR analysis of aortic miR-217 and Sirt1 (silent information regulator 1) expression in control and endothelium-specific miR-217–overexpressing miR-217KI mice 12 wk post-TMX injection. Each dot represents an individual mouse. CD indicates cluster of differentiation; GFP, green fluorescent protein; IRES, internal ribosome entry site; and qPCR, quantitative polymerase chain reaction. *P<0.05, **P<0.01 by unpaired t test.
To assess the impact of miR-217 endothelial expression on cardiovascular physiology in vivo, we generated an endothelium-specific and inducible miR-217 knock-in mouse model by crossing Rosa26 miR-217 knock-in mice (Rosa26miR217ki)15 with the endothelium-specific tamoxifen-inducible VE-Cad CreERT2TG line16 (Figure 1B). Examination after tamoxifen administration verified GFP and miR-217 expression in the endothelium of VE-Cad CreERT2TG/+; Rosa26miR217ki/+ mice (hereafter called miR-217KI mice) and its absence in VE-Cad CreERT2TG/+; Rosa26+/+ control mice (Figure 1C and 1D; Figure IB in the Data Supplement). Analysis of Sirt1 expression showed that the increased endothelial expression of miR-217 in miR-217KI mice reduced Sirt1 expression in vivo (Figure 1D).
These results demonstrate that miR-217 expression increases during physiological aging and that endothelial miR-217 expression downregulates the metabolic sensor Sirt1 in vivo.
Endothelial miR-217 Expression Leads to Endothelial Dysfunction
To assess the pathophysiological consequences of endothelial miR-217 expression, we tested the endothelial function of miR-217KI mice in the apoE−/− proatherogenic background. Control apoE−/− mice (VE-Cad CreERT2TG/+; Rosa26miR217+/+; apoE−/−) or miR-217KI apoE−/− mice (VE-Cad CreERT2TG/+; Rosa26miR217KI/+; apoE−/−) were injected with tamoxifen to induce Cre-dependent endothelial expression and fed an HFD to promote endothelial stress. Analysis of miR-217KI apoE−/− mouse aorta revealed a reduction in endothelial Sirt1 expression (Figure IC in the Data Supplement) similar to that observed in the wild-type background (Figure 1D). Since Sirt1 is a regulator of eNOS activity in endothelial cells12 and eNOS-derived NO is essential for vascular function, we analyzed NO production and the contractile and relaxant responses of miR-217KI apoE−/− mouse arteries. We examined the aorta as a conductance artery in which endothelium-induced relaxation is highly NO dependent and the mesenteric arteries as an example of resistance vessels in which NO makes a minor contribution.21,22 Aortic overexpression of endothelial miR-217 resulted in reduced eNOS activity, as shown by reduced acetyl choline–induced NO production (Figure 2A) and impaired endothelium-dependent vasodilation (Figure 2B). Moreover, vasodilation in response to the NO donor diethylamine NONOate was similar in control apoE−/− and miR-217KI apoE−/− mice (Figure 2B), suggesting that the endothelial dysfunction in miR-217KI apoE−/− mouse aorta is due to deficient NO production by endothelium rather than altered NO signaling in the underlying smooth muscle. Interestingly, neither mouse group showed differences in vasoconstrictor responses (Figure ID in the Data Supplement). Analysis of vascular function in mesenteric resistance arteries revealed similar acetylcholine-induced relaxation in arteries from both mouse groups (Figure IE in the Data Supplement); however, phenylephrine-induced mesenteric artery vasoconstriction was significantly enhanced in mice overexpressing miR-217 in the endothelium (Figure 2C). These results indicate that endothelial miR-217 expression causes deficient NO production and endothelial dysfunction.
Figure 2.
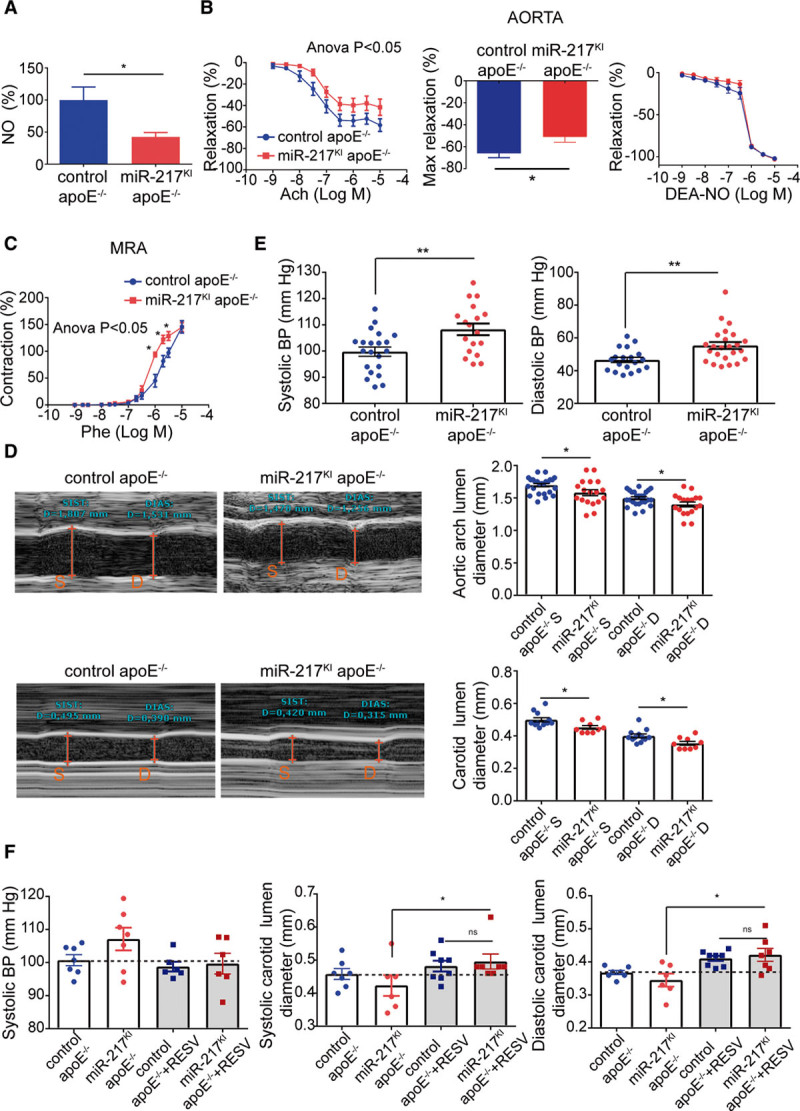
miR-217 expression impairs endothelial physiology. A, Acetylcholine (ACh)-induced NO production in aortas from male control apoE−/− mice (miR-217+/+ VE-Cad CreERT2TG/+ apoE−/−) and miR-217KI apoE−/− mice (miR-217KI/+ VE-Cad CreERT2TG/+ apoE−/−) fed with HFD for 12 wk. B, Concentration-response curves to ACh or the NO donor diethylamine NONOate (DEA-NO) and maximal relaxation to ACh of aorta from male control apoE−/− mice and miR-217KI apoE−/− mice. C, Concentration-response curve to phenylephrine (Phe) of mesenteric resistance arteries, from male control apoE−/− mice and miR-217KI apoE−/− mice. *P<0.05 vs control apoE−/− mice by 2-way ANOVA followed by Bonferroni post-test. Maximal relaxation was analyzed by the Mann-Whitney U test. D, Echocardiography assessment of systolic (S) and diastolic (D) aortic arch lumen diameter (top) and right carotid lumen diameter (bottom) in control and miR-217 apoE KO (knockout) mice fed with HFD for 12 wk. E, Systolic and diastolic blood pressure (BP) in control apoE−/− and miR-217KI apoE−/− mice fed with HFD for 10 to 11 wk. F, BP and carotid diameters in control apoE−/− and miR-217KI apoE−/− mice fed an HFD before and 1 wk after resveratrol (RESV) administration. Each dot represents an individual mouse. Systolic BP unpaired t test P (miR-217KI apoE−/− vs control apoE−/−+resveratrol P=0.059; miR-217KI apoE−/− vs miR-217KI apoE−/−+resveratrol P=0.14; control apoE−/−+resveratrol vs miR-217KI apoE−/−+resveratrol P=0.8) *P<0.05, **P<0.01, ns (not significant) by unpaired t test (A–E). *P<0.05 by paired t test (F). apoE indicates apolipoprotein E; and MRA, mesenteric resistance arteries.
To determine whether miR-217 expression affects vessel size in vivo, we measured the lumen diameters of arteries by ultrasound imaging. Compared with vessels from control animals, vessels of miR-217KI apoE−/− mice had smaller lumen diameters during systole and diastole (Figure 2D; Figure IF in the Data Supplement). In addition, miR-217 overexpression in HFD-fed apoE−/− mice resulted in higher systolic and diastolic BPs (Figure 2E), most likely as a consequence of impaired endothelial function.
To assess whether miR-217–induced vascular dysfunction could be reverted with a vasoprotective treatment, we analyzed BP and carotid lumen diameter in control apoE−/− and miR-217KI apoE−/− mice after administration of resveratrol—a widely used multifunctional compound that stimulates Sirt1 and eNOS (Figure IG in the Data Supplement).23–25 Resveratrol reverted the miR-217–induced BP increase and carotid vasoconstriction in apoE−/− mice (Figure 2F).
Overall, these results show that increased expression of miR-217 in the endothelium under proatherogenic stress conditions disturbs endothelium-derived NO production and impairs vascular physiology in vivo.
Endothelial miR-217 Expression Accelerates Atherosclerosis and Coronary Lesion Development
To study the role of endothelial miR-217 expression in atherogenesis, we analyzed the presence and quantity of lipid-rich atherosclerotic plaques in miR-217KI apoE−/− and control apoE−/− mice in aortic root cross sections. Endothelial miR-217 overexpression in apoE−/− mice increased the size of atherosclerotic plaques in the aortic root (Figure 3A and 3B; Figure IIA in the Data Supplement). Plaques in miR-217KI apoE−/− mice showed no significant changes in macrophage content (Figure 3C; Figure IIB and IIC in the Data Supplement) but were more advanced and had larger necrotic cores and lower collagen content than plaques in control apoE−/− mice (Figure 3D). An increased size of the plaque necrotic core can be associated with high rates of apoptosis,26 and we found significantly elevated active caspase-3 staining in plaques isolated from miR-217KI apoE−/− mice (Figure 3E). Moreover, miR-217KI apoE−/− mice had significant atherosclerosis in the coronary arteries (Figure 3E), whereas coronary artery atherosclerosis was minimal in control apoE−/− mice (Figure 3F), as reported in previous studies of HFD-fed apoE−/− mice.27,28 miR-217KI apoE−/− and control apoE−/− mice showed no significant differences in the aortic expression of the proinflammatory molecules Icam, IL1β, IL6, and TNFα (Figure IID in the Data Supplement), the expression of endothelial activation molecules Vcam-1 and Icam-1 in atheroma plaques (Figure IIE in the Data Supplement), or the content of circulating lipids and cholesterol (Figure IIF in the Data Supplement). These results suggest that endothelial miR-217 expression does not alter endothelial cell activation or promote changes in lipid biosynthesis or retention mechanisms.
Figure 3.
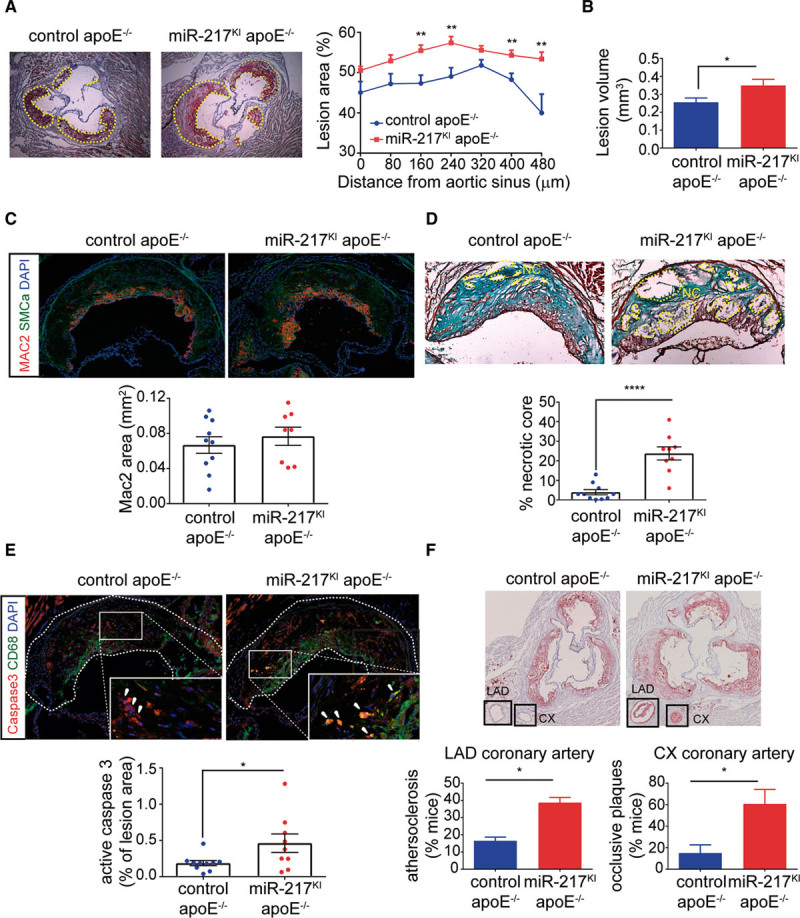
Endothelial miR-217 expression promotes atherosclerosis and coronary artery disease. A and B, Quantification of atherosclerotic lesions by red oil staining of aortic sinus and proximal aorta heart sections in control apoE−/− mice (miR-217+/+ VE-Cad CreERT2TG/+ apoE−/−, n=12) and miR-217KI apoE−/− mice (miR-217 KI/+ VE-Cad CreERT2TG/+ apoE−/−, n=9) fed an HFD for 7 wk. A, Representative red oil–stained aortic sinus cross sections from control apoE−/− and miR-217KI apoE−/− HFD-fed mice (left) and quantification of atherosclerotic lesion area through the aortic sinus and ascending aorta (right). Each data point represents mean lesion area at the specified distance from the aortic sinus. B, Total atherosclerotic lesion volume in aortic sinus of control apoE−/− and miR-217KI apoE−/− mice, determined by calculating the area under the curve for each mouse from lesion area graphs (mm2; Figure II in the Data Supplement). C, Macrophage infiltration area quantified by Mac2 immunofluorescence in atherosclerotic lesions from control apoE−/− and miR-217KI apoE−/− HFD-fed mice. Each data point represents total Mac2+ area in the aortic sinus cross section of an individual mouse. D, Necrotic core (NC) content of atherosclerotic lesions determined by Masson trichrome staining on aortic sinus cross sections in HFD-fed control apoE−/− (n=9) and miR-217KI apoE−/− (n=12) mice. Top, Representative images. Each data point represents an individual mouse. E, Cellular apoptosis in atheroma plaques quantifed by active caspase-3 immunofluorescence in aortic sinus cross sections from control apoE−/− and miR-217KI apoE−/− HFD-fed mice. Each data point represents the proportion of active caspase-3 area per lesion in an individual mouse. Green, CD68; red, caspase-3. Top, Representative images of the quantification shown in the lower graphs. F, Atheroma plaque quantification in the left coronary artery by red oil staining of aortic sinus cross sections in 3 independent experiments (total n=26 control apoE−/− and n=29 miR-217KI apoE−/− mice). Top, Representative images. Charts show the proportion of mice with atherosclerosis (≥20% of plaque in lumen) in the left anterior descending (LAD) coronary artery (left) and the proportion with occlusive plaques (≥50% of plaque in lumen) in the circumflex (CX) coronary artery. Error bars show the SD in the quantification between experiments. *P<0.05, **P<0.01, ****P<0.0001 by unpaired t test (A–D). apoE indicates apolipoprotein E; CD, cluster of differentiation; DAPI, 4′,6-diamidino-2-phenylindole; MAC2, galectin 3; and SMCa, smooth muscle cell actin.
Endothelial miR-217 expression thus aggravates atherogenesis development in apoE−/− mice, resulting in the generation of larger, more advanced aortic atherosclerotic plaques and the development of coronary atherosclerosis.
Endothelial miR-217 Expression Triggers Impaired Left Ventricular Function
To determine whether endothelial dysfunction and coronary artery disease caused by endothelial miR-217 expression compromise cardiac performance, we conducted echocardiographic studies of HFD-fed control and miR-217KI apoE−/− mice. miR-217–overexpressing mice had altered left ventricular diastolic function, characterized by reduced left ventricular relaxation. Transmitral Doppler echocardiography revealed a reduced ratio of peak velocity flow in early diastole (E wave) to the peak velocity flow in late diastole caused by atrial contraction (A wave; E/A ratio; Figure 4A), whereas pulmonary venous Doppler echocardiography detected reduced diastolic (D2 wave) pulmonary venous flow (Figure 4B). Moreover, miR-217KI apoE−/− mice also showed left ventricular systolic dysfunction, revealed by a reduced left ventricular ejection fraction in M-mode long-axis echocardiography studies (Figure 4C; Movie I in the Data Supplement), consistent with a reduction in left ventricular contractile function in these mice. The reduced contractility in miR-217KI apoE−/− mice was independent of changes in the left ventricular diastolic diameter, wall thickness (posterior wall thickness), and interventricular wall septal thickness (not shown).
Figure 4.
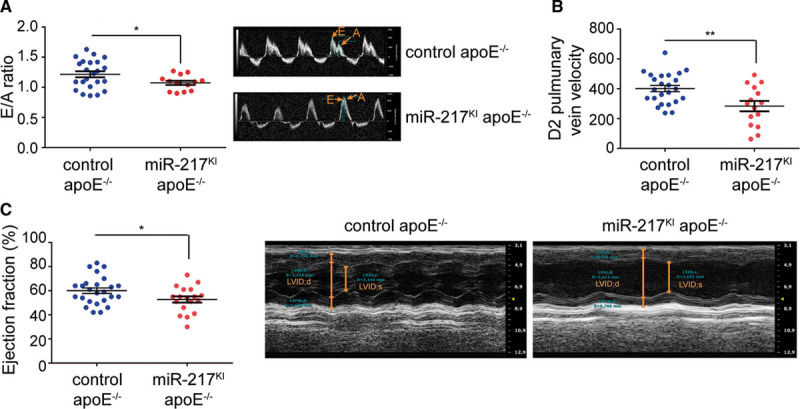
miR-217 expression promotes left ventricular dysfunction. A, Left ventricular diastolic function assessed as the ratio of Doppler-detected peak early diastolic velocity (E′) to peak late diastolic velocity (A′) in control apoE−/− mice (miR-217+/+ VE-Cad CreERT2TG/+ apoE−/−) and miR-217KI apoE−/− mice (miR-217KI/+ VE-Cad CreERT2TG/+ apoE−/−) fed an HFD for 8 wk. Right, Representative echocardiography images. B, D2 pulmonary vein velocity in control apoE−/− and miR-217KI apoE−/− mice fed an HFD for 8 wk. C, Left ventricular ejection fraction in control apoE−/− and miR-217KI apoE−/− mice determined by echocardiography after 10 wk on an HFD. Data are from 2 independent experiments. Each dot represents an individual mouse. *P<0.05, **P<0.01 by unpaired t test (A–C). apoE indicates apolipoprotein E; LVID;d, left ventricular internal diameter in diastole; and LVID;s, left ventricular internal diameter in systole.
miR-217 Inhibition Improves Vascular Contractile Function and Reduces Atherosclerotic Development in Proatherogenic Mice
The ability of endogenous miR-217 inhibition to improve endothelial function in proatherogenic conditions was assessed in miR-217 loss-of-function assays in HFD-fed apoE−/− mice. For this analysis, we generated miR-217SPG (Figure 5A), in which miR-217–complementary sequences were cloned in tandem within the 3′UTR (the three prime untranslated region) of a reporter gene, thus functioning as competitive inhibitors for miR-217 binding to its endogenous binding sites.15,29 The aorta was transduced by injection of control or miR-217SPG lentivirus into the jugular vein of apoE−/− mice (Figure 5B),18,30 and aortic contractile properties were assessed in isometric tension studies. miR-217 inhibition by miR-217SPG slightly increased endothelium-dependent vasodilation and significantly reduced phenylephrine-induced aortic contraction, without modifying diethylamine NONOate–induced relaxation (Figure 5C and 5D). The NOS inhibitor L-NAME increased phenylephrine responses in arteries from both miR-217SPG–transduced and control lentivirus-transduced mice (Figure 5D). The effect of L-NAME was greater in miR-217SPG–transduced aortas than in controls, as shown by the greater L-NAME–induced increase in Emax (control, 123.1±9.2%; miR-217SPG, 170.8±18%; P<0.05), indicating more NO availability. In addition, we found that transduction of apoE−/− mice with miR-217SPG lentivirus reduced HFD-induced atherosclerotic plaque development in the aortic root (Figure 5E).
Figure 5.
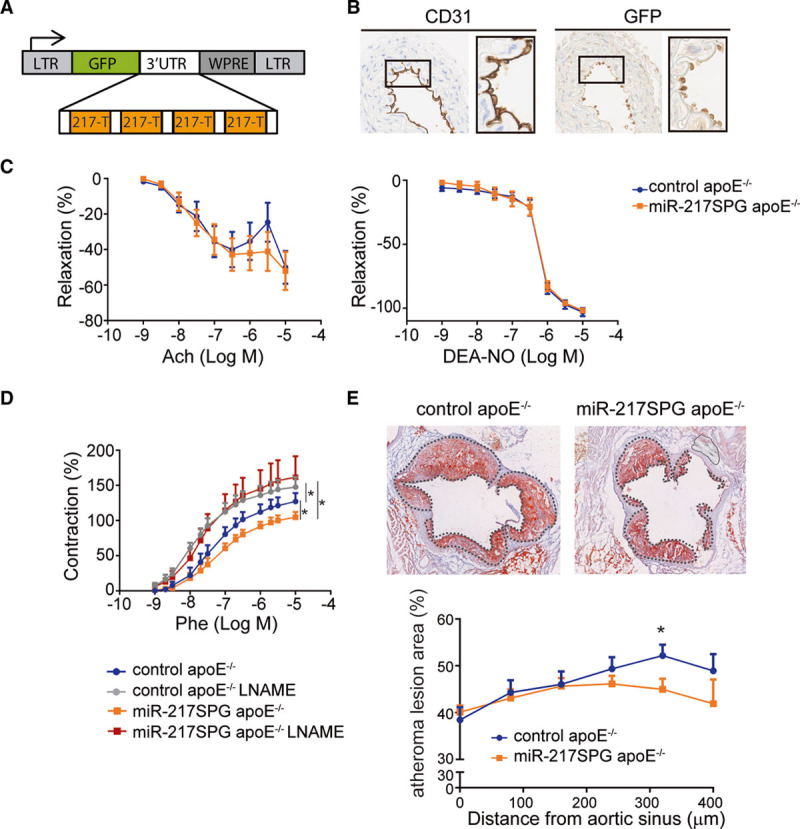
Inhibition of endogenous miR-217 improves vascular contractile function and reduces atherosclerosis development. A, miR-217-Sponge (miR-217SPG) construct used for vascular miR-217 inhibition. Four miR-217 complementary sites, separated by 4-nt spacers, were placed downstream of GFP in the pHRSIN lentiviral vector. B, CD31 and GFP immunostaining in ascending aorta sections of apoE−/− mice inoculated through the jugular vein with control or miR-217SPG pHRSIN lentivirus. C, Concentration-response curves to acetylcholine (left) and the NO donor diethylamine NONOate (DEA-NO; right) and (D) effect of L-NAME on the concentration-response curve to phenylephrine (Phe), in aortic segments from male control and miR-217SPG–infected apoE−/− mice fed HFD for 12 wk. E, Red oil staining analysis of atherosclerotic lesion area in aortic sinus and ascending aorta sections from control and miR-217SPG–infected apoE−/− mice fed an HFD for 12 wk. Left, Representative red oil–stained cross sections 320 µm from the aortic sinus in control and miR-217SPG–infected mice. *P<0.05 by unpaired t test or ANOVA. 3'UTR indicates the three prime untranslated region; Ach, acetylcholine; apoE, apolipoprotein E; CD, cluster of differentiation; GFP, green fluorescent protein; L-NAME, N (gamma)-nitro-L-arginine methyl ester; LTR, long terminal repeat; and WPRE, woodchuck hepatitis virus post-transcriptional regulatory element.
These results thus show that inhibition of miR-217 in the vasculature improves vascular function and reduces atherosclerotic development, revealing the therapeutic potential of endothelial miR-217 inhibition for the treatment of aging-related CVD.
miR-217 Expression Inhibits eNOS Stimulatory Pathways in Endothelial Cells
To gain mechanistic insight into the function of miR-217 in endothelial gene expression, we performed RNA sequencing transcriptome analysis on endothelial cells from miR-217KI apoE−/− and control apoE−/− mice (Table IA in the Data Supplement; Figure IIIA in the Data Supplement). miR-217 expression altered the vascular function signature, with changes in gene expression that promote decreased arterial relaxation and increased BP (Figure 6A; Table ID and IE in the Data Supplement; Figure IIIB in the Data Supplement). Detailed pathway enrichment analysis showed that miR-217 modifies gene pathways important in endothelial biology such as cell signaling, leukocyte adhesion and endothelial permeability, and extracellular matrix and collagen-mediated pathways (Figure 6B; Table IB and IC in the Data Supplement). Moreover, we found that miR-217 inhibits signaling pathways that activate eNOS (APLNR [apelin receptor] signaling, VEGF signaling [VEGFR (VEGF receptor) and VEGF], adenylyl cyclase, and the lysophosphatidic acid receptor [LPAR]31,32; Figure 6C; Table IB in the Data Supplement) through direct and secondary target gene regulation (Figure IIIC and IIID in the Data Supplement; Table IG through II in the Data Supplement). To validate these transcriptome results, we analyzed the expression of key eNOS stimulatory genes by quantitative real-time polymerase chain reaction in MLECs isolated from miR-217KI apoE−/− or control apoE−/− mice. This analysis confirmed robust downregulation in miR-217KI apoE−/− MLECs of multiple genes, including the APLNR (an eNOS activator via AKT) and VEGFR1 (Flt1; the receptor for VEGFA, VEGFB, and placental growth factor and an initiator of the PI3K-Akt-eNOS signaling pathway). We also detected modest downregulation of genes of other eNOS-activating pathways, including LPAR, ADCY2, and Sirt1. Endothelial cells from miR-217KKI apoE−/− mice also showed significant downregulation in the expression of Nos3—the gene encoding eNOS (Figure 6D). Consistent with these results, we found that miR-217SPG–lentivirus-mediated inhibition of miR-217 in apoE−/− MLECs resulted in the upregulation of eNOS activator genes in the apelin and VEGF signaling pathways (Figure IIIE in the Data Supplement).
Figure 6.
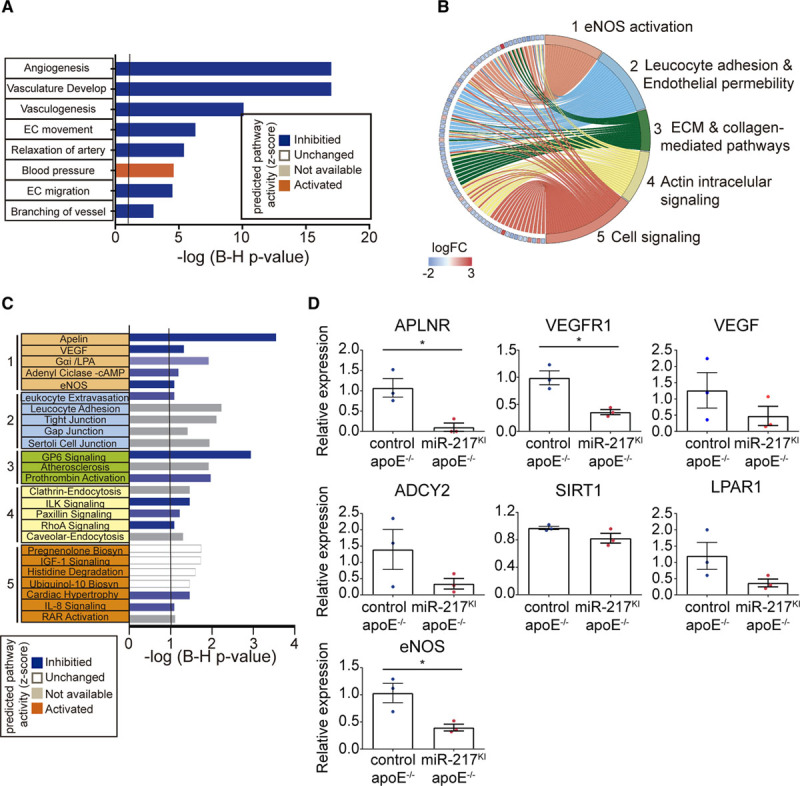
Effect of miR-217 on endothelial transcriptome. Study of gene networks regulated by miR-217 in primary mouse endothelial cells. A, Pathway activity prediction in significantly changed Cardiovascular Development and Functions analysis of RNA sequencing (RNA-Seq) gene expression quantification in endothelial cells isolated from lungs (MLECs) of miR-217KI apoE−/− or control apoE−/− mice. Pathway activity prediction is shown with a color code (blue, inhibited; orange, activated). B, Circus plot showing the fold change expression (logFC; left) of genes of significantly changed cardiovascular-related pathways (1–5; right). Gene expression was quantified by RNA-Seq in MLECs derived from miR-217KI apoE−/− or control apoE−/− mice. C, Ingenuity canonical pathway analysis from significantly changed pathways grouped in the 5 general cardiovascular-related categories shown in B. Pathway activity prediction is shown with a color code (blue, inhibited; orange, activated). P values were corrected for multiple testing using the Benjamini-Hochberg (B–H) false discovery rate. D, qPCR analysis of gene expression in control apoE−/− and miR-217KI apoE−/− MLECs. Expression was normalized to GAPDH. ADCY2 indicates adenylate cyclase 2; APLNR, apelin receptor; apoE, apolipoprotein E; EC, endothelial; ECM, extracellular matrix; eNOS, endothelial NO synthase; LPAR1, lysophosphatidic acid receptor-1; qPCR, quantitative polymerase chain reaction; SIRT1, silent information regulator 1; VEGF, vascular endothelial growth factor; and VEGFR, vascular endothelial growth factor receptor.
miR-217 thus regulates a hub of endothelial physiology pathways and triggers a coordinated downregulation of eNOS stimulatory genes, resulting in eNOS pathway inhibition in endothelial cells.
Plasma miR-217 Is a Biomarker of Cardiovascular Aging in Humans
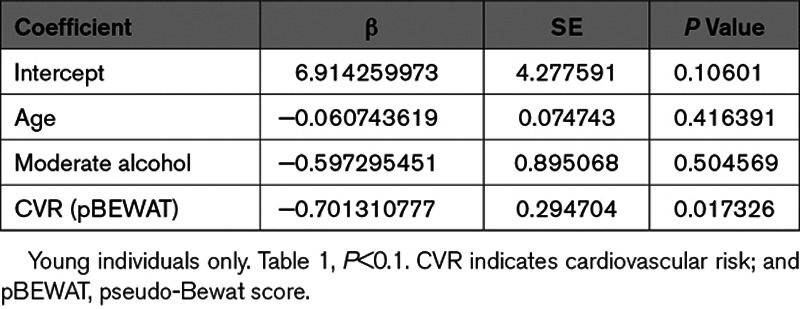
To determine whether miR-217 is associated with human vascular aging, we analyzed miR-217 expression in the context of some of the best characterized cardiovascular risk factors, including age, BMI, and lifestyle risks. Analysis of miR-217 expression in a collection of plasma samples from nonagenarian and young (<50 years old) individuals revealed a higher proportion of individuals expressing miR-217 among the nonagenarian group (Figure 7A), indicating that plasma miR-217 expression is an aging-associated biomarker in humans. Analysis of the association between plasma miR-217 lifestyle variables revealed a lower rate of miR-217 expression among individuals with lower risk habits (physical activity, moderate alcohol consumption, and health dietary habits such as weekly consumption of fruit and vegetables and low intake of processed meat; Figure 7B). Moreover, individuals with detectable plasma miR-217 tended to have a higher BMI (Figure 7C), whereas mean BMI did not differ significantly between young individuals and nonagenarians (P=0.46; Figure IVA in the Data Supplement). These results are consistent with the association of increased vascular miR-217 expression with age (Figure 1B) and HFD (Figure IVB in the Data Supplement) in mice, and we found that increased vascular miR-217 expression in mice was associated with the presence of miR-217 in serum (Figure IVC in the Data Supplement). Univariate logistic regression to identify variables associated with plasma miR-217 expression in the young and nonagenarian cohorts identified a negative correlation between plasma miR-217 expression and the pseudo-BEWAT cardiovascular prediction score (Table 1). A subsequent multivariate logistic regression model based on noncollinear variables (Tables 2 and 3) revealed that young individuals without miR-217 had higher pseudo-BEWAT risk scores, which translates into better cardiovascular health (b=−0.7, P=0.017; Table 3). In agreement with these results and with recent reports,33 we found that plasma miR-217 levels are increased in individuals with coronary atherosclerosis (Figure 7D).
Figure 7.
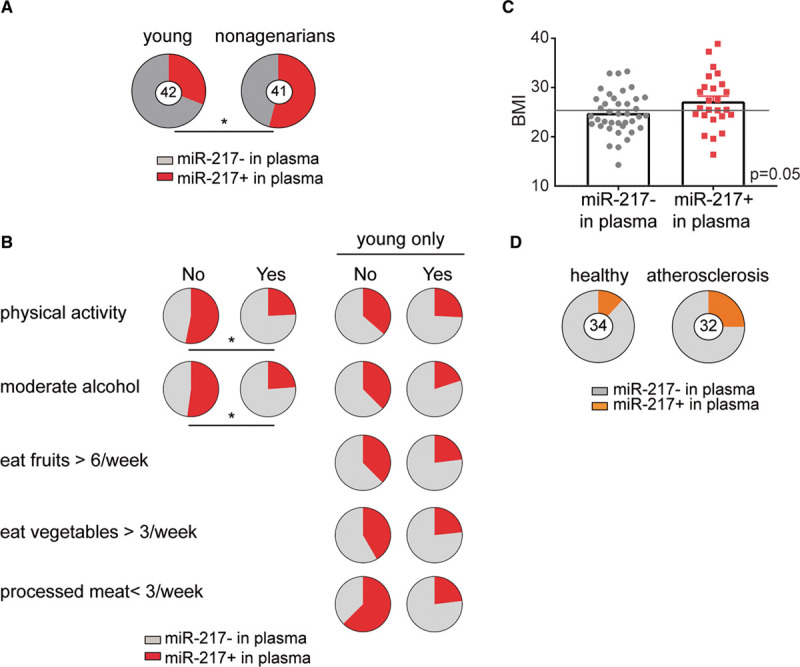
Plasma miR-217 is a biomarker of aging associated with cardiovascular risk in humans. A, Plasma miR-217 is a biomarker of human aging. The proportion of individuals with detectable plasma miR-217 was determined by qPCR in samples from young healthy control individuals (<50 y old; n=42) and nonagenarian individuals (n=41). B, miR-217 is a biomarker for cardiovascular aging. The proportion of individuals with detectable miR-217 in plasma stratified by lifestyle habits in young control and nonagenarian cohorts or only in the young cohort. C, Plasma miR-217 is associated with increased body mass index (BMI). BMI values are shown for individuals with detectable or no detectable plasma miR-217 in cohorts of sex-matched young healthy controls (<50 y old) and nonagenarian women. The gray line indicates overweight (BMI, ≥25). Unpaired t test P=0.05. D, Serum miR-217 is a biomarker of atherosclerosis in humans. The chart shows the proportions of healthy controls (n=34) and individuals with coronary atherosclerosis (n=32) with detectable miR-217 in plasma determined by quantitative real-time polymerase chain reaction. qPCR indicates quantitative polymerase chain reaction. *P<0.05 by Fisher exact test.
Table 1.
Univariate Logistic Regression Model for All Lifestyle and Cardiovascular Risk Factors Available in the Study
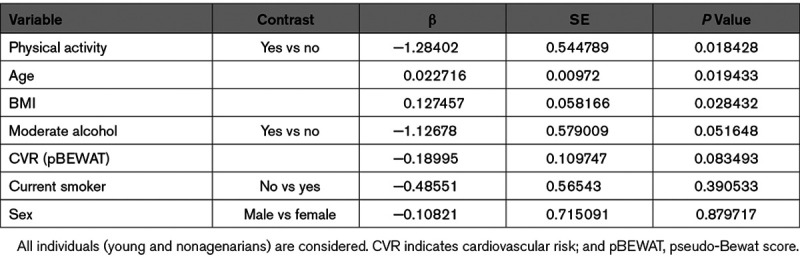
Table 2.
Lifestyle Multivariate Logistic Regression Model for Nonredundant Variables Significantly Associated With the Presence of miR-217 in Plasma in the Univariate Analysis
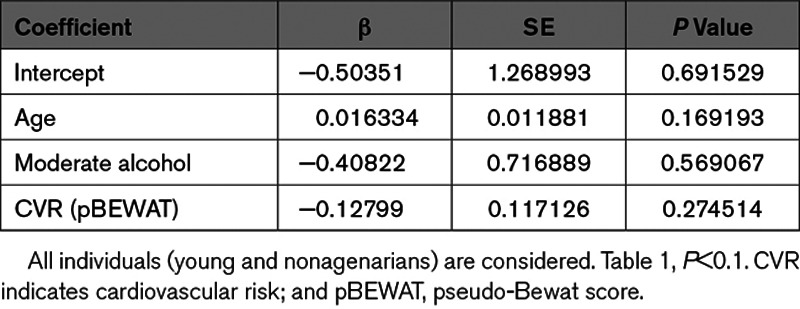
Table 3.
Multivariate Logistic Regression Model for Nonredundant Variables Significantly Associated With the Presence of miR-217 in Plasma in the Univariate Analysis
Overall, our results show that the presence of miR-217 in plasma is a biomarker of aging and a predictor of cardiovascular risk in humans.
Discussion
In this study, we have generated the first animal model assessing the role of an aging-induced miRNA in endothelial pathophysiology. Previous reports identified miR-217 as the most highly induced miRNA in human endothelial cells during in vitro aging.10 Our results show that miR-217 expression increases during physiological endothelial cell aging and demonstrate that plasma miR-217 is a biomarker of human aging associated with elevated cardiovascular risk. It remains uncertain whether the aging-related increase of plasma miR-217 in nonagenarian individuals is exclusively due to increased endothelial cell expression; however, we found that endothelium-specific miR-217 expression in mice results in increased serum miR-217. The cellular mechanisms responsible for circulating miR-217, whether involving exosomal or other paracrine secretion routes, remain to be explored.
In agreement with previous reports in human vein endothelial cells,10 we found that miR-217 downregulates Sirt1 expression in vivo. However, the global impact of miR-217 on the endothelial transcriptome had not been addressed before. We found that miR-217 expression disrupts endothelial physiology, dysregulating the expression of various signaling pathways important for endothelial function. Remarkably, we found concurrent downregulation of various signaling pathways leading to eNOS activation, including the apelin and VEGF pathways,31,32 and of eNOS expression. In agreement with our transcriptome data and the key role of endothelial-derived NO production in the vasculature,13,14 we found that endothelial miR-217 overexpression had a notable impact on vascular homeostasis.34–38 Consistent with diminished eNOS activity and expression, endothelial-dependent NO production was reduced in the aortas of miR-217KI apoE−/− mice, and this was accompanied by parallel functional changes. Nevertheless, our transcriptomic data suggest that in addition to the defects caused by a diminished eNOS expression and activity, miR-217 regulates through direct targing a hub of endothelial functions (including extracellular matrix, collagen-mediated, and actin intracellular signaling pathways) that are likely to contribute to the observed phenotype.15 Further research is warranted into how these miR-217 targets contribute to the impact of miR-217 expression on endothelial physiology and CVD.
Endothelial miR-217 expression resulted in several clinically relevant cardiovascular disorders in apoE−/− mice, including endothelial dysfunction, increased BP, exacerbated atherosclerosis development, coronary artery disease, and left ventricular systolic and diastolic dysfunction. Diminished eNOS expression promotes endothelial dysfunction,12,39 which is causally linked to reduced endothelial NO production40,41; we, therefore, postulate that endothelial dysfunction in miR-217 mice is directly caused by miR-217–induced eNOS-NO pathway downregulation (see Graphic Abstract). Interestingly, the mesenteric arteries of miR-217KI apoE−/− mice showed no alteration in vascular endothelial function but had an enhanced contractile response to phenylephrine; this is likely related to the minor contribution of NO to endothelium-dependent relaxation in this vascular bed. Our data further show that administration of resveratrol—a multifunctional vasoprotective treatment that stimulates Sirt1 and eNOS,23 reverts the vasoconstriction and increased BP found in miR-217 mice, revealing a protective effect of resveratrol on aging-related miR-217–induced vascular dysfunctions.
Most cardiovascular abnormalities seen in miR-217KI apoE−/− mice (high BP, atherosclerosis, and left ventricular diastolic dysfunction) are closely interrelated40,42 and can be triggered by endothelial dysfunction. We, therefore, propose that miR-217–induced endothelial dysfunction is the first step in a chain of events leading to atherosclerosis and CVD (see Graphic Abstract). Previous research has shown that apoE−/− mice develop endothelial and cardiac dysfunction phenotypes with age.43 Endothelial expression of the aging-induced miRNA miR-217 thus recapitulates various cardiovascular phenotypes associated with aging in the apoE-deficient proatherogenic mouse model.
Although miR-217–induced endothelial aging and dysfunction can lead to atherosclerosis, raised BP, and left ventricular diastolic dysfunction, endothelial dysfunction is unlikely to be the primary cause of the left ventricular systolic dysfunction found in miR-217KI apoE−/− mice. A more likely cause is the development of significant coronary atherosclerosis, which, together with increased BP, is one of the main causes of cardiac systolic dysfunction.44,45 Interestingly, apoE−/− mice are generally resistant to coronary artery atherosclerosis, but the combination of apoE deficiency with additional genetic alterations has enabled the generation of mouse models of coronary artery disease. This is the case for mutations affecting HDL (high-density lipoprotein) signaling via SR-B1 (scavenger receptor, class B type 1), which lead to eNOS activation.27,46 It is thus likely that deficient eNOS activation contributes to coronary atherosclerosis in miR-217KI apoE−/− mice, and we, therefore, propose that left ventricular systolic dysfunction in miR-217KI apoE−/− mice is secondary to coronary artery disease and increased BP in this proatherogenic mouse model (see Graphic Abstract).
The major implication of left ventricular dysfunction in heart failure42,47 underscores the clinical implications of our findings on the contribution of miR-217 expression to the development of CVD. Interestingly, miR-217 is among the most upregulated miRNAs in the hearts of chronic heart failure patients,33 and overexpression of miR-217 in mouse hearts aggravates aortic constriction–induced cardiac hypertrophy and dysfunction.48 Together, these results suggest that the increased expression of miR-217 in vascular and cardiac tissues is deleterious to cardiovascular function.
By establishing that miR-217 inhibition in the vasculature of proatherogenic mice partially protects vascular function and reduces atherosclerosis, our results suggest that modulation of miR-217 expression or activity has therapeutic potential. Further studies of miR-217 inhibition in other animal models of CVD will be required to establish whether the inhibition of miR-217 in the vasculature has therapeutic potential for the treatment of left ventricular systolic or diastolic dysfunction.
Our analysis of human cohorts reveals a remarkable association of plasma miR-217 with well-established cardiovascular risk factors, including low physical activity, poor dietary habits, and high BMI. This trend is reflected in a significant association of plasma miR-217 with a validated cardiovascular risk factor score predictor in healthy control individuals, establishing miR-217 as a biomarker of cardiovascular aging in humans.
Overall, our results reveal the role of an aging-induced miRNA in CVD and provide a rationale for the design of therapeutic strategies for aging-related cardiovascular disorders based on the inhibition of endothelial miR-217 expression.
Acknowledgments
We thank all members of the B Cell Biology Lab, Jacob Bentzon, Daniel Morales Cano, Sara Martínez-Martínez, Paula Yunes, Miguel R. Campanero, Gabriela Guzmán, Antonio Fernandez Ortiz, and Vicente Andrés, for scientific discussion. We also thank Lorena Flores and Ana Vanesa Alonso, the Centro Nacional de Investigaciones Cardiovasculares (CNIC) Imaging and Cardiovascular Physiology Unit, the CNIC Genomics Unit, Verónica Labrador, and Antonio de Molina for technical advice; Ralf Adams for VE-Cad (VE-cadherin) CreERT2 mice; and Simon Bartlett for English editing. V.G. de Yébenes, A.M. Briones, J. Oller, F. Bilal, M. González-Amor, S.M. Mur, I. Martos-Folgado, and N. Méndez-Barbero performed experiments. J. Carlos Silla-Castro, and F. Sánchez-Cabo performed statistical analysis of the human data. V.G. de Yébenes and A.M. Briones analyzed and interpreted the data. L.J. Jiménez-Borreguero, H. Bueno, M. Salaices, and J. Miguel Redondo contributed to the experimental design. V.G. de Yébenes and A.R. Ramiro conceived the overall strategy and wrote the manuscript. All authors revised and contributed to the final manuscript.
Sources of Funding
V.G. de Yébenes was supported by Ramón y Cajal grant RYC-2009-04503 and Asociación Española contra el Cáncer foundation grant INVES18013GARC and by the Universidad Complutense de Madrid. S.M. Mur and A.R. Ramiro are supported by Centro Nacional de Investigaciones Cardiovasculares (CNIC) funding. A.R. Ramiro was supported by the Spanish Ministerio de Ciencia e Innovación (PID2019-107551RB-I00), the Spanish Ministerio de Economía, Industria y Competitividad (SAF2013-42767-R and SAF2016-75511-R), and the European Research Council StG ID-207844/BCLYM. M. Salaices was supported by the Ministerio de Ciencia e Innovación (SAF2016-80305P) and with J. Miguel Redondo by Instituto de Salud Carlos III (CIBER de Enfermedades Cardiovasculares, CB16/11/00286 and CB16/11/00264) and Comunidad de Madrid (B2017/BMD-3676). V.G. de Yébenes was supported by Ministerio de Ciencia e Innovación (PID2019-107551RB-I00). Further support was provided by the European Social Fund and the European Regional Development Fund “A Way to Build Europe.” The CNIC is supported by Ministerio de Ciencia, Innovacion y Universidades, and the Pro CNIC Foundation and is a Severo Ochoa Center of Excellence (SEV-2015-0505).
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- ADCY2
- adenylate cyclase 2
- APLNR
- apelin receptor
- BMI
- body mass index
- BP
- blood pressure
- CVD
- cardiovascular disease
- eNOS
- endothelial NO synthase
- HDL
- high-density lipoprotein
- HFD
- high-fat diet
- IL
- interleukin
- KHS
- Krebs-Henseleit solution
- LPAR
- lysophosphatidic acid receptor
- miR-217SPG
- miR-217 sponge construct
- miRNA
- microRNA
- MLEC
- mouse lung endothelial cell
- NOS
- NO synthase
- Sirt1
- silent information regulator 1
- TNFα
- tumor necrosis factor-alpha
- VE-Cad
- VE-cadherin
- VEGF
- vascular endothelial growth factor
- VEGFR
- vascular endothelial growth factor receptor
For Sources of Funding and Disclosures, see page 2423.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.314333.
Current address for N. Méndez-Barbero: Vascular Research Lab, FIIS-Fundacion Jimenez Díaz, Autonoma University, Madrid, Spain.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, et al. ; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. 2012;110:1097–1108. doi: 10.1161/CIRCRESAHA.111.246876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gimbrone MA, Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hata A. Functions of microRNAs in cardiovascular biology and disease. Annu Rev Physiol. 2013;75:69–93. doi: 10.1146/annurev-physiol-030212-183737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lucas T, Bonauer A, Dimmeler S. RNA Therapeutics in cardiovascular disease. Circ Res. 2018;123:205–220. doi: 10.1161/CIRCRESAHA.117.311311 [DOI] [PubMed] [Google Scholar]
- 6.Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of dicer and drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007;101:59–68. doi: 10.1161/CIRCRESAHA.107.153916 [DOI] [PubMed] [Google Scholar]
- 7.Suárez Y, Fernández-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–1173. doi: 10.1161/01.RES.0000265065.26744.17 [DOI] [PubMed] [Google Scholar]
- 8.Suárez Y, Fernández-Hernando C, Yu J, Gerber SA, Harrison KD, Pober JS, Iruela-Arispe ML, Merkenschlager M, Sessa WC. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc Natl Acad Sci USA. 2008;105:14082–14087. doi: 10.1073/pnas.0804597105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernández-Hernando C, Suárez Y. MicroRNAs in endothelial cell homeostasis and vascular disease. Curr Opin Hematol. 2018;25:227–236. doi: 10.1097/MOH.0000000000000424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menghini R, Casagrande V, Cardellini M, Martelli E, Terrinoni A, Amati F, Vasa-Nicotera M, Ippoliti A, Novelli G, Melino G, et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation. 2009;120:1524–1532. doi: 10.1161/CIRCULATIONAHA.109.864629 [DOI] [PubMed] [Google Scholar]
- 11.Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J. 2015;36:3404–3412. doi: 10.1093/eurheartj/ehv290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532 [DOI] [PubMed] [Google Scholar]
- 14.Daiber A, Xia N, Steven S, Oelze M, Hanf A, Kroller-Schon S, Munzel T, Li H. New therapeutic implications of endothelial nitric oxide synthase (eNOS) function/dysfunction in cardiovascular disease. Int J Mol Sci. 2019;20:187 doi: 10.3390/ijms20010187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Yébenes VG, Bartolomé-Izquierdo N, Nogales-Cadenas R, Pérez-Durán P, Mur SM, Martínez N, Di Lisio L, Robbiani DF, Pascual-Montano A, Cañamero M, et al. miR-217 is an oncogene that enhances the germinal center reaction. Blood. 2014;124:229–239. doi: 10.1182/blood-2013-12-543611 [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002 [DOI] [PubMed] [Google Scholar]
- 17.Venegas-Pino DE, Banko N, Khan MI, Shi Y, Werstuck GH. Quantitative analysis and characterization of atherosclerotic lesions in the murine aortic sinus. J Vis Exp. 2013;82:50933 doi: 10.3791/50933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oller J, Méndez-Barbero N, Ruiz EJ, Villahoz S, Renard M, Canelas LI, Briones AM, Alberca R, Lozano-Vidal N, Hurlé MA, et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease adamts1 and in a mouse model of Marfan syndrome. Nat Med. 2017;23:200–212. doi: 10.1038/nm.4266 [DOI] [PubMed] [Google Scholar]
- 19.Gómez-Pardo E, Fernández-Alvira JM, Vilanova M, Haro D, Martínez R, Carvajal I, Carral V, Rodríguez C, de Miguel M, Bodega P, et al. A Comprehensive lifestyle peer group-based intervention on cardiovascular risk factors: the randomized controlled fifty-fifty program. J Am Coll Cardiol. 2016;67:476–485. doi: 10.1016/j.jacc.2015.10.033 [DOI] [PubMed] [Google Scholar]
- 20.Fernández-Alvira JM, Fuster V, Pocock S, Sanz J, Fernández-Friera L, Laclaustra M, Fernández-Jiménez R, Mendiguren J, Fernández-Ortiz A, Ibáñez B, et al. Predicting subclinical atherosclerosis in low-risk individuals: ideal cardiovascular health score and fuster-BEWAT score. J Am Coll Cardiol. 2017;70:2463–2473. doi: 10.1016/j.jacc.2017.09.032 [DOI] [PubMed] [Google Scholar]
- 21.Leung SW, Vanhoutte PM. Endothelium-dependent hyperpolarization: age, gender and blood pressure, do they matter? Acta Physiol (Oxf). 2017;219:108–123. doi: 10.1111/apha.12628 [DOI] [PubMed] [Google Scholar]
- 22.Ellis A, Pannirselvam M, Anderson TJ, Triggle CR. Catalase has negligible inhibitory effects on endothelium-dependent relaxations in mouse isolated aorta and small mesenteric artery. Br J Pharmacol. 2003;140:1193–1200. doi: 10.1038/sj.bjp.0705549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Xia N, Hasselwander S, Daiber A. Resveratrol and vascular function. Int J Mol Sci. 2019;20:2155 doi: 10.3390/ijms20092155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35:146–154. doi: 10.1016/j.tips.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cantó C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev. 2012;64:166–187. doi: 10.1124/pr.110.003905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tabas I. Macrophage apoptosis in atherosclerosis: consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid Redox Signal. 2009;11:2333–2339. doi: 10.1089/ars.2009.2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trigatti BL, Fuller M. HDL signaling and protection against coronary artery atherosclerosis in mice. J Biomed Res. 2016;30:94–100. doi: 10.7555/JBR.30.20150079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Getz GS, Reardon CA. Do the apoe-/- and Ldlr-/- mice yield the same insight on atherogenesis? Arterioscler Thromb Vasc Biol. 2016;36:1734–1741. doi: 10.1161/ATVBAHA.116.306874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esteban V, Méndez-Barbero N, Jiménez-Borreguero LJ, Roqué M, Novensá L, García-Redondo AB, Salaices M, Vila L, Arbonés ML, Campanero MR, et al. Regulator of calcineurin 1 mediates pathological vascular wall remodeling. J Exp Med. 2011;208:2125–2139. doi: 10.1084/jem.20110503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quillon A, Fromy B, Debret R. Endothelium microenvironment sensing leading to nitric oxide mediated vasodilation: a review of nervous and biomechanical signals. Nitric Oxide. 2015;45:20–26. doi: 10.1016/j.niox.2015.01.006 [DOI] [PubMed] [Google Scholar]
- 32.Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247–260. doi: 10.1016/j.cardiores.2007.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu K, Xuekelati S, Zhou K, Yan Z, Yang X, Inayat A, Wu J, Guo X. Expression profiles of six atherosclerosis- associated microRNAs that cluster in patients with hyperhomocysteinemia: a clinical study. DNA Cell Biol. 2018;37:189–198. doi: 10.1089/dna.2017.3845 [DOI] [PubMed] [Google Scholar]
- 34.Stein S, Schäfer N, Breitenstein A, Besler C, Winnik S, Lohmann C, Heinrich K, Brokopp CE, Handschin C, Landmesser U, et al. SIRT1 reduces endothelial activation without affecting vascular function in ApoE-/- mice. Aging (Albany NY). 2010;2:353–360. doi: 10.18632/aging.100162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stein S, Lohmann C, Schäfer N, Hofmann J, Rohrer L, Besler C, Rothgiesser KM, Becher B, Hottiger MO, Borén J, et al. SIRT1 decreases lox-1-mediated foam cell formation in atherogenesis. Eur Heart J. 2010;31:2301–2309. doi: 10.1093/eurheartj/ehq107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villarroya J, Redondo-Angulo I, Iglesias R, Giralt M, Villarroya F, Planavila A. Sirt1 mediates the effects of a short-term high-fat diet on the heart. J Nutr Biochem. 2015;26:1328–1337. doi: 10.1016/j.jnutbio.2015.07.029 [DOI] [PubMed] [Google Scholar]
- 37.Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, Sadoshima J. PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 2011;14:598–611. doi: 10.1016/j.cmet.2011.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren NSX, Ji M, Tokar EJ, Busch EL, Xu X, Lewis D, Li X, Jin A, Zhang Y, Wu WKK, et al. Haploinsufficiency of SIRT1 enhances glutamine metabolism and promotes cancer development. Curr Biol. 2017;27:483–494. doi: 10.1016/j.cub.2016.12.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf). 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x [DOI] [PubMed] [Google Scholar]
- 41.Heiss C, Rodriguez-Mateos A, Kelm M. Central role of eNOS in the maintenance of endothelial homeostasis. Antioxid Redox Signal. 2015;22:1230–1242. doi: 10.1089/ars.2014.6158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marti CN, Gheorghiade M, Kalogeropoulos AP, Georgiopoulou VV, Quyyumi AA, Butler J. Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol. 2012;60:1455–1469. doi: 10.1016/j.jacc.2011.11.082 [DOI] [PubMed] [Google Scholar]
- 43.Vasquez EC, Peotta VA, Gava AL, Pereira TM, Meyrelles SS. Cardiac and vascular phenotypes in the apolipoprotein E-deficient mouse. J Biomed Sci. 2012;19:22 doi: 10.1186/1423-0127-19-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savarese G, Lund LH. Global public health Burden of heart failure. Card Fail Rev. 2017;3:7–11. doi: 10.15420/cfr.2016:25:2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Armstrong PW. Left ventricular dysfunction: causes, natural history, and hopes for reversal. Heart. 2000;84suppl 1i15–7:discussion i50. doi: 10.1136/heart.84.suppl_1.i15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399 [DOI] [PubMed] [Google Scholar]
- 47.Gaasch WH, Zile MR. Left ventricular diastolic dysfunction and diastolic heart failure. Annu Rev Med. 2004;55:373–394. doi: 10.1146/annurev.med.55.091902.104417 [DOI] [PubMed] [Google Scholar]
- 48.Nie X, Fan J, Li H, Yin Z, Zhao Y, Dai B, Dong N, Chen C, Wang DW. miR-217 promotes cardiac hypertrophy and dysfunction by targeting PTEN. Mol Ther Nucleic Acids. 2018;12:254–266. doi: 10.1016/j.omtn.2018.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.