

Abstract
Purpose
In 2007 the two senior authors wrote a review on the structure and function of the endothelial glycocalyx layer (Weinbaum in Annu Rev Biomed Eng 9:121–167, 2007). Since then there has been an explosion of interest in this hydrated gel-like structure that coats the luminal surface of endothelial cells that line our vasculature due to its important functions in (A) basic vascular physiology and (B) vascular related diseases. This review will highlight the major advances that have occurred since our 2007 paper.
Methods
A literature search mainly focusing on the role of the glycocalyx in the two major areas described above was performed using electronic databases.
Results
In part (A) of this review, the new formulation of the century old Starling principle, now referred to as the Michel–Weinbaum glycoclayx model or revised Starling hypothesis, is described including new subtleties and physiological ramifications. New insights into mechanotransduction and release of nitric oxide due to fluid shear stress sensed by the glycocalyx are elaborated. Major advances in understanding the organization and function of glycocalyx components, and new techniques for measuring both its thickness and spatio-chemical organization based on super resolution, stochastic optical reconstruction microscopy (STORM) are presented. As discussed in part (B) of this review, it is now recognized that artery wall stiffness associated with hypertension and aging induces glycocalyx degradation, endothelial dysfunction and vascular disease. In addition to atherosclerosis and cardiovascular diseases, the glycocalyx plays an important role in lifestyle related diseases (e.g., diabetes) and cancer. Infectious diseases including sepsis, Dengue, Zika and Corona viruses, and malaria also involve the glycocalyx. Because of increasing recognition of the role of the glycocalyx in a wide range of diseases, there has been a vigorous search for methods to protect the glycocalyx from degradation or to enhance its synthesis in disease environments.
Conclusion
As we have seen in this review, many important developments in our basic understanding of GCX structure, function and role in diseases have been described since the 2007 paper. The future is wide open for continued GCX research.
Keywords: Endothelial glycocalyx, Mechanotransduction, Revised Starling principle, Michel–Weinbaum model, Cancer cell glycocalyx
Introduction
In 2007 the two senior authors and ER Damiano wrote a highly cited extensive review220 summarizing what was known at the time of the structure and function of the endothelial glycocalyx (GCX), the hydrated gel-like structure that coats the luminal surface of the endothelial cells (EC) that line our vasculature. This surface matrix is comprised of sulfated proteoglycans—the transmembrane syndecans and the membrane bound glypicans, with their covalently bound glycosaminoglycans (GAGs)—heparan sulfate (HS) and chondroitin sulfate (CS) and the non-sulfated GAG hyaluronic acid (HA), as well as glycoproteins and plasma proteins (see Fig. 1). In the past decade there has been an explosion of interest in this hydrated gel-like structure due to its important functions in (A) basic vascular physiology and (B) vascular related diseases. In Part A we describe how the GCX has led to a new formulation of the century old Starling principle195 now referred to as the Michel–Weinbaum glycoclayx model134,219 or the revised Starling hypothesis for microvascular fluid exchange. There have been new insights into mechanotransduction and the release of nitric oxide (NO) due to fluid shear stress (FSS),50,60,231 major advances in our understanding of the structure and function of its various components,159,236 and new techniques for measuring both its thickness and structural organization based on confocal,232 rapid freeze substitution51 and super resolution stochastic optical reconstruction microscopy (STORM).57
Figure 1.

Simplified schematic of the glycocalyx: cell-surface core proteins and their associated GAGs. Reprinted from Ref. 12.
In Part B we emphasize that it is now recognized that artery wall stiffness associated with hypertension and aging induces GCX degradation, endothelial dysfunction and vascular disease.120 In addition to atherosclerosis and cardiovascular diseases, the GCX plays an important role in lifestyle related diseases (e.g., diabetes) and cancer.200 Infectious diseases including sepsis, Dengue, Zika and Corona viruses, and malaria also involve the GCX.85,136,167 Because of increasing recognition of the role of the GCX in a wide range of diseases, there has been a vigorous search for methods to protect the GCX from degradation or to enhance its synthesis in disease environments.200
This review will highlight the major advances outlined above that have occurred since the 2007 paper.
Part A: The Role of the Glycocalyx in Vascular Physiology
The Revised Starling Principle: Michel–Weinbaum Model for Microvascular Fluid Exchange
It is relatively rare that a widely accepted textbook description of a basic microcirculatory phenomenon should be challenged after nearly a century of use. However, this is indeed what happened with the classical Starling hypothesis. While the basic ideas for the revised Starling hypothesis were introduced by Michel134 and Weinbaum219 more than 20 years ago and the experimental verification of the Michel–Weinbaum model confirmed in mesentery venules in frog by Hu et al.77 and rat by Adamson et al.1 more than a decade ago, the widespread acceptance of the revised hypothesis had not occurred when our 2007 review paper on the GCX was published. Two new contributions greatly changed this. In 2010 Levick and Michel108 wrote a Spotlight Review in Cardiovascular Research which emphasized the clinical importance of the revised principle and more recently Tom Woodcock, an English clinician, prepared a 27 min video emphasizing the clinical applications of the revised hypothesis. This video is the lead entry if you do a Google search for the Michel–Weinbaum model which currently has > 100,000 listings.
In the present review, after a brief summary of the basic differences between the classic and revised Starling hypothesis, we will highlight the fundamental differences between one and two-dimensional models and the role that trapped microdomains within the intercellular cleft and at the cleft exit play in the time dependent response that leads to steady state behavior.
Classic Starling principle
From classic thermodynamic principles one can write the volume filtration rate per unit area of endothelial surface or fluid flux Jv/A as a balance between two forces, the difference in hydrostatic pressures across a membrane PL − Pi and the difference in oncotic pressures πp − πi where the subscripts L and i refer to the lumen of the microvessel and the interstitial fluid (ISF) in the tissue and πP is the oncotic pressure of plasma. If this was a simple membrane with pores one could write the expression for Jv/A as
| 1 |
where Lp and σ are the filtration coefficient (or hydraulic conductivity) and osmotic reflection coefficient respectively. The value for σ varies between 0 and 1 where 0 would be for a solute much smaller than the membrane pores and 1 for a plasma protein that was too large to pass through the pore. Starling realized that the plasma proteins in the blood and, in particular the most abundant plasma protein, albumin, would be greatly hindered and could have a value of σ that would be close to one creating a colloidal oncotic pressure (COP) for fluid absorption.
For many years it was widely believed that the pores in the EC monolayer were the intercellular clefts between adjacent ECs and the constricting pores were either a matrix in the cleft or associated with the junction strands between the ECs which served as the molecular filter for the plasma proteins. The other basic assumption in the original hypothesis was that the COP in the plasma, which could be measured, was typically 25–28 mmHg and the interstitial COP was close to zero, though the techniques for measuring the latter were not developed until the 1980s. Using classic micropipette measuring techniques developed by Landis101 and Pappenheimer and Soto-Rivera,161 one observed that at heart level the typical hydrostatic pressure on the arterial side of capillaries was 35 mmHg whereas on the venous side a typical value was 15 mmHg. Thus, when the capillary pressure PL was > 25 mmHg, water would leave the capillary. When PL was < 25 mmHg water would be reabsorbed. Thus, several generations of medical students were taught using Eq. (1) that the plasma that filtered out of our capillaries when PL was > πP, and was reabsorbed on the venous side because the COP of our plasma proteins πP was > PL. This was often referred to as the non-steady state absorption rule applied at heart level.
With the advent of reliable techniques for measuring the COP of the interstitial space it was discovered that πi was not close to zero but typically 40% of πP. This extraordinary observation was first reported in 1991 by Levick107 who showed that in the 14 tissues, including muscle, skin and lung, where πi had been measured there was no venous absorption. The only exceptions were the renal cortex, where fluid is continuously taken up by the peritubular capillaries, and the ascending vasa recta of the intestinal mucosa where protein free fluid is continuously secreted by neighboring epithelia, see Levick and Michel.108
Revised Starling Principle
In 1996 Levick and Michel organized a centennial meeting in London to celebrate the 100th anniversary of Starling’s classic 1896 paper. The highlight of this meeting was Levick’s presentation summarizing the results of his 1991 paper,107 Fig. 2b in Ref. 108. showing the imbalance of the four classical Starling pressures when all four terms are measured in the same tissue and species. On the last day of the meeting Levick, Michel, Curry and Weinbaum met to try and understand how it was possible that the predictions of Eq. (1), the classic Starling force balance, could be so far off in nearly all tissues where these forces had been measured. This situation was further complicated by Michel and Phillips now classic experiment135 shown in Fig. 2 using the Landis technique applied to single perfused frog mesentery capillaries where the fluid exchange is measured immediately after raising or lowering the capillary pressure Pc (transient state) and after Pc has been held constant for 2–5 min (steady state). The transient state clearly satisfied the classic Starling force balance when Pc was less than πL whereas in the steady state Jv/A always returned to a very low rate of filtration and the curve had the characteristic shape of a hockey stick. The latter behavior is described in detail in a recent review by Curry.37
Figure 2.
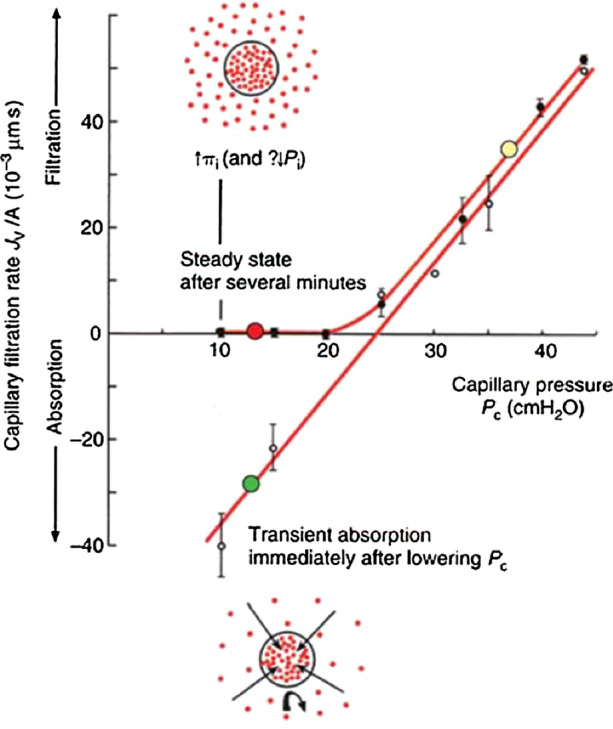
Water flux Jv/A of frog mesentery capillary as a function of capillary pressure in single perfused frog mesentery capillary. Note the transient state in which measurements are performed in the first 15 s after the perfusion pressure is fixed fall along a straight line in which absorption does occur for pressures below 25 cm H2O, whereas steady-state measurements performed 2–5 min after perfusion pressure is fixed show no absorption, but a low level of filtration at low capillary pressures. Reprinted from Ref. 108.
The shape of the steady state curve in Fig. 2 can be predicted from a standard one-dimensional convection–diffusion equation in which one requires that in the far field at steady state the tissue concentration Ci is equal to the ratio of the solute influx from all sources Js to the total water flux Jv or Ci = Js/Jv. The solute crossing the endothelium can come not only from fluid flow though the intercellular clefts but also from vesicular transport or back loading and Ci varies inversely with Jv. Michel133 realized that as Jv decreased for the steady state curve in Fig. 2, the concentration in the tissue would have to approach the concentration in the plasma and reabsorption would vanish. The relation between Jv and Pc is non-linear since Jv depends on the extracapillary protein concentration. The remarkable feature of Michel’s transport equation is that it could reproduce the shape of the steady state curve in Fig. 2, with its hockey stick like bend, by varying the value of a single parameter, the permeability coefficient PA for albumin in the expression for the Peclet number (Jv/A)(1 − σ)/PA without ever including the cleft or its tight junction strand or the mixing of the ISF at the cleft exit. This unusual behavior will be explained with the aid of the two-dimensional model in Fig. 3 where these features are included.
Figure 3.

Schematic of idealized mathematical model showing surface matrix layer, cleft with junction strand, and tissue regions B and C describing mixing at cleft exit. Dimensions shown are typical of frog mesentery capillary. Reprinted from Ref. 219.
What Michel and Weinbaum independently realized after the 1996 meeting was that the molecular sieve for plasma proteins, and in particular albumin, did not lie within structures within the cleft itself but the GCX at the surface of the endothelium which clearly covered the cleft entrance. This new view was presented by Michel in his 1997 review paper summarizing the results of the Starling centennial meeting134 and Weinbaum in his 1997 Whitaker Distinguished Lecture at the annual Meeting of BMES in La Jolla.219 Michel intuitively realized that the protein concentration gradients within the cleft would be small if the GCX was a formidable barrier to diffusion and his conceptual model also included alternate non-convective pathways for larger molecules, such as vesicular transport which bypassed the clefts entirely. Weinbaum, on the other hand, shared Michel’s view of the GCX, as the primary molecular barrier, but was also concerned about the role of the tight junction strands, since if the breaks in the strands were small and widely spaced the convective transport of proteins through the breaks could greatly impede back diffusion of proteins from the tissue. If this happened the concentration of the proteins behind the GCX could differ greatly from the ISF. Such non-uniformity in behavior would need to be described by a two-dimensional transport equation in the cleft.
Weinbaum’s 2-D Transport Model
A sketch of the two-dimensional transport model used in Weinbaum219 is shown in Fig. 3. Instead of Eq. (1) one applies the Starling force balance just across the GCX
| 2 |
Here the subscript G refers to the protein concentration just behind the GCX (x = 0 in Fig. 3) and one allows both the pressure and concentration to vary with lateral position y behind the GCX. As shown in Fig. 3 the breaks are periodically arranged along the length of the junction strand at a distance 2D = 4320 nm, where y = 0 is at the center of each break whose width 2d is 150 nm. These dimensions are typical for frog mesentery venules. The model has four regions, (1) a GCX of thickness 150 nm, (2) the cleft itself whose depth is 400 nm with a junction strand at x = 200 nm, (3) a mixing region B of radius 5 µm, the half distance for the separation of the clefts, and (4) a mixing region C of width 100 µm, the half distance between adjacent venules. The concentration in region C is set at the value Ci = CA = Js/Jv as proposed in Michel or Ci = 0.4 CL as observed in Levick.107 A unique feature of the model is that the flow in the cleft is treated as a 2-D Hele-Shaw flow so that the detailed pressure distribution and velocity field within the cleft can be examined and this 2-D velocity field used in the transport equation for the solute concentration.
We first examine the results of this 2-D model for Michel and Phillips steady state experiments in Fig. 2 for two cases, Pc = 43 cm H2O, a high filtration state, and Pc = 15 cm H2O, a value significantly below plasma oncotic pressure, Figs. 4 and 5 respectively. There is a small negative swelling pressure in the tissue of − 2 mmHg.68 In Fig. 4a we see that at the high pressure, 43 cm H2O, nearly half of the pressure drop occurs across the GCX and there is a significant additional drop in pressure as the water is funneled into the break in the junction strand. The pressure drop across the junction strand at x = 200 nm is nearly the same as the pressure drop across the GCX. The breadth of the spread in pressure on the GCX side of the break is wider than on the tissue side leading to a narrower water jet on the exit side than the entrance side (velocity profiles not shown). More striking in Fig. 4b is the fact that the concentration profile behind the GCX at x = 0 is nearly identical to that at the cleft exit. The albumin diffusion coefficient in the cleft is assumed to be that of free diffusion D∞ whereas that in the GCX DG is 1000 times smaller. This value of DG provides for a very close fit of the bend in the curve shown in Fig. 2 for Michel and Phillips steady state experiments. This observation, that the concentration in the cleft is nearly uniform, is central to Michel’s133 simplified one-dimensional model. Note also that in this high filtration limit C at the cleft exit has the limiting value (1 − σ)CL for pure filtration across the GCX.
Figure 4.
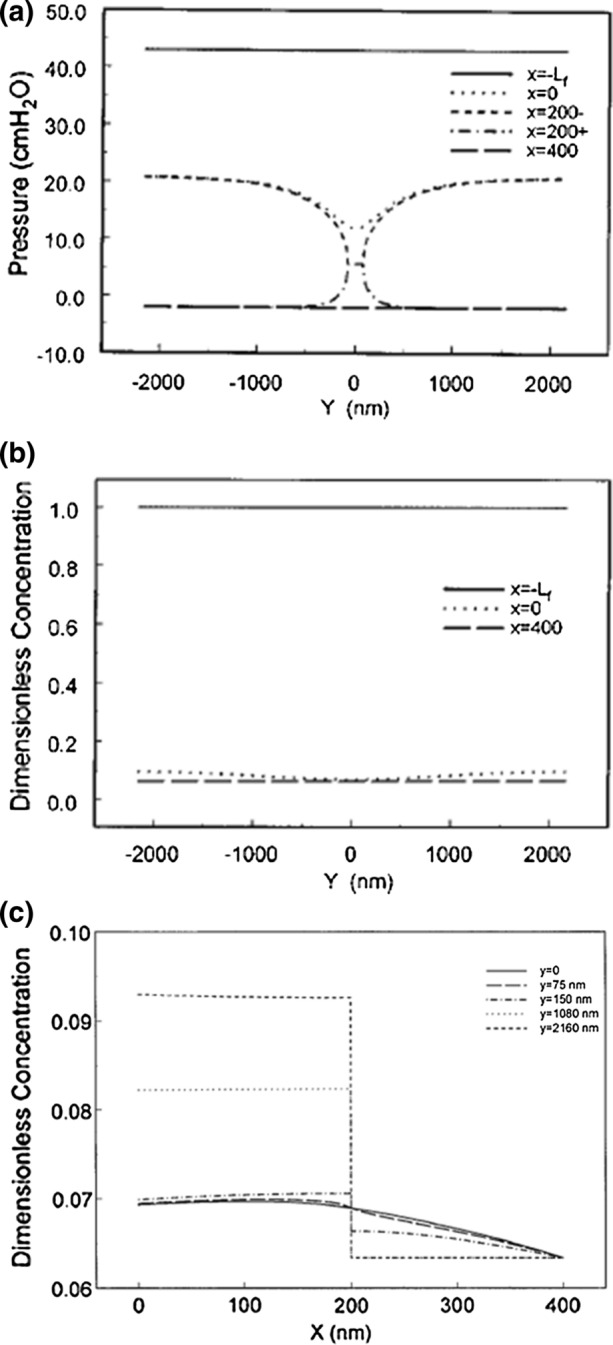
Predictions of theoretical model for Pc = 43 cm H2O for pressure (a), protein concentration profiles at various locations (b, c) in cleft identified in Fig. 3. (a) and (b) reprinted from Ref. 219; (c) reprinted from Ref. 78.
Figure 5.
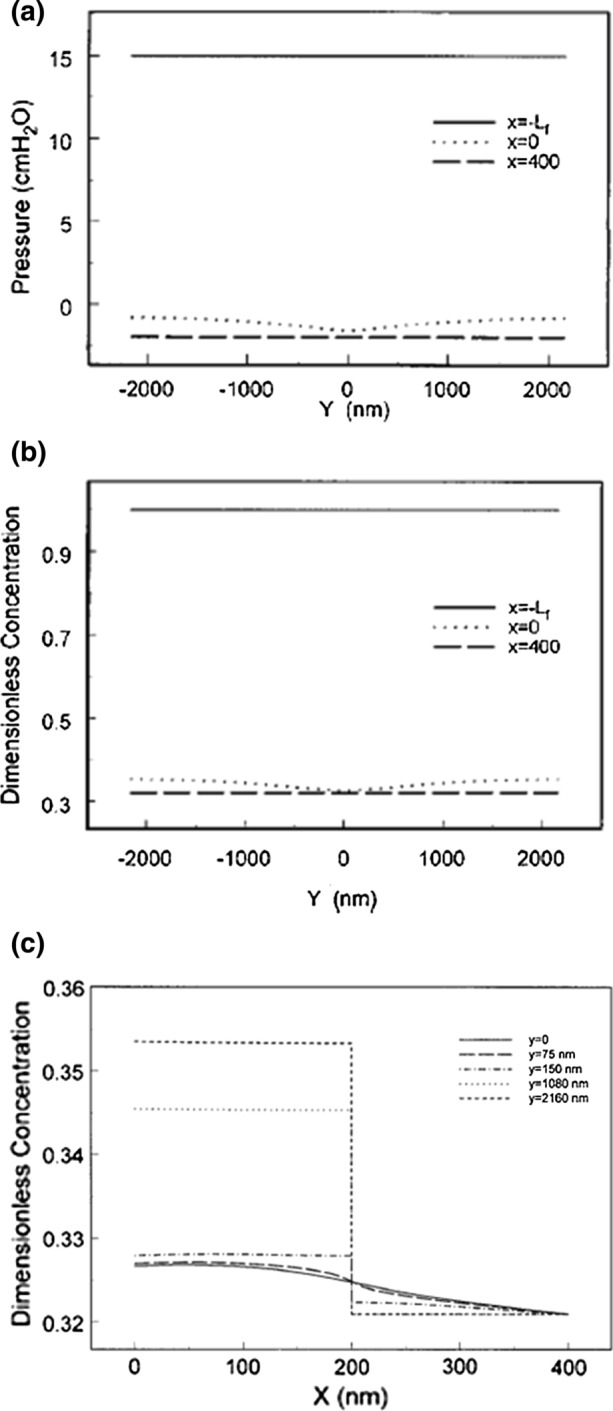
Predictions of theoretical model for Pc = 15 cm H2O for pressure (a), protein concentration profiles at various locations (b, c) in cleft identified in Fig. 3. (a) and (b) Reprinted from Ref. 219; (c) reprinted from Ref. 78.
The near uniformity in concentration at the cleft exit in Fig. 4b may seem surprising since there is a spike in the velocity profiles (not shown to save space) downstream of the break in the junction strand. The detailed concentration profiles at different y locations show why this is the case. Along y = 0, the cleft centerline, the concentration does drop continuously along the cleft centerline from a value 0.07 to near 0.06 close to the convection limit since σ = 0.94. One notes that there is a small but not insignificant drop in concentration, roughly 3%, across the junction strand at larger values of y due to the diffusional spreading in the y direction. Convection is not important since there is hardly any fluid motion in the y direction except in the immediate vicinity of the junction strand breaks. Michel’s133 model does not consider any mixing processes at the cleft exit but only requires that far from the cleft exit one has to achieve a uniform concentration Ci = CA = Js/Jv since there is no region B in Fig. 3. In Weinbaum219 and Hu et al.78 a simple realistic model is developed for this mixing process in region B. The flow at the cleft exit is treated as a line source whose velocity and concentration vary in the y direction and transport equations for the radial and axial spread of the cleft exit jets are developed which satisfy appropriate matching conditions with the two-dimensional solutions in the cleft.
In Fig. 5a we see the steady state pressure distribution for the case where Pc = 15 cm H2O and a small negative tissue pressure of − 2 mmHg due to tissue matrix expansion. The pressure profiles behind the GCX at x = 0 and the cleft exit x = 0 differ only slightly indicating that that there is hardly any water flux in the cleft except for a very low filtration since the pressure at x = 0 is slightly higher than the cleft exit. The concentration profiles in Fig. 5b look very similar to those for PC = 43 cm H20 in Fig. 4b with the important exception that the dimensionless cleft exit concentration at x = 400 nm is now close to 0.32. The drop in concentration across the junction strand more distant from the junction strand breaks is again about 3%. Since the experiment starts with the tissue side in pure Ringer solution it is evident that the transition to steady state, which takes several minutes, occurs through a process where initially after the rapid drop in pressure from 43 cmH2O to 15 cm H2O there is reabsorption which brings water into cleft where the GCX acts as a molecular sieve causing the protein concentration behind the GCX to increase until there is a near equilibrium of hydrostatic and oncotic pressures across the GCX. This is clearly seen in Fig. 5c where the concentration across the cleft is shown when this equilibrium is achieved. One notes that along the center line of the junction strand break there is a small positive gradient directed toward x = 0. In the plasma the sum of the COP 26 cm H2O and hydrostatic pressures 15 cm H2O is a net 9 cm H2O whereas behind the GCX at x = 0 the average oncotic pressure is roughly 0.34 πp or 9.5 cm H2O providing for a very small filtration through the junction strand breaks.
In summary, the steady state solutions at Pc = 43 cm H2O and Pc = 15 cm H2O in Figs. 4 and 5 show that concentration gradients behind the GCX within the cleft are very small because the diffusional resistance in the GCX overwhelms that in the cleft itself and at the low filtration pressure a steady state reabsorption cannot be achieved because the back flow from the tissue will cause the concentration behind the GCX to rise to a value that is nearly equal to that in the lumen and a very low level of positive filtration is achieved.
Why Does Michel’s 1-D Model Work?
The results in Figs. 4 and 5 raise the critical question. Why does the simple one-dimensional model proposed in Michel133 provide such good agreement in the steady state data in Fig. 2 when it neither describes the transport in the cleft or the mixing at the cleft exit? The answer to this is that the model contains one degree of freedom, the permeability coefficient Ps that appears in the Peclet number Pe = (Jv/A)(1 − σ)/Ps in Michel’s equation
| 3 |
By choosing the desired value of Ps or Peclet number Pe one can obtain an optimum fit for the steady state curve in Fig. 2. In essence this one parameter captures the combined resistance of the GCX, the cleft and the mixing region B in Fig. 3 of the two-dimensional model in Weinbaum219 and Hu et al.78
What Happens If the Tissue is Back Loaded or There is a Vesicular Pathway?
While Michel’s transient and steady state experiments in Fig. 2 are very insightful they were performed in only one species, frog, and in one tissue, mesentery. In marked contrast the in vivo results in Levick,107 where the classic Starling principle was being challenged, included skin, muscle and lung tissue in humans and several animal species. For many of these tissues the solute concentration in the far field in region C in Fig. 3 is typically 0.4 CL. The solutions for the concentration profiles within the cleft for this case are shown in Fig. 6a and 6b. While the cleft and junction strand geometry is typical of frog mesenteric venules, the only available cleft geometry at the time, subsequent detailed reconstructions of rat mesenteric venules by Adamson et al.1 revealed a quite similar structure. The results in Fig. 6a and 6b are for a high filtration state where PL = 43 cm H2O.
Figure 6.
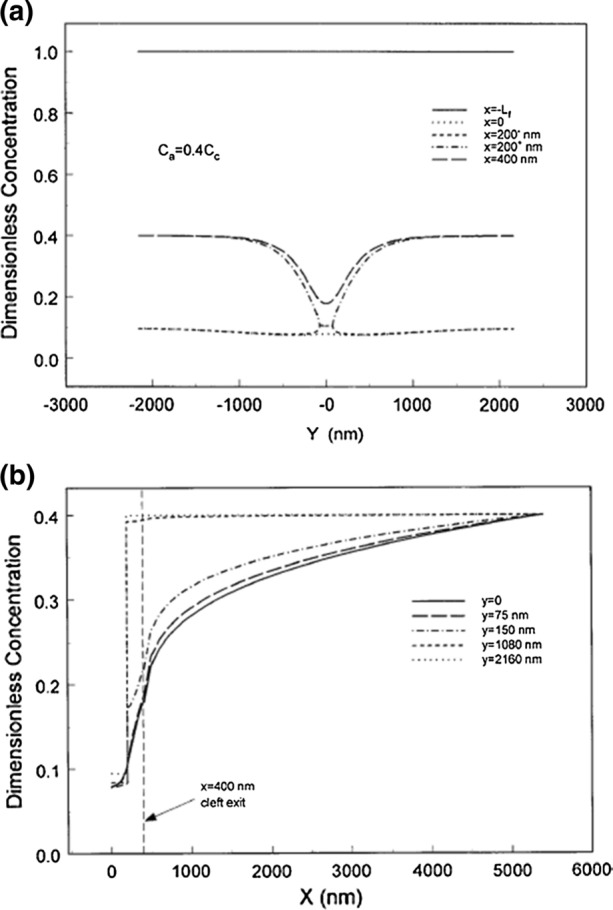
Predictions of theoretical model for Pc = 43 cm H2O for concentration profiles in the cleft along x (a) and along y (b) when there is a parallel non-convective pathway for albumin which increases the tissue concentration at the edge of region B, Ca, to 0.4Cc. Flow geometry is shown in Fig. 3. Plasma oncotic pressure is 26 cm H2O and σ = 0.94. Reprinted from Ref. 78.
The results in Fig. 6a differ greatly from the concentration profiles in Fig. 4b where at the same lumen pressure the concentration is nearly uniform throughout the cleft and satisfies the convective limit (1 − σ)CL. The basic difference is that all the solute passes through the breaks in the junction strand in Fig. 4b whereas in the present case there is another transendothelial pathway for large solutes which is non-convective, e.g. vesicular transport. There is a large increase in concentration across the junction strand and the concentration at the cleft exit is close to 0.4 CL except in the vicinity of the junction strand break. Here the convective flow through the break greatly impedes back diffusion from the tissue into the protected region between the GCX and the front part of the junction strand. Because of the large difference in concentration across the junction strand the concentration behind the GCX can differ greatly from that in the tissue and the classical Starling Eq. (1) no longer applies.
These results are more easily understood if one looks at the concentration profiles in Fig. 6b at fixed values of y from x = 0 to the far field at x = 5 µm, the edge of region B where the concentration is 0.4 CL. For values of y removed from the junction strand break the concentration behind the GCX is uncoupled from that in the tissue and on the centerline of the break, y = 0, where the C profile is continuous, the dimensionless concentration increases from 0.08 at x = 0 to 0.18 at the cleft exit. This same behavior will be observed if the tissue is back loaded as we discuss next.
The behavior just described was convincingly demonstrated in both frog77 and rat mesenteric venules1 where the tissue was back loaded so that the lumen and tissue concentration were both 50 mg/mL. In addition, serial section microscopy was performed to determine the location and spacing of the junction strand breaks so that the 2D model in Weinbaum219 could be rigorously applied. In both experiments the lumen was perfused with albumin and the exterior of the mesentery was superfused with saline or the same albumin solution as in the lumen. The experiments for rat mesentery are shown in Fig. 7.
Figure 7.
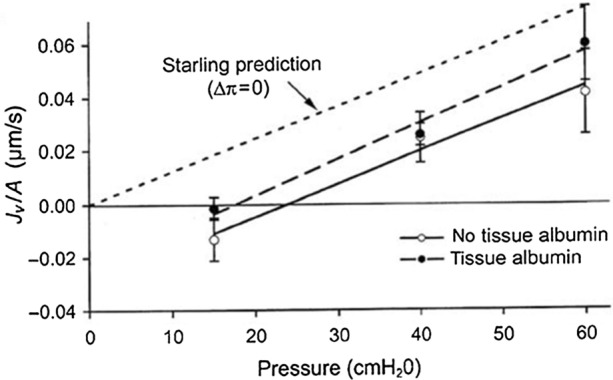
Fluid exchange in single rat mesenteric venules at controlled microvascular pressure, measured by the modified Landis red cell method (transient state). The lumen was perfused with albumin solution and the exterior was superfused with saline (‘no tissue albumin’, open symbols) or the same albumin solution as in the lumen (‘tissue albumin’, filled symbols). Short dashes show the expected increase in filtration rate for the latter according to the classic Starling principle. The much smaller observed response was as predicted by the glycocalyx–cleft model. Reprinted from Ref. 108.
The short dashed curved represents the classical Starling prediction when πL = πi. The longer dashed curve represents the results with tissue albumin at the same concentration as the lumen. For frog mesentery77 there was very little change in the filtration rate in marked contrast to the classical Starling prediction but in good agreement with the two-dimensional Weinbaum model in Hu et al.78 In the refined series of experiments with rat mesenteric venules shown in Fig. 7 (Fig. 4b in Levick and Michel108) the increase in Jv due to raising πi from zero to plasma level, was only 30% of that predicted by the classic Starling equation. Measurement of filtration across confluent cultured bovine endothelium in a Ussing chamber where the oncotic pressure was either the same or different in the luminal and abluminal reservoirs by Pang and Tarbell160 have given results very similar to those just described. At the time these tissue culture experiments were performed the role of heparinase in compromising the GCX had not been performed.
Pericytes and Enclosed Microdomains
We close this section with what is probably the most important mystery in the Michel and Phillips135 paper, the time scale over which the transition from the transient to the steady curves in Fig. 2 occurs. This transition was completed in a time that was typically less than 2 min. Using the simplified 1D model in Zhang et al.,241 one can estimate both Jv/A and JS and calculate how long it would take the tissue concentration Ci to change. The velocity in the transient state at the cleft exit when the pressure is dropped from 43 to 15 cm H2O at the start of reabsorption is − 5.6 µm/s and slowly rises to 0.2 µm/s when the new steady state of low filtrations is achieved (see Fig. 1 in Zhang et al.242). There is a 500 fold expansion in area from the cleft exit to the surrounding tissue so the average velocity in the tissue surrounding the cleft exit is roughly 0.002 times the cleft exit velocity or ~ 6 nm/s. The tissue itself is filled with water that has entered during the high filtration state and its concentration is close to the convective limit which we showed in Fig. 4 is 0.07CL. The half spacing of the mesenteric venules is typically 100 µm and it is this tissue volume which needs to be drained to achieve the new steady state of slow filtration. Using the above estimate of the water velocity in the tissue this would take approximately 5 h and not two minutes and yet the concentration in the cleft has risen from 0.07 CL to 0.32 CL at the cleft exit. A similar calculation for the solute would show that it would take 6 days for the solute flux from the cleft to raise the tissue concentration from 3 to 25.6 mg/mL. This is impossible unless something fundamental has been left out of the analysis. This basic paradox was finally explained in Zhang et al.242
In the same way that there is a small protected region or enclosed microdomain between the rear of the GCX and the front of the tight junction strand, there is a similar enclosed microdomain at the cleft exits of rat mesenteric venules as shown in Fig. 8. These venules which are typically about 40 µm in diameter have typically 7–8 cleft exits along a vessel cross-section. Approximately 85% of these cleft exits are covered by pericytes (PC) which cover much of the surface of these venules. As shown in the enlarged image in Fig. 8b there is an enclosed trapped microdomain (TM) formed by the pericytes surrounding the cleft exits whose thickness can vary between 100 and 500 nm depending on the state of hydration. At the edge of the TM there is an attachment region or adherens junction between the EC membrane and the edge of the PCs. The TM region is very similar in structure to the region between the EC layer in aortic endothelium and the internal elastic lamina which also has been shown to vary in thickness by about fivefold depending on the pressure in the lumen and state of hydration.80,81
Figure 8.
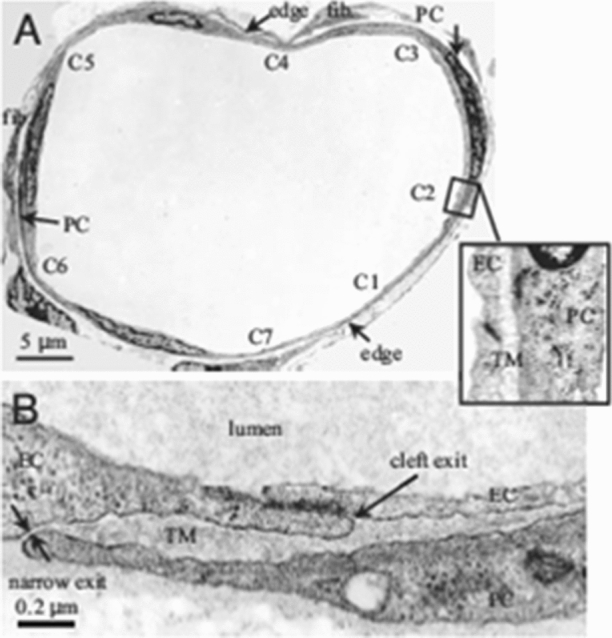
Electron microscope images of a rat venular microvessel (VM). (a) Cross-section of a VM in rat mesentery. There are seven endothelial cell (EC) clefts, C1–C7, with all of the cleft exits covered by neighboring pericytes (PCs). PCs are immediately underneath ECs. There are two fibroblasts (fib.) outside PCs. The blowout shows C2 covered by a PC with a trapped microdomain (TM) of uniform thickness in between. (b) An enlarged view showing a cleft exit covered by a PC, creating a TM of roughly uniform thickness except at the narrow exit. Reprinted from Ref. 242.
Zhang et al.242 have developed an elastohydrodynamic model for the TM region observed in Fig. 8. The key results in this model are shown in Fig. 9a and 9b. One observes in Fig. 9a how the fluid drains from the TM in the case when PL has been quickly dropped from 60 cmH2O to 15 cmH2O and a new steady state allowed to develop. One notes for this case that the cleft exit velocity increases from − 7.4 µm/s reabsorption to 0.2 µm/s, a slow rate of steady filtration. In Fig. 9b we see how the concentration in the TM increases from 3 mg/mL to 25.6 mg/mL as the fluid drains out of the TM and the change in TM thickness W from 500 nm to roughly 100 nm due to this fluid reabsorption. The key observation is that the time scale of this transition process is typically one minute, very similar to the classic paper of Michel and Phillips,135 and not hours. The experiments in Michel’s paper were conducted in frog mesenteric venules whereas the model results in Fig. 9 were based on the rat mesenteric venules shown in Fig. 8.
Figure 9.

Time-dependent change in v, C, and w at the cleft exit. Velocity v (a) and concentration C (b Left) and thickness w of region TM (b right) as functions of time after PL is dropped from 60 to 15 cm H2O. Reprinted from Ref. 242.
The model just described applies to tissues where a large fraction of the EC clefts are covered by pericytes and filtration occurs primarily through the clefts. However, the model can also be applied to skeletal muscle if there are small regions where water can accumulate. Clinically it has been observed that reabsorption can continue in human skeletal muscle for 20–30 min after a sudden drop in PL as would occur after hemorrhage.107 Typical reabsorption when PL was reduced 5 mmHg was 15–30 mL per kg of tissue in 30 min. Thus, reabsorption of between 0.3 and 0.6 L is expected from 20 kg of human muscle in 20–30 min. This reabsorption would produce a 15–20% increase in interstitial concentration. This is enough to slow but not stop further reabsorption and is shorter than values of an hour or more that would be required to produce changes in the total ISF volume.
In summary, we have tried to highlight in this section the fundamental differences between the one-dimensional model in Michel133 and the two-dimensional model in Weinbaum219 and Hu et al.78 where the detailed effect of the cleft and its junction strand can be examined as well as the mixing process at the cleft exit. We have also tried to explain why Michel’s one-dimensional model works so well in fitting the steady state results in Michel and Phillips135 in Fig. 2. The mixing process at the cleft exit is of particular significance when the tissue concentration is raised by a non-convective parallel pathway. We also explored the role that pericytes at the cleft exit play in determining the time scale of the transition from transient to steady state behavior, as might occur in muscle tissue when there is a sudden drop in capillary pressure due to hemorrhage, if there are local regions of trapped water in the vicinity of the microvessels.
This opening section of Part A, on the revised Starling Principle, has begun to reveal how vascular physiology is influenced by the GCX. This theme continues in the next section on Mechanotransduction with its emphasis on the GCX as a mechanosensor for shear-induced nitric oxide production and substrate stiffness-induced endothelial dysfunction associated with loss of GCX. The final section of Part A describes new insights into GCX structure obtained through high resolution microscopy that will contribute to future advances in cardiovascular physiology.
Mechanotransduction
An important physiological function of the GCX is to serve as a mechanosensor and transducer.61,69,200–202,220 It is well known that a dysfunctional endothelium is an early manifestation of atherosclerosis234 and that in disease prone locations such as branches and curvatures, the flow departs from pulsatile, unidirectional shear stress to create flow separation zones that include low wall shear stress, flow reversal, oscillatory and multi-directional shear stress, and sometimes turbulence (chaotic flow).39,41,247 In contrast, in adjacent, non-diseased regions, shear stress is higher and flow is unidirectional with minimal reversal. The GCX plays an important role in EC mechanotransduction of shear stress that mediates localization of atherosclerosis. Weinbaum et al.221 pointed out that the existence of the GCX could reduce the fluid shear stress on the vascular EC surface to a negligible level, while Secomb et al.185 described the transfer of fluid mechanical shear stress at the interface between the fluid and the outer regions of the GCX, to solid mechanical stress within the matrix that is transmitted to the cell body. Thi et al.205 further showed that the GCX is required for the EC cytoskeleton to respond to shear stress.
It is well established that after 24 h of laminar flow, ECs align in the direction of flow and their proliferation is suppressed. Yao et al.229 removed HS from EC in vitro using the specific enzyme heparinase III and observed that EC no longer aligned under flow after 24 h and they proliferated as if there were no flow present. Subsequently, Baeyens et al.7 reported that EC in the thoracic aortas of syndecan 4 (SDC4) null mice were poorly aligned in the direction of the flow. They found that depletion of SDC4 in human umbilical vein ECs (HUVECs) using shRNA also inhibited flow-induced alignment in vitro, which was rescued by re-expression. In hypercholesterolemic mice, they observed that deletion of SDC4 drastically increased atherosclerotic plaque burden with the appearance of plaque in normally resistant locations.
Shear-Induced Nitric Oxide Production
Effects of Enzyme Degradation of GCX on NO Production
Florian et al.60 showed that enzymatic removal of HS from the surface of cultured EC in vitro with heparinase III completely blocked shear-induced NO production in both steady flow (15 dyn/cm2) and reversing oscillatory flow (10 ± 15 dyn/cm2) out to 3 h exposure. These experiments included both the early phase (seconds to minutes after a step increase in shear) that is calcium and G-protein dependent as well as the later phase (minutes to hours) that is independent of calcium and G-protein.98 Yen et al.231 later demonstrated in vivo that degradation of HS in the GCX of rat mesenteric microvessels greatly inhibited NO production in response to an increase in flow. Pahakis et al.159 showed that shear-induced NO production was blocked by enzymes removing HS and HA, but not CS and that these treatments had no effect on the shear-induced production of the important vasodilator and antiplatelet agent, prostacyclin (PGI2).
In related work, Dull et al.48 examined the transmural pressure vs. hydraulic conductivity (Lp) relationship in lung microvascular ECs and demonstrated that HS participates in pressure-sensitive mechanotransduction that results in EC transport barrier dysfunction. Note that increased transmural pressure drives increased flow through intercellular junctions and over the nearby EC surfaces. Pretreatment of lung capillary EC with heparinase III completely abolished the pressure-induced increase in Lp and reduced intracellular NO detected by a fluorescent dye, suggesting that cell-surface HS directly participates in mechanotransduction resulting in NO production and increased Lp. In a related in vivo study Dull et al.47 examined the roles of HS and NO in pressure induced alterations of the capillary filtration coefficient, K(fc), and found that heparinase III attenuated the effects of increased capillary pressure on K(fc).
GCX Core Proteins Involved in NO Production
Because the proteoglycan core protein glypican-1 (GPC1) only binds HS and not CS and the dominant apical syndecan (syndecan-1; SDC1) binds both HS and CS, it was hypothesized that GPC1 is the core protein that transmits the fluid shear force sensed by the HS to the cell surface where it is ultimately transduced via the phosphorylation of endothelial nitric oxide synthase (eNOS) into NO. This hypothesis was tested by Ebong et al.50 who showed that knockdown of GPC1 using shRNA completely blocked shear-induced phosphorylation of eNOS, whereas knockdown of SDC1 had no effect. Because knockdown of genes by shRNA can have off-target effects, the hypothesis was further tested using atomic force microscopy (AFM) with cantilevers coated with specific antibodies to HS, GPC1, and SDC1.12 Force applied to HS or GPC1 resulted in NO production, whereas force applied to SDC1 did not. These experiments were carried out at 10 and 30 min and did not specifically interrogate the early G-protein-dependent phase of activation. Zeng and Liu237 found that shear stress has a dual role in eNOS activation: 4 dyn/cm2 shear stress inhibited the activation of eNOS, and 15 dyn/cm2 shear stress induced it. Removal of GPC1 by phosphatidylinositol phospholipase C (PI-PLC) significantly suppressed the shear stress-induced eNOS activity at 15 dyn/cm2 shear and further reduced the 4 dyn/cm2-inhibited eNOS activity.237
Glypican-1 and PECAM-1 Interact in NO Production
Recent work has shown that ECs transfected with platelet endothelial cell adhesion molecule-1 (PECAM-1) siRNA lose their inherent sensitivity to flow in the production of NO.59,226 In both bovine aortic ECs (BAECs) and HUVECs, the absence of PECAM-1 protein decreased the phosphorylation of eNOS at ser1177 and production of NO after 30 min of flow.59,226 Xu et al.226 found that flow-mediated eNOS phosphorylation in vivo induced by voluntary wheel running was reduced in PECAM-1 knockout mice. These findings implicate PECAM-1 as an important regulator of shear-induced NO release in ECs, although they do not clarify whether PECAM-1 directly senses flow to initiate these processes or responds to an upstream sensor of fluid shear stress.
In many cases, PECAM-1 works in conjunction with other mechanisms to sense changes in extracellular fluid flow.43,125,208 HS and GPC1 cluster via mobile lipid rafts at cell–cell junctions, where the majority of PECAM-1 is located, after 30 min of fluid shear stress application.239 A HS-to-PECAM-1 binding complex has been shown to exist29,33 and participate in early phase endothelial mechanotransduction.43 Human coronary artery ECs (HCAECs) treated with heparinase III or reconstituted with PECAM-1 mutants lacking HS binding domains had inhibited (early phase) shear-induced signaling, specifically Akt phosphorylation.43
In addition to forming binding complexes with its extracellular Ig-like domains, PECAM-1 can also participate in signaling cascades by activation of its intracellular tail. PECAM-1 has two highly conserved tyrosines, 686 and 663, that become phosphorylated in response to mechanical force, including physiologic levels of fluid shear stress in ECs.125,157 PECAM-1 phosphorylation is mediated by Src family kinases, specifically c-Src.59,125,157 PECAM-1 activation is also regulated by recruitment of the protein tyrosine phosphatase SHP-2, which dephosphorylates PECAM-1, allowing cyclic signaling waves to stem from continuous fluid shear stress application.79,125 Importantly, blocking PECAM-1 phosphorylation using the Src kinase inhibitor PP1 completely blocks downstream shear-induced eNOS phosphorylation,59 clarifying that PECAM-1 activation is a critical step in shear-induced NO production via the phosphorylation of eNOS.
Recently, Zeng et al.240 described the relationship between GPC1 and PECAM-1 in shear-induced production of NO. Using AFM methods on HUVECs and rat fat pad ECs (RFPECs) in vitro for 30 min, they showed that force applied directly to GPC1 induces NO production while force applied directly to PECAM-1 does not. When PECAM-1 was knocked down by siRNA, force applied directly to GPC1 did not induce NO production. Furthermore, using GPC1 knockout mice, Bartosch (unpublished data) demonstrated lower NO production than the wild type mice under chronic flow conditions with no differences in PECAM-1 expression. Under acutely increased flow stimulated by administration of dobutamine that elevates heart rate, there was an upregulation of NO production in the wild type mice after 30 min, but no increase in the knockout mice. These results demonstrate that GPC1 is the upstream shear sensor and PECAM-1 is the downstream effector.
But how does GPC1 communicate with PECAM-1? In Zeng et al.240 it was shown that blocking interactions between the HS GAG on GPC1 and the extracellular domain of PECAM-1 had no effect on signaling, while enzymatic removal of HS from GPC1 blocked phosphorylation of the intracellular domain of PECAM-1 and downstream phosphorylation of eNOS, the precursor to NO formation. The overall mechanism suggested by these studies is summarized in Fig. 10.
Figure 10.

Shear-Induced Nitric Oxide Production via Heparan Sulfate and Glypican-1 Mechanotransduction and PECAM-1 Phosphorylation leading to eNOS activation and NO synthesis. Fluid shear stress is initially sensed by heparan sulfate proteoglycan glypican-1. That signal is transduced to the intracellular tail of PECAM-1 protein. PECAM-1 becomes activated by tyrosine phosphorylation. PECAM-1 activation triggers phosphorylation of eNOS and NO production increases. Reprinted from Ref. 240.
Shear-Induced PGI2 is Mediated by PECAM-1
To follow-up on the observation that GAG removal by enzymes did not block shear induced PGI2 production,159 Russell-Puleri et al.176 investigated primary cilia, SDC4, and PECAM-1, as potential mechanosensors for PGI2 production. Primary cilia are localized to the apical surface of EC and SDC4 to the basal surface. In confluent ECs PECAM-1 localizes at intercellular junctions where it regulates homophilic binding between cells. Separate knockdown of the three putative mechanosensors in cultured cells revealed that only PECAM-1 deletion blocked shear-induced PGI2 production and upregulation of the enzyme cyclooxygenase 2 (COX-2) by a mechanism involving inside-out activation α5β1 integrin. The role of PECAM-1 in shear-induced PGI2 was further confirmed in a PECAM-1 knockout mouse model.176 Conway et al.32 using a fluorescence resonance energy transfer (FRET) tension sensor, showed that shear stress increased tension across PECAM-1 at intercellular junctions, suggesting a mechanism by which shear stress activates PECAM-1.
Substrate Stiffness Suppresses the Glycocalyx
With increased age, or as a result of hypertension, arteries lose elasticity and thicken, giving rise to a stiffened arterial wall and substrate for ECs.140,217 Arterial stiffness is a key underlying risk factor for, and is a hallmark of, age-related cardiovascular diseases such as atherosclerosis and stroke.141 EC dysfunction, characterized by increased proliferation, permeability, and inflammation is at the heart of the mechanisms driving these age-mediated vascular diseases. Arterial stiffness is a key trigger for EC dysfunction to occur.49,124
In a recent study, Mahmoud et al.120 showed that EC grown on polyacrylamide (PA) gels of substrate stiffness 10 kPa (mimicking the subendothelial stiffness of aged, unhealthy arteries) displayed a significant inhibition of GCX expression compared to cells cultured on softer PA gels (2.5 kPa, mimicking the subendothelial stiffness of young, healthy arteries). The effect of stiffness on HS immunostaining is displayed in Fig. 11. Gene and protein screens revealed that a GCX core protein, GPC1, was inhibited in cells cultured on stiff gels.
Figure 11.

Substrate stiffness inhibits the glycocalyx GAG heparan sulfate. HUVECs were cultured on polyacrylamide gels of stiffness ranging from 2.5 kPa (soft gel) through 10 kPa (stiff gel) up to 100 kPa. Heparan Sulfate (green) expression was assessed by immunostaining. Cell nuclei were identified using DAPI. Reprinted from Ref. 120.
Cells grown on 10 kPa gels had enhanced EC dysfunction as determined by enhanced cell inflammation (enhanced inflammatory gene expression, monocyte adhesion and inhibited eNOS phosphorylation), proliferation and endothelial mesenchymal transition (EndMT). Removal of GPC1 using gene-specific silencing with siRNA or gene overexpression using a plasmid revealed that GPC1 is required to protect against stiffness-mediated EC dysfunction. Consistent with this, using a model of age-mediated stiffness, older mice exhibited a reduced expression of GPC1 and enhanced EC dysfunction compared to young mice. EC dysfunction was exacerbated in young, but not aged, GPC1 knockout mice.
Structural Mechanism-Ultrastructure and Organization of Endothelial GCX
In addition to the above summarized molecular mechanisms by which GCX plays its role as a mechano-sensor and transducer, an organized structure of endothelial GCX at the molecular level is necessary. Due to the limitation of optical and electron microscopy, the ultra-structure of endothelial GCX has not been revealed until the recent development of super resolution stochastic optical reconstruction microscopy, STORM.14,225 Fan et al.57 used two photo-switchable dyes, ATTO488 and AF647, to label HS and HA of GCX on bEnd3 (mouse brain microvascular EC) monolayers, and revealed their organization and ultra-structure by N-STORM (Fig. 12). Figure 12 demonstrates that HA is a long molecule weaving into a network which covers the endothelial luminal surface. In contrast, HS is a shorter molecule, perpendicular to the cell surface. HA and HS are partially overlapped with each other at the endothelial luminal surface. These findings suggest that HS plays a major role in mechanosensing and HA plays a major role in the molecular sieve.
Figure 12.

Illustrations of STORM images for HS and HA from the cell luminal surface. In the illustrations, the second column is the HS image (green), the third column is the HA image (red) and the first column is the overlay of the second and the third columns. The first row is the 2D top view and the second row is the 3D view of the field of ~ 34 μm × 34 μm. The third and fourth rows are the enlarged 2D and 3D views from the region with the green dashed line in the first row. The color bar at the left of the 3D view for HS (second column) and HA (third column) represents the depth (length) scale. Reprinted from Ref. 57.
Part B: The Role of the Glycocalyx in Vascular Related Diseases
Infectious Diseases
Since the GCX is at the interface of the circulating blood and vessel wall, in addition to experiencing the forces generated by the blood flow, it is constantly affected by the blood-borne substances including toxins from infectious diseases.
Sepsis
Sepsis is a serious medical condition. It is caused by an overwhelming immune response to infection. The body releases immune chemicals (cytokines) into the blood to combat the infection. The cytokines trigger widespread inflammation, which leads to blood clots and leaky blood vessels. As a result, blood flow is impaired, and that deprives organs of nutrients and oxygen and leads to organ damage. In severe cases, one or more organs fail. In the worst cases, blood pressure drops, the heart weakens, and the patient spirals toward septic shock. Once this happens, multiple organs, lungs, kidneys, brain, may quickly fail, and the patient can die. Sepsis is a major challenge in hospitals, where it is one of the leading causes of death. It afflicts > 750,000 individuals annually in the United States alone and has mortality rates of 40–60%.63
Sepsis occurs unpredictably and can progress rapidly. Many types of microbes can cause sepsis, including bacteria, fungi, and viruses. However, bacteria are the most common cause. One of the key molecular causes of Gram-negative bacteria induced septicemia is endotoxin that consists of lipopolysaccharides (LPSs) bound with high affinity to LPS-binding glycoprotein. Complex LPS-binding glycoprotein is recognized by cognate receptor Toll-like receptor 4 (TLR4) and co-receptor CD14 on monocytes/macrophages and ECs to induce the secretion of proinflammatory cytokines.3 Considering the systemic nature of septicemia, vascular endothelium represents the first line of exposure to bacterial endotoxins. The role of microcirculation and microvascular endothelium in the pathogenesis of septic multiorgan failure has been known for a long time,3 but recent studies have identified endothelial GCX in manifestations of endothelial dysfunction in sepsis.182,194,248
Among the earliest responses of activated ECs to endotoxin are exocytosis of Weibel-Palade bodies (WPBs) and secretory lysosomes. WPBs are rod-shaped members of lysosome-related organelles (0.2 μm by 2 to 3 μm) characteristic to ECs and containing an array of proteins, peptides and cytokines, which can be released quickly on urgent demand. The endothelial lysosomes contain acid/secretory sphingomyelinase, glycohydrolases, cathepsins, fucosidase, phosphatases, HS sulfatase, among others. Some of these enzymes can degrade the endothelial GCX. Upon application of LPS for minutes to simulate the onset of sepsis, confocal and STORM microscopy confirmed the patch loss of GCX simultaneously with the exocytosis of lysosome-related organelles and WPBs in cultured ECs and mouse aorta248 (Fig. 13). During sepsis, the GCX becomes thinner, inducing tissue edema.25,209 In mice, intravenous LPS injection results in elevated levels of tumor necrosis factor (TNF)-α and IL-6.58 Elevated levels of TNF-α contribute to GCX damage via increased matrix metalloproteinase (MMP) activity and syndecan loss.173 In human, low-dose endotoxin resulted in a significant reduction in the depth of the sublingual vessel GCX and a concurrent elevation in plasma HA, suggesting GCX shedding.151 Circulating levels of HA and HS have also been increased fourfold in patients with sepsis, being higher in those who did not survive 90 days.149 Therefore, multiple circulating biomarkers of GCX shedding have been studied in humans as markers of sepsis, including SDC1, HS and HA.15,36,118,227
Figure 13.

Representative STORM-acquired images of anti-HS labeled EG and vWF-labeled WPBs distribution in control and 10 min post-LPS. The left panel shows 2D images of a region of ~ 40 µm x 40 µm at the apical surface of cultured mouse brain microvascular endothelial cell (EC). An enlarged top view of a yellow box (~ 2 µm x 2 µm) in the left panel is shown in the middle panel and its side view in the right panel. The EC surface is at the bottom of the side view. Upper panels are for the control and bottom ones for after 10 min LPS treatment. Reprinted from Ref. 248.
Sepsis, in general, can be characterized as a catabolic state of degradation of proteins, lipids and carbohydrates.215 Recent advanced metabolic gene expression analysis of LPS-induced endothelial dysfunction found an increase in glycan production to accompany GCX loss in ECs and an increase in endothelial permeability.129 Findings in patients with sepsis showed that upregulation of glycan synthesis was among the worst-affected areas of metabolism, being most severe in non-survivors. These observations are consistent with a view of compensatory induction of synthesis of structural components of GCX, upon application of GCX-degrading stressors such as LPS, suggesting a tight feedback control mechanism coupling its synthesis and degradation. That the degradation of GCX in sepsis prevails over increased synthesis of glycans, major components of GCX, suggests an enhanced enzymatic degradation, which is carried out by sheddases, including a disintegrin and metalloproteinases (ADAMs), MMPs, heparanases and hyaluronidases.63 While ADAM 10 is expressed constitutively, ADAM 17 is inducible, as it is converted to an active form upon cell stimulations.137 In addition to turning TNF-α into a soluble form, ADAM 17 cleaves SDC4 while ADAM 10 sheds SDC1 and ADAM15 cleaves CD44.227 MMPs induced in sepsis activate TNF-α, interfere in CD44-HA binding and shed HS, CS, and the SDC1 ectodomain.244
The wall of the cerebral microvessel is named the blood–brain barrier (BBB) which is formed by ECs, pericytes, basement membrane and astrocyte foot processes. The GCX on ECs is the most luminal layer of the BBB. During sepsis, patients develop cognitive impairment. Although the pathogenesis of this condition is complicated, current studies suggest a role for cytokines (IL-1, IL-6 and TNF-α) penetrating the BBB and activating astrocytes.165 Studies also suggest that during sepsis HS fragments penetrate the BBB in the hippocampus to inhibit long-term potentiation, the process responsible for memory formation.76
Increased pulmonary vascular permeability occurs early in sepsis128 and may be due to GCX damage. Although a study using bovine lung microvascular ECs in vitro suggested that HS of GCX contributed to the barrier function, a study using ex vivo rat lungs did not.47,48 One explanation for this discrepancy is that the enzyme degrading HS completely compromised the barrier function of GCX for the in vitro lung microvascular ECs formed monolayer, but only partially compromised that of GCX in the ex vivo rat lung microvessels. In addition to HS, there is significant HA in the GCX of microvessels. As predicated by a transport model for the paracellular pathway (inter-endothelial cleft) of the microvessel wall,21 as long as there is 50% GCX left that covers the inter-endothelial cleft, the hydraulic conductivity and permeability to a large sized molecule would not have a significant increase. For a rat mesenteric microvessel, the inter-endothelial cleft occupies about 0.2% of a vessel lumen area.1 Even though 99.8% GCX is degraded from the vessel lumen, as long as 0.2% GCX still covers the inter-endothelial cleft, no significant increase in the vascular permeability would be observed. This explanation also applies to the recent finding by Guerci et al.64 that GCX degradation is independent of vascular barrier permeability increases in nontraumatic hemorrhagic shock in rats.
However, if that 0.2% GCX covering the inter-endothelial cleft is damaged, vascular permeability will be enhanced. If other structural components, such as tight and adherens junctions inside the inter-endothelial cleft and the width of the cleft are disrupted, large increases in the vascular permeability will occur. For example, a study by Schmidt et al.182 shows that immune cell extravasation and inflammation during sepsis can alter the pulmonary GCX, resulting in pulmonary edema.
Using in vivo multiphoton microscopy in mice, Butler et al.20 observed a continuous GCX (identified by fluorescently labeled wheat germ agglutinin bound to sialic acid) at glomerular capillaries that contributes to the restriction of macromolecules and water passage from the glomerulus into the Bowman space. In a rat model, sepsis at its initial phase alters glomerular GCX and induces albuminuria (albumin leakage into urine).2 Taking these together, sepsis first destroys the frontline of the vascular barrier, the GCX, eventually resulting in multiorgan failure including brain, lungs and kidneys, to name a few.
Dengue Virus
Dengue is the most prevalent arboviral disease in humans and a major public health problem worldwide. Systemic plasma leakage, leading to hypovolemic shock and potentially fatal complications, is a critical determinant of dengue severity. A novel pathogenic effect of the secreted dengue virus (DENV) non-structural protein 1 (NS1) is triggering hyper-permeability of human ECs in vitro and systemic vascular leakage in vivo. It has been demonstrated that DENV NS1 disrupts the GCX on human pulmonary microvascular ECs, inducing degradation of sialic acid and shedding of HS proteoglycans by a mechanism involving NS1-induced expression of sialidases and heparanase.168 The data suggest an important role for GCX shedding in Dengue disease.
In a related study of 103 Dengue-infected patients, elevated levels of SDC1 and CS were strongly associated with plasma leakage, while elevated levels of SDC1 and claudin-5 were strongly associated with severe plasma leakage.196
Flaviviruses
Flaviviruses cause systemic or neurotropic-encephalitic pathology in humans. The flavivirus nonstructural protein 1 (NS1) is a secreted glycoprotein involved in viral replication, immune evasion, and vascular leakage during Dengue virus infection. In an extension of their dengue virus studies, Puerta-Guardo et al.167 investigated the contribution of secreted NS1 from related flaviviruses and demonstrated that NS1 from Zika, West Nile, Japanese encephalitis, and yellow fever viruses selectively binds to and alters permeability of human ECs from lung, dermis, umbilical vein, brain, and liver in vitro and causes tissue specific vascular leakage in mice, reflecting the pathophysiology of each flavivirus. Mechanistically, the observed hyperpermeability in vitro is related to disruption of the GCX components sialic acid, HS, and SDC1 following upregulation of sialidases as well as activity and subsequent expression of heparanase, enzymes known to contribute to the degradation of cellular GAGs and proteoglycans in the GCX.196,199
For the common influenza A virus (IAV) that has caused the Spanish flu (1918), Asian flu (1957), Hong Kong flu (1967) and bird flu (2004), multivalent binding to the host cell surface is a common strategy for infection. IAV binds to the host cell surface via its trimeric spike protein hemagglutinin, which recognizes sialic acids on the GCX of cellular membranes. Despite the low binding affinity of individual hemagglutinin receptor binding sites to sialic acid, a stable adhesion originates from multiple ligand–receptor binding events between the virus and the sialic acid glycoconjugates on the cell surface.105
Human Corona Viruses
(HCoV-NL63) is an alpha coronavirus that was first identified in 2004. Previous studies showed that HCoV-NL63 and severe acute respiratory syndrome (SARS)-CoV (2002) employ the same receptor for host cell entry, angiotensin converting enzyme 2 (ACE2), but it is largely unclear whether ACE2 interactions are sufficient to allow HCoV-NL63 binding to cells. Milewska et al.136 showed that directed expression of ACE2 on cells previously resistant to HCoV-NL63 renders them susceptible, showing that ACE2 protein acts as a functional receptor and that its expression is required for infection. However, competition experiments using a range of soluble elements of cellular membrane-associated components revealed that HS proteoglycans (HSPGs) constitute HCoV-NL63 adhesion receptors. Binding of HCoV-NL63 to heparan sulfates was required for viral attachment and infection of target cells.
Lang et al.103 showed that the efficient entry of SARS-CoV into the infected cells requires the involvement of HSPGs in concert with ACE2. Increased infectivity of SARS pseudovirus was associated with binding to HS. Elimination of cell-surface HS by heparinase or the addition of exogenous heparin reduced the ability of the virus to bind to the cell surface and increased cellular resistance to infection (Fig. 14).
Figure 14.
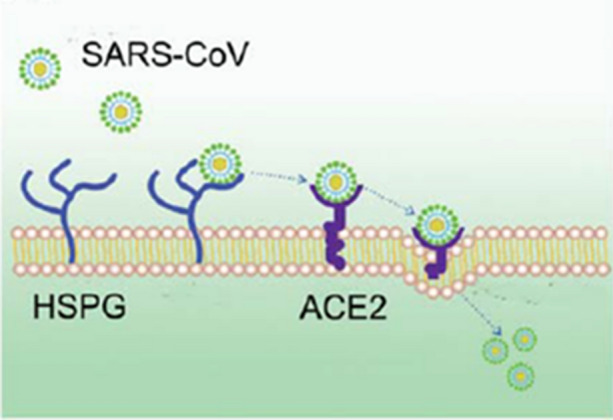
HSPGs play an important role in the process of SARS-CoV cell entry. The anchoring sites provided by HSPGs permit initial contact between SARS-CoV and host cells and the concentration of virus particles on cell surface. SARS-CoV rolls onto the cell membrane by binding to HSPGs and scans for specific entry receptors, which leads to subsequent cell entry. Reprinted from Ref. 103.
In a recent review paper, Sardu et al. 180 emphasized that to access host cells, SARS-CoV-2 (COVID-19) uses a surface glycoprotein known as spike to engage the ACE2 receptor for coronavirus entry. Therefore, the density of ACE2 in each tissue may correlate with the severity of the disease in that tissue. ACE2 is known to be well expressed by ECs. Hypertension, thrombosis, kidney failure, and diabetes all show strong co-morbidities with COVID-19 and are known to be associated with endothelial dysfunction, suggesting that COVID-19 is in a certain sense an endothelial disease.
Cardiac injury is a prevalent complication in severe COVID-19 patients, exacerbating the disease severity. ACE2, the key host cellular receptor of SARS-CoV-2, has been identified in multiple organs, and its cellular distribution in human heart was illuminated recently by Chen et al.27 They developed a single cell atlas of adult human heart tissue, and revealed that pericytes with high expression of ACE2 might act as the target cardiac cell of SARS-CoV-2. Pericyte injury due to virus infection may result in capillary EC dysfunction, inducing microvascular dysfunction.
Malaria
It has been proposed that a breakdown of the endothelial GCX plays a key role in severe malaria. In a study by Introini et al.,85 the interactions between Plasmodium falciparum infected red blood cells (PfRBCs) and GCX were investigated in vitro. Impairment of the GCX was obtained by enzymatic removal of sialic acid residues, which, due to their terminal location and net negative charge, are implicated in the initial interactions with contacting cells. There was a more than two fold increase of PfRBC adhesion to ECs upon enzymatic treatment, relative to untreated ECs. As a control, no effect of enzymatic treatment on healthy red blood cell adhesion was found. These results provide evidence of the increased cytoadherence of PfRBCs to GCX impaired vascular endothelium, thus supporting the role of GCX disruption in the pathogenesis of this disease.
Biochemical assessment of the luminal surface of ECs from cerebral malaria (CM) mice revealed malaria-induced loss of SDC1, SDC4, HS and CS.75 This loss was mirrored in increases of the same components in peripheral blood samples. Corticosteroid treatment protected against CM, reduced inflammation, and prevented GCX loss. Adjunctive antithrombin-3 also prevented GCX loss and significantly reduced CM-associated mortality. Thus, GCX loss may be associated with malaria pathogenesis.
Yeo et al.233 prospectively enrolled Indonesian inpatients (≥ 18 years old) with severe (SM) or moderately-severe falciparum malaria (MSM) and healthy controls (HCs). GCX breakdown products were measured in samples of urine and plasma. SDC1 and GAGs were significantly increased in MSM relative to HC patients and even more so in SM patients. GCX degradation was positively associated with parasite biomass in MSM and SM patients and inversely associated with endothelial NO bioavailability. This study further supports the view that the GCX is protective against malaria.
Cardiovascular and Related Diseases
Atherosclerosis
Our 2016 review paper200 traced the observations of diminished vascular GCX in atheroprone locations in arteries from the study by Lewis et al.109 in the coronary arteries of White Carneau pigeons in the early 1980s, through the study by van den Berg et al.212 20 years later showed that GCX thickness was significantly reduced in the disease-prone sinus region of the mouse internal carotid artery compared to non-diseased regions in the common carotid artery. Subsequently, van den Berg et al.213 observed that intimal accumulation of low density lipoprotein (LDL) was enhanced in the internal carotid branch of mice where the GCX was thinner than in adjacent regions of the common carotid. It was suggested that the enhanced LDL accumulation was due to impaired GCX barrier properties. In a related study, Nagy et al.148 observed that inhibition of HA synthesis in a mouse model facilitated leucocyte adhesion, subsequent inflammation and progression of atherosclerosis.
More recently, Cancel et al.22 examined the GCX in the brachiocephalic artery bifurcation of APOE−/− mice fed a high cholesterol diet for 10 weeks and observed a greatly diminished GCX on the intact ECs on plaque surfaces of the outer wall compared to the plaque free opposite wall and plaque-free descending aorta. The plaque surface was also shown to be a region of elevated EC apoptosis compared to the non-plaque regions. LDL accumulation was enhanced where the endothelial GCX was thinner and apoptosis higher than in adjacent regions of the common carotid. It was suggested that in concert with endothelial apoptosis that increases lipid permeability, glycocalyx shedding initiated by inflammation facilitates monocyte adhesion and macrophage infiltration that promote lipid retention and the development of atherosclerotic plaques.
Recent studies from H. Jo’s group192 using an APOE−/− mouse model with partial ligation of the left carotid artery showed rapid development of atherosclerotic plaques (2 weeks) due to a mechanism involving oscillatory shear-induced upregulation of microRNA 712 that suppresses tissue inhibitor of metalloproteinase 3. This allowed enhanced activity of MMPs that are known to degrade the GCX.235 We have previously stained samples from these partial ligation experiments192 and observed a complete removal of HA in the atherosclerotic vessels.200
Recent findings indicate that SDC1 is elevated in serum, indicative of the degradation of GCX, in patients with ischemic heart disease or heart failure.94 GCX shedding markers have been examined in the pathophysiology of heart failure with reduced ejection fraction (HFrEF). HA levels and SDC1 levels were measured in HFrEF patients. HA was significantly elevated in HFrEF patients and an independent predictor for worse clinical outcome, whereas SDC1 elevation was not significant.152
Recent work with 4- to 8-week-old apolipoprotein E/low-density lipoprotein receptor deficient mice, at the stage before development of atherosclerotic plaques, displayed increased endothelial stiffness, reduced GCX coverage and thickness, reduced NO production in the aorta, impaired acetylcholine-induced vasodilation, and increased endothelial permeability.9 These observations are consistent with independent studies reviewed in the sections on mechanotransduction and sepsis.
Harding et al.,74 using cultured EC in vitro and a mouse model, have demonstrated that pro-atherosclerotic endothelial dysfunction occurs as a result of disturbed flow-induced reduction in GCX expression and impaired endothelial sensitivity to flow. Results confirmed that disturbed flow down-regulates caveolin-1 mechanosignaling, as indicated by its reduced colocalization with phosphorylated eNOS (peNOS-serine1177), a vasoregulatory signaling molecule producing NO that is activated by shear stress through the GCX. In the absence of HS, uniform flow-conditioned ECs exhibited disturbed flow-like caveolin-1 expression, localization, and colocalization with peNOS.
Stroke
Ueno et al.210 characterized the GCX in capillaries of the frontal cortex and hypothalamus of stroke-prone spontaneously hypertensive (SHRSP) rats. They observed decreased GCX abundance in the hypothalamus and frontal cortex of SHRSP rats compared to control Wistar Kyoto (WKY) rats. Similar observations of decreased GCX abundance were made in the hippocampus of SHRSP compared to WKY rats.210
The GCX is partially accessible to flowing red blood cells at its luminal side in the so called “perfused boundary region” (PBR). GCX damage results in increased PBR, which can be measured in the sublingual microvasculature. Martens et al.94 tested whether PBR is increased in patients with lacunar stroke, and further distinguished patients with white matter lesions as a sign of extensive cerebral small vessel disease. They observed that white matter lesions are associated with an increase in the red blood cell permeable part of the sublingual microvascular GCX, and this implicates a compromised GCX.
Elevated systemic haematocrit increases the risk of stroke and myocardial infarction. Richter et al.175 used an erythropoietin overexpressing transgenic mouse line (tg6) with a haematocrit of 0.85 to assess the effects of elevated haematocrit and observed that the GCX is nearly abolished in tg6 mice. This suggests that the pathological effects of elevated haematocrit in these mice, and possibly in polycythemic humans, relates to a reduced GCX thickness and the consequent alteration in the blood–endothelium interface.
Sehba et al.186 developed a rat model to study the mechanisms underlying subarachnoid haemorrhage. This model was used to examine the fate of microvascular GCX in the brain 1 h after the imposed haemorrhage. It was shown that there is a robust GCX as characterized by HA staining in the control vessels that is completely degraded in the subarachnoid haemorrhage model.200
Fihlo et al.206 emphasized that the GCX is an integral constituent of the endothelial barrier, and an increasing number of studies suggest that recovery and stabilization of the microcirculation, particularly the damaged GCX, is a major element of a successful resuscitation regimen for haemorrhagic shock.207
Chronic Venous Disease
Ligi et al.114 recently reviewed the role of the GCX in chronic venous disorders underlying common vascular pathologies—varicose veins, edema, venous ulcerations- of great medical and socioeconomic impact. The predominant pathophysiological mechanisms of chronic venous disease (CVD) start from the development of venous hypertension and reflux, leading to endothelial dysfunction, GCX degradation and venous wall dilatation. The altered hemodynamics (disturbed flow) and loss of GCX lead to altered mechanotransduction upregulating harmful bio-molecular pathways, creating a vicious cycle among shear stress, proteolytic remodeling, and inflammatory processes.
Hypertension and Aging
In our previous review22 we described studies showing a loss of GCX in the blood–brain barrier of spontaneously hypertensive rats,210,211 the retinal and choroidal capillaries of rats with diabetes and hypertension,99 as well as reduced GCX thickness associated with elevated salt concentration in blood that reduces the salt sequestering capacity of the GCX leading to salt overload and hypertension.100,155 It was also shown that RBCs from hypertensive rats are subject to enhanced GCX cleavage compared to the RBCs of the normotensive rats due to the action of blood-borne MMPs.155
More recently, Schierke et al.181 have shown that high plasma Na + concentration associated with hypertension stiffens the endothelial cortex and diminishes the GCX leading to decreased NO while inducing endothelial release of pro-inflammatory cytokines promoting monocyte adhesion. Expression of GCX markers in plasma, including HS, HA, and SDC1, was detected in monocrotaline-induced pulmonary artery hypertension in rats indicative of endothelial GCX shedding.66 Endothelial GCX is impaired in untreated hypertensive patients and is related to arterial stiffness and coronary, and myocardial dysfunction.84 Preeclampsia related hypertension also leads to GCX degradation leading to endothelial dysfunction, reduced microvascular perfusion and vascular injury.222
In a study of the GCX in microvessels of young and old male mice using intravital microscopy and transmission electron microscopy and in human participants using intravital microscopy, GCX thickness in mesenteric and skeletal muscle microvessels was 51–54% lower in old compared with young mice. A 33% lower GCX thickness in the sublingual microcirculation of humans was observed in advanced age. These data provide evidence that a diminished GCX is present in advanced age and is accompanied by markers of impaired microvascular perfusion.119
Alzheimers and Dementia
Nagga et al.147 have demonstrated changes in the levels of HA in cerebrospinal fluid (CSF) in patients with Alzheimer’s disease (AD) with documented vascular alterations. To further investigate if the level of HA in CSF can be used as a clinical diagnostic biomarker to identify vascular pathology, a study of patients with vascular dementia (VaD) (n = 46), AD (n = 45), and controls without dementia (n = 26) was undertaken. They found significantly increased levels of HA in CSF from patients with VaD compared with controls, whereas the levels of HA in patients with AD were found to be unaltered compared with controls and patients with VaD. They also found a significant positive correlation between the levels of HA and the CSF/serum albumin ratio, an indicator of blood–brain barrier integrity, in patients with VaD and AD, supporting the role of HA in vascular changes in the brain.
Diabetes
Hyperglycaemia that is linked with diabetes has been shown in vitro to lead to loss of HS and associated shear stress-induced phosphorylation of eNOS116 and elongation of ECs in the direction of shear17 that are caused by mechanotransduction mediated by HS. In diabetic individuals, it has been observed that atherosclerosis is distributed more uniformly in arteries, not following the typical association with regions of low shear or disturbed flow near bifurcations and curvatures.40 In addition, vasodilation is altered in diabetes.174 All of these observations imply a fundamental difference in mechanotransduction by the ECs of diabetic individuals that may be explained in part by alterations in the GCX.
Endothelial dysfunction and increased risk for cardiovascular morbidity and mortality are associated with renal failure. Vlahu et al.216 demonstrated that dialysis patients shed HA and SDC1 into their blood and had an impaired GCX barrier. Salmon et al.179 reviewed the role of dysfunction of the GCX in the glomerulus leading to albuminuria and systemic increases in microvascular permeability.
Padberg et al.158 examined whether renal failure correlated with structural changes in the GCX using nephrectomized rats and patients with chronic kidney disease (CKD). The physical properties of the GCX were studied in rats using AFM. The results showed that CKD is associated with a damaged GCX and that increased GCX breakdown products correlate with the physical stability and thickness of the GCX.
Further evidence that HSPGs are shed in diabetes was provided by Ramnath et al.173 They demonstrated that the inflammatory cytokine TNF-α which induces disruption of the glomerular GCX in diabetes, reduced cell surface SDC4 and HS significantly. The mechanism involved MMP 9-mediated shedding of SDC4/HS. In addition, Neves et al.150 evaluated the relationship between plasma SDC1 levels and renal dysfunction in patients with acute decompensated heart failure. They determined that plasma SDC1 levels predicted increased risk of developing acute kidney injury.
Patients with type 1 diabetes present increased plasma levels of both HA and hyaluronidase 1 (HYAL1) that degrades HA. Dogne et al. 44,45 investigated the role of HYAL1 in the development of diabetes-induced endothelial dysfunction, endothelial-dependent vasodilation, arteriolar GCX size, and glomerular barrier properties in wild-type and HYAL1 knockout mice with or without streptozotocin (STZ)-induced diabetes. Four weeks after STZ injections, the lack of HYAL1 prevented diabetes-induced increases in soluble P-selectin concentrations, limited the impact of the disease on vasorelaxation, increased GCX thickness, maintained GCX structure and HA content during diabetes, and prevented diabetes-induced glomerular barrier dysfunction—all suggesting that HYAL1 contributes to endothelial and GCX dysfunction induced by diabetes.
Leskova et al.106 investigated the effects of diabetes and HYAL1 on the thickness of the GCX in the mouse retina. The retinal circulation of diabetic Ins2 (Akita) mice and their nondiabetic littermates were observed via intravital microscopy. In nondiabetic C57BL/6 mice, the GCX thickness also was evaluated prior to and following infusion of HYAL1. Both diabetes and hyaluronidase infusion significantly reduced the thickness of the GCX in retinal arterioles (but not in venules), and hyaluronidase increased retinal microvascular leakage. The findings demonstrate that diabetes and hyaluronidase reduce the thickness of the retinal GCX, in which HA plays a significant role in barrier function.
The mechanical properties of the GCX and endothelium in ex vivo mouse aortas were determined simultaneously in indentation experiments with AFM for diabetic and control mice at ages of 11–19 weeks.203 In diabetic mice, marked endothelial stiffening and reduced GCX coverage were present already in 11-week-old mice and persisted in older animals. In contrast, reduction of GCX length was progressive and was most pronounced in 19-week-old diabetic mice. The reduction of GCX length correlated with an increasing level of glycated haemoglobin and decreased endothelial NO production secondary to loss of mechanotransduction through the GCX. The stiffening of ECs and diminished GCX coverage occurred in early diabetes and were followed by the reduction of the GCX length that correlated with diabetes progression.
Exercise and Endurance Training
A recent study evaluated whether oxidative stress and inflammation induced by strenuous exercise affect GCX integrity and endothelial function in healthy young men. No significant changes in serum SDC1 and HS concentrations were observed, and it was concluded that a single bout of severe-intensity exercise is well accommodated by endothelium in young, healthy men as it neither results in evident GCX disruption nor in the impairment of NO or prostacyclin production.121 In a follow up study, Majerczak et al.122 found that moderate-intensity endurance training exerts a pronounced protective effect on endothelial GCX integrity at rest and during exercise. Training led to a significant decrease in basal and end-exercise concentrations of blood markers of GCX damage (SDC1 and HS). They suggested this was the result of an improvement of antioxidant defense that may represent the vasoprotective mechanism highly responsive to moderate-intensity training.
High-intensity interval training (HIIT) has been proposed to exert vasculoprotective effects. A study by Schmitz et al.183 evaluated whether HIIT affects the microvasculature, including the endothelial GCX barrier. Sidestream dark-field imaging was performed at baseline and follow-up to detect changes of the sublingual microvasculature including the endothelial GCX thickness. At baseline, a significant correlation between maximal exercise performance capacity and glycocalyx thickness was detected. Increased exercise performance at follow-up also correlated with GCX thickness. Moreover, increased baseline microRNA-143 (miR-143) levels predicted increased GCX thickness at follow-up. miR-143 may represent a tool for monitoring early vasculoprotective adaptations to physical activity.
Trauma
Endothelial dysfunction has emerged as a critical component associated with hemorrhagic shock and trauma.90 The endothelial GCX is an integral constituent of the endothelial protective barrier, and an increasing number of studies suggest that recovery and stabilization of the microcirculation is associated with GCX recovery.206,207
Johansson et al.90 have reported in trauma patients, that among the 17 markers of GCX degradation and endothelial damage, high circulating SDC1 was associated with increased mortality in patients with penetrating injury and severe head injury. Chignalia et al.28 have reviewed the GCX in lung injury and emphasized that the respiratory epithelial GCX is a target in bacterial pneumonia with emphasis on SDC1 and hyaluronan.
Cancer (Cancer Cell Glycocalyx)
The cancer cell GCX mediates the interactions between a tumor and its microenvironment and profoundly affects cancer cell signaling, migration, invasion, and metastasis.18 Paszek et al.163 observed that circulating tumor cells (CTCs) from patients with aggressive breast cancers overexpressed bulky glycoproteins. To test whether bulky glycoproteins could facilitate survival at distant sites, they incorporated a long glycoprotein mimetic into non-malignant mammary epithelial cells and found cells remained viable when plated on soft hydrogels that mimic sites of colonization, whereas untreated cells were not viable. The bulky GCX promoted growth and survival by increasing integrin adhesion and signaling. Woods et al.224 coated tumor cells with synthetic glycopolymers of various thicknesses and found that longer, bulkier GCX promoted cell survival and proliferation thus increasing their metastatic potential in a mouse model.
Altered expression of the GAG components of the cancer cell GCX, and their cell surface core proteins and receptors, has long been linked to cancer progression. Increased HA expression is consistently found in many types of cancers. Increases in tumor cell associated HA is seen in tumors arising from simple epithelia such as ovarian, colorectal, and gastric cancer, while increases in HA expression in the tumor stroma are observed in, for example, breast, prostate, and bladder carcinomas, and lung adenocarcinomas. In either case, increased HA expression is associated with tumor progression and poor survival.198 HA interactions with its cell surface receptors activate signaling pathways that modulate cell motility, proliferation, differentiation, and invasiveness.92,139
CD44 is the major cell surface receptor for HA. Due to alternative splicing and post-translational modifications, multiple variants are generated (CD44v). While the standard form of CD44 (CD44s) is expressed in most cells, CD44v are expressed in tumor cells and have been shown to promote tumor progression and metastatic potential in lung, breast, and colon cancer.139 Another HA receptor, RHAMM, is upregulated in various cancers and its expression is associated with poor prognosis in lung, breast, colorectal, gastric, pancreatic, and ovarian cancers.26 The role of HA-CD44 and HA-RHAMM interactions in cancer has been reviewed in detail.138
HS interacts with a wide range of growth factors and chemokines making it essential to many aspects of cell regulation and function.62 Modification of HS by enzymes such as heparanase, sulf-1, and sulf-2 plays an important role in regulating cancer growth35 and altered expression of these enzymes is found in many types of cancers.71,190 Jung et al.91 found that heparanase degrades HS on SDC1 exposing a binding site that couples vascular endothelial growth factor receptor-2 to α4β1 integrin in multiple myeloma, leading to tumor cell migration and invasion. Shinyo et al.189 found reduced expression of HS was associated with the progression of cervical intraepithelial neoplasms to invasive cancer. Through a different mechanism, HS on the cancer cell surface mediates flow-induced migration and metastasis in renal carcinoma cells (discussed below).169,170 Thus the role of HS in tumor progression may be context and cell type specific.
The syndecan and glypican proteoglycans interact, directly or through their HS chains, with an array of extracellular matrix proteins that mediate cell-signaling events during carcinogenesis86 (see Table 1). As with HS, the role of SDC1 may depend on cell type and disease stage. Loss of SDC1 appears to be a key step in regulating epithelial to mesenchymal transition.86 Recently, Kind et al.95 examined tissue microarrays for SDC1 expression and found it was highest in squamous cell carcinomas such as esophagus, cervix, and lung and in adenocarcinomas in lung and colorectal cancer. Expression was low in germ cell tumors, sarcomas, endocrine, and neuroendocrine tumors. Barbareschi et al.10 reported that SDC1 was highly expressed and associated with aggressive phenotype in breast carcinoma. By contrast, SDC1 was downregulated in invasive cervical cancer189 and multiple myeloma.91
Table 1.
Syndecans and glypicans in cancer.
| Component | Expression in cancer/possible mechanisms | References |
|---|---|---|
| Syndecans | ||
| Syndecan-1 | Upregulated in squamous cell carcinomas, aggressive breast carcinomas; downregulated in sarcomas, germ cell, and endocrine tumors. Regulates epithelial to mesenchymal transition | 10, 86, 91, 95, 189 |
| Syndecan-2 | Upregulated in highly metastatic Lewis lung carcinoma; promotes tumor progression in breast and colon carcinomas | 31, 42, 86, 115, 162, 191 |
| Syndecan-4 | Upregulated in renal cell carcinoma; downregulated in invasive colon carcinoma. Mediates cell adhesion to extracellular matrix | 52, 86 |
| Glypicans | ||
| Glypican-1 | Upregulated in lymphoma, breast, pancreatic, and cervical cancer. Modulates mitogenic response | 19, 46, 54, 72, 97, 110, 111, 117, 126, 127, 131, 166, 177, 178, 218 |
| Glypican-3 | Upregulated in hepatocellular, gastric, ovarian and lung squamous cell carcinoma; downregulated in breast, lung, ovary, and mesothelium cancers | 67, 86, 153 |
Syndecan-2 plays an important role in mediating cell shape, adhesion and migration.86 Syndecan-2 overexpression is found in low metastatic clones of Lewis lung carcinoma, and upregulation of syndecan-2 in highly metastatic clones inhibits their metastasis.191 In colon31,162 and breast115 carcinoma cell lines, syndecan-2 promotes tumor progression by mediating cell adhesion and enhancing migration. Shed syndencan-2 inhibits angiogenesis which affects tumor growth.42
SDC4 is expressed in most tumors and mediates cell adhesion to the extracellular matrix. SCD4 is downregulated in invasive colon carcinoma,86 but upregulated in the renal cell carcinoma tissue samples.52 The specific roles SDC4 plays in tumor progression and metastasis may depend on cell type and other factors in the tumor microenvironment86 and more studies are needed to advance our knowledge in this area.
High levels of GPC1 have been reported in lymphoma,54 breast,126 pancreatic,46,97,117 esophageal,72 and cervical cancer.127 In breast and pancreatic cancer, GPC1 was shown to modulate the mitogenic response of cancer cells to heparin-binding growth factors.97,126 GPC1 expression was also a predictor of perineural invasion46 and poor prognosis117 in patients with pancreatic cancer. Knockdown of GPC1 in human colon carcinoma cell lines resulted in a decrease in invasion and migration.111 In patients with glioblastoma, elevated GPC1 expression was correlated with tumor dissemination and shorter overall survival.178 The use of GPC1 antibodies as a diagnostic tool to detect circulating tumor cells and exosomes, as well as labeling of cancer cells in situ, is an active area of investigation.19,110,131,166,177,218
Glypican-3 is found to be downregulated in breast, lung, ovary, and mesothelium cancers.86 By contrast, glypican-3 is overexpressed in hepatocellular, gastric, ovarian, and lung squamous cell carcinomas, and melanomas.153 In hepatocellular carcinoma, glypican-3 is actively investigated as a tool for diagnosis and treatment.67
Cancer Cell Mechanotransduction
Interstitial flow (IF) is enhanced in tumors due to the leaky nature of tumor vasculature and increased interstitial pressure in the tumor microenvironment.146 This enhanced flow has been shown to alter the migration and invasiveness of several types of cancers.70,82,145,169,171,188 The role of the GCX as a mechanosensor for IF has only recently begun to be investigated. Our previous review200 looked at the in vitro work of Qazi et al.169 showing IF significantly increased migration of a highly metastatic kidney cell line (SN12L1). The same levels of flow did not increase migration in a low metastatic kidney carcinoma cell line (SN12C). The effect was completely inhibited by removal of either HS with heparinase or HA with hyaluronidase. This work was recently extended to an animal model to investigate the possibility that cancer cell GCX components mediate metastasis. Qazi et al.170 implanted SN12L1 cells in the renal capsule of mice and assessed metastatic colonization to distant organs. They used shRNA to knockdown NDST1 in SN12L1 cells thus suppressing synthesis of HS. This resulted in a 95% reduction in metastasis of tumors formed by knockdown cells compared to tumors formed by unmodified cells (wild type). In addition, they found that HS deficient tumors had well-defined boundaries and did not invade the surrounding normal tissue, while wild type tumors had poorly defined margins and aggressively invaded the surrounding tissue.
Kingsmore et al.96 used patient-derived xenograft models to determine the effect of high flow on invasiveness of glioblastoma stem cells. They observed increased invasion into high flow regions compared to low flow regions in two of the three patient-derived cell lines. Examining the cell lines in vitro, they found that the flow response could be occurring through two mechanisms: autologous chemotaxis mediated by CXCR4/CXCL2, or mechanotransduction mediated by CD44. Characterization of each cell line revealed different percentages of CXCR4, CXCL2, and CD44 expressing cells indicating that subpopulations of glioblastoma stem cells could respond to flow via either mechanism within a single patient.
Moran et al.142 investigated the specific proteins that link surface GAGs that sense IF to the cancer cell body where mechanotransduction resulting in increased migration occurs. They measured the expression of GPC1, SDC1, and CD44 in high (SN12L1) and low (SN12C) metastatic kidney cell lines and found significantly higher GPC1 levels in the high metastatic cell line. Knockdown of GPC1 by siRNA resulted in significant inhibition of flow-induced migration in the SN12L1 cell line. While the baseline (static) protein expression of CD44 was not different between SN12L1 and SN12C cells, this group has previously shown that 4 h of flow significantly increased CD44 gene expression in SN12L1 cells and that blocking CD44 expression resulted in blocking flow-induced migration.169 Recently, they investigated the possibility that the other HA surface receptor, RHAMM, could also act as the mechanical link between HA and the cell. They found increased levels of RHAMM in SN12L1 compared to SN12C, and knockdown of RHAMM by siRNA significantly reduced flow-induced migration in the SN12L1 cell line (unpublished data). A recent study by Mele et al.130 found that RHAMM knockdown slowed tumor growth and inhibited metastasis of colorectal cancer cells in a mouse model.
In summary, differential expression and modification of GCX components appear to play key roles in cancer progression. Cell-associated and extracellular matrix HA and its cell-surface receptors CD44 and RHAMM are upregulated in most cancers and are associated with poor prognosis. Heparanase mediated shedding of HS on SDC1 enables growth factor in the tumor microenvironment to access receptors and activate cell signaling that fosters tumor growth and progression. On the other hand, HS on GPC1 acts as a mechanosensor for IF that alters migration and invasiveness in some types of cancer. Finally, abnormal glycosylation18,143 and GAG modification by enzymes35,71 (e.g. Heparanase, sulf-1) are actively investigated as key mechanisms in tumor growth and metastasis.
Cancer Metastasis (Endothelial Cell Glycocalyx)
The major cancer metastatic steps from a solid tumor include migration of tumor cells from the original site to nearby microvessels, intravasation into the blood circulation through the microvessel wall, arrest and adhesion to the microvasculature at a distant organ, interaction with the ECs forming the microvessel wall and extravasation from the leaky endothelial barrier to the surrounding tissue, survival, proliferation and formation of a new colony. While cancer cell GCX enhances cancer cell migration in the tissue during its metastasis, damage of endothelial GCX has been found during tumor cell adhesion and extravasation. An in vivo study by Cai et al.21 showed that adhesion of malignant breast cancer cell MDA-MB-231 to microvessel walls degrades endothelial GCX in a timely manner (Fig. 15). Preservation of the GCX either by a plasma glycoprotein orosomucoid21 or sphingosine-1-phosphate (S1P),245 and inhibition of VEGF receptor (KDR/Flk-1)187 can reduce the tumor cell adhesion. In vitro studies also found that breast cancer cell adhesion to and transmigration across an in vitro blood–brain barrier significantly destroy the endothelial GCX but not the cancer cell GCX.56 Although the mechanism by which cancer cells degrade the endothelial GCX is not completely understood, integrin β4 signaling65 enhanced ErbB2-dependent expression of VEGF by tumor cells55 is a likely reason. VEGF can degrade GCX21,56 to expose more cell and extracellular matrix adhesion molecules, e.g. E-selectin,172 at the endothelium and underneath the basement membrane, e.g. laminin,55 for tumor cell adhesion and transmigration.
Figure 15.

Endothelial surface glycocalyx labeled with FITC-anti-HS (green) on a rat mesenteric microvessel (left panel) without breast cancer cell adhesion (left panels), at initial adhesion (middle panel, red spots are cancer cells) and after 45 min perfusion (right panel). Reprinted from Ref. 21.
Drugs and Strategies to Rescue the Glycocalyx
A previous review200 summarized a number of emerging approaches to rescue and/or stabilize the GCX. Below we update with recent developments on these and newer approaches. These are summarized in Table 2. Save for a few exceptions, the effects of the drugs discussed below on the GCX have not yet been tested in human trials and the results derive from in vitro and/or animal studies.
Table 2.
Drugs that rescue the GCX.
| Compound | Possible mechanisms | References |
|---|---|---|
| Albumin/S1P | Regenerate GCX, inhibit shedding | 4, 11, 132, 235, 238 |
| Sulodexide | Supplement GCX, inhibit degrading enzymes | 30, 112, 123, 193, 194, 214 |
| Rhamnan Sulfate | Enhances HS synthesis | 23, 156, 197 |
| Sevoflurane | Inhibit shedding, increase GCX regeneration | 5, 24, 93, 113 |
| Biomimetic Nanoparticles | Replace shed GCX | 223, 243 |
| ALM BHB/M P-188 | Inhibit shedding | 206 |
| Blueberry Metabolites | Regenerate GCX | 13, 16, 38 |
| Empagliflozin | Regenerate GCX | 34, 83 |
| Metformin | Regenerate GCX | 8, 53, 102, 204, 214 |
S1P sphingosine-1 phosphate, ALM adenosine + lidocaine + Mg, BHB/M beta-hydroxybutryrate + melatonin, P-188 poloxamer 188
Albumin/S1P
The administration of plasma (albumin) is an established practice in the treatment of massive hemorrhage and trauma. A recent review summarizes the role of plasma in protecting the endothelium.11 A key mediator in the effect of plasma on the endothelial GCX may be albumin-bound S1P, which has been shown to inhibit the shedding of SDC1 and regenerate the GCX.235,238 Recently, Mensah et al.132 combined S1P with exogenous HS to restore expression of Connexin 43 and interendothelial communication after GCX degradation with heparinese III in vitro. While both S1P and exogenous HS restored HS after degradation, only the combination of both agents was found to restore interendothelial communication. Alves et al.4 used an in vivo rat model of hemorrhagic shock and resuscitation (HSR) to show that S1P reduces microvascular leakage induced by HSR and protects against GCX degradation. In vitro, they also show that S1P protects against mitochondrial dysfunction-induced endothelial barrier dysfunction by acting through mitochondrial complex III.
Sulodexide
Sulodexide is a highly purified GAG extracted from the porcine intestinal mucosa. It is composed of 80% fast-moving heparin (a HS mimetic) and 20% dermatan sulfate and is available for oral, intramuscular, and intravenous administration. Sulodexide has been applied in clinical trials for the treatment of CVD and diabetic nephropathy.30 Song and Goligorsky193 hypothesize that the superior effects of sulodexide are due to its dual action: supplementation of GCX components, and inhibition of GCX degrading enzymes. Fast-moving heparin inhibits heparanase while dermatan sulfate blocks the pro-MMP-9 molecule from changing into the active form.
The effect of sulodexide on endothelial function was recently assessed by Li et al.112 in the balloon-injury carotid artery model. Electron microscopy showed that sulodexide reconstituted the GCX structure thus preserving endothelial function. The authors found that sulodexide treatment increased eNOS levels, reduced endothelial hyperplasia, inhibited platelet aggregation, decreased expression of CD31, ICAM-1, and VCAM-1, and reduced the infiltration of CD68-positive cells.
Van Haare et al.214 studied the effects of a 4-week sulodexide treatment on microvascular perfusion capacity of high fat diet-fed rats. They showed that sulodexide restored the hyperemic response to adenosine in myocardial perfusion, which was blunted by the high fat diet in control animals.
Degradation of the GCX is an early event in sepsis which contributes to high mortality. Song et al.194 used sulodexide to accelerate GCX restoration which resulted in reduced vascular permeability and improved animal survival in a mouse model of sepsis.
CVD patients treated with sulodexide show significant improvements in peripheral venous pressure, patient-assessed quality of life scores, and ulcer healing, along with reductions plasma concentrations of MMP-9, IL-6, and MCP-1, and reductions in other clinical signs and symptoms.123
Rhamnan Sulfate
Suzuki and Terasawa197 recently reviewed the many beneficial biological activities of rhamnan sulfate (RS), a sulfated polysaccharide extracted from the green algae M. nitidum. RS is found to have antiviral and antithrombotic activities. It mitigates the high-fat diet induced elevation of LDL cholesterol, and the postprandial increase in blood glucose level in healthy human subjects. RS also has anti-inflammatory effects on endothelial cells: it suppresses the elevation of tissue factor and vWF in TNF-α or thrombin-induced inflammation of ECs.156 We have previously shown that RS enhances the GCX and reduces LDL permeability of cultured ECs.23 Recently, Aaron Baker’s lab studied the effects of oral rhamnan sulfate on a mouse model of atherosclerosis and found significant reductions in vascular inflammation and atherosclerotic plaque formation (unpublished data).
Sevoflurane
Sevoflurane, a volatile anesthetic, has been shown to protect the GCX during ischemia and reperfusion (IR). Annecke et al.5 first showed that ischemia led to shedding of HS and SDC1, and degradation of the GCX in isolated guinea pig hearts. Treatment with sevoflurane before or after IR was found to attenuate shedding of HS and SDC1. Treatment pre- plus post-IR with sevoflurane was found to also preserve the GCX structure. Chappell et al.24 showed sevoflurane pretreatment also reduced leukocyte and platelet adhesion after IR. In a study on Sprague–Dawley rats, Li et al.113 showed sevoflurane reduced the shedding of HS and SDC1, and preserved the endothelial GCX in the liver following hepatic ischemia. The mechanisms by which sevoflurane inhibitis GCX shedding in IR are not well understood. Annecke et al.5 found that sevoflurane treatment reduced the release of cathepsin B, a protease stored in lysosomes and associated with degradation of extracellular matrix and inflammatory diseases. A recent study on rat aortas found that sevoflurane promoted regeneration of the GCX by upregulating the sialytransferase ST6Gal-1.93
Biomimetic Nanoparticles and Surfaces
Wodicka et al.223 developed a coating composed of a dermatan sulfate backbone containing selectin-binding peptides to target endothelium with exposed adhesion molecules due to loss of the GCX. The coating was shown to reduce platelet activation and binding to ECs in vitro. In an in vivo mouse model of deep vein thrombosis the coating reduced thrombus formation without decreasing the whole blood clotting time, suggesting its potential to overcome the negative side effects of standard anticoagulant therapies such as heparin.
Liposomal nanocarriers of preassembled GCX have been developed by the Goligorsky Lab.243 The nanocarriers restored mechanically-induced NO production in cultured ECs pretreated with heparinase III. The flow-induced vasodilatation of mesenteric arteries with degraded GCX was also restored after perfusion with the nanocarriers.
Others
Because of increased recognition of the role of the GCX as a key mediator in vascular diseases, drugs and natural compounds previously shown to have beneficial properties are now being investigated for their effects on the GCX. Below we summarize some recent developments.
Torres Filho et al.206 tested three drugs previously shown to improve outcomes after ischemia and hemorrhage for their effect on the GCX in a rat model of hemorrhagic shock and resuscitation. They found that all three compounds tested, adenosine + lidocaine + Mg (ALM), beta-hydroxybutryrate + melatonin (BHB/M), and poloxamer 188 (P-188) significantly restored the GCX thickness compared to resuscitation with lactated Ringer’s solution. Lower plasma SDC1 levels were also found with all three drugs. While the mechanisms by which these drugs restore the GCX are unknown, it is suggested that they may counteract the production of reactive oxygen species and oxidative stress that leads to GCX shedding.
Blueberry metabolites have been shown to attenuate endothelial dysfunction in an in vitro model of lipoxocity,16 and clinical studies support a cardio-protective role of blueberries in individuals with metabolic syndrome.13 Recently, Cutler et al.38 showed that blueberry metabolites restore GAGs, and reduce gene expression of inflammatory markers and monocyte binding on human aortic ECs derived from diabetic patients, suggesting that some of the beneficial effects of blueberries are due to protection and restoration of the GCX.
Empagliflozin is a sodium-glucose co-transporter-2 used to treat type 2 diabetes mellitus shown in clinical trials to significantly reduce cardiovascular events in addition to improving glycemic control. Cooper et al.34 recently showed that empagliflozin restores the GCX on human abdominal aortic ECs (HAAECs) after degradation with heparinase III. In addition, empagliflozin restored the flow-induced elongation, and inhibited leukocyte cell adhesion to HAAECs with degraded GCX. A new clinical trial83 found that treatment with empagliflozin showed greater improvement of vascular markers and endothelial GCX thickness than insulin treatment in patients with type 2 diabetes mellitus.
Metformin is the most popular anti-diabetic drug in the United States and has been shown to have vasculoprotective8 and anticancer102 properties in type 2 diabetes patients independent of its glucose lowering effects. Eskens et al.53 subjected a mouse model of type 2 diabetes to a 2-week metformin treatment and found improved endothelial GCX barrier properties compared to the untreated group. Notably, this improvement occurred without any effect on blood glucose levels. In the same sulodexide study cited above, Van Haare et al.214 showed that metformin was as effective as sulodexide in restoring the hyperemic response to adenosine in myocardial perfusion in high-fat fed rats. More recently, Targosz-Korecka et al.204 found in an in vitro study that metformin treatment restores the GCX, downregulates surface expression of adhesion molecules ICAM and E-selectin, and reduces stiffness of ECs grown in high glucose. In addition, they found that treatment with metformin reduced the adhesive interaction between ECs and human lung carcinoma cells. It is suggested that both the regeneration of the endothelial GCX and downregulation of adhesion molecules may contribute to the antimetastatic effects of metformin.
Drugs and Strategies to Target the Glycocalyx for Cancer Treatment
Carrier-Mediated Drug Delivery Systems
Because many cancer cells overexpress HA receptors CD44 and RHAMM, targeting of these using the HA molecule itself has been used to deliver cytotoxic agents directly to cancer cells. For example, nimesulide is a hydrophobic COX-2 inhibitor anti-inflammatory known to also inhibit tumor growth in various types of cancer by inducing apoptosis. Jian et al.89 used an HA-nimesulide conjugate to target CD44-overexpressing HT-29 colorectal cancer cells in a mouse model improving the drug’s selectivity and solubility. In vitro HA-nimesulide increased apoptosis in HT-29 cells to 47.1%, compared to 2.9% in untreated cells. Tumor growth was inhibited by 82.3% in mice treated with HA-nimesulide, compared to control, whereas it was inhibited by 55.9% in mice treated with free nimesulide. Yang et al.228 used HA conjugated with folic acid to form micelles that were then loaded with doxorubicin (DOX). In vitro cytotoxicity tests revealed that the DOX-loaded micelles were less toxic to tumor cells than free DOX, likely because free DOX could diffuse quickly into the cells while the micelles had to be internalized via endocytosis, delaying the release of DOX into the cells. However, when the micelles were used in nude mice bearing human liver cancer cells tumors, which express CD44, they resulted in improved targetability of DOX to the tumors. Zhong et al.246 developed HA micelles loaded with paclitaxel to successfully inhibit tumor growth of MCF-7 human breast tumor xenografts in mice.
Antibody-Based Therapies
Antibody–drug conjugates (ADCs) are a targeted therapy wherein a cytotoxic agent is linked to an antibody. A number of ADCs have been developed that target GCX components that are overexpressed in cancer cells. Matsuzaki et al.127 developed a novel anti-GPC1 antibody and conjugated it to the cytotoxic agent monomethyl auristatin F (MMAF) for the treatment of uterine cervical cancer. In preclinical studies, the GPC1-ADC was internalized and was cytotoxic in HeLa and ME180, two uterine cervical cancer cell lines that overexpress GPC1, while it had no effect on an ovarian cancer cell line that did not express GPC1. The GPC1-ADC also inhibited tumor growth in a murine xenograph model. The same group developed a different GPC1-ADC, also conjugated to MMAF, which showed tumor growth inhibition in patient-derived tumor models of pancreatic cancer.154
Indatuximab ravtansine is an ADC that links an anti-SDC1 antibody to the cytotoxic agent maytansine. It was recently shown to strongly inhibit tumor growth in a high SDC1-expressing xenograph model of triple-negative breast cancer.184 The first clinical study on patients with relapsed and/or refractory multiple myeloma showed over 75% of patients achieved stable disease.87
Antibody-dependent cellular cytotoxicity (ADCC) is a mechanism of immune defense in which natural killer cells lyse target cells whose surface antigens have been bound by specific antibodies. Several therapeutic antibodies against glypican-3 have been developed for treatment of hepatocelullar carcinoma which features overexpression of glypican-3.230 The recombinant monoclonal antibody GC33 (codrituzumab) binds to the juxtamembrane domain of glypican-3 and induces ADCC. It was well tolerated and had antitumor effects in phase I trials. Although GC33 did not show benefits in a phase II trial, a phase I trial of the combination of GC33 and an immune checkpoint targeting antibody is currently in progress.153 Harada et al.73 recently developed an anti-GPC1 antibody for the treatment of esophageal squamous cell carcinoma. The antibody inhibited tumor growth via ADCC and decreased angiogenesis in patient derived tumor xenograft models.
Heparanase Inhibitors
Heparanase is upregulated in all major types of human cancer and is associated with increased tumor size, tumor angiogenesis, and metastasis.164 This has stimulated efforts to identify or design heparanase inhibitors for use in cancer therapy. Heparin derivatives and HS mimetics constitute a major class of heparanase inhibitors (recently reviewed by Lanzi and Cassinelli104) and several of these have progressed into clinical trials. The synthesis of small molecule heparanase inhibitors with high selectivity is also an active area of research.6,88
Specific Editing of the Glycan Structure
An emerging strategy is the design of tools that allow for precise editing of the GCX by targeting the glycan structure. These include small molecule inhibitors of glycan biosynthesis, modification of glycoproteins, and CRISPR-based editing of glycosyltransferases. Buffone and Weaver18 recently reviewed these new strategies for GCX editing. Munkley and Scott reviewed the targeting of glycan sialylation which is upregulated in cancer.144
Future Directions
As we have seen in this review, many important developments in our basic understanding of GCX structure, function and role in diseases have been described since the 2007 review.220 The future is wide open for continued GCX research. A few suggestions derived from this review are outlined below:
The implications of the Michel–Weinbaum revised Starling hypothesis for controlling fluid balance in perioperative settings where acute normovolemic hemodilution is required to compensate for blood loss or hypovolemia should be pursued as well as the role of trapped microdomains in muscle tissue.
The importance of HSPG GPC1 as the upstream mechanosensor in shear-induced NO production after longer exposures to shear (minutes to hours) has been reviewed, but the role of the GCX in NO production after acute (seconds to minutes) changes in shear has not been investigated.
The mechanism by which substrate stiffness suppresses GPC1 expression leading to endothelial dysfunction has not been thoroughly investigated.
While it has been shown that GPC1 is suppressed in a mouse model of aging, a hypertensive mouse or rat model has not been considered and these questions have not been addressed in human subjects.
STORM imaging of the GCX on ECs should be extended to examine additional GAGs and proteoglycans as well as a variety of cell types both in vitro and in vivo.
The role of GCX components in infectious diseases is understudied. A question related to the recent SARS COV-2 (COVID -19) virus is whether GAGs/proteoglycans mediate binding of the virus to the host cell as has been shown for SARS COV-1. There are many other questions.
Does restoration of the GCX on ECs in inflammatory environments really limit or reverse atherosclerosis and related cardiovascular diseases? This should be tested extensively in animals and, if promising, in humans.
An important role for IF in cancer metastasis has been suggested by recent studies. Because mechanotransduction of IF by the cancer cell GCX is involved, it will be important to determine which cancers are sensitive to IF and which GCX components are the sensors and transducers of IF.
Degradation of the endothelial GCX has been suggested by recent studies as a requirement for cancer cell extravasation from the vasculature during metastasis. More extensive animal studies are required to demonstrate the importance of this mechanism.
While there has been extensive recent research to develop GCX preserving or enhancing agents for treatment of vascular and related diseases, there is a great need for additional testing in animal models of diseases as pre-clinical preparation for clinical trials.
Funding
Research supported by National Institutes of Health Grant RO1CA204949.
Conflict of interest
Author SB, Author LMC, Author BF, and Author JMT declare that they have no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Adamson RH, Lenz JF, Zhang X, Adamson GN, Weinbaum S, Curry FE. Oncotic pressures opposing filtration across non-fenestrated rat microvessels. J. Physiol. 2004;557(Pt 3):889–907. doi: 10.1113/jphysiol.2003.058255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adembri C, Sgambati E, Vitali L, Selmi V, Margheri M, Tani A, et al. Sepsis induces albuminuria and alterations in the glomerular filtration barrier: a morphofunctional study in the rat. Crit. Care. 2011;15(6):R277. doi: 10.1186/cc10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101(10):3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 4.Alves NG, Trujillo AN, Breslin JW, Yuan SY. Sphingosine-1-phosphate reduces hemorrhagic shock and resuscitation-induced microvascular leakage by protecting endothelial mitochondrial integrity. Shock. 2019;52(4):423–433. doi: 10.1097/SHK.0000000000001280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Annecke T, Chappell D, Chen C, Jacob M, Welsch U, Sommerhoff CP, et al. Sevoflurane preserves the endothelial glycocalyx against ischaemia-reperfusion injury. Br. J. Anaesth. 2010;104(4):414–421. doi: 10.1093/bja/aeq019. [DOI] [PubMed] [Google Scholar]
- 6.Baburajeev CP, Mohan CD, Rangappa S, Mason DJ, Fuchs JE, Bender A, et al. Identification of novel class of triazolo-thiadiazoles as potent inhibitors of human heparanase and their anticancer activity. BMC Cancer. 2017;17(1):235. doi: 10.1186/s12885-017-3214-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baeyens N, Mulligan-Kehoe MJ, Corti F, Simon DD, Ross TD, Rhodes JM, et al. Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc. Natl. Acad. Sci. U S A. 2014;111(48):17308–17313. doi: 10.1073/pnas.1413725111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey CJ. Metformin: effects on micro and macrovascular complications in type 2 diabetes. Cardiovasc. Drugs Ther. 2008;22(3):215–224. doi: 10.1007/s10557-008-6092-0. [DOI] [PubMed] [Google Scholar]
- 9.Bar A, Targosz-Korecka M, Suraj J, Proniewski B, Jasztal A, Marczyk B, et al. Degradation of glycocalyx and multiple manifestations of endothelial dysfunction coincide in the early phase of endothelial dysfunction before atherosclerotic plaque development in apolipoprotein E/low-density lipoprotein receptor-deficient mice. J. Am. Heart Assoc. 2019;8(6):e011171. doi: 10.1161/JAHA.118.011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbareschi M, Maisonneuve P, Aldovini D, Cangi MG, Pecciarini L, Angelo Mauri F, et al. High syndecan-1 expression in breast carcinoma is related to an aggressive phenotype and to poorer prognosis. Cancer. 2003;98(3):474–483. doi: 10.1002/cncr.11515. [DOI] [PubMed] [Google Scholar]
- 11.Barelli S, Alberio L. The role of plasma transfusion in massive bleeding: protecting the endothelial glycocalyx? Front. Med.. (Lausanne). 2018;5:91. doi: 10.3389/fmed.2018.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartosch AMW, Mathews R, Tarbell JM. Endothelial glycocalyx-mediated nitric oxide production in response to selective AFM pulling. Biophys. J. 2017;113(1):101–108. doi: 10.1016/j.bpj.2017.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basu A, Du M, Leyva MJ, Sanchez K, Betts NM, Wu M, et al. Blueberries decrease cardiovascular risk factors in obese men and women with metabolic syndrome. J. Nutr. 2010;140(9):1582–1587. doi: 10.3945/jn.110.124701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317(5845):1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Becker BF, Jacob M, Leipert S, Salmon AH, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br. J. Clin. Pharmacol. 2015;80(3):389–402. doi: 10.1111/bcp.12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bharat D, Cavalcanti RRM, Petersen C, Begaye N, Cutler BR, Costa MMA, et al. Blueberry metabolites attenuate lipotoxicity-induced endothelial dysfunction. Mol. Nutr. Food Res. 2018 doi: 10.1002/mnfr.201700601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brower JB, Targovnik JH, Caplan MR, Massia SP. High glucose-mediated loss of cell surface heparan sulfate proteoglycan impairs the endothelial shear stress response. Cytoskeleton (Hoboken). 2010;67(3):135–141. doi: 10.1002/cm.20430. [DOI] [PubMed] [Google Scholar]
- 18.Buffone A, Weaver VM. Don’t sugarcoat it: how glycocalyx composition influences cancer progression. J. Cell Biol. 2020;219(1):e201910070. doi: 10.1083/jcb.201910070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buscail E, Alix-Panabieres C, Quincy P, Cauvin T, Chauvet A, Degrandi O, et al. High clinical value of liquid biopsy to detect circulating tumor cells and tumor exosomes in pancreatic ductal adenocarcinoma patients eligible for up-front surgery. Cancers (Basel). 2019;11(11):1656. doi: 10.3390/cancers11111656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butler MJ, Ramnath R, Kadoya H, Desposito D, Riquier-Brison A, Ferguson JK, et al. Aldosterone induces albuminuria via matrix metalloproteinase-dependent damage of the endothelial glycocalyx. Kidney Int. 2019;95(1):94–107. doi: 10.1016/j.kint.2018.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai B, Fan J, Zeng M, Zhang L, Fu BM. Adhesion of malignant mammary tumor cells MDA-MB-231 to microvessel wall increases microvascular permeability via degradation of endothelial surface glycocalyx. J. Appl. Physiol. 2012;113(7):1141–1153. doi: 10.1152/japplphysiol.00479.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancel LM, Ebong EE, Mensah S, Hirschberg C, Tarbell JM. Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis. 2016;252:136–146. doi: 10.1016/j.atherosclerosis.2016.07.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancel LM, Tarbell JM. Rhamnan sulfate enhances the endothelial glycocalyx and decreases the LDL permeability of human coronary artery endothelial cells in vitro. FASEB J. 2013;27:7. doi: 10.1096/fasebj.27.1_supplement.896.3. [DOI] [Google Scholar]
- 24.Chappell D, Heindl B, Jacob M, Annecke T, Chen C, Rehm M, et al. Sevoflurane reduces leukocyte and platelet adhesion after ischemia-reperfusion by protecting the endothelial glycocalyx. Anesthesiology. 2011;115(3):483–491. doi: 10.1097/ALN.0b013e3182289988. [DOI] [PubMed] [Google Scholar]
- 25.Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit. Care. 2015;19(1):26. doi: 10.1186/s13054-015-0741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YT, Chen Z, Du YN. Immunohistochemical analysis of RHAMM expression in normal and neoplastic human tissues: a cell cycle protein with distinctive expression in mitotic cells and testicular germ cells. Oncotarget. 2018;9(30):20941–20952. doi: 10.18632/oncotarget.24939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020;116(6):1097–1100. doi: 10.1093/cvr/cvaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chignalia AZ, Yetimakman F, Christiaans SC, Unal S, Bayrakci B, Wagener BM, et al. The glycocalyx and trauma: a review. Shock. 2016;45(4):338–348. doi: 10.1097/SHK.0000000000000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chistiakov DA, Orekhov AN, Bobryshev YV. Endothelial PECAM-1 and its function in vascular physiology and atherogenic pathology. Exp. Mol. Pathol. 2016;100(3):409–415. doi: 10.1016/j.yexmp.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 30.Coccheri S, Mannello F. Development and use of sulodexide in vascular diseases: implications for treatment. Drug Des. Devel. Ther. 2013;8:49–65. doi: 10.2147/DDDT.S6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Contreras HR, Fabre M, Granes F, Casaroli-Marano R, Rocamora N, Herreros AG, et al. Syndecan-2 expression in colorectal cancer-derived HT-29 M6 epithelial cells induces a migratory phenotype. Biochem. Biophys. Res. Commun. 2001;286(4):742–751. doi: 10.1006/bbrc.2001.5459. [DOI] [PubMed] [Google Scholar]
- 32.Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr. Biol. 2013;23(11):1024–1030. doi: 10.1016/j.cub.2013.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coombe DR, Stevenson SM, Kinnear BF, Gandhi NS, Mancera RL, Osmond RI, et al. Platelet endothelial cell adhesion molecule 1 (PECAM-1) and its interactions with glycosaminoglycans: 2. Biochemical analyses. Biochemistry. 2008;47(17):4863–4875. doi: 10.1021/bi7024595. [DOI] [PubMed] [Google Scholar]
- 34.Cooper S, Teoh H, Campeau MA, Verma S, Leask RL. Empagliflozin restores the integrity of the endothelial glycocalyx in vitro. Mol. Cell. Biochem. 2019;459(1–2):121–130. doi: 10.1007/s11010-019-03555-2. [DOI] [PubMed] [Google Scholar]
- 35.Couchman JR, Multhaupt H, Sanderson RD. Recent insights into cell surface heparan sulphate proteoglycans and cancer. F1000Res. 2016 doi: 10.12688/f1000research.8543.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui N, Wang H, Long Y, Su L, Liu D. Dexamethasone suppressed LPS-induced matrix metalloproteinase and its effect on endothelial glycocalyx shedding. Mediators Inflamm. 2015;2015:912726. doi: 10.1155/2015/912726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Curry FE. The molecular structure of the endothelial glycocalyx layer (EGL) and surface layers (ESL) modulation of transvascular exchange. Adv. Exp. Med. Biol. 2018;1097:29–49. doi: 10.1007/978-3-319-96445-4_2. [DOI] [PubMed] [Google Scholar]
- 38.Cutler BR, Gholami S, Chua JS, Kuberan B, Anandh Babu PV. Blueberry metabolites restore cell surface glycosaminoglycans and attenuate endothelial inflammation in diabetic human aortic endothelial cells. Int. J. Cardiol. 2018;261:155–158. doi: 10.1016/j.ijcard.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dabagh M, Jalali P, Butler PJ, Randles A, Tarbell JM. Mechanotransmission in endothelial cells subjected to oscillatory and multi-directional shear flow. J. R. Soc. Interface. 2017;14(130):20170185. doi: 10.1098/rsif.2017.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danese C, Vestri AR, D’Alfonso V, Deriu G, Dispensa S, Baldini R, et al. Do hypertension and diabetes mellitus influence the site of atherosclerotic plaques? Clin. Ter. 2006;157(1):9–13. [PubMed] [Google Scholar]
- 41.Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009;6(1):16–26. doi: 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Rossi G, Evans AR, Kay E, Woodfin A, McKay TR, Nourshargh S, et al. Shed syndecan-2 inhibits angiogenesis. J. Cell Sci. 2014;127(Pt 21):4788–4799. doi: 10.1242/jcs.153015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.dela Paz NG, Melchior B, Shayo FY, Frangos JA. Heparan sulfates mediate the interaction between platelet endothelial cell adhesion molecule-1 (PECAM-1) and the Gαq/11 subunits of heterotrimeric G proteins. J. Biol. Chem. 2014;289(11):7413–7424. doi: 10.1074/jbc.M113.542514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dogné S, Flamion B, Caron N. Endothelial glycocalyx as a shield against diabetic vascular complications: involvement of hyaluronan and hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 2018;38(7):1427–1439. doi: 10.1161/ATVBAHA.118.310839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dogné S, Rath G, Jouret F, Caron N, Dessy C, Flamion B. Hyaluronidase 1 deficiency preserves endothelial function and glycocalyx integrity in early streptozotocin-induced diabetes. Diabetes. 2016;65(9):2742–2753. doi: 10.2337/db15-1662. [DOI] [PubMed] [Google Scholar]
- 46.Duan L, Hu XQ, Feng DY, Lei SY, Hu GH. GPC-1 may serve as a predictor of perineural invasion and a prognosticator of survival in pancreatic cancer. Asian J Surg. 2013;36(1):7–12. doi: 10.1016/j.asjsur.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Dull RO, Cluff M, Kingston J, Hill D, Chen H, Hoehne S, et al. Lung heparan sulfates modulate K(fc) during increased vascular pressure: evidence for glycocalyx-mediated mechanotransduction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012;302(9):L816–L828. doi: 10.1152/ajplung.00080.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dull RO, Mecham I, McJames S. Heparan sulfates mediate pressure-induced increase in lung endothelial hydraulic conductivity via nitric oxide/reactive oxygen species. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292(6):L1452–L1458. doi: 10.1152/ajplung.00376.2006. [DOI] [PubMed] [Google Scholar]
- 49.Duprez Daniel A. Arterial stiffness and endothelial function. Hypertension. 2010;55(3):612–613. doi: 10.1161/HYPERTENSIONAHA.109.144725. [DOI] [PubMed] [Google Scholar]
- 50.Ebong EE, Lopez-Quintero SV, Rizzo V, Spray DC, Tarbell JM. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr Biol (Camb). 2014;6(3):338–347. doi: 10.1039/c3ib40199e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ebong EE, Macaluso FP, Spray DC, Tarbell JM. Imaging the endothelial glycocalyx in vitro by rapid freezing/freeze substitution transmission electron microscopy. Arterioscler. Thromb. Vasc. Biol. 2011;31(8):1908–1915. doi: 10.1161/ATVBAHA.111.225268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Erdem M, Erdem S, Sanli O, Sak H, Kilicaslan I, Sahin F, et al. Up-regulation of TGM2 with ITGB1 and SDC4 is important in the development and metastasis of renal cell carcinoma. Urol. Oncol. 2014;32(1):25. doi: 10.1016/j.urolonc.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 53.Eskens BJ, Zuurbier CJ, van Haare J, Vink H, van Teeffelen JW. Effects of two weeks of metformin treatment on whole-body glycocalyx barrier properties in db/db mice. Cardiovasc. Diabetol. 2013;12:175. doi: 10.1186/1475-2840-12-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Estep RD, Govindan AN, Manoharan M, Li H, Fei SS, Park BS, et al. Molecular analysis of lymphoid tissue from rhesus macaque rhadinovirus-infected monkeys identifies alterations in host genes associated with oncogenesis. PLoS ONE. 2020;15(2):e0228484. doi: 10.1371/journal.pone.0228484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fan J, Cai B, Zeng M, Hao Y, Giancotti FG, Fu BM. Integrin β4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann. Biomed. Eng. 2011;39(8):2223–2241. doi: 10.1007/s10439-011-0321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fan J, Fu BM. Quantification of malignant breast cancer cell MDA-MB-231 transmigration across brain and lung microvascular endothelium. Ann. Biomed. Eng. 2016;44(7):2189–2201. doi: 10.1007/s10439-015-1517-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fan J, Sun Y, Xia Y, Tarbell JM, Fu BM. Endothelial surface glycocalyx (ESG) components and ultra-structure revealed by stochastic optical reconstruction microscopy (STORM) Biorheology. 2019;56(2–3):77–88. doi: 10.3233/BIR-180204. [DOI] [PubMed] [Google Scholar]
- 58.Fink MP. Animal models of sepsis. Virulence. 2014;5(1):143–153. doi: 10.4161/viru.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J. Cell Sci. 2005;118(Pt 18):4103–4111. doi: 10.1242/jcs.02541. [DOI] [PubMed] [Google Scholar]
- 60.Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ. Res. 2003;93(10):e136–e142. doi: 10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 61.Fu BM, Tarbell JM. Mechano-sensing and transduction by endothelial surface glycocalyx: composition, structure, and function. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013;5(3):381–390. doi: 10.1002/wsbm.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gallagher J. Fell-Muir lecture: heparan sulphate and the art of cell regulation: a polymer chain conducts the protein orchestra. Int. J. Exp. Pathol. 2015;96(4):203–231. doi: 10.1111/iep.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goligorsky MS, Sun D. Glycocalyx in endotoxemia and sepsis. Am. J. Pathol. 2020;190(4):791–798. doi: 10.1016/j.ajpath.2019.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guerci P, Ergin B, Uz Z, Ince Y, Westphal M, Heger M, et al. Glycocalyx degradation is independent of vascular barrier permeability increase in nontraumatic hemorrhagic shock in rats. Anesth. Analg. 2019;129(2):598–607. doi: 10.1213/ANE.0000000000003918. [DOI] [PubMed] [Google Scholar]
- 65.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004;5(10):816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 66.Guo J, Yang ZC, Liu Y. Attenuating pulmonary hypertension by protecting the integrity of glycocalyx in rats model of pulmonary artery hypertension. Inflammation. 2019;42(6):1951–1956. doi: 10.1007/s10753-019-01055-5. [DOI] [PubMed] [Google Scholar]
- 67.Guo M, Zhang H, Zheng J, Liu Y. Glypican-3: a new target for diagnosis and treatment of hepatocellular carcinoma. J. Cancer. 2020;11(8):2008–2021. doi: 10.7150/jca.39972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guyton HG, Decker HM. Respiratory protection provided by five new contagion masks. Appl. Microbiol. 1963;11(1):66–68. doi: 10.1128/am.11.1.66-68.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haeren RH, van de Ven SE, van Zandvoort MA, Vink H, van Overbeeke JJ, Hoogland G, et al. Assessment and imaging of the cerebrovascular glycocalyx. Curr. Neurovasc. Res. 2016;13(3):249–260. doi: 10.2174/1567202613666160504104434. [DOI] [PubMed] [Google Scholar]
- 70.Haessler U, Teo JC, Foretay D, Renaud P, Swartz MA. Migration dynamics of breast cancer cells in a tunable 3D interstitial flow chamber. Integr. Biol. (Camb). 2012;4(4):401–409. doi: 10.1039/c1ib00128k. [DOI] [PubMed] [Google Scholar]
- 71.Hammond E, Khurana A, Shridhar V, Dredge K. The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Front. Oncol. 2014;4:195. doi: 10.3389/fonc.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hara H, Takahashi T, Serada S, Fujimoto M, Ohkawara T, Nakatsuka R, et al. Overexpression of glypican-1 implicates poor prognosis and their chemoresistance in oesophageal squamous cell carcinoma. Br. J. Cancer. 2016;115(1):66–75. doi: 10.1038/bjc.2016.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harada E, Serada S, Fujimoto M, Takahashi Y, Takahashi T, Hara H, et al. Glypican-1 targeted antibody-based therapy induces preclinical antitumor activity against esophageal squamous cell carcinoma. Oncotarget. 2017;8(15):24741–24752. doi: 10.18632/oncotarget.15799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harding IC, Mitra R, Mensah SA, Herman IM, Ebong EE. Pro-atherosclerotic disturbed flow disrupts caveolin-1 expression, localization, and function via glycocalyx degradation. J. Transl. Med. 2018;16(1):364. doi: 10.1186/s12967-018-1721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hempel C, Sporring J, Kurtzhals JAL. Experimental cerebral malaria is associated with profound loss of both glycan and protein components of the endothelial glycocalyx. FASEB J. 2019;33(2):2058–2071. doi: 10.1096/fj.201800657R. [DOI] [PubMed] [Google Scholar]
- 76.Hippensteel JA, Anderson BJ, Orfila JE, McMurtry SA, Dietz RM, Su G, et al. Circulating heparan sulfate fragments mediate septic cognitive dysfunction. J Clin Invest. 2019;129(4):1779–1784. doi: 10.1172/JCI124485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu X, Adamson RH, Liu B, Curry FE, Weinbaum S. Starling forces that oppose filtration after tissue oncotic pressure is increased. Am. J. Physiol. Heart Circ. Physiol. 2000;279(4):H1724–H1736. doi: 10.1152/ajpheart.2000.279.4.H1724. [DOI] [PubMed] [Google Scholar]
- 78.Hu X, Weinbaum S. A new view of Starling’s hypothesis at the microstructural level. Microvasc. Res. 1999;58(3):281–304. doi: 10.1006/mvre.1999.2177. [DOI] [PubMed] [Google Scholar]
- 79.Hua CT, Gamble JR, Vadas MA, Jackson DE. Recruitment and activation of SHP-1 protein-tyrosine phosphatase by human platelet endothelial cell adhesion molecule-1 (PECAM-1). Identification of immunoreceptor tyrosine-based inhibitory motif-like binding motifs and substrates. J. Biol. Chem. 1998;273(43):28332–28340. doi: 10.1074/jbc.273.43.28332. [DOI] [PubMed] [Google Scholar]
- 80.Huang Y, Jan KM, Rumschitzki D, Weinbaum S. Structural changes in rat aortic intima due to transmural pressure. J. Biomech. Eng. 1998;120(4):476–483. doi: 10.1115/1.2798017. [DOI] [PubMed] [Google Scholar]
- 81.Huang Y, Rumschitzki D, Chien S, Weinbaum S. A fiber matrix model for the filtration through fenestral pores in a compressible arterial intima. Am. J. Physiol. 1997;272(4 Pt 2):H2023–H2039. doi: 10.1152/ajpheart.1997.272.4.H2023. [DOI] [PubMed] [Google Scholar]
- 82.Huang YL, Tung CK, Zheng A, Kim BJ, Wu M. Interstitial flows promote amoeboid over mesenchymal motility of breast cancer cells revealed by a three dimensional microfluidic model. Integr. Biol. (Camb). 2015;7(11):1402–1411. doi: 10.1039/c5ib00115c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ikonomidis I, Pavlidis G, Thymis J, Birba D, Kalogeris A, Kousathana F, et al. Effects of glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter-2 inhibitors, and their combination on endothelial glycocalyx, arterial function, and myocardial work index in patients with type 2 diabetes mellitus after 12-month treatment. J. Am. Heart Assoc. 2020;9(9):e015716. doi: 10.1161/JAHA.119.015716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ikonomidis I, Voumvourakis A, Makavos G, Triantafyllidi H, Pavlidis G, Katogiannis K, et al. Association of impaired endothelial glycocalyx with arterial stiffness, coronary microcirculatory dysfunction, and abnormal myocardial deformation in untreated hypertensives. J. Clin. Hypertens. (Greenwich). 2018;20(4):672–679. doi: 10.1111/jch.13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Introini V, Carciati A, Tomaiuolo G, Cicuta P, Guido S. Endothelial glycocalyx regulates cytoadherence in Plasmodium falciparum malaria. J. R. Soc. Interface. 2018;15(149):20180773. doi: 10.1098/rsif.2018.0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell Mol. Med. 2011;15(5):1013–1031. doi: 10.1111/j.1582-4934.2010.01236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jagannath S, Heffner LT, Jr, Ailawadhi S, Munshi NC, Zimmerman TM, Rosenblatt J, et al. Indatuximab ravtansine (BT062) monotherapy in patients with relapsed and/or refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2019;19(6):372–380. doi: 10.1016/j.clml.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 88.Jia L, Ma S. Recent advances in the discovery of heparanase inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2016;121:209–220. doi: 10.1016/j.ejmech.2016.05.052. [DOI] [PubMed] [Google Scholar]
- 89.Jian YS, Chen CW, Lin CA, Yu HP, Lin HY, Liao MY, et al. Hyaluronic acid-nimesulide conjugates as anticancer drugs against CD44-overexpressing HT-29 colorectal cancer in vitro and in vivo. Int. J. Nanomed. 2017;12:2315–2333. doi: 10.2147/IJN.S120847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann. Surg. 2011;254(2):194–200. doi: 10.1097/SLA.0b013e318226113d. [DOI] [PubMed] [Google Scholar]
- 91.Jung O, Trapp-Stamborski V, Purushothaman A, Jin H, Wang H, Sanderson RD, et al. Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: prevention by novel synstatins. Oncogenesis. 2016;5:e202. doi: 10.1038/oncsis.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karousou E, Misra S, Ghatak S, Dobra K, Gotte M, Vigetti D, et al. Roles and targeting of the HAS/hyaluronan/CD44 molecular system in cancer. Matrix Biol. 2017;59:3–22. doi: 10.1016/j.matbio.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 93.Kazuma S, Tokinaga Y, Kimizuka M, Azumaguchi R, Hamada K, Yamakage M. Sevoflurane promotes regeneration of the endothelial glycocalyx by upregulating sialyltransferase. J. Surg. Res. 2019;241:40–47. doi: 10.1016/j.jss.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 94.Kim YH, Nijst P, Kiefer K, Tang WH. Endothelial glycocalyx as biomarker for cardiovascular diseases: mechanistic and clinical implications. Curr. Heart Fail. Rep. 2017;14(2):117–126. doi: 10.1007/s11897-017-0320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kind S, Merenkow C, Buscheck F, Moller K, Dum D, Chirico V, et al. Prevalence of Syndecan-1 (CD138) expression in different kinds of human tumors and normal tissues. Dis. Markers. 2019;2019:4928315. doi: 10.1155/2019/4928315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kingsmore KM, Logsdon DK, Floyd DH, Peirce SM, Purow BW, Munson JM. Interstitial flow differentially increases patient-derived glioblastoma stem cell invasion via CXCR4, CXCL12, and CD44-mediated mechanisms. Integr. Biol. (Camb). 2016;8(12):1246–1260. doi: 10.1039/c6ib00167j. [DOI] [PubMed] [Google Scholar]
- 97.Kleeff J, Ishiwata T, Kumbasar A, Friess H, Buchler MW, Lander AD, et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J. Clin. Invest. 1998;102(9):1662–1673. doi: 10.1172/JCI4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuchan MJ, Frangos JA. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am. J. Physiol. 1994;266(3 Pt 1):C628–C636. doi: 10.1152/ajpcell.1994.266.3.C628. [DOI] [PubMed] [Google Scholar]
- 99.Kumase F, Morizane Y, Mohri S, Takasu I, Ohtsuka A, Ohtsuki H. Glycocalyx degradation in retinal and choroidal capillary endothelium in rats with diabetes and hypertension. Acta Med. Okayama. 2010;64(5):277–283. doi: 10.18926/AMO/40502. [DOI] [PubMed] [Google Scholar]
- 100.Kusche-Vihrog K, Oberleithner H. An emerging concept of vascular salt sensitivity. Biol. Rep. 2012;4:20. doi: 10.3410/B4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Landis HR. Laboratory and clinical methods. Trans. Am. Climatol. Clin. Assoc. 1927;43:134–143. [PMC free article] [PubMed] [Google Scholar]
- 102.Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010;33(2):322–326. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lang J, Yang N, Deng J, Liu K, Yang P, Zhang G, et al. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE. 2011;6(8):e23710. doi: 10.1371/journal.pone.0023710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lanzi C, Cassinelli G. Heparan sulfate mimetics in cancer therapy: the challenge to define structural determinants and the relevance of targets for optimal activity. Molecules. 2018;23(11):2915. doi: 10.3390/molecules23112915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lauster D, Glanz M, Bardua M, Ludwig K, Hellmund M, Hoffmann U, et al. Multivalent peptide-nanoparticle conjugates for influenza-virus inhibition. Angew. Chem. Int. Ed. Engl. 2017;56(21):5931–5936. doi: 10.1002/anie.201702005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leskova W, Pickett H, Eshaq RS, Shrestha B, Pattillo CB, Harris NR. Effect of diabetes and hyaluronidase on the retinal endothelial glycocalyx in mice. Exp. Eye Res. 2019;179:125–131. doi: 10.1016/j.exer.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Levick JR. Capillary filtration-absorption balance reconsidered in light of dynamic extravascular factors. Exp. Physiol. 1991;76(6):825–857. doi: 10.1113/expphysiol.1991.sp003549. [DOI] [PubMed] [Google Scholar]
- 108.Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 2010;87(2):198–210. doi: 10.1093/cvr/cvq062. [DOI] [PubMed] [Google Scholar]
- 109.Lewis JC, Taylor RG, Jones ND, St Clair RW, Cornhill JF. Endothelial surface characteristics in pigeon coronary artery atherosclerosis. I. Cellular alterations during the initial stages of dietary cholesterol challenge. Lab Invest. 1982;46(2):123–138. [PubMed] [Google Scholar]
- 110.Li J, Chen Y, Guo X, Zhou L, Jia Z, Peng Z, et al. GPC1 exosome and its regulatory miRNAs are specific markers for the detection and target therapy of colorectal cancer. J. Cell Mol. Med. 2017;21(5):838–847. doi: 10.1111/jcmm.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li J, Li B, Ren C, Chen Y, Guo X, Zhou L, et al. The clinical significance of circulating GPC1 positive exosomes and its regulative miRNAs in colon cancer patients. Oncotarget. 2017;8(60):101189–101202. doi: 10.18632/oncotarget.20516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li T, Liu X, Zhao Z, Ni L, Liu C. Sulodexide recovers endothelial function through reconstructing glycocalyx in the balloon-injury rat carotid artery model. Oncotarget. 2017;8(53):91350–91361. doi: 10.18632/oncotarget.20518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li J, Yuan T, Zhao X, Lv GY, Liu HQ. Protective effects of sevoflurane in hepatic ischemia-reperfusion injury. Int. J. Immunopathol. Pharmacol. 2016;29(2):300–307. doi: 10.1177/0394632016638346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ligi D, Croce L, Mannello F. Chronic venous disorders: the dangerous, the good, and the diverse. Int. J. Mol. Sci. 2018;19(9):2544. doi: 10.3390/ijms19092544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lim HC, Couchman JR. Syndecan-2 regulation of morphology in breast carcinoma cells is dependent on RhoGTPases. Biochim. Biophys. Acta. 2014;1840(8):2482–2490. doi: 10.1016/j.bbagen.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 116.Lopez-Quintero SV, Cancel LM, Pierides A, Antonetti D, Spray DC, Tarbell JM. High glucose attenuates shear-induced changes in endothelial hydraulic conductivity by degrading the glycocalyx. PLoS ONE. 2013;8(11):e78954. doi: 10.1371/journal.pone.0078954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu H, Niu F, Liu F, Gao J, Sun Y, Zhao X. Elevated glypican-1 expression is associated with an unfavorable prognosis in pancreatic ductal adenocarcinoma. Cancer Med. 2017;6(6):1181–1191. doi: 10.1002/cam4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lygizos MI, Yang Y, Altmann CJ, Okamura K, Hernando AA, Perez MJ, et al. Heparanase mediates renal dysfunction during early sepsis in mice. Physiol. Rep. 2013;1(6):e00153. doi: 10.1002/phy2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Machin DR, Bloom SI, Campbell RA, Phuong TTT, Gates PE, Lesniewski LA, et al. Advanced age results in a diminished endothelial glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2018;315(3):H531–H539. doi: 10.1152/ajpheart.00104.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mahmoud M, Mayer M, Cancel LM, Bartosch AM, Mathews R, Tarbell JM. The Glycocalyx core protein Glypican 1 protects vessel wall endothelial cells from stiffness-mediated dysfunction and disease. Cardiovasc. Res. 2020;20:10. doi: 10.1093/cvr/cvaa201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Majerczak J, Duda K, Chlopicki S, Bartosz G, Zakrzewska A, Balcerczyk A, et al. Endothelial glycocalyx integrity is preserved in young, healthy men during a single bout of strenuous physical exercise. Physiol. Res. 2016;65(2):281–291. doi: 10.33549/physiolres.933049. [DOI] [PubMed] [Google Scholar]
- 122.Majerczak J, Grandys M, Duda K, Zakrzewska A, Balcerczyk A, Kolodziejski L, et al. Moderate-intensity endurance training improves endothelial glycocalyx layer integrity in healthy young men. Exp. Physiol. 2017;102(1):70–85. doi: 10.1113/EP085887. [DOI] [PubMed] [Google Scholar]
- 123.Mansilha A, Sousa J. Pathophysiological mechanisms of chronic venous disease and implications for venoactive drug therapy. Int. J. Mol. Sci. 2018;19(6):1669. doi: 10.3390/ijms19061669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Maruhashi T, Soga J, Fujimura N, Idei N, Mikami S, Iwamoto Y, et al. Endothelial dysfunction, increased arterial stiffness, and cardiovascular risk prediction in patients with coronary artery disease: FMD-J (Flow-Mediated Dilation Japan) Study A. J. Am. Heart Assoc. 2018;7(14):e008588. doi: 10.1161/JAHA.118.008588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Masuda M, Osawa M, Shigematsu H, Harada N, Fujiwara K. Platelet endothelial cell adhesion molecule-1 is a major SH-PTP2 binding protein in vascular endothelial cells. FEBS Lett. 1997;408(3):331–336. doi: 10.1016/s0014-5793(97)00457-2. [DOI] [PubMed] [Google Scholar]
- 126.Matsuda K, Maruyama H, Guo F, Kleeff J, Itakura J, Matsumoto Y, et al. Glypican-1 is overexpressed in human breast cancer and modulates the mitogenic effects of multiple heparin-binding growth factors in breast cancer cells. Cancer Res. 2001;61(14):5562–5569. [PubMed] [Google Scholar]
- 127.Matsuzaki S, Serada S, Hiramatsu K, Nojima S, Ueda Y, Ohkawara T, et al. Anti-glypican-1 antibody-drug conjugate exhibits potent preclinical antitumor activity against glypican-1 positive uterine cervical cancer. Int. J. Cancer. 2018;142(5):1056–1066. doi: 10.1002/ijc.31124. [DOI] [PubMed] [Google Scholar]
- 128.Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers. 2019;5(1):18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McGarrity S, Anuforo Ó, Halldórsson H, Bergmann A, Halldórsson S, Palsson S, et al. Metabolic systems analysis of LPS induced endothelial dysfunction applied to sepsis patient stratification. Sci. Rep. 2018;8(1):6811. doi: 10.1038/s41598-018-25015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mele V, Sokol L, Kolzer VH, Pfaff D, Muraro MG, Keller I, et al. The hyaluronan-mediated motility receptor RHAMM promotes growth, invasiveness and dissemination of colorectal cancer. Oncotarget. 2017;8(41):70617–70629. doi: 10.18632/oncotarget.19904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mensah SA, Cheng MJ, Homayoni H, Plouffe BD, Coury AJ, Ebong EE. Regeneration of glycocalyx by heparan sulfate and sphingosine 1-phosphate restores inter-endothelial communication. PLoS ONE. 2017;12(10):e0186116. doi: 10.1371/journal.pone.0186116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Michel, C. C. Fluid movement through capillary walls. Handbook of Physiology: Sect 2. Philadelphia: American Physiological Society, 1984.
- 134.Michel CC. Starling: the formulation of his hypothesis of microvascular fluid exchange and its significance after 100 years. Exp. Physiol. 1997;82(1):1–30. doi: 10.1113/expphysiol.1997.sp004000. [DOI] [PubMed] [Google Scholar]
- 135.Michel CC, Phillips ME. Steady-state fluid filtration at different capillary pressures in perfused frog mesenteric capillaries. J. Physiol. 1987;388:421–435. doi: 10.1113/jphysiol.1987.sp016622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Milewska A, Zarebski M, Nowak P, Stozek K, Potempa J, Pyrc K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 2014;88(22):13221–13230. doi: 10.1128/JVI.02078-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mishra HK, Ma J, Walcheck B. Ectodomain Shedding by ADAM17: its role in neutrophil recruitment and the impairment of this process during sepsis. Front Cell Infect. Microbiol. 2017;7:138. doi: 10.3389/fcimb.2017.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front Immunol. 2015;6:201. doi: 10.3389/fimmu.2015.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Misra S, Heldin P, Hascall VC, Karamanos NK, Skandalis SS, Markwald RR, et al. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 2011;278(9):1429–1443. doi: 10.1111/j.1742-4658.2011.08071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mitchell GF. Arterial stiffness and hypertension: chicken or egg? Hypertension. 2014;64(2):210–214. doi: 10.1161/HYPERTENSIONAHA.114.03449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mitchell GF, Hwang S-J, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121(4):505–511. doi: 10.1161/CIRCULATIONAHA.109.886655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Moran H, Cancel LM, Mayer MA, Qazi H, Munn LL, Tarbell JM. The cancer cell glycocalyx proteoglycan Glypican-1 mediates interstitial flow mechanotransduction to enhance cell migration and metastasis. Biorheology. 2019;56(2–3):151–161. doi: 10.3233/BIR-180203. [DOI] [PubMed] [Google Scholar]
- 143.Munkley J, Elliott DJ. Hallmarks of glycosylation in cancer. Oncotarget. 2016;7(23):35478–35489. doi: 10.18632/oncotarget.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Munkley J, Scott E. Targeting aberrant sialylation to treat cancer. Medicines (Basel). 2019;6(4):102. doi: 10.3390/medicines6040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Munson JM, Bellamkonda RV, Swartz MA. Interstitial flow in a 3D microenvironment increases glioma invasion by a CXCR4-dependent mechanism. Cancer Res. 2013;73(5):1536–1546. doi: 10.1158/0008-5472.CAN-12-2838. [DOI] [PubMed] [Google Scholar]
- 146.Munson JM, Shieh AC. Interstitial fluid flow in cancer: implications for disease progression and treatment. Cancer Manag. Res. 2014;6:317–328. doi: 10.2147/CMAR.S65444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nägga K, Hansson O, van Westen D, Minthon L, Wennström M. Increased levels of hyaluronic acid in cerebrospinal fluid in patients with vascular dementia. J. Alzheimers Dis. 2014;42(4):1435–1441. doi: 10.3233/JAD-141200. [DOI] [PubMed] [Google Scholar]
- 148.Nagy N, Freudenberger T, Melchior-Becker A, Röck K, Ter Braak M, Jastrow H, et al. Inhibition of hyaluronan synthesis accelerates murine atherosclerosis: novel insights into the role of hyaluronan synthesis. Circulation. 2010;122(22):2313–2322. doi: 10.1161/CIRCULATIONAHA.110.972653. [DOI] [PubMed] [Google Scholar]
- 149.Nelson A, Berkestedt I, Bodelsson M. Circulating glycosaminoglycan species in septic shock. Acta Anaesthesiol. Scand. 2014;58(1):36–43. doi: 10.1111/aas.12223. [DOI] [PubMed] [Google Scholar]
- 150.Neves FM, Meneses GC, Sousa NE, Menezes RR, Parahyba MC, Martins AM, et al. Syndecan-1 in acute decompensated heart failure-association with renal function and mortality. Circ. J. 2015;79(7):1511–1519. doi: 10.1253/circj.CJ-14-1195. [DOI] [PubMed] [Google Scholar]
- 151.Nieuwdorp M, Meuwese MC, Mooij HL, van Lieshout MH, Hayden A, Levi M, et al. Tumor necrosis factor-alpha inhibition protects against endotoxin-induced endothelial glycocalyx perturbation. Atherosclerosis. 2009;202(1):296–303. doi: 10.1016/j.atherosclerosis.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 152.Nijst P, Cops J, Martens P, Swennen Q, Dupont M, Tang WHW, et al. Endovascular shedding markers in patients with heart failure with reduced ejection fraction: results from a single-center exploratory study. Microcirculation. 2018;25:2. doi: 10.3390/jcm7110400. [DOI] [PubMed] [Google Scholar]
- 153.Nishida T, Kataoka H. Glypican 3-targeted therapy in hepatocellular carcinoma. Cancers (Basel). 2019;11(9):1339. doi: 10.3390/cancers11091339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nishigaki T, Takahashi T, Serada S, Fujimoto M, Ohkawara T, Hara H, et al. Anti-glypican-1 antibody-drug conjugate is a potential therapy against pancreatic cancer. Br. J. Cancer. 2020;122(9):1333–1341. doi: 10.1038/s41416-020-0781-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Oberleithner H. Vascular endothelium: a vulnerable transit zone for merciless sodium. Nephrol. Dial. Transplant. 2014;29(2):240–246. doi: 10.1093/ndt/gft461. [DOI] [PubMed] [Google Scholar]
- 156.Okamoto T, Akita N, Terasawa M, Hayashi T, Suzuki K. Rhamnan sulfate extracted from Monostroma nitidum attenuates blood coagulation and inflammation of vascular endothelial cells. J. Nat. Med. 2019;73(3):614–619. doi: 10.1007/s11418-019-01289-5. [DOI] [PubMed] [Google Scholar]
- 157.Osawa M, Masuda M, Harada N, Lopes RB, Fujiwara K. Tyrosine phosphorylation of platelet endothelial cell adhesion molecule-1 (PECAM-1, CD31) in mechanically stimulated vascular endothelial cells. Eur. J. Cell Biol. 1997;72(3):229–237. [PubMed] [Google Scholar]
- 158.Padberg JS, Wiesinger A, di Marco GS, Reuter S, Grabner A, Kentrup D, et al. Damage of the endothelial glycocalyx in chronic kidney disease. Atherosclerosis. 2014;234(2):335–343. doi: 10.1016/j.atherosclerosis.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 159.Pahakis MY, Kosky JR, Dull RO, Tarbell JM. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 2007;355(1):228–233. doi: 10.1016/j.bbrc.2007.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pang Z, Tarbell JM. In vitro study of Starling’s hypothesis in a cultured monolayer of bovine aortic endothelial cells. J. Vasc. Res. 2003;40(4):351–358. doi: 10.1159/000072699. [DOI] [PubMed] [Google Scholar]
- 161.Pappenheimer JR, Soto-Rivera A. Effective osmotic pressure of the plasma proteins and other quantities associated with the capillary circulation in the hindlimbs of cats and dogs. Am. J. Physiol. 1948;152(3):471–491. doi: 10.1152/ajplegacy.1948.152.3.471. [DOI] [PubMed] [Google Scholar]
- 162.Park H, Kim Y, Lim Y, Han I, Oh ES. Syndecan-2 mediates adhesion and proliferation of colon carcinoma cells. J. Biol. Chem. 2002;277(33):29730–29736. doi: 10.1074/jbc.M202435200. [DOI] [PubMed] [Google Scholar]
- 163.Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature. 2014;511(7509):319–325. doi: 10.1038/nature13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pisano C, Vlodavsky I, Ilan N, Zunino F. The potential of heparanase as a therapeutic target in cancer. Biochem. Pharmacol. 2014;89(1):12–19. doi: 10.1016/j.bcp.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Piva S, McCreadie VA, Latronico N. Neuroinflammation in sepsis: sepsis associated delirium. Cardiovasc. Hematol. Disord.: Drug Targets. 2015;15(1):10–18. doi: 10.2174/1871529x15666150108112452. [DOI] [PubMed] [Google Scholar]
- 166.Polikarpov D, Liang L, Care A, Sunna A, Campbell D, Walsh B, et al. Functionalized upconversion nanoparticles for targeted labelling of bladder cancer cells. Biomolecules. 2019;9(12):820. doi: 10.3390/biom9120820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Puerta-Guardo H, Glasner DR, Espinosa DA, Biering SB, Patana M, Ratnasiri K, et al. Flavivirus NS1 triggers tissue-specific vascular endothelial dysfunction reflecting disease tropism. Cell Rep. 2019;26(6):1598–1613. doi: 10.1016/j.celrep.2019.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Puerta-Guardo H, Glasner DR, Harris E. Dengue virus NS1 disrupts the endothelial glycocalyx, leading to hyperpermeability. PLoS Pathog. 2016;12(7):e1005738. doi: 10.1371/journal.ppat.1005738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Qazi H, Palomino R, Shi ZD, Munn LL, Tarbell JM. Cancer cell glycocalyx mediates mechanotransduction and flow-regulated invasion. Integr. Biol. (Camb). 2013;5(11):1334–1343. doi: 10.1039/c3ib40057c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Qazi H, Shi ZD, Song JW, Cancel LM, Huang P, Zeng Y, et al. Heparan sulfate proteoglycans mediate renal carcinoma metastasis. Int. J. Cancer. 2016;139(12):2791–2801. doi: 10.1002/ijc.30397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Qazi H, Shi ZD, Tarbell JM. Fluid shear stress regulates the invasive potential of glioma cells via modulation of migratory activity and matrix metalloproteinase expression. PLoS ONE. 2011;6(5):e20348. doi: 10.1371/journal.pone.0020348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rai S, Nejadhamzeeigilani Z, Gutowski NJ, Whatmore JL. Loss of the endothelial glycocalyx is associated with increased E-selectin mediated adhesion of lung tumour cells to the brain microvascular endothelium. J. Exp. Clin. Cancer Res. 2015;34:105. doi: 10.1186/s13046-015-0223-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ramnath R, Foster RR, Qiu Y, Cope G, Butler MJ, Salmon AH, et al. Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor α: a contributor to endothelial cell glycocalyx dysfunction. FASEB J. 2014;28(11):4686–4699. doi: 10.1096/fj.14-252221. [DOI] [PubMed] [Google Scholar]
- 174.Reyes-Soffer G, Holleran S, Di Tullio MR, Homma S, Boden-Albala B, Ramakrishnan R, et al. Endothelial function in individuals with coronary artery disease with and without type 2 diabetes mellitus. Metabolism. 2010;59(9):1365–1371. doi: 10.1016/j.metabol.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Richter V, Savery MD, Gassmann M, Baum O, Damiano ER, Pries AR. Excessive erythrocytosis compromises the blood-endothelium interface in erythropoietin-overexpressing mice. J. Physiol. 2011;589(Pt 21):5181–5192. doi: 10.1113/jphysiol.2011.209262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Russell-Puleri S, Dela Paz NG, Adams D, Chattopadhyay M, Cancel L, Ebong E, et al. Fluid shear stress induces upregulation of COX-2 and PGI(2) release in endothelial cells via a pathway involving PECAM-1, PI3K, FAK, and p38. Am. J. Physiol. Heart Circ. Physiol. 2017;312(3):H485–H500. doi: 10.1152/ajpheart.00035.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rzhevskiy AS, Razavi Bazaz S, Ding L, Kapitannikova A, Sayyadi N, Campbell D, et al. Rapid and label-free isolation of tumour cells from the urine of patients with localised prostate cancer using inertial microfluidics. Cancers (Basel). 2019;12(1):81. doi: 10.3390/cancers12010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Saito T, Sugiyama K, Hama S, Yamasaki F, Takayasu T, Nosaka R, et al. High expression of glypican-1 predicts dissemination and poor prognosis in glioblastomas. World Neurosurg. 2017;105:282–288. doi: 10.1016/j.wneu.2017.05.165. [DOI] [PubMed] [Google Scholar]
- 179.Salmon AH, Ferguson JK, Burford JL, Gevorgyan H, Nakano D, Harper SJ, et al. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J. Am. Soc. Nephrol. 2012;23(8):1339–1350. doi: 10.1681/ASN.2012010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sardu C, Gambardella J, Morelli MB, Wang X, Marfella R, Santulli G. Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J. Clin. Med. 2020;9(5):1417. doi: 10.3390/jcm9051417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Schierke F, Wyrwoll MJ, Wisdorf M, Niedzielski L, Maase M, Ruck T, et al. Nanomechanics of the endothelial glycocalyx contribute to Na(+)-induced vascular inflammation. Sci. Rep. 2017;7:46476. doi: 10.1038/srep46476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012;18(8):1217–1223. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schmitz B, Niehues H, Lenders M, Thorwesten L, Klose A, Krüger M, et al. Effects of high-intensity interval training on microvascular glycocalyx and associated microRNAs. Am. J. Physiol. Heart Circ. Physiol. 2019;316(6):H1538–H1551. doi: 10.1152/ajpheart.00751.2018. [DOI] [PubMed] [Google Scholar]
- 184.Schonfeld K, Herbener P, Zuber C, Hader T, Bernoster K, Uherek C, et al. Activity of indatuximab ravtansine against triple-negative breast cancer in preclinical tumor models. Pharm. Res. 2018;35(6):118. doi: 10.1007/s11095-018-2400-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Secomb TW, Hsu R, Pries AR. Effect of the endothelial surface layer on transmission of fluid shear stress to endothelial cells. Biorheology. 2001;38(2–3):143–150. [PubMed] [Google Scholar]
- 186.Sehba FA, Ding WH, Chereshnev I, Bederson JB. Effects of S-nitrosoglutathione on acute vasoconstriction and glutamate release after subarachnoid hemorrhage. Stroke. 1999;30(9):1955–1961. doi: 10.1161/01.str.30.9.1955. [DOI] [PubMed] [Google Scholar]
- 187.Shen S, Fan J, Cai B, Lv Y, Zeng M, Hao Y, et al. Vascular endothelial growth factor enhances cancer cell adhesion to microvascular endothelium in vivo. Exp. Physiol. 2010;95(2):369–379. doi: 10.1113/expphysiol.2009.050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell. 2007;11(6):526–538. doi: 10.1016/j.ccr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 189.Shinyo Y, Kodama J, Hasengaowa T, Kusumoto T, Hiramatsu Y. Loss of cell-surface heparan sulfate expression in both cervical intraepithelial neoplasm and invasive cervical cancer. Gynecol. Oncol. 2005;96(3):776–783. doi: 10.1016/j.ygyno.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 190.Simizu S, Ishida K, Osada H. Heparanase as a molecular target of cancer chemotherapy. Cancer Sci. 2004;95(7):553–558. doi: 10.1111/j.1349-7006.2004.tb02485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Soares MA, Teixeira FC, Fontes M, Areas AL, Leal MG, Pavao MS, et al. Heparan sulfate proteoglycans may promote or inhibit cancer progression by interacting with integrins and affecting cell migration. Biomed. Res. Int. 2015;2015:453801. doi: 10.1155/2015/453801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Son DJ, Kumar S, Takabe W, Kim CW, Ni CW, Alberts-Grill N, et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat. Commun. 2013;4:3000. doi: 10.1038/ncomms4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Song JW, Goligorsky MS. Perioperative implication of the endothelial glycocalyx. Korean J. Anesthesiol. 2018;71(2):92–102. doi: 10.4097/kjae.2018.71.2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Song JW, Zullo JA, Liveris D, Dragovich M, Zhang XF, Goligorsky MS. Therapeutic restoration of endothelial glycocalyx in sepsis. J. Pharmacol. Exp. Ther. 2017;361(1):115–121. doi: 10.1124/jpet.116.239509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Starling EH. On the absorption of fluids from the connective tissue spaces. J. Physiol. 1896;19(4):312–326. doi: 10.1113/jphysiol.1896.sp000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Suwarto S, Sasmono RT, Sinto R, Ibrahim E, Suryamin M. Association of endothelial glycocalyx and tight and adherens junctions with severity of plasma leakage in dengue infection. J. Infect. Dis. 2017;215(6):992–999. doi: 10.1093/infdis/jix041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Suzuki K, Terasawa M. Biological activities of rhamnan sulfate extract from the green algae Monostroma nitidum (Hitoegusa) Mar. Drugs. 2020;18(4):228. doi: 10.3390/md18040228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tammi RH, Kultti A, Kosma VM, Pirinen R, Auvinen P, Tammi MI. Hyaluronan in human tumors: pathobiological and prognostic messages from cell-associated and stromal hyaluronan. Semin. Cancer Biol. 2008;18(4):288–295. doi: 10.1016/j.semcancer.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 199.Tang TH, Alonso S, Ng LF, Thein TL, Pang VJ, Leo YS, et al. Increased serum hyaluronic acid and heparan sulfate in dengue fever: association with plasma leakage and disease severity. Sci. Rep. 2017;7:46191. doi: 10.1038/srep46191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tarbell JM, Cancel LM. The glycocalyx and its significance in human medicine. J. Intern. Med. 2016;280(1):97–113. doi: 10.1111/joim.12465. [DOI] [PubMed] [Google Scholar]
- 201.Tarbell JM, Ebong EE. The endothelial glycocalyx: a mechano-sensor and -transducer. Sci. Signal. 2008;1(40):8. doi: 10.1126/scisignal.140pt8. [DOI] [PubMed] [Google Scholar]
- 202.Tarbell JM, Pahakis MY. Mechanotransduction and the glycocalyx. J. Intern. Med. 2006;259(4):339–350. doi: 10.1111/j.1365-2796.2006.01620.x. [DOI] [PubMed] [Google Scholar]
- 203.Targosz-Korecka M, Jaglarz M, Malek-Zietek KE, Gregorius A, Zakrzewska A, Sitek B, et al. AFM-based detection of glycocalyx degradation and endothelial stiffening in the db/db mouse model of diabetes. Sci. Rep. 2017;7(1):15951. doi: 10.1038/s41598-017-16179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Targosz-Korecka M, Malek-Zietek KE, Kloska D, Rajfur Z, Stepien EL, Grochot-Przeczek A, et al. Metformin attenuates adhesion between cancer and endothelial cells in chronic hyperglycemia by recovery of the endothelial glycocalyx barrier. Biochim. Biophys. Acta Gen. Subj. 2020;1864(4):129533. doi: 10.1016/j.bbagen.2020.129533. [DOI] [PubMed] [Google Scholar]
- 205.Thi MM, Tarbell JM, Weinbaum S, Spray DC. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a “bumper-car” model. Proc. Natl. Acad. Sci. USA. 2004;101(47):16483–16488. doi: 10.1073/pnas.0407474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Torres Filho IP, Torres LN, Salgado C, Dubick MA. Novel adjunct drugs reverse endothelial glycocalyx damage after hemorrhagic shock in rats. Shock. 2017;48(5):583–589. doi: 10.1097/SHK.0000000000000895. [DOI] [PubMed] [Google Scholar]
- 207.Tuma M, Canestrini S, Alwahab Z, Marshall J. Trauma and endothelial glycocalyx: the microcirculation helmet? Shock. 2016;46(4):352–357. doi: 10.1097/SHK.0000000000000635. [DOI] [PubMed] [Google Scholar]
- 208.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 209.Uchimido R, Schmidt EP, Shapiro NI. The glycocalyx: a novel diagnostic and therapeutic target in sepsis. Crit. Care. 2019;23(1):16. doi: 10.1186/s13054-018-2292-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ueno M, Sakamoto H, Liao YJ, Onodera M, Huang CL, Miyanaka H, et al. Blood-brain barrier disruption in the hypothalamus of young adult spontaneously hypertensive rats. Histochem. Cell Biol. 2004;122(2):131–137. doi: 10.1007/s00418-004-0684-y. [DOI] [PubMed] [Google Scholar]
- 211.Ueno M, Sakamoto H, Tomimoto H, Akiguchi I, Onodera M, Huang CL, et al. Blood-brain barrier is impaired in the hippocampus of young adult spontaneously hypertensive rats. Acta Neuropathol. 2004;107(6):532–538. doi: 10.1007/s00401-004-0845-z. [DOI] [PubMed] [Google Scholar]
- 212.van den Berg BM, Spaan JA, Rolf TM, Vink H. Atherogenic region and diet diminish glycocalyx dimension and increase intima-to-media ratios at murine carotid artery bifurcation. Am. J. Physiol. Heart. Circ. Physiol. 2006;290(2):H915–H920. doi: 10.1152/ajpheart.00051.2005. [DOI] [PubMed] [Google Scholar]
- 213.van den Berg BM, Spaan JA, Vink H. Impaired glycocalyx barrier properties contribute to enhanced intimal low-density lipoprotein accumulation at the carotid artery bifurcation in mice. Pflugers Arch. 2009;457(6):1199–1206. doi: 10.1007/s00424-008-0590-6. [DOI] [PubMed] [Google Scholar]
- 214.van Haare J, Kooi ME, van Teeffelen JW, Vink H, Slenter J, Cobelens H, et al. Metformin and sulodexide restore cardiac microvascular perfusion capacity in diet-induced obese rats. Cardiovasc. Diabetol. 2017;16(1):47. doi: 10.1186/s12933-017-0525-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Van Wyngene L, Vandewalle J, Libert C. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last. EMBO Mol. Med. 2018;10(8):7812. doi: 10.15252/emmm.201708712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Vlahu CA, Lemkes BA, Struijk DG, Koopman MG, Krediet RT, Vink H. Damage of the endothelial glycocalyx in dialysis patients. J. Am. Soc. Nephrol. 2012;23(11):1900–1908. doi: 10.1681/ASN.2011121181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wagenseil JE, Mecham RP. Elastin in large artery stiffness and hypertension. J. Cardiovasc. Transl. Res. 2012;5(3):264–273. doi: 10.1007/s12265-012-9349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang S, Qiu Y, Bai B. The expression, regulation, and biomarker potential of glypican-1 in cancer. Front. Oncol. 2019;9:614. doi: 10.3389/fonc.2019.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Weinbaum S. 1997 Whitaker distinguished lecture: models to solve mysteries in biomechanics at the cellular level; a new view of fiber matrix layers. Ann. Biomed. Eng. 1998;26(4):627–643. doi: 10.1114/1.134. [DOI] [PubMed] [Google Scholar]
- 220.Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 2007;9:121–167. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- 221.Weinbaum S, Zhang X, Han Y, Vink H, Cowin SC. Mechanotransduction and flow across the endothelial glycocalyx. Proc. Natl. Acad. Sci. USA. 2003;100(13):7988–7995. doi: 10.1073/pnas.1332808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Weissgerber TL, Garcia-Valencia O, Milic NM, Codsi E, Cubro H, Nath MC, et al. Early onset preeclampsia is associated with glycocalyx degradation and reduced microvascular perfusion. J. Am. Heart Assoc. 2019;8(4):e010647. doi: 10.1161/JAHA.118.010647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wodicka JR, Chambers AM, Sangha GS, Goergen CJ, Panitch A. Development of a glycosaminoglycan derived, selectin targeting anti-adhesive coating to treat endothelial cell dysfunction. Pharmaceuticals (Basel). 2017;10(2):36. doi: 10.3390/ph10020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Woods EC, Kai F, Barnes JM, Pedram K, Pickup MW, Hollander MJ, et al. A bulky glycocalyx fosters metastasis formation by promoting G1 cell cycle progression. Elife. 2017;6:e25752. doi: 10.7554/eLife.25752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Xia Y, Fu BM. Investigation of endothelial surface glycocalyx components and ultrastructure by single molecule localization microscopy: stochastic optical reconstruction microscopy (STORM) Yale J. Biol. Med. 2018;91(3):257–266. [PMC free article] [PubMed] [Google Scholar]
- 226.Xu S, Ha CH, Wang W, Xu X, Yin M, Jin FQ, et al. PECAM1 regulates flow-mediated Gab1 tyrosine phosphorylation and signaling. Cell Signal. 2016;28(3):117–124. doi: 10.1016/j.cellsig.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Yang X, Meegan JE, Jannaway M, Coleman DC, Yuan SY. A disintegrin and metalloproteinase 15-mediated glycocalyx shedding contributes to vascular leakage during inflammation. Cardiovasc. Res. 2018;114(13):1752–1763. doi: 10.1093/cvr/cvy167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Yang Y, Zhao Y, Lan J, Kang Y, Zhang T, Ding Y, et al. Reduction-sensitive CD44 receptor-targeted hyaluronic acid derivative micelles for doxorubicin delivery. Int. J. Nanomed. 2018;13:4361–4378. doi: 10.2147/IJN.S165359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yao Y, Rabodzey A, Dewey CF., Jr Glycocalyx modulates the motility and proliferative response of vascular endothelium to fluid shear stress. Am. J. Physiol. Heart Circ. Physiol. 2007;293(2):H1023–H1030. doi: 10.1152/ajpheart.00162.2007. [DOI] [PubMed] [Google Scholar]
- 230.Yao M, Wang L, Fang M, Zheng W, Dong Z, Yao D. Advances in the study of oncofetal antigen glypican-3 expression in HBV-related hepatocellular carcinoma. Biosci. Trends. 2016;10(5):337–343. doi: 10.5582/bst.2016.01176. [DOI] [PubMed] [Google Scholar]
- 231.Yen W, Cai B, Yang J, Zhang L, Zeng M, Tarbell JM, et al. Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS ONE. 2015;10(1):e0117133. doi: 10.1371/journal.pone.0117133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yen WY, Cai B, Zeng M, Tarbell JM, Fu BM. Quantification of the endothelial surface glycocalyx on rat and mouse blood vessels. Microvasc. Res. 2012;83(3):337–346. doi: 10.1016/j.mvr.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yeo TW, Weinberg JB, Lampah DA, Kenangalem E, Bush P, Chen Y, et al. Glycocalyx breakdown is associated with severe disease and fatal outcome in Plasmodium falciparum malaria. Clin. Infect. Dis. 2019;69(10):1712–1720. doi: 10.1093/cid/ciz038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Yurdagul A, Jr, Finney AC, Woolard MD, Orr AW. The arterial microenvironment: the where and why of atherosclerosis. Biochem. J. 2016;473(10):1281–1295. doi: 10.1042/BJ20150844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zeng Y, Adamson RH, Curry FR, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am. J. Physiol. Heart. Circ. Physiol. 2014;306(3):H363–H372. doi: 10.1152/ajpheart.00687.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zeng Y, Ebong EE, Fu BM, Tarbell JM. The structural stability of the endothelial glycocalyx after enzymatic removal of glycosaminoglycans. PLoS ONE. 2012;7(8):e43168. doi: 10.1371/journal.pone.0043168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zeng Y, Liu J. Role of glypican-1 in endothelial NOS activation under various steady shear stress magnitudes. Exp. Cell Res. 2016;348(2):184–189. doi: 10.1016/j.yexcr.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 238.Zeng Y, Liu XH, Tarbell J, Fu B. Sphingosine 1-phosphate induced synthesis of glycocalyx on endothelial cells. Exp. Cell Res. 2015;339(1):90–95. doi: 10.1016/j.yexcr.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 239.Zeng Y, Waters M, Andrews A, Honarmandi P, Ebong E, Rizzo V, et al. Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am. J. Physiol. Heart Circ. Physiol. 2013;305:811–820. doi: 10.1152/ajpheart.00764.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Zeng Y, Zhang XF, Fu BM, Tarbell JM. The role of endothelial surface glycocalyx in mechanosensing and transduction. Adv. Exp. Med. Biol. 2018;1097:1–27. doi: 10.1007/978-3-319-96445-4_1. [DOI] [PubMed] [Google Scholar]
- 241.Zhang X, Adamson RH, Curry FR, Weinbaum S. A 1-D model to explore the effects of tissue loading and tissue concentration gradients in the revised Starling principle. Am. J. Physiol. Heart Circ. Physiol. 2006;291(6):H2950–H2964. doi: 10.1152/ajpheart.01160.2005. [DOI] [PubMed] [Google Scholar]
- 242.Zhang X, Adamson RH, Curry FE, Weinbaum S. Transient regulation of transport by pericytes in venular microvessels via trapped microdomains. Proc. Natl. Acad. Sci. U S A. 2008;105(4):1374–1379. doi: 10.1073/pnas.0710986105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Zhang X, Sun D, Song JW, Zullo J, Lipphardt M, Coneh-Gould L, et al. Endothelial cell dysfunction and glycocalyx: a vicious circle. Matrix Biol. 2018;71–72:421–431. doi: 10.1016/j.matbio.2018.01.026. [DOI] [PubMed] [Google Scholar]
- 244.Zhang L, Zeng M, Fan J, Tarbell JM, Curry FR, Fu BM. Sphingosine-1-phosphate maintains normal vascular permeability by preserving endothelial surface glycocalyx in intact microvessels. Microcirculation. 2016;23(4):301–310. doi: 10.1111/micc.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Zhang L, Zeng M, Fu BM. Sphingosine-1-phosphate reduces adhesion of malignant mammary tumor cells MDA-MB-231 to microvessel walls by protecting endothelial surface glycocalyx. Cell Mol. Biol. 2017;63(4):16–22. doi: 10.14715/cmb/2017.63.4.3. [DOI] [PubMed] [Google Scholar]
- 246.Zhong Y, Goltsche K, Cheng L, Xie F, Meng F, Deng C, et al. Hyaluronic acid-shelled acid-activatable paclitaxel prodrug micelles effectively target and treat CD44-overexpressing human breast tumor xenografts in vivo. Biomaterials. 2016;84:250–261. doi: 10.1016/j.biomaterials.2016.01.049. [DOI] [PubMed] [Google Scholar]
- 247.Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014;34(10):2191–2198. doi: 10.1161/ATVBAHA.114.303422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zullo JA, Fan J, Azar TT, Yen W, Zeng M, Chen J, et al. Exocytosis of endothelial lysosome-related organelles hair-triggers a patchy loss of glycocalyx at the onset of sepsis. Am. J. Pathol. 2016;186(2):248–258. doi: 10.1016/j.ajpath.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]