

SUMMARY
Anaphase-promoting complex/cyclosome (APC/C) is a well-characterized E3 ligase that couples with UBE2C and UBE2S E2s for substrate ubiquitylation by the K11 linkage. Our recent data show that SAG/RBX2/ROC2, a RING component of Cullin-RING E3 ligase, also complexes with these E2s for K11-linked substrate polyubiquitylation. Whether these two E3s cross-talk with each other was previously unknown. Here, we report that SAG competes with APC2 for UBE2C/UBE2S binding to act as a potential endogenous inhibitor of APC/C, thereby regulating the G2-to-M progression. As such, SAG knockdown triggers premature activation of APC/C, leading to mitotic slippage and resistance to anti-microtubule drugs. On the other hand, SAG itself is a substrate of APC/CCDH1 for targeted degradation at the G1 phase. The degradation-resistant mutant of SAG-R98A/L101A accelerates the G1-to-S progression. Our study reveals that the negative cross-talk between SAG and APC/C is likely a mechanism to ensure the fidelity of cell cycle progression.
Graphical Abstract
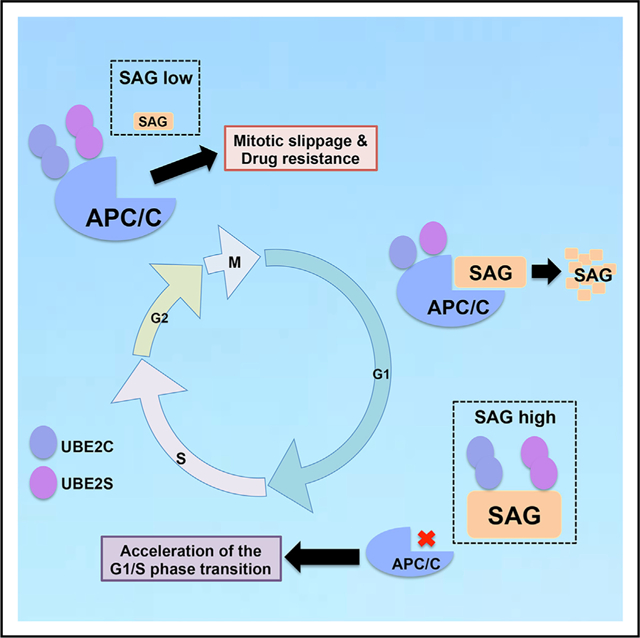
In Brief
Zhang et al. provide a mechanistic insight of how negative cross-talk between E3 ligases SAG and APC/C ensures proper cell cycle progression. SAG knockdown prematurely activates APC/C to promote mitotic progression and trigger anti-microtubule drugs resistance, whereas SAG degradation by APC/CCDH1 mainly occurs in G1 phase for proper G1-to-S transition.
INTRODUCTION
Ubiquitin-mediated proteasomal degradation is an irreversible process catalyzed by a sequential enzymatic reaction by E1 ubiquitin activating enzyme, E2 ubiquitin conjugating enzyme, and E3 ubiquitin ligase, leading to attachment of the ubiquitin to a lysine residue on a substrate protein. Multiple rounds of this reaction cause substrate poly-ubiquitylation, which is then recognized by the 26S proteasome for targeted degradation (Husnjak and Dikic, 2012). In humans, there are two E1 enzymes, about 38 E2 enzymes and over 600 E3 enzymes controlling the ubiquitin modification of thousands of protein substrates (Ciechanover, 2015). The RING-finger-type E3 ligases are the largest E3 ubiquitin ligases, including the CRL (Cullin-RING ligase) and APC/C (anaphase-promoting complex/cyclosome); both are crucial for cell cycle progression and tumorigeneses (Nakayama and Nakayama, 2006; Zhang et al., 2014a).
The CRL consists of four components: cullin, as a scaffold subunit, binds to an adaptor protein and a substrate-recognizing F-box protein at the N terminus and a RING protein at the C terminus (Deshaies, 1999; Zheng et al., 2002). There are two family members of the RING component, namely, RBX1/ROC1 and RBX2/ROC2 (also known as Sensitive to Apoptosis Gene [SAG]) (Duan et al., 1999; Sun et al., 2001), that complex with cullins to form the core ligase activity of CRLs (Zhang and Sun, 2020; Zhao and Sun, 2013) for substrate ubiquitylation, and cullin neddylation fully activates the ligase activity of CRLs (Zhao et al., 2014). Significantly, CRLs are responsible for ubiquitylation of ~20% of cellular proteins doomed for proteasome degradation (Soucy et al., 2009), thus regulating multiple key cellular processes (Nakayama and Nakayama, 2006; Zhao and Sun, 2013).
APC/C E3 contains at least 15 different subunits. Among them, APC2 and APC11 are regarded as core components because the heterodimeric complex of APC2 and APC11 possesses the ubiquitin ligase activity and is sufficient to catalyze the ubiquitylation of substrates (Tang et al., 2001). The activity of APC/C largely depends on the association with the two activator proteins CDC20 and CDH1, which are substrate-recognizing subunits. In general, APC/CCDC20 regulates the initiation of anaphase by primarily targeting securin and cyclins, whereas APC/CCDH1 is activated in late mitosis and throughout the subsequent G1 phase (Peters, 2006; Thornton and Toczyski, 2006).
Previous studies have revealed negative cross-talks between the CRL and APC/C E3s, mainly focusing on mutual degradation of the E3 components. For example, F-box protein SKP2 is subjected to ubiquitylation and degradation by APC/CCDH1 in the G1 phase of cell cycle, which prevents the unscheduled degradation of SCFSkp2substrates, such as p21 and p27, to maintain the G1 state (Bashir et al., 2004; Wei et al., 2004). APC/CCDH1 also promotes ubiquitylation and degradation of the F-box protein NIPA (Nuclear Interaction Partner of Alk kinase) in late mitosis (Klitzing et al., 2011). On the other hand, SCF E3, particularly SCFb-TRCP, is responsible for targeted ubiquitylation and degradation of CDH1 during the S phase (Benmaamar and Pagano, 2005; Fukushima et al., 2012). SCFFBXW7controls CDH1 activity in a cyclin-E-dependent manner, and the loss of FBXW7 leads to an aberrant increase in the levels of both SCFFBXW7 and APCCDH1 substrates (Lau et al., 2013). A recent study showed a mutual degradation of SCFCyclinF and APC/CCDH1 to precisely regulate the S-phase entry (Choudhury et al., 2016). Furthermore, the SCF and APC/C E3s even work together for ubiquitylation and degradation of the S-phase cyclin Clb6 during the transition from G1 to S phase (Wu et al., 2016). However, it is completely unknown whether and how the negative cross-talks also exist between SAG, a CRL E3, and APC/C E3, particularly at the level for E2 competition.
We recently reported that unlike RBX1, which binds to CDC34 or UBCH5C E2s to assemble uniform Lys48-linked ubiquitin polymers on substrates (Petroski and Deshaies, 2005), SAG prefers to bind to UBE2C and UBE2S E2s to promote substrate polyubiquitylation by the K11 linkage (Kuang et al., 2016; Zhou et al., 2017b). Given that APC/C also couples with UBE2C (to initiate) and UBE2S (to extend) to promote polyubiquitylation of a substrate by the K11 linkage (Meyer and Rape, 2014), we hypothesized that SAG and APC/C may negatively regulate each other by competing for UBE2C/2S E2s binding. In this study, we report that SAG and APC2 indeed compete for UBE2C/2S binding to negatively regulate the activity of each other, ensuring a precise control of cell cycle progression from the G2/M to the G1 phase of the next cell cycle. SAG inactivation triggers resistance to anti-microtubule drugs by premature activation of APC/C to induce mitotic slippage. SAG is also a substrate of APC/CCDH1, being degraded mainly in the G1 phase, and a SAG degradation-resistant mutant promoted the G1-to-S progression and cell survival. Our study provides a mechanistic insight of fine regulation of two important E3 ligases to ensure proper cell cycle progression.
RESULTS
SAG Competes with APC/C for Binding with UBE2C or UBE2S
Because UBE2C and UBE2S couple with both APC/C and SAG E3 ligases for K11-linked ubiquitylation (Brown et al., 2016; Kuang et al., 2016; Zhang et al., 2014a), we hypothesized that SAG and APC/C E3 might compete with each other for UBE2C/UBE2S binding. Indeed, immunoprecipitation (IP)-based pull-down performed using antibodies against UBE2C or UBE2S revealed that SAG knockdown significantly enhanced the binding between APC2, a scaffold component of APC/C, and UBE2C (Figures 1A and S1A) or UBE2S (Figures 1B and S1B), whereas ectopic SAG expression reduced their interaction (Figures 1C, 1D, S1C, and S1D). Likewise, SAG binding with UBE2C or UBE2S at the endogenous level was also enhanced upon APC2 knockdown (Figures 1E, 1F, S1E, and S1F). Knockdown of APC11, a RING component of APC/C enhanced the binding of SAG-UBE2C, but not SAG-UBE2S (Figures S1E and S1F). Notably, decreased binding between APC2 and UBE2C/S upon SAG overexpression was largely rescued when either UBE2C or UBE2S was overexpressed (Figures S1G and S1H). Collectively, these data indicated that SAG and APC2 compete with each other for UBE2C/2S binding under the physiological conditions.
Figure 1. SAG Competes with APC/C for UBE2C or UBE2S Binding to Affect APC/C Activity.
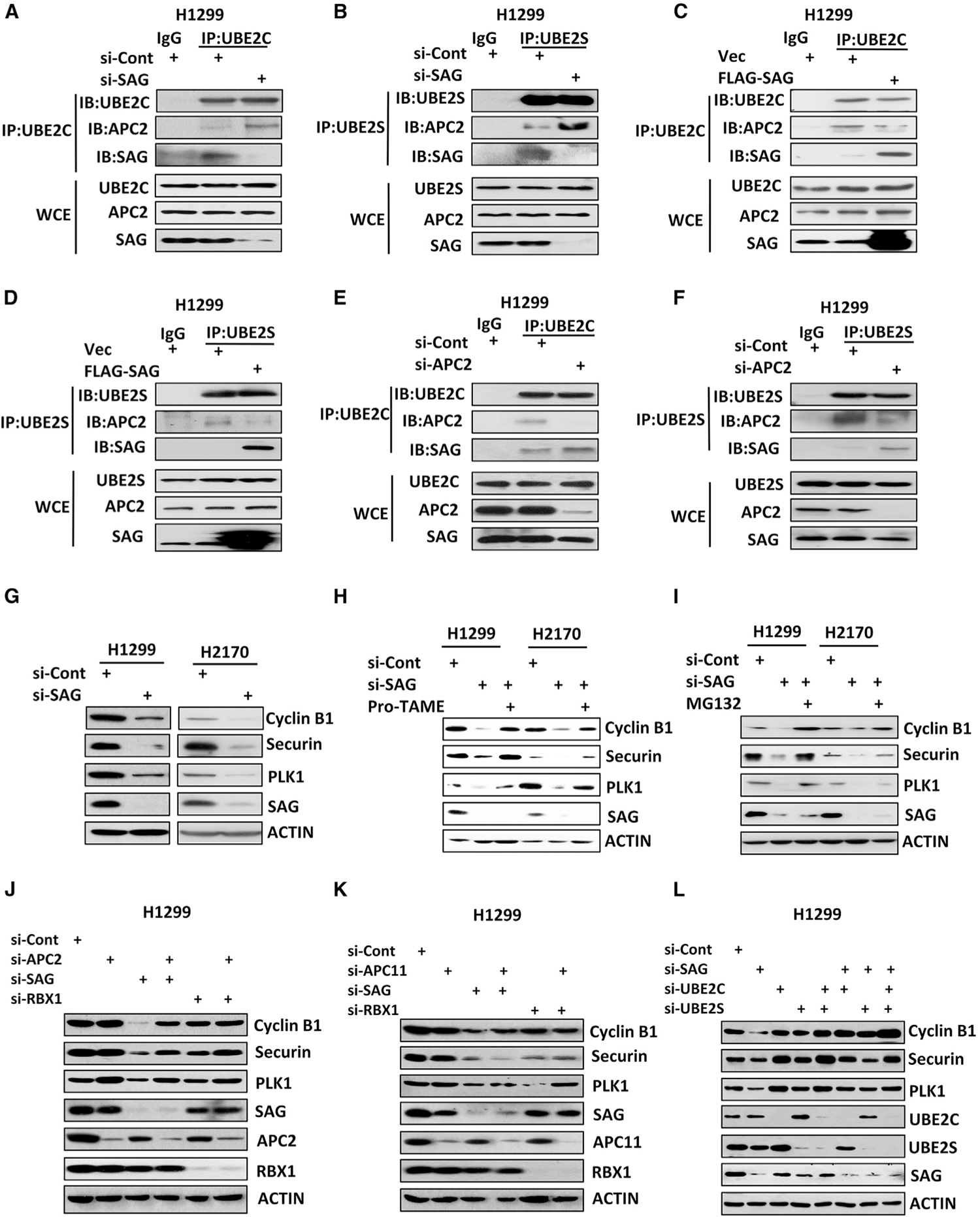
(A–D) H1299 cells were transfected with siRNA targeting SAG or control siCont (A and B) or a plasmid expressing FLAG-tagged SAG or vector control (C and D) for 48 h with last 8-h treatment of 10 μM MG132 before harvesting. Cell lysates were prepared for immunoprecipitation (IP) with immunoglobulin G (IgG) control or antibodies (Abs) against UBE2C (A and C) or UBE2S (B and D), followed by immunoblot (IB) with indicated Abs. WCE, whole-cell extract.
(E and F) H1299 cells were transfected with siRNA targeting APC2 or control siCont for 48 h with last-8 h treatment of 10 μM MG132 before harvesting. Cell lysates were prepared for IP with IgG control or Abs against UBE2C (E) or UBE2S (F), followed by IB with indicated Abs.
(G–I) H1299 or H2170 cells were transfected with siRNA targeting SAG or control siCont for 48 h, followed by 24-h treatment of 12 uM pro-TAME (H) or 10 μM MG132 (I) in indicated samples before harvesting for IB analysis with indicated Abs.
(J–L) H1299 cells were transfected with various siRNA targeting indicated mRNAs, along with control siCont for 48 h. Cell lysates were prepared for IB analysis with indicated Abs.
Also see Figures S1 and S2.
SAG Knockdown Activates APC/C E3 Ligase Activity
APC/C is a large multi-subunit E3 ubiquitin ligase responsible for targeted ubiquitylation and degradation of a variety of cell cycle proteins, particularly those involved in G2/M progression, such as cyclin B1, securin, and PLK1 (Nakayama and Nakayama, 2006). Given that SAG knockdown enhanced the binding of APC2-UBE2C/2S, we next determined if such an enhancement would reflect an enhanced ligase activity of APC/C toward its substrates in unsynchronized cells. Indeed, SAG knockdown significantly reduced the protein levels of APC/C substrates, including cyclin B1, securin, and PLK1 (Figure 1G), whereas SAG ectopic expression slightly increased the protein levels of several APC/C substrates (e.g., cyclin B1 and securin) (Figure S1I). Moreover, when SAG was overexpressed in SAG low expressing cells (A549 and A427), the levels of APC/C substrates (cyclin B1 and securin) were increased more obviously (Figure S1J). Interestingly, manipulation of APC11 had a minimal, if any, effect on APC/C substrates (Figure S1I). We confirmed the reduced levels of APC/C substrates were due to enhanced degradation by APC/C proteasome system because it can be abrogated by both an APC/C inhibitor, pro-TAME (Zeng et al., 2010), and a proteasome inhibitor, MG132 (Figures 1H and 1I).
We further examined whether reduction of APC/C substrates upon SAG knockdown was indeed attributable to enhanced APC/C activity by simultaneous knockdown of APC/C components APC2 or APC11. Again, knockdown of SAG, but not of RBX1, which is incapable of binding with UBE2C/2S (Kuang et al., 2016), reduced the levels of cyclin B1, securin, and PLK1, which is rescued by simultaneous knockdown of APC2 (Figures 1J and S1K), but not of APC11 (Figures 1K and S1L), consistent with the lack of competition between APC11 and SAG for UBE2C/2S binding. Finally, we showed that the effect of SAG knockdown on APC/C substrates was indeed mediated by UBE2C or UBE2S because it can be completely rescued by simultaneous knockdown of either E2s (Figures 1L and S1M). Collectively, these results demonstrated that SAG knockdown could enhance APC/C activity by competing for UBE2C/2S E2 binding.
We next systematically examined whether this SAG-knockdown effect occurs in a cell-cycle-dependent manner. Using the synchronized cells, we found that SAG knockdown enhanced the UBE2C/2S binding with APC2 mainly occurring in the M phase, but not in the G1 phase of the cell cycle (Figures S2A and S2B). Consistently, a greater decrease of APC/C substrates (cyclin B1 and securin) was observed in the M phase cells upon SAG knockdown (Figure S2C). Significantly, the effect of SAG knockdown on APC/C substrates can be rescued by simultaneous knockdown of either E2s or APC2, which also mainly occurred in the M phase (Figures S2D–S2I). We then determined the dynamic interaction between APC2 and UBE2C/2S during cell cycle by monitoring cells from a nocodazole-induced metaphase blockage. We found an enhanced binding between APC2 and UBE2C/2S in SAG knockdown cells upon nocodazole treatment or release up to 1 h, whereas in si-Cont cells, the APC2-UBE2C/2S binding was only seen at 1 h after nocodazole release (Figure 2A), strongly suggesting that the lack of SAG enhanced the binding between APC/C-UBE2C/2S.
Figure 2. SAG Knockdown Prematurely Activates APC/C to Degrade Its Substrates.
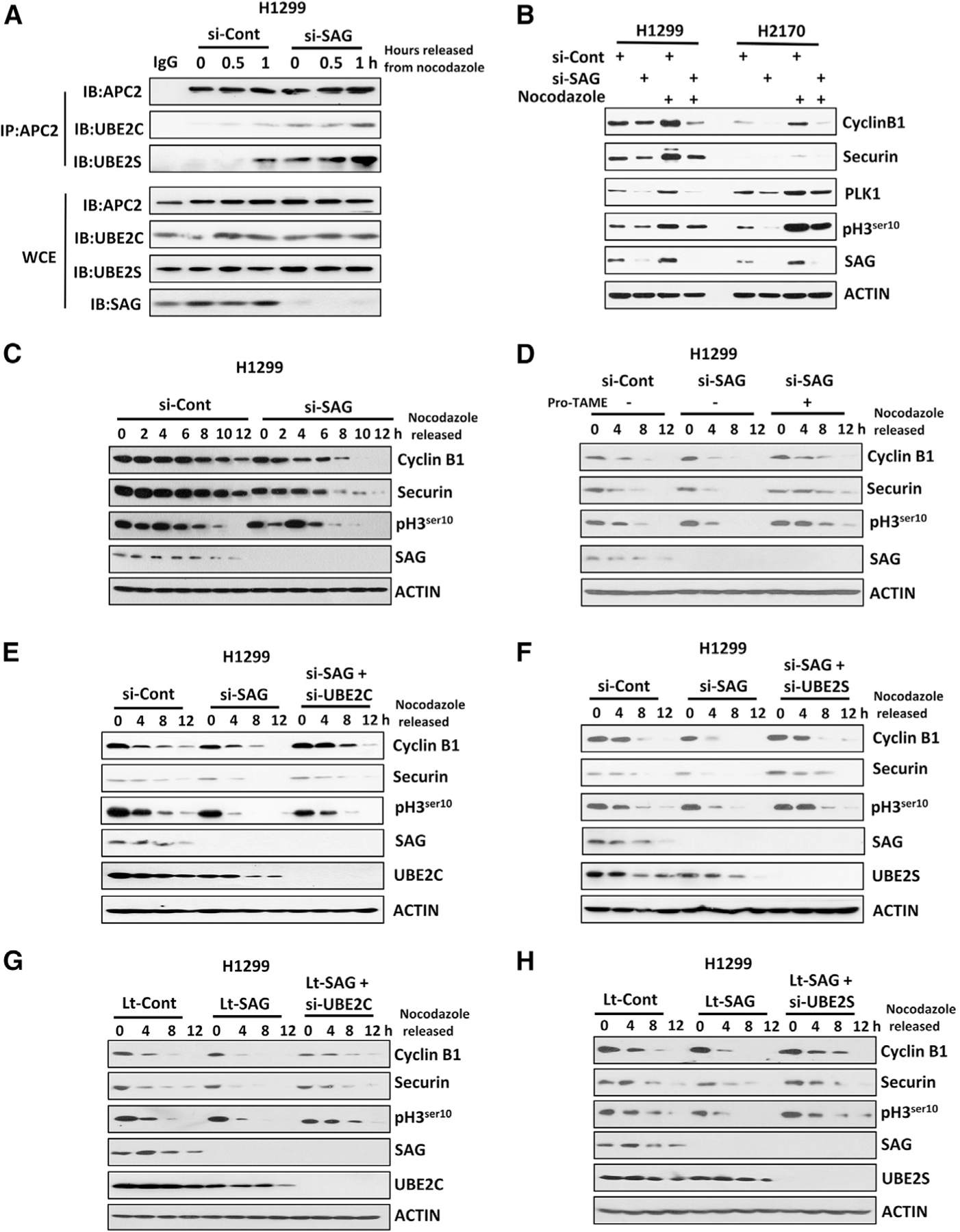
(A) H1299 cells were transfected with siRNA targeting SAG or control siCont for 48 h, then treated with nocodazole for 16 h, and released from nocodazole block for indicated time points. Cell lysates were prepared for IP with IgG control or Ab against APC2, followed by IB with indicated Abs.
(B) H1299 and H2170 cells were transfected with siRNA targeting SAG or control siCont for 48 h. The indicated cells were then treated with nocodazole for 24 h before harvesting for IB analysis with indicated Abs.
(C) H1299 cells were transfected with siRNA targeting SAG or control siCont for 24 h, synchronized at M phase by nocodazole blockage, and then released for indicated time points. Cell lysates were prepared for IB analysis with indicated Abs.
(D) H1299 cells were transfected with siRNA targeting SAG or control siCont for 24 h and synchronized at M phase by nocodazole blockage. Cells were then released in the absence or presence of pro-TAME for indicated time points before harvesting for IB using indicated Abs.
(E and F) H1299 cells were transfected with various siRNA targeting indicated mRNAs, along with control siCont for 24 h and synchronized at M phase by nocodazole blockage. Cells were then released for indicated time points before harvesting for IB using indicated Abs.
(G and H) H1299 cells with lentivirus-mediated SAG knockdown (Lt-SAG) and its vector control (Lt-Cont) were synchronized at the M phase by nocodazole blockage. Cells were then released for indicated time points before harvesting for IB using indicated Abs.
Also see Figure S3.
We next determined whether enhanced E2-E3 binding would activate APC/C E3 ligase activity in synchronized cells. Indeed, SAG knockdown reduced the levels of APC/C substrates, including cyclin B1, securin, and PLK1, as well as pH3ser10, a mitotic biomarker, particularly in nocodazole-treated cells (Figure 2B). SAG knockdown also triggered a faster degradation of APC/C substrates cyclin B1 and securin and an earlier appearance of pH3ser10 upon thymidine release of the S phase blockage (Figure S3A). The opposite effect was seen upon ectopic expression of SAG, although to a lesser extent (Figure S3B). Likewise, the time course experiments showed that after releasing from M phase arrest, SAG-depleted cells exhibited a quicker degradation of cyclin B1 and securin and dephosphorylation of pH3ser10 (Figures 2C and S3C). Importantly, this SAG-knockdown effect can be largely rescued by pro-TAME (Figures 2D and S3D) or by small interfering RNA (siRNA)-based depletion of either UBE2C or UBE2S (Figures 2E, 2F, S3E, and S3F), indicating a causal role played by APC/C and UBE2C/2S. To exclude potential off-target effects of siRNA, we used lentivirus-based sh-SAG vectors targeting different regions of SAG mRNA to repeat these experiments and obtained consistent results (Figures 2G, 2H, and S3G). Collectively, these results demonstrated that SAG knockdown promotes premature activation of APC/C in the M phase to degrade its substrates in a manner largely dependent of UBE2C/2S.
SAG Knockdown Promotes Mitotic Progression
Given that SAG regulates the biochemical activity of APC/C, a prominent E3 ubiquitin ligase that plays an essential role in promoting mitotic progression (Zhang et al., 2014a), we next investigated the biological consequence of APC/C activation upon SAG knockdown. In unsynchronized cells, SAG knockdown reduced the cell population in the G2/M phase with a corresponding increase in the G1 phase. Similar results were observed in nocodazole-treated cells (Figures 3A and S4A), indicating that SAG knockdown promotes mitotic progression and earlier entrance of the G1 phase of next cell cycle. These SAG-knockdown effects were indeed caused by APC/C activation because the population decrease in the G2/M and increase in the G1 phase can be largely rescued by APC2 knockdown or pro-TAME treatment (Figures 3B and S4B) or by knockdown of UBE2C or UBE2S (Figures 3C and S4C).
Figure 3. SAG Knockdown Promotes Mitotic Progression.
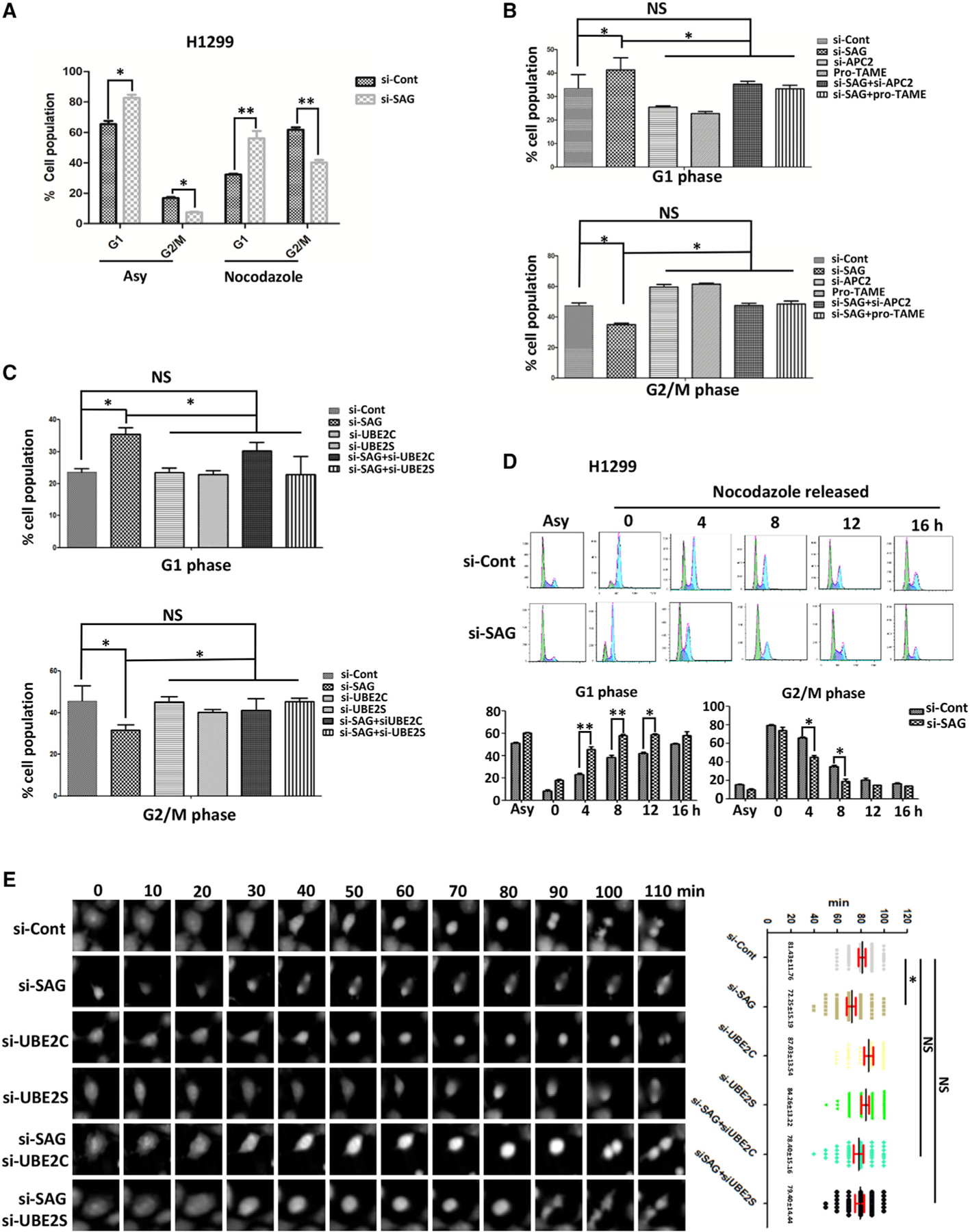
(A) H1299 cells were transfected with siRNA targeting SAG or control siCont for 48 h; cells were then treated with or without nocodazole for 24 h, followed by FACS analysis. The percentage of cell populations at the G1 and G2/M phase were plotted. Shown are mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01.
(B) H1299 cells were transfected with siRNA targeting SAG or APC2 or control siCont for 24 h. Cells were then treated with nocodazole for 48 h in the presence or absence of pro-TAME, followed by FACS analysis. The percentage of cell populations at the G1 or G2/M phase was plotted. Shown are mean ± SEM from three independent experiments. *p < 0.05; NS, not significant.
(C) H1299 cells were transfected with various siRNA targeting indicated mRNAs, along with control siCont for 24 h. Cells were then treated with nocodazole for 48 h, followed by FACS analysis. The percentage of cell populations at the G1 and G2/M phases were plotted. Shown are mean ± SEM from three independent experiments. *p < 0.05; NS, not significant.
(D) H1299 cells were transfected with siRNA targeting SAG or control siCont for 24 h; cells were then blocked at the M phase by nocodazole treatment, and then released from the M phase for indicated time points for FACS analysis. The percentage of cell populations at the G1 and G2/M phases were plotted. Shown are mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01.
(E) H1299 cells were transfected with various siRNA targeting indicated mRNAs, along with control siCont for 24 h, followed by G2 arrest by RO3306 for 24 h. Cells were then released from G2/M phase at the indicated time points when the photos of individual cells were taken (left). The time required for the separation into two daughter cells (mitotic time) was recorded and plotted (right). The results were derived from three independent experiments. *p < 0.01; NS, not significant.
Also see Figure S4.
We next monitored dynamic changes of the G2/M and G1 population after release from mitotic arrest by nocodazole for various time points up to 16 h. SAG knockdown triggered a significant decrease of the M cell population and a corresponding increase of the G1 cell population at 4 and 8 h after release, respectively (Figures 3D and S4D). Moreover, a time-lapse video of transfected cells after releasing from G2 arrest by R03306 treatment revealed that the overall mitotic time was significantly shortened in SAG knockdown cells compared to the control cells (81.43 ± 11.76 min versus 72.25 ± 15.19 min) (Figure 3E). Again, this SAG-knockdown effect can be largely rescued by simultaneous depletion of either UBE2C or UBE2S (Figure 3E). Moreover, we used yet another siRNA oligonucleotide (siSAG2) targeting a different region of SAG mRNA in H1299 cells, and we confirmed that SAG-knockdown cells underwent a shorter overall mitotic time after releasing from G2 arrest, which could be completely rescued by simultaneous depletion of either E2s (Figure S4E). Taken together, these results demonstrated that SAG knockdown prematurely activates APC/C activity, leading to accelerated mitotic progression.
SAG Knockdown Triggers Mitotic Slippage and Confers Resistance to Apoptosis Induced by Anti-microtubule Drugs
It was previously reported that continued exposure of cells to anti-microtubule drugs would trigger mitotic slippage even when the spindle assembly checkpoint (SAC) is not satisfied if cyclin B1 is progressively degraded by APC/C (Brito and Rieder, 2006). Given that APC/C is prematurely activated upon SAG knockdown, we investigated the possibility of mitotic slippage by continuous exposure to nocodazole. Indeed, SAG knockdown triggered premature degradation of cyclin B1 and reduction of mitotic marker pH3ser10 when cells were continuously exposed to nocodazole for various time points up to 24 h (Figures 4A and S5A), implying a the presence of a biochemical process that triggers mitotic slippage. This SAG-knockdown effect was largely rescued by pro-TAME (Figures 4A and S5A), indicating a causal role of APC/C. Biologically, SAG knockdown caused a significant increase in bi-nucleated cells upon the continuous exposure of nocodazole (Figures 4B and S5B), strongly indicating the induction of mitotic slippage, which gave rise to undivided tetraploid cells due to premature mitotic exit without chromosome segregation. Furthermore, fluorescence-activated cell sorting (FACS) analysis confirmed that SAG knockdown caused a 2- to 3-fold increase in polyploidy cell population (>4N DNA content), coinciding with decreased apoptotic cell population (sub-G1 population) after treatment with nocodazole/taxol for various time periods up to 96 h (Figures 4C and S5C). Apoptosis reduction in SAG knockdown cells upon the treatment of nocodazole or taxol was further confirmed by decreased levels of cleaved Poly (ADP-ribose) polymerase (PARP) and caspase-3, two well-established apoptotic biochemical markers (Figures 4D and S5D–S5G). Significantly, the increased mitotic slippage and decreased apoptosis induction in SAG knockdown cells after continuous exposure to nocodazole were largely rescued by pro-TAME or simultaneous knockdown of UBE2C or UBE2S, as evidenced by a reduced population of polyploidy cells and increased sub-G1 population in SAG-silencing cells (Figures 4E, 4F, S6A, and S6B), further indicating a causal role of UBE2C/2S-APC/C in the process.
Figure 4. SAG Knockdown Triggers Mitotic Slippage and Confers Apoptosis Resistance.
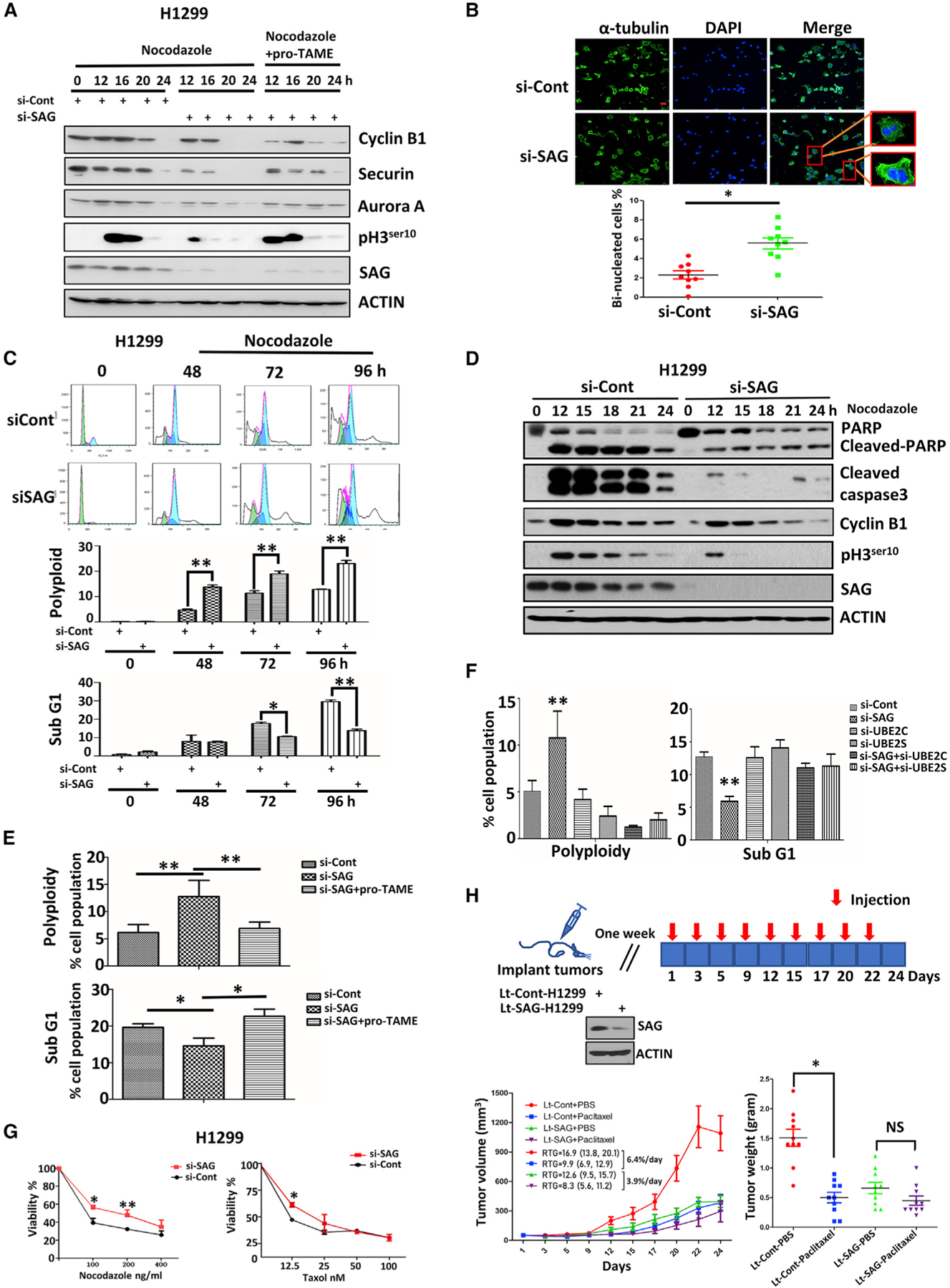
(A) H1299 cells were transfected with siRNA targeting SAG or control siCont for 24 h. Cells were then exposed to nocodazole in the presence or absence of pro-TAME for the indicated time points and then harvested for IB analysis with indicated Abs.
(B) H1299 cells were transfected with siRNA targeting SAG or control siCont for 48 h, followed by nocodazole treatment for 24 h. Cells were then stained with α-tubulin or DAPI and examined under a confocal microscope for photos. Representative images of binucleated cells are shown (top), and the percentage of binucleated cells were counted from at least 5 fields and plotted (bottom). The results were derived from three independent experiments. *p < 0.05. Scale bar, 20 μm.
(C) H1299 cells were transfected with siRNA targeting SAG or control siCont for 48 h; then cells were treated with continuous exposure of nocodazole for up to 96 h before being subjected to FACS analysis (left). The percentage of polyploidy and sub-G1 populations were recorded and plotted (right). Shown are mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01.
(D) H1299 cells were transfected with siRNA targeting SAG or control siCont for 48 h; then cells were treated by the continuous exposure of nocodazole for indicated times. Cell lysates were prepared for IB analysis with indicated Abs.
(E and F) H1299 cells were transfected with siRNA targeting SAG or control siCont (E) or various siRNAs targeting mRNAs encoding indicated proteins (F) for 24 h. Cells were then treated with nocodazole in the presence or absence of pro-TAME (as indicated in E) for 48 h and then subjected to FACS analysis. The percentages of polyploidy and sub-G1 populations were recorded and plotted. Shown are mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01.
(G) H1299 cells were transfected with siRNA targeting SAG or control siCont for 48 h; then cells were treated with indicated concentrations of nocodazole or taxol, followed by ATP-lite assay. Shown are mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01.
(H) Three million of Lt-Cont-H1299 and Lt-SAG-H1299 cells were inoculated subcutaneously into both flanks of SCID mice. The mice were randomized and the treatment started at 7 days after inoculation. Animals were dosed as indicated for 3 weeks. The growth of tumors was measured each time before injection. Tumors were harvested at the end and weighed with results plotted. RTG, rate of tumor growth; *p < 0.05; NS, not significant.
Also see Figures S5 and S6.
A previous study showed that in response to anti-microtubule drugs, such as nocodazole and taxol, a portion of cancer cells escaped from mitotic-arrest-induced apoptosis by undergoing mitotic slippage, thereby causing drug resistance (Gascoigne and Taylor, 2008). Given SAG knockdown triggered mitotic slippage and apoptosis protection, we finally determined whether these phenotypical changes led to drug resistance. Indeed, SAG knockdown conferred resistance to nocodazole/taxol, as measured by both in vitro cell proliferation and clonogenic survival assays (Figures 4G, S6C, and S6D). We further extended this cell culture observation to an in vivo xenograft tumor model. Specifically, H1299 cells with lentivirus-mediated SAG knockdown (Lt-SAG-H1299) and its vector control (Lt-Cont-H1299) were subcutaneously injected into two flanking sites of severe combined immunodeficiency (SCID) mice, followed by treatment with PBS or paclitaxel (Figure 4H). Consistent with our previous results in pancreatic cancer cells (Jia et al., 2010), SAG knockdown dramatically suppressed tumor growth in vivo (Lt-Cont-PBS versus Lt-SAG-PBS) (Figures 4H and S4H). Interestingly, the rate of tumor growth in Lt-Cont groups was significantly inhibited by paclitaxel (Lt-Cont-PBS versus Lt-Cont-paclitaxel), indicating the cells were paclitaxel sensitive. In contrast, paclitaxel only had minimal, if any, effect on the rate of tumor growth in Lt-SAG groups (Lt-SAG-PBS versus Lt-SAG-paclitaxel) (Figures 4H and S6E). The statistical analysis revealed that paclitaxel reduced the tumor growth rate by 6.4%/ day or 3.9%/day in the Lt-Cont group or in the Lt-SAG group, respectively, reflecting that suppression of the tumor growth by paclitaxel in the Lt-SAG group is 39.1% less effective than that in the Lt-Cont group (p < 0.001) (Figure 4H), which indicated paclitaxel resistance upon SAG depletion in this in vivo tumor xenograft model. Finally, the dosage of paclitaxel used here was non-toxic to animals, as judged by a minimal loss of body weight (Figure S6E). Taken together, our results show that, under continuous exposure to anti-microtubule drugs, SAG depletion causes mitotic slippage, apoptosis protection, and eventually drug resistance both in in vitro cell culture and in vivo xenograft tumor models (Figure S6F).
SAG Is Subjected to Cell Cycle Fluctuation in a CDH1-Dependent Manner
Given that SAG negatively regulated the APC/C activity and that the SAG levels progressively decreased after cells releasing from nocodazole blockage (Figures 2C and S3C), we wondered whether SAG itself is subjected to cell cycle regulation. To this end, we arrested cells at the G0/G1 stage by serum starvation and then released cells to the cell cycle by serum addition and collected cells at various time points afterward. The levels of SAG, but not RBX1, indeed fluctuated during cell cycle progression, with the lowest level in the G1 and highest in the S (Figures 5A and 5B). Similar SAG fluctuation was also observed in cells synchronized and released by nocodazole block (Figures S7A and S7B). A similar decrease was observed in two APC/C substrates, cyclin B1 and Aurora A, with a corresponding increase of CDH1 (Figure 5A) in the second cycle of the G1, suggesting that SAG could be a substrate of APC/CCDH1.
Figure 5. SAG Binds to CDH1 at the G1 Phase and Acts as a Novel Substrate of CDH1 for Ubiquitylation and Degradation.
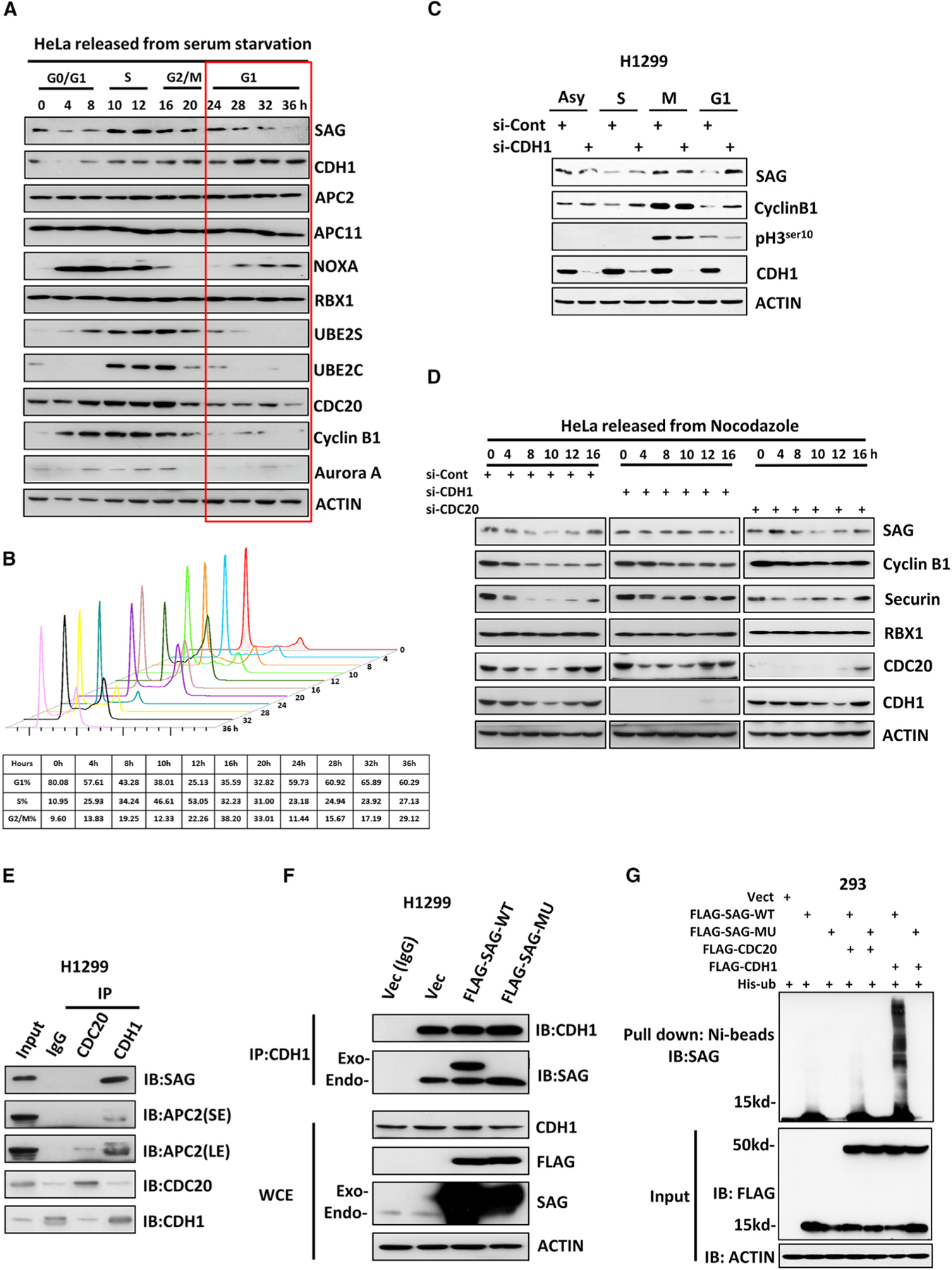
(A and B) HeLa cells were serum starved for 24 h and then released by serum addition. Cells were harvested at indicated time points thereafter and subjected to IB analysis (A) or FACS analysis (B).
(C) H1299 cells were transfected with siRNA targeting CDH1 or control siCont for 24 h and then a
rested at the different phases of cell cycle before being harvested for IB analysis with indicated Abs.
(D) HeLa cells were transfected with siRNA targeting CDC20 or CDH1, along with control siCont for 24 h; then cells were synchronized at the M phase by nocodazole block and released for indicated time points. Cells were harvested for IB analysis with indicated Abs.
(E) H1299 cells were pretreated with 10 μM MG132 for 8 h before harvesting for IP assay using anti-CDC20 or anti-CDH1 Abs, along with IgG control, followed by IB analysis with indicated Abs.
(F) H1299 cells were transfected with a plasmid expressing FLAG-tagged SAG-WT or SAG-MU, along with the vector control for 48 h with the last 8-h treatment of 10 μM MG132 before harvesting. Cell lysates were prepared for IP with IgG control or anti-CDH1 Ab (D), followed by IB analysis with indicated Abs.
(G) The 293 cells were transfected with the indicated combination of plasmids for 48 h. In the last 8 h, 10 μM MG132 was added. Cell lysates were prepared for Ni-bead pull down in 8 M urea, and washed beads were subjected to PAGE, blotted with anti-SAG Ab.
Also see Figures S7 and S8.
We then determined whether an inverse relationship exists between CDH1 and SAG and found that CDH1 knockdown indeed caused SAG accumulation, mainly at the G1 phase (Figures 5C and S7C). SAG fluctuation after nocodazole arrest/release was completely abrogated upon knockdown of CDH1, but not of CDC20 (Figures 5D and S7D). Furthermore, SAG levels were also gradually decreased upon continuous exposure to nocodazole for 20–24 h (Figures 4A and S5A), which appeared to be mediated by APC/CCDH1 because CDH1 knockdown stabilized the SAG levels at these late time points (Figures S7E and S7F). Thus, it appeared that the SAG level is controlled by APC/CCDH1 and that SAG is stabilized in CDH1-depleted cells at the G1 phase of cell cycle or upon mitotic slippage to the G1 phase.
CDH1 Promotes SAG Ubiquitylation in a D-Box-Dependent Manner
To define the molecular basis of SAG degradation by CDH1, we searched the SAG protein sequence and identified an evolutionarily conserved D-box (RXXLXXXXD/E/D), which is known to be a substrate recognition motif of CDH1 or CDC20 (Harper et al., 2002; He et al., 2013; Figure S8A). The subsequent IP-based pull-down assay showed that the endogenous CDH1, but not CDC20, complexed with endogenous SAG (Figures 5E and S8B). Importantly, the CDH1-SAG binding can only be detected in cells at the G1 phase after releasing from nocodazole arrest (Figures S8C and S8D). To determine whether the CDH1-SAG binding is dependent of the D-box motif on SAG, we generated a R98A/L101A double mutant to abrogate key residues of Arg-98 and Leu-101 in the D-box motif (SAG-MU) (Figure S8A). Notably, the CDH1 antibody was able to pull down endogenous SAG, as well as exogenous wild-type (WT) SAG, but not SAG-MU (Figure 5F), indicating a D-box-dependent binding between CDH1 and SAG. A reciprocal coIP assay confirmed that WT SAG, but not SAG-MU, bound with CDH1, although SAG-MU was able to bind to both E2s (Figure S8E). Furthermore, compared with WT-SAG, ectopically expressed SAG-MU was much more stable in cells releasing from G1 phase blocked by palbociclib (Figure S8F), suggesting that this D-box SAG mutant is largely resistant to CDH1-mediated destruction at the G1 phase of the cell cycle. Finally, we performed an in vivo ubiquitylation assay and showed that ectopically expressed CDH1, but not CDC20, significantly promoted polyubiquitylation of SAG-WT, but not SAG-MU (Figure 5G). Taken together, these results showed that SAG is a potential substrate of APC/CCDH1 E3 ligase complex, subjected to its ubiquitylation and degradation at the G1 phase in a manner dependent of the D-box motif (Figure S8G).
SAG Mutant Promotes the G1-to-S Transition and Cell Survival
APC/CCDH1represents a major class of ubiquitin ligase whose activity is thought to regulate primarily at the G1-to-S transition. Cells with ectopic expression of a non-phosphorylatable CDH1 mutant delayed the entry into the S phase (Pagano et al., 1992), whereas CDH1 depletion by siRNA or neutralizing antibody promoted the G1-to-S transition (Wei et al., 2004). We therefore determined whether SAG manipulation would affect the G1-to-S transition by cross-talking with APC/CCDH1 by monitoring the G1-to-S transition in cells released from palbociclib-induced G1 arrest. Indeed, ectopic expression of SAG, especially SAG-MU, triggered an earlier S phase entry. Specifically, the FACS profiling showed that compared to the vector control, WT-SAG transfection decreased the G1 population from 71.74% to 63.47% (8 h) or 60.22% to 52.83% (12 h) and increased the S population from 22.66% to 29.19% (8 h) or 34.46% to 41.63% (12 h), respectively. An even bigger decrease in the G1 (71.74% to 60.87% [8 h] or 60.22% to 49.47% [12 h]) and an increase in the S population (22.66% to 32.16% [8 h] or 34.46% to 48.13% [12 h]) were observed upon SAG-MU transfection (Figure 6A). Biochemically, increased levels of cyclin A and decreased levels of p27 were observed upon transfection of WT-SAG or SAG-MU (Figures 6B, 6C, S9A, and S9B). A similar promotion of the G1-to-S progression by SAG transfection was also observed in nocodazole released cells (Figure S9C). We reasoned that this effect was likely due to SAG-mediated inactivation of CDH1 because the binding of CDH1 with UBE2C/S was dramatically reduced upon SAG transfection (Figure 6D). To determine the causal role of CDH1, we simultaneously co-transfected CDH1 with SAG-WT or SAG-MU. Indeed, CDH1 co-transfection rescued the effect on both accelerated G1-to-S progression and altered levels of cyclin A and p27, which was induced only by SAG-WT, but not by degradation-resistant SAG-MU (Figures 6A–6C, S9A, and S9B). Consistently, siRNA-based knockdown of endogenous SAG triggered an opposite effect, as evidenced by reduced cyclin A and increased p27 levels, which was accompanied by higher G1 and lower S populations (Figures S9D–S9F). Taken together, these results strongly suggested that ectopic expression of SAG, particularly degradation-resistant SAG-MU, impairs the APC/CCDH1 activity in the G1 phase by competitively binding with UBE2C/2S, thereby promoting the G1-to-S progression.
Figure 6. SAG Degradation-Resistant Mutant Promotes the G1-to-S Transition and Cell Survival.
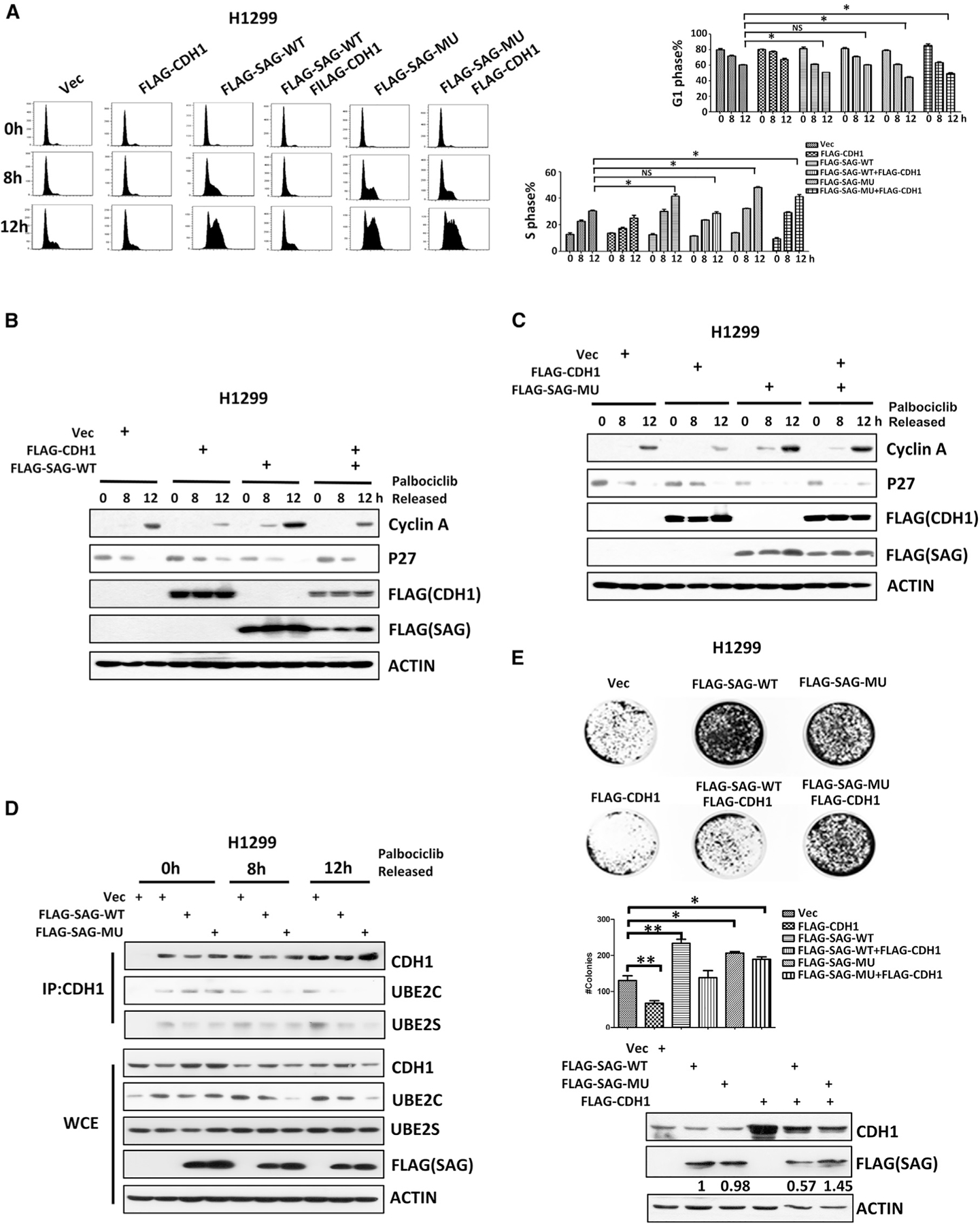
(A) H1299 cells were transfected with indicated plasmids for 24 h. Cells were then arrested at the G1 phase by palbociclib and harvested at 8 or 12 h after release for FACS analysis (left). The percentage of the cell population at the G1 or S phase was plotted (right). Shown is mean ± SEM from three independent experiments. *p < 0.05; NS, not significant.
(B and C) H1299 cells were transfected with various indicated plasmids for 24 h, synchronized at G1 phase by palbociclib block, then released for indicated time points, and followed by IB analysis with indicated Ab.
(D) H1299 cells were transfected with indicated plasmids for 24 h and released from palbociclib block for indicated times. Cell lysates were prepared for IP with anti-CDH1 Ab, followed by IB with indicated Abs.
(E) H1299 cells expressing the indicated plasmids were subjected to clonogenic survival assays. The plates were stained and photographed (top). The colonies with greater than 50 cells were counted and plotted (bottom). Shown are mean ± SEM from three independent experiments. *p < 0.05; **p < 0.01.
Also see Figure S9.
Previous studies have shown that SAG has oncogenic activity, whereas CDH1 is likely to be a tumor suppressor (García-Higuera et al., 2008; Jia et al., 2010; Sun and Li, 2013). We, therefore, determined the biological consequence of the SAG-CDH1 cross-talk by performing a clonogenic survival assay in lung cancer cells. Indeed, transfection of SAG (either WT or MU) promoted clonogenic survival, whereas transfection of CDH1 suppressed it. Significantly, CDH1 co-transfection rescued the effect of SAG-WT, but not of SAG-MU (Figure 6E). Thus, the SAG mediated pro-proliferative effect in lung cancer cells was controlled, at least in part, by APC/CCDH1, which is abrogated by a SAG-MU mutant resistant to CDH1 binding and degradation.
DISCUSSION
In this study, we report an interesting and important finding with mechanistic elucidation that two major E3 ubiquitin ligases, SAG E3 and APC/C, negatively cross-talk with each other in the regulation of cell cycle progression, mitotic slippage, and drug resistance by competing for the same E2s. Specifically, SAG, on one hand, inhibits the APC/C activity most likely by competing with APC/C for UBE2C/2S binding. On the other hand, SAG itself is a substrate of APC/C for destruction at the G1 phase through a D-box-dependent ubiquitylation. Biologically, our finding revealed few previously unrealized functions and regulations of SAG. First, SAG is a critical cell cycle regulator by inhibiting APC/C activity; second, SAG with oncogenic functions is subjected to degradation by tumor suppressive APC/CCDH1 E3; third, SAG, which was previously identified as an anti-apoptotic protein (Sun and Li, 2013), can also act as a pro-apoptotic protein at the M phase because SAG knockdown confers lung cancer cells resistance to anti-microtubule chemo-drugs by premature activation of APC/C to trigger mitotic slippage.
UBE2C and UBE2S are two canonical E2s that are known to couple with APC/C for poly-ubiquitination of the substrates by the K11 linkage (Meyer and Rape, 2014). Our recent studies showed that SAG E3 is capable of doing the same by complexing with UBE2C/2S (Kuang et al., 2016; Zhou et al., 2017b). In the present study, we revealed that these two E3s would negatively regulate each other by competing for E2s binding. Specifically, SAG and APC2 compete with each other for UBE2C/2S binding under the physiological conditions. As a result, SAG knockdown frees up UBE2C/2S for enhancing APC2 binding, leading to premature activation of APC/C. Several previous studies have demonstrated that APC/C ligase is essential for the orderly mitotic progression, whereas overexpression of UBE2C or UBE2S promotes mitosis and mitotic exit (Cardozo and Pagano, 2004; Fujioka et al., 2018; Garnett et al., 2009; Peters, 2006). We found that SAG knockdown indeed mimicked the effect of UBE2C/2S overexpression. At the biochemical levels, SAG knockdown reduced the levels of APC/C substrates, particularly those involved in the G2-to-M transition and mitotic progression, including cyclin B1, securin, and PLK1, due to enhanced degradation. At biological levels, SAG knockdown accelerated mitosis progression and triggered mitotic slippage, as evidenced by the decrease or increase of the M or G1 phase populations, respectively, after releasing from the M phase arrest, along with an increased population of polyploidy upon continuous exposure of nocodazole/taxol. Both biochemical and biological alterations can be largely rescued by simultaneous knockdown of UBE2C/ 2S or by treatment with a APC/C inhibitor, pro-TAME, indicating a causal role of the UBE2C/2S-APC/C axis in the process. However, we cannot completely exclude the possibility that reduced levels of APC/C substrates upon SAG depletion could be affected by other cell-cycle-related factors, in addition to the loss of UBE2C/S competition.
Anti-microtubule drugs are extensively used in the treatment of several types of human cancers (Jordan and Wilson, 2004) by inducing a prolonged mitotic arrest in the presence of activated SAC and therefore inducing cell apoptosis to kill cancer cells (Bekier et al., 2009). However, a small proportion of cancer cells adapted to the checkpoint and triggered drug resistance by premature mitotic exit due to rapid mitotic slippage (Huang et al., 2009), which has occurred following the proteolysis of cyclinB1 mediated by APC/C even when SAC is not satisfied (Brito and Rieder, 2006). The cell fate in response to anti-microtubule chemo-drugs is dictated by two competing networks, namely, mitotic slippage and death in mitosis. Specifically, if the cyclin B1 degradation signal is activated first to reduce cyclin B1 levels below the mitotic exit threshold, cells would undergo the slippage. Otherwise, cells undergo apoptosis in mitosis if the death threshold is breached first (Gascoigne and Taylor, 2008). Although some slippage cells are doomed to die in the subsequent cell cycles, contributing to enhanced cytotoxicity of antimitotic drugs (Giovinazzi et al., 2013), there are cells, which acquired from disordered genome by slippage, that survive and lead to drug resistance (Mittal et al., 2017; Zhang et al., 2014b). Inhibition of mitotic slippage by inactivation of APC/C by RNAi, Apcin, or pro-TAME may increase the sensitivity of tumor cells to anti-microtubule agents (Huang et al., 2009; Raab et al., 2019; Sackton et al., 2014). In this study, we found that cells upon SAG knockdown had reduced levels of mitotic-regulation proteins, such as cyclinB1 and Aurora A, an observation similar to cells with developed paclitaxel resistance (Chong et al., 2018). Consequently, these SAG knockdown cells become resistance to apoptosis induced by nocodazole/taxol. The fact that the phenotypes of mitotic slippage and apoptosis resistance can be abrogated by UBE2C/2S knockdown or pro-TAME treatment strongly indicated that increased APC/C-UBE2C/2S binding and subsequent APC/C activation play a causal role to trigger mitotic slippage by accelerating cyclin B1 proteolysis upon SAG depletion. We have previously demonstrated that SAG could couple with UBE2F E2 to induce CUL5 neddylation, leading to activation of CRL5 E3 to promote NOXA poly-ubiquitylation and proteasomal degradation, thus protecting cancer cells from apoptosis and promoting cell growth (Jia et al., 2010; Zhou et al., 2017a). In this study, we showed yet another characteristic of SAG, in which SAG knockdown leads to taxol resistance by enhancing APC/C-mediated mitotic slippage. In this sense, targeting SAG may eventually trigger the resistance to antimitotic drugs, which is an unfavorable condition for anti-cancer therapy by using this class of agents.
The ordered dynamic regulation of APC/C activity is important for cell cycle progression in the time between metaphase and the end of the next G1 phase. Although APC/CCDC20 is mainly active during early mitosis, APC/CCDH1 is activated in anaphase and remains active during G1 phase to ensure a stable G1 environment by continuously degrading a variety of proteins involved in DNA replication and mitosis (Bassermann et al., 2014; Peters, 2002; Zhang et al., 2014a), as well as the G1-to-S transition promoting factor, such as SKP2 (Wei et al., 2004). Our previous studies have shown that SAG, under overexpression conditions, promotes the G1-to-S promotion by promoting p27 degradation (Duan et al., 2001; He et al., 2008). Here, we showed that SAG levels fluctuate during the cell cycle, with the high levels at the M phase and the lowest levels at the G1 phase. The biological implications are that high SAG levels are needed in the M phase to keep APC/C activity under the check for proper mitotic progression, whereas low SAG levels in the G1 phase are controlled by APC/CCDH1, which is known to be active in the G1 phase (Kramer et al., 2000) to ensure proper G1-to-S progression when cells are ready. We further defined that APC/CCDH1-mediated SAG degradation requires a consensus D box sequence on SAG and the mutation on the D box renders SAG resistance to CDH1 binding and subsequent ubiquitylation and degradation. Biologically, this degradation-resistant SAG mutant promotes the G1-to-S transition by inactivating APC/CCDH1 to confer cell survival. Previous studies have shown that to allow cells entry into S phase, APC/CCDH1 is inactivated by CDH1 phosphorylation by cyclin A/Cdk2 or c-Jun N-terminal kinase (JNK) by Emil binding and by its own degradation in late G1 (Bassermann et al., 2014; Listovsky et al., 2004). Our study shown here indicates that APC/CCDH1 can also be inactivated by SAG by competing for UBE2C/2S binding. Taken together, our study further revealed the important role of APC/CCDH1 in regulation of the G1-to-S transition by promoting ubiquitylation of (1) SKP2 as previously reported (Wei et al., 2004) and of (2) SAG in this study, which both allow p27 accumulation for G1 arrest (Duan et al., 2001; He et al., 2008; Kossatz et al., 2004).
How could SAG be a substrate of APC/CCDH1, if it impairs the binding of E2s to APC? Given that SAG expression fluctuates during cell cycle progression, with the lowest level at the G1 phase, and CDH1 knockdown caused SAG accumulation mainly at the G1 phase, SAG is likely targeted by APC/CCDH1 at the G1 phase. It is reasonable to propose that the competition between SAG and APC2 for UBE2C/S binding is a dynamic process. At the G1 phase, where CDH1 is expressed at the highest level (Figure 5A), APC/CCDH1 would be activated to trigger initial SAG degradation, which would then minimize SAG-E2 competition to further activate APC/CCDH1, thus establishing a feed-forward loop to ensure full activity of APC/CCDH1. Furthermore, we indeed found a D box in the SAG protein sequence (Figure S8A), which is required for CDH1 binding and for SAG ubiquitylation by APC/CCDH1 (Figures 5F and 5G), thus providing the structure basis. On the other hand, SAG binds to CDH1 and UBE2C/S both by its RING domain, and it is likely that a dynamic competition also exists between CDH1 and UBE2C/S for SAG binding. Again, at the G1 phase of the cell cycle, when CDH1 levels are relatively higher, CDH1 “wins” the competition for SAG binding with UBE2C/2S and subsequently promotes SAG ubiquitylation and degradation to reduce SAG levels. Taken together, our results suggest that SAG by competitive binding with UBE2C/S E2 acts as a dual molecule of being either an inhibitor or a substrate of APC/C in a cell-cycle-dependent manner to ensure precise cell cycle progression. Finally, we have previously shown that SAG is a substrate of NEDD4 E3 ligase under physiological conditions (Zhou et al., 2014). Identification of APC/CCDH1 as a new SAG E3 in controlling the G1-to-S transition further suggested that SAG is a biologically significant protein that demands multiple E3s to precisely regulate its levels under various physiological and stressed conditions.
In summary, our study fits the following working model. Under normal growth conditions, CDH1 binds to SAG at the G1 phase to promote SAG ubiquitylation and degradation for ensuring the G1-to-S transition when cells are ready; the physiological level of SAG will also ensure proper APC/C activation for proper mitotic progression. Under a condition of high SAG (e.g., overexpression in cancer cells), SAG binds to UBE2C/2S to inactivate CDH1 to accelerate the G1-to-S transition for enhanced proliferation. The high level of SAG would also, in theory, inactivates APC/C to cause G2/M arrest. However, such an effect is moderate (Figure S3B), likely due to the fact that UBE2C/2S levels are at the highest at the G2/M phase, and when the UBE2C/2S are saturated, the effect of SAG (due to competition of UBE2C/2S) would be minimized. Thus, the major effect of SAG overexpression would be at the level to promote the G1-to-S transition. Under a condition of low SAG (e.g., SAG inactivation by drug or siRNA), APC/C is prematurely activated to promote mitotic progression and slippage, leading to drug resistance (Figure 7).
Figure 7. Working Model.
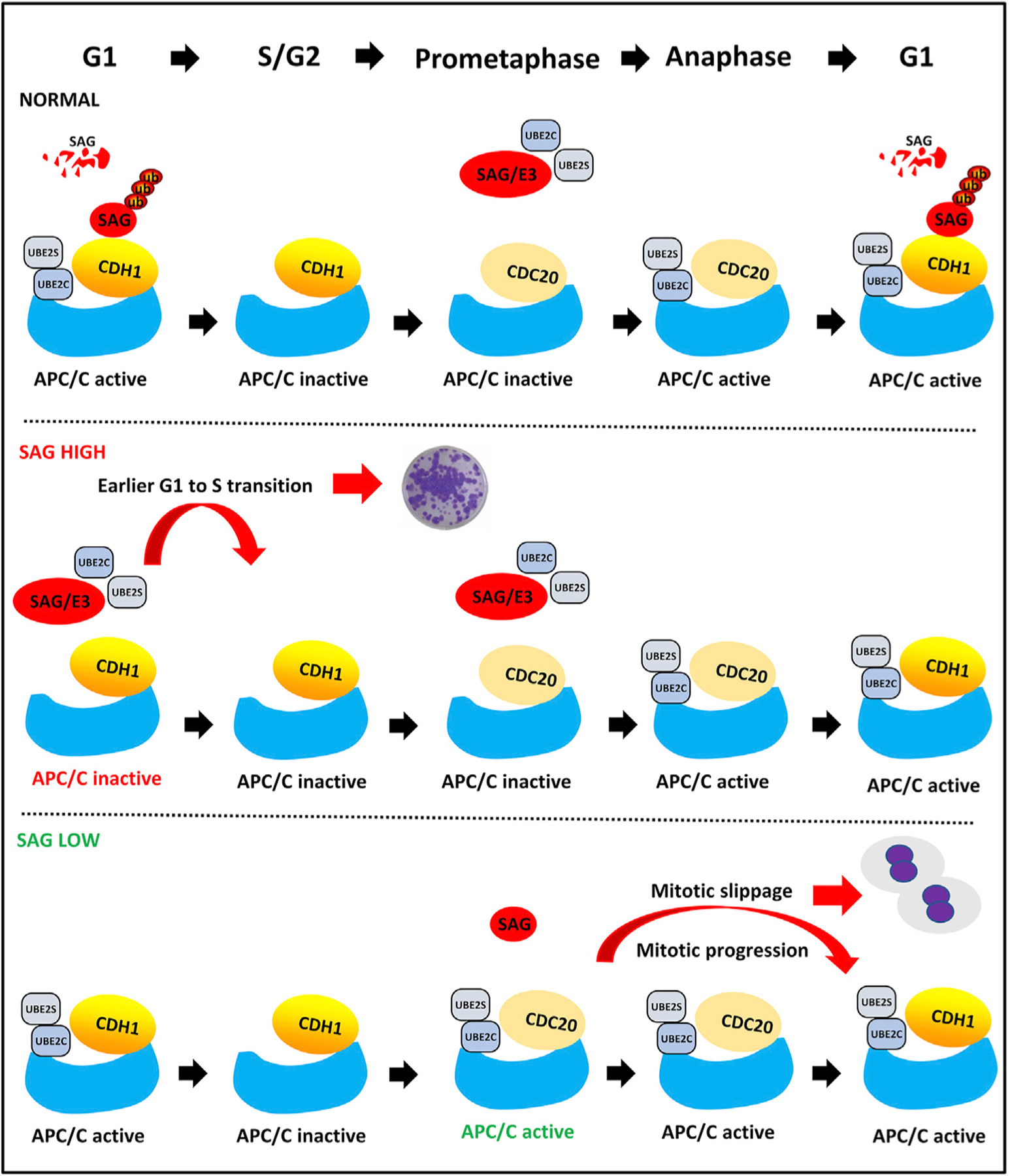
Under normal conditions (normal), SAG is degraded by CDH1 at the G1 phase but inhibits APC/C at the M phase to ensure a timely progression of the cell cycle. Under a condition of high SAG (e.g., overexpression in cancer cells), SAG escapes from CDH1 degradation to accelerate the G1-to-S transition. Under a condition of low SAG (e.g., SAG inactivation by drug or siRNA), APC/C is prematurely activated to promote mitotic progression and slippage, leading to drug resistance.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yi Sun (yisun@zju.edu.cn).
Materials Availability
This study did not generate any new reagents.
Data and Code Availability
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/8c6gygxngj.1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Six- to seven-week-old female severe combined immunodeficient (SCID) mice were inbred at the University of Michigan animal facility. Animal experimentation was approved by the University of Michigan Committee on the Use and Care of Animals. Animal care was provided in accordance with the principles and procedures outlined in the National Research Council Guide for the Care and Use of Laboratory Animals.
Cell culture
H1299, H2170, A549, A427, HEK293 and HeLa cells were purchased from the American Type Culture Collection (ATCC). A427, HEK293 and HeLa cells were grown in Dulbecco’s modified Eagle’s medium, and H1299and H2170 cells were maintained in RPMI-1640 Medium. A549 cells were grown in ATCC-formulated F-12K Medium (30–2004). All media were supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, and cells were cultured in a 37°C humidified incubator with 5% CO2.
METHOD DETAILS
Cell synchronization and live cell imaging
Cell cycle synchronization was performed as described previously (Wan et al., 2014; Wei et al., 2004). Briefly, for G1 phase synchronization, cells were exposed to a CDK4/6 inhibitor, Palbociclib (Selleckchem S1116) for 24 hr. For S phase synchronization, cells were treated by 2mM thymidine (Sigma T1895) for 14 hr and released for 9 hr, then treated with 2 mM thymidine for another 14 hr. For M phase synchronization, cells were treated with 2 mM thymidine for 20 hr first, then released into thymidine-free media for 3 hr and treated by 330 nM nocodazole (Sigma M1404) for 16 hr. For anti-microtubule drug treatment, cells were treated with thymidine (2 mM) for 20 hr first, released into fresh media for 3 hr, then treated with nocodazole (330 nM) or Taxol (12.5 nM, Selleckchem S115010) for different time periods. For live cells imaging, the cells were blocked in G2/M by 24 hr treatment of RO3306 (10mM, R&D systems 4181) and recorded by live imaging using the Operetta CLS (PerkinElmer, USA), equipped with an environmental chamber maintained at 35°C-37°C and 5% CO2.
Ectopic expression and siRNA knockdown
Various FLAG-tagged constructs were made in pcDNA-3 vector by standard cloning techniques, and transfected into cells as described as previously (Li et al., 2014). The Lenti-shSAG, along with lenti-shCont, was made as described previously (Gu et al., 2007). The siRNAs were purchased from Genepharma and transfected into sub-confluent cells using Genemute (Invitrogen) according to the manufacturer’s instructions.
FACS analysis
Cells with different pretreated were collected at the indicated time points by trypsinization. After fixation in 75% ethanol, cells were stained with propidium iodide, and analyzed by Cytoflex flow cytometer (Beckman).
ATP-lite assay
Cells were infected with indicated siRNAs for 48 h, then split and seeded into96-well plates with 3,000 cells per well in quadruplicate. After treating with nocodazole for 24 hours, cell viability assay using the ATP-lite 1 step luminescence ATP detection assay system (Perkin-Elmer) was performed according to the manufacturer’s instruction.
Clonogenic survival assay
Five-hundreds cells were seeded into 6 cm plates in triplicate. After adherence and growth for 7 days, cells were treated with PBS and Taxol as indicated, followed by incubation for 7 days with drug-containing fresh medium replacement every other day. The colonies were fixed, stained and counted.
Immuno-blotting and co-immunoprecipitation (Co-IP)
Immune-blotting and Co-IP with appropriate antibodies in whole cell extracts were performed as previously described (Zhou et al., 2016). Briefly, to make whole cell extracts, cells were lysed in lysis buffer containing 50 mM Tris-HCl (pH7.5), 150 mM NaCl, 1% Triton X-100, followed by centrifugation to collect supernatants for direct western blotting. For IP, various primary antibodies (2 μg) were added into supernatant (1 mg), and incubated at 4°C overnight, followed by incubation with the protein G Sepharose-TM 4 fast flow (GE Healthcare, Muchen, Germany) for 3 hr. The beads were then washed with lysis buffer, and the immunoprecipitation complexes were subjected to SDS-PAGE.
Immunocytochemistry
Cells were grown on coverslips in 12-well dishes for 24 hours. Following various treatments, cells were fixed with 5% formaldehyde solution and permeabilized with 0.2% Triton X-100. The fixed cells were stained with primary antibodies for overnight at 4°C as previously described (Zhang et al., 2016), then incubated with fluorescence-conjugate secondary antibody for 1 hour at room temperature, DAPI (Molecular probes, D3571) was used for DNA counterstaining. Samples were imaged and examined under a confocal microscope (Nikon A1-Ti, Tokyo, Japan).
The in vivo ubiquitylation
The plasmids encoding SAG (WT) or SAG mutant (R98A/L101A) were co-transfected with ubiquitin construct. Cells were then lysed in 6 M guanidinium denaturing solution, as described previously (Kuang et al., 2016). Polyubiquitylated substrates were pull-downed by Ni-bead and detected by IB using respective antibodies.
The in vivo animal experiments
Animal studies were conducted and processed according to the guidelines established by the University Committee on Use and Care of Animals. Construction of lentivirus-based siRNA knockdown of SAG (Lt-SAG), along with scrambled siRNA control (Lt-Cont) was performed as previously described (Tan et al., 2016). H1299 cells (3.0 × 106) stably infected with Lt-Cont or Lt-SAG were suspended in 100 μL PBS mixed with 100 μL Matrigel (Corning) and inoculated subcutaneously in both franks into female SCID mice (6–7 weeks).
When the tumor size reached ~50 mm3 at 7 days after inoculation, the mice were randomized and treated with PBS or paclitaxel (FRESENIUS KABI, 760305) at a dose of 15 mg/kg for intraperitoneal injection three times per week for three weeks. Mice were monitored and weighted each time before the injection. Tumor sizes were captured by a caliper gauge, as estimated from the formula (L × W2)/2. At the end of in vivo experiment, tumors were harvested, weighed, and photographed.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical significance of the difference was determined by one-way and repeated-measures ANOVA with the Tukey post-comparison test in GraphPad PRISM version 5 (GraphPad). The results were expressed as the mean ± SEM from at least three independent experiments. For in vivo xenograft experiment, the group-wise difference in tumor weights were tested using Wilcoxon rank sum test for clustered data (Rosner et al., 2006). The tumor growth rates were assessed using a linear mixed regression model, in which the nested random effects were used to account for the repeated-measurements of two tumors within each mouse. Interactions between time and SAG knockdown as well as paclitaxel were also included in the regression model to assess their effects on tumor growth rate. For all analyses, two-sided P values of < 0.05 were considered statistically significant and values < 0.1 were considered a marginal association. Analyses were performed using R (version 3.6.1).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SAG | home-made (Jia et al., 2010) | N/A |
| Rabbit Polyclonal anti-APC2 | Cell Signaling | Cat# 12301s; RRID:AB_2619505 |
| Rabbit monoclonal anti-APC11 | Cell Signaling | Cat# 14090s; RRID:AB_485228 |
| Rabbit monoclonal anti-CyclinB1 | Cell Signaling | Cat# 12231; RRID:AB_369073 |
| Rabbit monoclonal anti-Securin | Cell Signaling | Cat# 13445; RRID:AB_564009 |
| Rabbit monoclonal anti-Aurora A | Cell Signaling | Cat# 91590s; RRID:AB_451072 |
| Rabbit monoclonal anti-PLK1 | Cell Signaling | Cat# 4513; RRID:AB_613880 |
| Rabbit monoclonal anti-Phosphorylation- H3(ser10) | Cell Signaling | Cat# 3377p; RRID:AB_10688281 |
| Mouse monoclonal anti-Cyclin A | Cell Signaling | Cat# 4656P; RRID:AB_563675 |
| Mouse monoclonal anti-UBE2C | Santa Cruz | Cat# sc271050; RRID:AB_11215373 |
| Mouse monoclonal anti-CDC20 | Santa Cruz | Cat# sc13162; RRID:AB_2244441 |
| Rabbit polyclonal anti-UBE2S | Abcam | Cat# ab177508; RRID:AB_2211467 |
| Rabbit polyclonal anti-CDH1 | Abcam | Cat# ab217038; RRID:AB_10650758 |
| Mouse monoclonal anti-FLAG | Sigma | Cat# F1804–200UG; RRID:AB_1537400 |
| Mouse monoclonal anti-p27 | BD Biosciences | Cat# BD554069; RRID:AB_563927 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MG132 | Sigma | Cat#: M7449 |
| Palbociclib | Selleckchem | Cat#: S1116 |
| Thymidine | Sigma | Cat#: T1895 |
| Nocodazole | Sigma | Cat#: M1404 |
| Taxol | Selleckchem | Cat#: S115010 |
| RO3306 | R&D systems | Cat#: 4181 |
| Paclitaxel | FRESENIUS KABI, | Cat#: 760305 |
| Pro-TAME | R&D systems | Cat#: I-440–01M |
| Critical Commercial Assays | ||
| ATPlite Luminescence Assay Kit | PerkinElmer | Cat#: 6016943 |
| Experimental Models: Cell Lines | ||
| H1299 | American Type Culture Collection (ATCC) | Cat# NCI-H1299 ATCC® CRL-5803 |
| H2170 | ATCC | NCI-H2170 [H2170] ATCC® CRL-5928 |
| HEK293 | ATCC | Cat# 293 [HEK293] ATCC® CRL-1573 |
| HeLa | ATCC | Cat# HeLa ATCC® CCL-2 |
| A549 | ATCC | Cat# A549 ATCC® CCL-185 |
| A427 | ATCC | Cat# A-427 ATCC® HTB-53 |
| H1299: lentivirus-based siRNA knockdown of SAG (Lt-SAG), along with scrambled siRNA control (Lt-Cont) | Tan et al., 2016 | N/A |
| Oligonucleotides | ||
| siRNA targeting sequence: APC2: TGCGCGGAGTCTTGTTCTTTA | Genepharma | N/A |
| siRNA targeting sequence: APC11: GACCATTCATGTGACCIIIII GG | Genepharma | N/A |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| siRNA targeting sequence: UBE2C: CCUGCAAGAAACCUACUCA | Genepharma | N/A |
| siRNA targeting sequence: UBE2S: CCTCCAACTCTGTCTCTAA | Genepharma | N/A |
| siRNA targeting sequence: SAG: CCTGTGGGTGAAACAGAACAA | Genepharma | N/A |
| siRNA targeting sequence: SAG#2: GAGGACUGUGUUGUGGUCU | Genepharma | N/A |
| siRNA targeting sequence: RBX1: GACTTTCCCTGCTGTTACCTAA | Genepharma | N/A |
| siRNA targeting sequence: CDH1: UGAGAAGUCUCCCAGUCAG | Genepharma | N/A |
| siRNA targeting sequence: CDC20: CGGCAGGACUCCGGGCCGA | Genepharma | N/A |
| Recombinant DNA | ||
| Plasmid: pcDNA3 | This paper | N/A |
| Plasmid: pcDNA3-Flag-SAG | This paper | N/A |
| Plasmid: pcDNA3-Flag-SAG (R98A/L101A) | This paper | N/A |
| Plasmid: pcDNA3-Flag-APC11 | This paper | N/A |
| Plasmid: pcDNA3-Flag-CDC20 | This paper | N/A |
| Plasmid: pcDNA3-Flag-CDH1 | This paper | N/A |
| Plasmid: pcDNA3-His-ubiqutin | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism software version 5.01 | GraphPad | https://www.graphpad.com/support/prism-5-updates/ |
| ImageJ | NIH Image | https://imagej.net/Download |
Highlights.
SAG competes with APC2 for binding with UBE2C/2S E2s
SAG inactivation activates APC/C to trigger mitotic slippage and drug resistance
SAG is a substrate of APC/CCDH1, being subjected to its degradation at the G1 phase
SAG mutant resistant to CDH1 degradation promotes the G1-to-S transition
ACKNOWLEDGMENTS
This work is supported in part by the National Key R&D Program of China 2016YFA0501800 (Yi Sun), the Chinese NSFC grants 81630076 (Yi Sun), the NIH (R01CA200651 to W.W.), and the Chinese Scholarship Fund 201906320389 (Z.Z.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108102.
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
REFERENCES
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, and Pagano M (2004). Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428, 190–193. [DOI] [PubMed] [Google Scholar]
- Bassermann F, Eichner R, and Pagano M (2014). The ubiquitin proteasome system - implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 1843, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekier ME, Fischbach R, Lee J, and Taylor WR (2009). Length of mitotic arrest induced by microtubule-stabilizing drugs determines cell death after mitotic exit. Mol. Cancer Ther 8, 1646–1654. [DOI] [PubMed] [Google Scholar]
- Benmaamar R, and Pagano M (2005). Involvement of the SCF complex in the control of Cdh1 degradation in S-phase. Cell Cycle 4, 1230–1232. [DOI] [PubMed] [Google Scholar]
- Brito DA, and Rieder CL (2006). Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol 16, 1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NG, VanderLinden R, Watson ER, Weissmann F, Ordureau A, Wu KP, Zhang W, Yu S, Mercredi PY, Harrison JS, et al. (2016). Dual RING E3 Architectures Regulate Multiubiquitination and Ubiquitin Chain Elongation by APC/C. Cell 165, 1440–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T, and Pagano M (2004). The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol 5, 739–751. [DOI] [PubMed] [Google Scholar]
- Chong T, Sarac A, Yao CQ, Liao L, Lyttle N, Boutros PC, Bartlett JMS, and Spears M (2018). Deregulation of the spindle assembly checkpoint is associated with paclitaxel resistance in ovarian cancer. J. Ovarian Res 11, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury R, Bonacci T, Arceci A, Lahiri D, Mills CA, Kernan JL, Branigan TB, DeCaprio JA, Burke DJ, and Emanuele MJ (2016). APC/C and SCF(cyclin F) Constitute a Reciprocal Feedback Circuit Controlling S-Phase Entry. Cell Rep. 16, 3359–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A (2015). The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol 16, 322–324. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ (1999). SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol 15, 435–467. [DOI] [PubMed] [Google Scholar]
- Duan H, Wang Y, Aviram M, Swaroop M, Loo JA, Bian J, Tian Y, Mueller T, Bisgaier CL, and Sun Y (1999). SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol. Cell. Biol 19, 3145–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan H, Tsvetkov LM, Liu Y, Song Y, Swaroop M, Wen R, Kung HF, Zhang H, and Sun Y (2001). Promotion of S-phase entry and cell growth under serum starvation by SAG/ROC2/Rbx2/Hrt2, an E3 ubiquitin ligase component: association with inhibition of p27 accumulation. Mol. Carcinog 30, 37–46. [DOI] [PubMed] [Google Scholar]
- Fujioka YA, Onuma A, Fujii W, Sugiura K, and Naito K (2018). Contributions of UBE2C and UBE2S to meiotic progression of porcine oocytes. J. Reprod. Dev 64, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima H, Matsumoto A, Inuzuka H, Zhai B, Lau AW, Wan L, Gao D, Shaik S, Yuan M, Gygi SP, et al. (2012). SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep. 1, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Higuera I, Manchado E, Dubus P, Cañamero M, Méndez J, Moreno S, and Malumbres M (2008). Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol 10, 802–811. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Mansfeld J, Godwin C, Matsusaka T, Wu J, Russell P, Pines J, and Venkitaraman AR (2009). UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat. Cell Biol 11, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, and Taylor SS (2008). Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122. [DOI] [PubMed] [Google Scholar]
- Giovinazzi S, Bellapu D, Morozov VM, and Ishov AM (2013). Targeting mitotic exit with hyperthermia or APC/C inhibition to increase paclitaxel efficacy. Cell Cycle 12, 2598–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Q, Tan M, and Sun Y (2007). SAG/ROC2/Rbx2 is a novel activator protein-1 target that promotes c-Jun degradation and inhibits 12-O-tetradeca-noylphorbol-13-acetate-induced neoplastic transformation. Cancer Res. 67, 3616–3625. [DOI] [PubMed] [Google Scholar]
- Harper JW, Burton JL, and Solomon MJ (2002). The anaphase-promoting complex: it’s not just for mitosis any more. Genes Dev. 16, 2179–2206. [DOI] [PubMed] [Google Scholar]
- He H, Gu Q, Zheng M, Normolle D, and Sun Y (2008). SAG/ROC2/RBX2 E3 ligase promotes UVB-induced skin hyperplasia, but not skin tumors, by simultaneously targeting c-Jun/AP-1 and p27. Carcinogenesis 29, 858–865. [DOI] [PubMed] [Google Scholar]
- He J, Chao WC, Zhang Z, Yang J, Cronin N, and Barford D (2013). Insights into degron recognition by APC/C coactivators from the structure of an Acm1-Cdh1 complex. Mol. Cell 50, 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Shi J, Orth JD, and Mitchison TJ (2009). Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell 16, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K, and Dikic I (2012). Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem 81, 291–322. [DOI] [PubMed] [Google Scholar]
- Jia L, Yang J, Hao X, Zheng M, He H, Xiong X, Xu L, and Sun Y (2010). Validation of SAG/RBX2/ROC2 E3 ubiquitin ligase as an anticancer and radiosensitizing target. Clin. Cancer Res 16, 814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, and Wilson L (2004). Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Klitzing C.v., Huss R, Illert AL, Fröschl A, Wötzel S, Peschel C, Bassermann F, and Duyster J (2011). APC/C(Cdh1)-mediated degradation of the F-box protein NIPA is regulated by its association with Skp1. PLoS One 6, e28998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossatz U, Dietrich N, Zender L, Buer J, Manns MP, and Malek NP (2004). Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 18, 2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, and Peters JM (2000). Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell 11, 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang P, Tan M, Zhou W, Zhang Q, and Sun Y (2016). SAG/RBX2 E3 ligase complexes with UBCH10 and UBE2S E2s to ubiquitylate b-TrCP1 via K11-linkage for degradation. Sci. Rep 6, 37441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AW, Inuzuka H, Fukushima H, Wan L, Liu P, Gao D, Sun Y, and Wei W (2013). Regulation of APC(Cdh1) E3 ligase activity by the Fbw7/cyclin E signaling axis contributes to the tumor suppressor function of Fbw7. Cell Res. 23, 947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tan M, Jia L, Wei D, Zhao Y, Chen G, Xu J, Zhao L, Thomas D, Beer DG, and Sun Y (2014). Inactivation of SAG/RBX2 E3 ubiquitin ligase suppresses KrasG12D-driven lung tumorigenesis. J. Clin. Invest 124, 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M, and Brandeis M (2004). Mammalian Cdh1/Fzr mediates its own degradation. EMBO J. 23, 1619–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HJ, and Rape M (2014). Enhanced protein degradation by branched ubiquitin chains. Cell 157, 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal K, Donthamsetty S, Kaur R, Yang C, Gupta MV, Reid MD, Choi DH, Rida PCG, and Aneja R (2017). Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer. Br. J. Cancer 116, 1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama KI, and Nakayama K (2006). Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381. [DOI] [PubMed] [Google Scholar]
- Pagano M, Pepperkok R, Verde F, Ansorge W, and Draetta G (1992). Cyclin A is required at two points in the human cell cycle. EMBO J. 11, 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM (2002). The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell 9, 931–943. [DOI] [PubMed] [Google Scholar]
- Peters JM (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol 7, 644–656. [DOI] [PubMed] [Google Scholar]
- Petroski MD, and Deshaies RJ (2005). Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell 123, 1107–1120. [DOI] [PubMed] [Google Scholar]
- Raab M, Sanhaji M, Zhou S, Rödel F, El-Balat A, Becker S, and Strebhardt K (2019). Blocking Mitotic Exit of Ovarian Cancer Cells by Pharmaceutical Inhibition of the Anaphase-Promoting Complex Reduces Chromosomal Instability. Neoplasia 21, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner B, Glynn RJ, and Lee ML (2006). Extension of the rank sum test for clustered data: two-group comparisons with group membership defined at the subunit level. Biometrics 62, 1251–1259. [DOI] [PubMed] [Google Scholar]
- Sackton KL, Dimova N, Zeng X, Tian W, Zhang M, Sackton TB, Meaders J, Pfaff KL, Sigoillot F, Yu H, et al. (2014). Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 514, 646–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736. [DOI] [PubMed] [Google Scholar]
- Sun Y, and Li H (2013). Functional characterization of SAG/RBX2/ROC2/ RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein Cell 4, 103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Tan M, Duan H, and Swaroop M (2001). SAG/ROC/Rbx/Hrt, a zinc RING finger gene family: molecular cloning, biochemical properties, and biological functions. Antioxid. Redox Signal 3, 635–650. [DOI] [PubMed] [Google Scholar]
- Tan M, Xu J, Siddiqui J, Feng F, and Sun Y (2016). Depletion of SAG/RBX2 E3 ubiquitin ligase suppresses prostate tumorigenesis via inactivation of the PI3K/AKT/mTOR axis. Mol. Cancer 15, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li B, Bharadwaj R, Zhu H, Ozkan E, Hakala K, Deisenhofer J, and Yu H (2001). APC2 Cullin protein and APC11 RING protein comprise the minimal ubiquitin ligase module of the anaphase-promoting complex. Mol. Biol. Cell 12, 3839–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton BR, and Toczyski DP (2006). Precise destruction: an emerging picture of the APC. Genes Dev. 20, 3069–3078. [DOI] [PubMed] [Google Scholar]
- Wan L, Tan M, Yang J, Inuzuka H, Dai X, Wu T, Liu J, Shaik S, Chen G, Deng J, et al. (2014). APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev. Cell 29, 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, and Kaelin WG Jr. (2004). Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428, 194–198. [DOI] [PubMed] [Google Scholar]
- Wu SY, Kuan VJ, Tzeng YW, Schuyler SC, and Juang YL (2016). The anaphase-promoting complex works together with the SCF complex for proteolysis of the S-phase cyclin Clb6 during the transition from G1 to S phase. Fungal Genet. Biol 91, 6–19. [DOI] [PubMed] [Google Scholar]
- Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh D-C, Hathaway N, Dimova N, Cuny GD, and King RW (2010). Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 18, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, and Sun Y (2020). Cullin RING Ligase 5 (CRL-5): Neddylation Activation and Biological Functions. Adv. Exp. Med. Biol 1217, 261–283. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wan L, Dai X, Sun Y, and Wei W (2014a). Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochim. Biophys. Acta 1845, 277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, and Liu J (2014b). Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 33, 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Karnak D, Tan M, Lawrence TS, Morgan MA, and Sun Y (2016). FBXW7 Facilitates Nonhomologous End-Joining via K63-Linked Polyubiquitylation of XRCC4. Mol. Cell 61, 419–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, and Sun Y (2013). Cullin-RING Ligases as attractive anti-cancer targets. Curr. Pharm. Des 19, 3215–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Morgan MA, and Sun Y (2014). Targeting Neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid. Redox Signal 21, 2383–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. (2002). Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416, 703–709 [DOI] [PubMed] [Google Scholar]
- Zhou W, Xu J, Zhao Y, and Sun Y (2014). SAG/RBX2 is a novel substrate of NEDD4–1 E3 ubiquitin ligase and mediates NEDD4–1 induced chemosensitization. Oncotarget 5, 6746–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Tan M, Nyati MK, Zhao Y, Wang G, and Sun Y (2016). Blockage of neddylation modification stimulates tumor sphere formation in vitro and stem cell differentiation and wound healing in vivo. Proc. Natl. Acad. Sci. USA 113, E2935–E2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W, Pan Z, and Sun Y (2017a). Neddylation E2 UBE2F Promotes the Survival of Lung Cancer Cells by Activating CRL5 to Degrade NOXA via the K11 Linkage. Clin. Cancer Res 23, 1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W, Pan Z, and Sun Y (2017b). Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin. Cancer Res 23, 1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/8c6gygxngj.1.