

Abstract
In Drosophila photoreceptors, the multivalent PDZ protein INAD interacts with multiple signaling components and localizes complexes to the rhabdomere, a subcellular compartment specialized for phototransduction. Since this localization is critical for signaling, we conducted a genetic screen of the third chromosome for mutations that result in mislocalization of an INAD-GFP fusion protein. We identified seven mutant lines that fall into two complementation groups, idl (INAD localization)-A and idl-B. We show that idl-A mutants fail to complement with chaoptic (chp) mutants. Since chaoptin is a structural component of the rhabdomere, mislocalization of INAD may be a secondary effect of the retinal degeneration in chp and idl-A mutants. Genetic complementation and DNA sequencing reveal that the two idl-B mutants represent new alleles of trp, a gene encoding the major light-activated channel. The molecular change in each allele affects a highly conserved residue in either an ankyrin domain on the N-terminus or in the S6 transmembrane domain of TRP. These changes lead to the loss of TRP protein. TRP has previously been shown to anchor INAD in the rhabdomeres, therefore the independent identification of two trp alleles validates our screen for INAD-GFP localization. One possibility is that a limited number of proteins are required for localizing INAD-signaling complexes. A similar screen of the X and second chromosomes may be required to find the remaining players involved.
Keywords: genetic screen, Drosophila, phototransduction, INAD, TRP channel, rhabdomere
Introduction
Intracellular organization of signaling proteins is important for fast and accurate signal transduction in many cell types. Drosophila photoreceptors have shown how a very high speed of signaling is achieved through proper subcellular organization of phototransduction proteins.1-4 Phototransduction occurs in the rhabdomere, a specialized subcellular compartment composed of ~60,000 microvilli that house all phototransduction signaling components. Upon light stimulation, the light-receptor rhodopsin photo-isomerizes to its active form, meta-rhodopsin, and activates the heterotrimeric Gq protein. The active Gqα subunit then stimulates the effector protein, phospholipase C (PLC). PLC catalyzes the breakdown of the minor membrane phospholipid, phosphatidylinositol-4,5-bisphosphate (PIP2) into two intracellular messengers: diacylglycerol (DAG) and inositol triphosphate (InsP3). Activation of PLC leads to the opening of at least two cationic channels, transient-receptor-potential (TRP) and TRP-like (TRPL), thereby generating a depolarizing membrane potential.1,5,6 Deactivation of the cascade occurs, at least in part, through activation of an eye-specific protein kinase C (eye-PKC) and calmodulin, as well as binding of an arrestin protein to rhodopsin.7-12
The organization of phototransduction components was found to be mediated by the scaffold protein INAD (inactivation-no-afterpotential-D), which is composed of five PDZ (postsynaptic density-95, discs-large, ZO-1) domains that each interact with a specific component of the phototransduction cascade. PDZ1 and specific component of the phototransduction cascade. PDZ1 and PDZ5 reportedly bind to two distinct sites on the C-terminus of the effector protein PLC,13-15 PDZ2 and PDZ4 have been shown to interact with eye-PKC,13,16 and PDZ3 binds near the extreme C-terminus of the light-activated TRP channel.13,17,18 Assembly of signaling complexes by INAD was shown to be essential for the localization of signaling components to the rhabdomeres of photoreceptor cells since inaD null photoreceptors displayed a complete mislocalization of PKC, PLC and TRP.13 Consequently, the mislocalization of INAD-signaling complexes resulted in severe signaling impairment, demonstrating the importance of localizing and organizing complexes in the rhabdomeres of photoreceptors.12,13 The formation and localization of macromolecular signaling complexes has emerged as a common cellular strategy underlying proper signal transduction.2,19,20
Little, however, is known about the mechanisms responsible for the assembly, targeting and anchoring of INAD-signaling complexes in photoreceptors. Previous studies have shown that the INAD-TRP interaction is required to anchor INAD-signaling complexes in the rhabdomere.18,21 Tsunoda et al. (2001) further hypothesized that additional anchors of INAD were likely to exist since ~25% of INAD protein remained localized in the rhabdomeres of trp null photoreceptors. In this study, we have taken a genetic approach to identify additional proteins involved in the rhabdomeric localization of INAD-signaling complexes.
We conducted a genetic screen for mutants that display mislocalization of an INAD-GFP fusion protein. After screening over 2,600 lines, we identified seven idl (INAD localization) mutant lines that displayed mislocalization of INAD. These mutants fall into two complementation groups, idl-A and idl-B. Complementation and immunolocalization assays revealed that idl-A encodes the protein chaoptin, which functions in the adhesion between microvilli in the rhabdomere.22,23 Thus, chaoptin is required for the structural integrity of the rhabdomere, and mislocalization of signaling proteins observed in idl-A photoreceptors may be due to the retinal degeneration seen in these mutants. Two independently isolated idl-B alleles display mislocalization of INAD to photoreceptor cell bodies and light-dependent retinal degeneration. Complementation analyses and DNA sequencing revealed that each idl-B allele contains a misense mutation in the trp gene: one in an N-terminal ankyrin domain and one in the S6 transmembrane domain of the channel. Both mutations result in a loss of TRP protein. Because only two complementation groups on the third chromosome were identified in our screen, we suggest that a limited number of proteins may be required for the rhabdomeric localization of INAD-signaling complexes. The independent identification of two new trp alleles validates our assay for INAD-GFP localization, and demonstrates the feasibility of a similar screen for the X and second chromosomes.
Results
Genetic screen for mutants displaying mislocalization of INAD-signaling complexes
To identify proteins involved in the rhabdomeric localization of INAD-signaling complexes, we conducted a forward genetic screen for mutants that display mislocalization of a GFP-tagged INAD fusion protein (INAD-GFP) in vivo. We first generated a transgenic fly line that expresses INAD-GFP in photoreceptor cells. To determine if INAD-GFP localizes and functions like wild-type INAD, we began by examining the localization of INAD-GFP in 1-μm-thick frozen retinal sections. We found that INAD-GFP was indeed localized exclusively to the rhabdomeres of photoreceptor cells (Fig. 1A). To examine if INAD-GFP was functional, the transgene was expressed in an inaD null background and analyzed by electroretinogram (ERG) recording. In inaD null flies, signaling complexes are not formed, and signaling is severely impaired.13 In contrast, when INAD-GFP was expressed in an inaD null background, a near wild-type ERG was restored to photoreceptors (Fig. 1B), suggesting that INAD-GFP assembles and localizes fully functional signaling complexes in photoreceptors. These results validate the use of INAD-GFP as a reporter for INAD localization.
Figure 1.

INAD-GFP protein expression, localization and function in the Drosophila retina. (A) Left, INAD-GFP fusion protein is expressed in photoreceptors, as seen in whole eyes of the fly. Right, 1-μm-thick retinal cross-sections show INAD-GFP is localized to the rhabdomeres of photoreceptor cells. The rhabdomere (R) and cell body (c) of a single photoreceptor cell are approximately outlined for illustration. (B) electroretinogram (eRG) recordings from wild-type, inaD1 null, and transgenic inaD1 null flies expressing INAD-GFP (P(inaD-GFP); inaD1). Flies were exposed to a 2 second flash of light (arrow). Note that P(inaD-GFP); inaD1 flies exhibit near complete rescue of signaling. (c) Visualization of INAD-GFP in intact eyes. shown are representative ommatidia expressing INAD-GFP in inaD1 (trp+) and inaD1; trp343 (trp-) backgrounds. INAD-GFP signal is seen in all seven rhabdomeres of each ommatidia in wild-type. For reference, seven rhabdomeres are numbered in a representative ommatidium. The trp null retina displayed diffuse localization of INAD-GFP localization pattern, especially in the six outer photoreceptors.
We next developed an assay to examine the localization of INAD-GFP in vivo. Live flies were secured and positioned on glass slides to examine one eye of each fly. Flies were overlaid with halocarbon oil, and imaged for INAD-GFP by standard epi-fluorescence at low magnification. In this manner, we were able to rapidly screen a large number of fly lines. In a wild-type background, every ommatidium in the field displayed INAD-GFP localized to the rhabdomeres of each of its seven visible photoreceptor cells (Fig. 1C). To test if mislocalization of INAD-GFP could be detected in our assay, we examined INAD-GFP in a trp null background. Since previous studies showed that TRP is required to localize INAD in the rhabdomeres,18,21 we expected the absence of TRP protein to result in clear INAD-GFP mislocalization. Indeed, when INAD-GFP was expressed in a trp background, GFP signal was nearly absent in the rhabdomeres of the six outer photoreceptor cells of each ommatidium (Fig. 1C). Thus, this assay was likely to detect mutations that disrupt the rhabdomeric localization of INAD.
We screened a collection of third chromosome viable mutants generated and maintained by the Zuker lab (the Zuker collection). These ~6,000 lines were generated by mutagenizing flies with ethyl methanesulfonate at a concentration predicted to result in at least seven mutant alleles in the collection for every nonessential gene.24 The Zuker collection has proven to be an invaluable resource for many genetic studies seeking nonessential mutants/genes involved in phototransduction,10,13,25,26 as well as a variety of other biological processes.24,27,28 To generate homozygous mutant flies with INAD-GFP expression, we designed a two-generation genetic scheme (Fig. 2). We first crossed P(GMR-inaD-GFP); bw; l29DTS4 D/TM6B female flies to third chromosome mutant males from the Zuker Collection, +; bw/CyO; st*/TM6B (* indicates the mutated chromosome). To bypass manual selection of F1 flies, we used the Dominant Temperature Sensitive 4 (l(3)DTS4) mutation to exclude unwanted progeny. To do this, we incubated the parental crosses at 29°C for 5 days. At this restrictive temperature, progeny carrying the DTS mutation would not survive past the embryonic/larval stage. Viable progeny, P(GMR-inaD-GFP)/+; bw/bw or bw/CyO; st*/TM6B, were then transferred to new vials and allowed to intermate (F1). Third chromosomes in the mutant collection are marked with a mutation in the scarlet (st) gene so that homozygous mutant lines (st*/st*) could be detected in a second chromosome bw/bw background by the presence of white eyes. We screened white-eyed F2 male and female flies (<24 hrs old) for localization of INAD-GFP.
Figure 2.

Genetic scheme for the INAD-GFP localization screen. The genetic scheme used to generate INAD-GFP expressing mutant lines for screening. Third chromosome eMs mutagenized males from the Zuker collection (* indicates the mutation on the third chromosome) and P(inaD-GFP); bw; DTS4 D/TM6B females were crossed at 29°c for five days. The non-permissive temperature of 29°c was used to eliminate all progeny containing DTS4 D in the F1 generation. All viable F1 males and females were inbred at 25°c. DTS4 escapers (not shown in the scheme) were identified by the presence of the dominant Dichete (D) mutation. F2 homozygous mutant flies (bw; st*) were identified by the presence of white eyes and screened for INAD-GFP localization.
The criterion for a wild-type INAD-GFP localization pattern was the presence of robust GFP signal in all seven rhabdomeres of all visible ommatidia. Mutants identified in our screen displayed fewer than seven distinct GFP-positive rhabdomeres in each ommatidium, or diffuse localization in cell bodies. From the 6,150 parental lines crossed, we were only able to screen sufficient numbers of white-eyed F2 flies from 2,624 F2 lines (Table 1). This unexpected reduction in the number of fly lines that could actually be screened was likely due to the accumulation of new mutations which have made the previously viable lines from the Zuker collection semi-lethal/lethal.24 From the 2,624 lines that we screened, 2,391 lines displayed wild-type localization of INAD-GFP and 233 fly lines were considered mutant with varying degrees of severity.
Table 1.
INAD-GFP localization screen sumary
| Parental Crosses | 6150 |
|
| |
| F1 Crosses | 5225 |
|
| |
| F2 Crosses | 4708 |
|
| |
| F2 Lines Screened | 2624 |
|
| |
| Primary Screen Mutants | 233 |
|
| |
| Seconadary Screen Mutants | 7 |
These 233 fly lines were then subjected to a secondary screen in which we examined the localization of endogenous INAD and rhodopsin (Rh1) in retinal tissue sections. Rh1 is the major light receptor in Drosophila photoreceptors R1-6, and represents a rhabdomeric protein that is not associated with the INAD-signaling complex. In addition to examining the specificity of INAD mislocalization, we also examined the structural integrity of retinal sections. This was important because apparent mislocalization of INAD-GFP could be the result of a structural or developmental defect affecting photoreceptor morphology. Indeed, a few mutants that were recovered from the primary screen were identified as having structural defects in our secondary screen. In one of these mutants (Z0189) the R7 cell is elongated while the second mutant (Z2528) lacks R7 altogether (data not shown). These structural mutants were not pursued further. Since many mutations affecting phototransduction also lead to retinal degeneration, we did not rule-out mutants that displayed potential degeneration. The majority of the fly lines re-screened, however, proved to be wild-type in the secondary screen. This may be due to the nature of the in vivo assay used. Physical damage to the eye, or extended exposure of the eye to fluorescent light stimulation, may have resulted in a misdiagnosis of INAD-GFP localization in the primary screen. From the 233 mutant fly lines re-screened, we identified seven lines, now referred to as idl (INAD localization) mutants, which appeared to display specific mislocalization of INAD. Intermating the seven idl mutant lines and examining the localization of INAD in their progeny revealed that these mutants fall into two complementation groups, idl-A and idl-B.
idl-A encodes the cell adhesion protein, chaoptin
The idl-A complementation group is comprised of five mutant alleles (Z3513, Z5240, Z4345, Z1062, Z2482) that all displayed mislocalization of INAD-GFP. idl-A mutant photoreceptors displayed mislocalization of INAD, but normal rhabdomeric localization of Rh1 (Fig. 3), suggesting that idl-A may play a role in the rhabdomeric localization of INAD. In characterizing idl-A mutants, we found that they also displayed retinal degeneration. Degeneration is largely light-independent since idl-A flies exhibit abnormal photoreceptor structure, with small and deteriorating rhabdomeres, regardless of light condition; prolonged light-exposure, however, did tend to enhance retinal degeneration (Fig. 3A).
Figure 3.

The idl-A mutants recovered from the screen are alleles of chaoptic. (A) shown are representative cross-sections from idl-A mutants that have been dark-raised (DARK) or light-exposed (LIGhT) for 10 days-post-eclosion, and then immunostained for INAD and Rh1. Wild-type control flies were dark-raised. A phase contrast image of each cross-section is shown to illustrate the photoreceptor structure. Wild-type retinal sections exhibited rhabdomeric localization of INAD and Rh1 in addition to robust structural integrity. Dark-raised idl-A mutant photoreceptors displayed retinal degeneration, depicted by smaller rhabdomeres, and mislocalization of INAD, but near normal localization of Rh1. With exposure to light, idl-A mutants showed a more severe loss of retinal integrity and mislocalization of both INAD and Rh1. (B) complementation analysis of idl-A and chaoptic (chp2) mutants. Retinal cross-sections from young (<2 day old), dark raised wild-type, idl-A, chp2 and idl-A/chp2 flies were immunostained for INAD and chaoptin. Wild-type photoreceptors exhibited normal retinal integrity, and rhabdomeric localization of both INAD and chaoptin. chp2, idl-A and idl-A/chp2 mutant photoreceptors all exhibited mislocalization of INAD, lack of chaoptin staining, and retinal degeneration.
Since light-independent retinal degeneration often results from mutations that affect the structural integrity of photoreceptors,23,30-32 we conducted complementation analyses with known third chromosome mutants affecting proteins such as chaoptin (chp) and Rh1 (ninaE). Chaoptin is an integral membrane protein that provides cell adhesion between microvilli of the rhabdomere.22,23 We show that idl-A mutants failed to complement with hypomorphic chaoptic alleles23 for INAD localization (Fig. 3B). Unfortunately, the available anti-chaoptin antibody (Developmental Hybridoma Studies Bank) did not work on immunoblots so we were not able to confirm complementation tests by immunoblot analysis. However, we were able to use the antibody on tissue sections, and we found that all idl-A alleles lacked chaoptin staining (Fig. 3B). Together, these results strongly suggest that our idl-A mutants are alleles of chaoptic. Although INAD is mislocalized in idl-A photoreceptors, we could not rule out the possibility that this phenotype is a secondary effect of the retinal degeneration caused by the loss of chaoptin.
idl-B mutants display INAD-signaling complex mislocalization and retinal degeneration
The idl-B complementation group is composed of two alleles, idl-B1 (Z5679) and idl-B2 (Z0838), isolated independently in the INAD-GFP mislocalization screen. To confirm that the INAD-signaling complex was specifically affected in idl-B mutant photoreceptors, we immunostained retinal tissue sections for PKC, TRPL and Rh1. Our results show that PKC is mislocalized to the cell body in both idl-B mutants (Fig. 4) while Rh1 and TRPL remain rhabdomeric (Fig. 4), suggesting that the displacement of signaling proteins in both idl-B mutants is specific to the INAD-signaling complex.
Figure 4.

INAD complexes are specifically mislocalized in idl-B mutant photoreceptor cells. shown are representative dark-raised wild-type, idl-B1 and idl-B2 retinal cross-sections (1-μm-thick) immunostained for INAD, pKc, TRpL and Rh1. Wild-type photoreceptors displayed rhabdomeric localization of INAD, pKc, TRpL and Rh1. Both idl-B alleles displayed mislocalization of INAD and pKc, but normal localization of TRpL and Rh1.
In addition to the mislocalization of INAD-signaling complexes, both idl-B mutants display retinal degeneration. Dark-raised idl-B mutants display an organized arrangement of intact ommatidia, while light-exposed flies 3 days post-eclosion (3 DPE) display retinal degeneration depicted by smaller or missing rhabdomeres, and an overall decreased organization within the retina (Fig. 5). This retinal degeneration seen in both idl-B mutants was light-dependent since 3 DPE dark-raised mutants displayed wild-type structure (Fig. 5).
Figure 5.
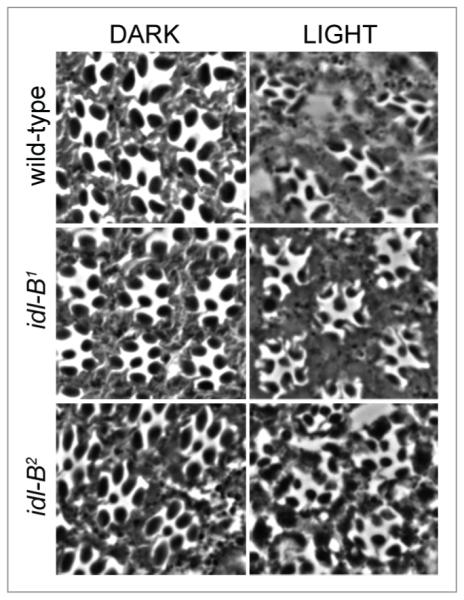
idl-B mutant photoreceptors display a light-dependant retinal degeneration. Representative retinal sections from wild-type, idl-B1 and idl-B2 flies that were dark-raised (DARK) or light-exposed (LIGHT) until 3 days-post-eclosion (3DPE ). All dark-raised flies exhibited wild-type photoreceptor structure. Light-exposure of both idl-B alleles resulted in retinal degeneration, especially depicted by smaller rhabdomeres.
idl-B mutants represent two new alleles of trp
Since previous studies have shown that trp mutants display both a mislocalization of INAD18,21 and light-dependent retinal degeneration,33,34 we investigated whether idl-B mutants have reduced levels of TRP protein. We found that idl-B1 and idl-B2 mutants lacked detectable TRP protein even in dark-raised flies (Fig. 6A). We also tested idl-B mutants by electroretinogram (ERG) recording. In wild-type flies, exposure to a light stimulus leads to a rapid response that is sustained for the duration of the light pulse. trp mutants, in contrast, lack this sustained response and display a transient response that decays during the light pulse (Fig. 7). We found that idl-B mutants display a transient response during a light pulse similar to trp mutants (Fig. 7A).
Figure 6.
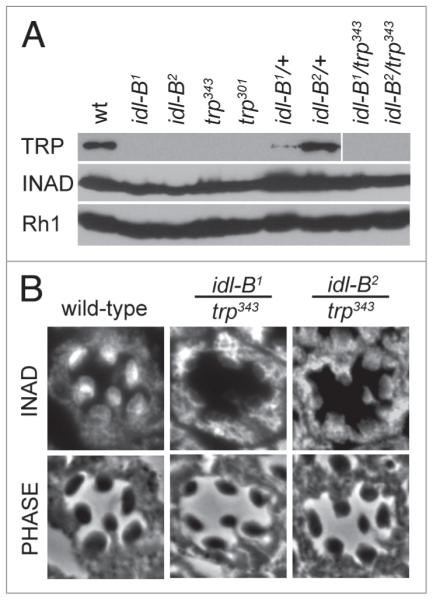
idl-B mutants lack TRp protein. (A) Representative immunoblots of wild-type (wt), idl-B alleles, trp alleles, idl-B heterozygotes and trans-heterozygous idl-B/trp flies. TRp protein was absent from all homozygous trp and idl-B alleles. The two idl-B alleles failed to complement with trp as depicted by the lack of TRp protein in the trans-heterozygous idl-B/trp samples. INAD and Rh1, in contrast, were present in all fly stocks. All samples contained 10 heads from darkraised flies. (B) Representative cross-sections of single ommatidia from dark-raised wild-type, idl-B1/trp343, and idl-B2/trp343 flies immunostained for INAD. Note that INAD is mislocalized in both idl-B1/trp343 and idl-B2/trp343, indicating that both idl-B alleles failed to complement with trp for INAD localization. phase-contrast images are shown to illustrate the wild-type structure of all three genotypes analyzed.
Figure 7.

electroretinogram recordings of idl-B mutants. (A) Representative eRG recordings from young, dark-raised wild-type and mutant flies. With white light (400–700 nm) at an intensity of 453 lux, both idl-B mutants showed a transient eRG response, similar to trp mutants. idl-B1/trp343 and idl-B2/trp343 trans-heterozygotes also displayed an eRG waveform closely resembling the trp mutants. 10 second light stimuli are depicted by a line above the eRG traces. (B) At a lower light intensity of 6.39 lux, idl-B1 responded more similar to wild-type then the trp343 null mutant. The trp301 hypomorph also responded more similar to wild-type at the lower light intensity.
To investigate if idl-B mutants are indeed trp alleles, we conducted complementation analyses with trp343 null flies. idl-B/trp343 progeny were analyzed for these three phenotypes: loss of TRP protein, mislocalization of INAD, and light-dependent retinal degeneration. We found that dark-raised idl-B/trp343 flies showed loss of TRP protein on immunoblots (Fig. 6A), mislocalization of INAD to the cell body of photoreceptors (Fig. 6B), and a transient ERG phenotype, all similar to the trp null mutant (Fig. 7A). Taken together, these results show that idl-B mutants are indeed alleles of trp.
To determine the molecular changes in idl-B1 and idl-B2 alleles, we sequenced the entire trp gene in both idl-B1 and idl-B2 mutants. Several overlapping ~1 kb genomic DNA fragments were isolated from both mutant lines and the wild-type parental strain, and sequenced using primers designed to cover the entire trp gene (5,770 bp). We found that idl-B1 contains a single base pair change that results in a leucine to phenylalanine change on the cytoplasmic N-terminus of TRP (L(74)F). Interestingly, L(74) F occurs in the second of the three conserved ankyrin repeats on the N-terminus of the TRP protein (Fig. 8B). The mutation in idl-B1 occurs in a leucine that is highly conserved in ankyrin repeats of the TRPC family (Fig. 8A) as well as among other ankyrin domains. Significantly, this leucine plays a role in the folding and maintenance of the ankyrin repeat shape.35-37 idl-B2 contains a mutation that results in an amino acid change (L(653) H) in the sixth transmembrane domain (S6) of TRP (Fig. 8B). The mutation in idl-B2 occurs in a highly conserved leucine in S6 of TRP channels. Both mutations lead to instability of the TRP protein, and subsequent mislocalization of INAD-signaling complexes.
Figure 8.

idl-B mutants encode new alleles of trp. (A) Ankyrin repeat comparison. sequence alignment of ankyrin repeats (AR) from various TRpc channels and AR containing proteins against the AR consensus sequence (ANK cONs, top). The well conserved amino acids within the ANK cONs are shown in bold.37 Drosophila melanogaster TRp protein (DTRp; p19334); Drosophila melanogaster TRpL (DTRpL; p48994); Mus musculus TRpc5 (MTRpc5; Q9QX29); Rattus norvegicus TRpc4 (RTRpc4; O35119); Homo sapiens TRpc1 (hTRpc1; p48995); Mus musculus TRpc7 (MTRpc7; Q9WVc5); Rattus norvegicus TRpc3 (RTRpc3; Q9JM19); Mus musculus TRpc6 (MTRpc6; Q61143). The accession numbers for each protein are shown in parenthesis. NcBI BLAsT was used for protein sequence comparison and the alignment was done via clustal2W, online program. Residues identical to the consensus sequence are shaded in black, residues similar to the consensus sequence are shaded gray. The asterisk indicates the leucine at position six of the AR that is mutated in idl-B1. Many of the TRp proteins from the TRpc family were found to contain this conserved leucine within the ankyrin repeat. (B) A cartoon representation of the Drosophila TRp protein illustrating the amino acid changes resulting in each idl-B mutant. shown are the six transmembrane domains (s1-s6), three ankyrin repeats on the N-terminus and the INAD binding site at the c-terminal end of the Drosophila TRp protein. The idl-B1 mutation is an amino acid change L(74)F in the second ankyrin domain on the cytoplasmic N-terminus of TRp. The idl-B2 mutation produces an amino acid change L(653)h in the sixth transmembrane domain (s6) of TRp.
To determine if there are any functional differences between the two idl-B mutants, we conducted ERG recordings using different light intensities. idl-B2 mutants display responses similar to trp null mutants at all light intensities. ERG responses of idl-B1 mutants were similar to trp null mutants at high light intensities, but interestingly, were similar to wild-type at lower light intensities (Fig. 7B). Previous studies have shown that low levels of TRP protein expressed under a heat shock promoter, 10% of wild-type levels, can produce a near wild-type ERG in trpl; trp null flies.34 Therefore, one possibility is that idl-B1 mutants have a small amount of TRP protein present that supports the light response at lower light intensities. To test this hypothesis, we loaded tissue from as many as ten idlB1 heads for immunoblot analysis, but were unable to detect any TRP protein present (Fig. 6), even after probing blots with two different anti-TRP antibodies (data not shown). Although we could not show that idlB1 flies express low levels of TRP, we noted that trp301, a reported severe hypomorph,38 also did not display TRP protein by similar immunoblot analysis. Since the trp301 mutant has been shown to be a hypomorph, we conducted ERG recordings to see if trp301 mutants display a similar ERG phenotype to idlB1 mutants. Indeed, trp301 flies displayed a transient response to pulses of higher light intensity and a wild-type response to pulses of lower light intensity similar to idl-B1 mutants (Fig. 7). These results suggest that idl-B2 is a trp null while idl-B1 may be a hypomorph of trp.
Discussion
The assembly of signaling proteins into macromolecular complexes by scaffold proteins like INAD has proven to be a general organizational strategy used in many systems and cell types. Here, we report a forward genetic screen for mutants that fail to localize a GFP-tagged INAD fusion protein in photoreceptors. The significance of localizing INAD-signaling complexes to the rhabdomeres of photoreceptors has previously been demonstrated in an inaD null mutant, which displays a mislocalization of binding proteins and a near-complete loss of signaling.13 Because previous evidence suggested that INAD-signaling complexes are pre-assembled and targeted as a whole to the rhabdomere,21 we envisioned that our screen would identify mutants with defects in many aspects of INAD-complex regulation, including assembly of complexes before localization, targeting and transport of complexes to the rhabdomeres, as well as anchoring of complexes in the rhabdomeric membrane. Understanding how INAD-signaling complexes are assembled, targeted and anchored will likely lend important insight into the mechanisms underlying similar processes for other macro-molecular complexes.
In an attempt to identify genes involved in these processes, we chose to screen mutant lines that were predicted to saturate non-essential genes of the third chromosome (the Zuker collection). The first problem we encountered was that we were only able to screen 43% of the mutant lines from the collection. This was due to an unexpected increase in the number of lines that have become lethal or semi-lethal in this collection; this is a problem that has been seen by multiple groups using the collection, and cannot be readily explained. Even so, with mutagenized chromosomes predicted to contain 5–6 “hits” each,24 we estimated that screening 2,624 lines would still be assaying most of the non-essential genes on the third chromosome. In the end, however, only two complementation groups were identified: (1) idl-A, which, based on complementation tests, but not sequence analysis, is likely to encode chaoptic, and (2) idl-B, which encodes trp. Both of these genes were previously identified.
Why were only two complementation groups identified? The independent identification of multiple alleles for each loci suggests that the portion of the collection we screened was likely to represent all of the third chromosome, and further, that our assay for INAD-GFP localization is reliable, and loss of chp or trp does indeed lead to mislocalization of INAD. Since chp mutants display rather severe retinal degeneration, mislocalization of INAD may be a secondary effect of the loss of structural integrity of the rhabdomere. Although rhodopsin localization was less affected than INAD, which could indicate that chaoptin does regulate INAD localization, this effect may not be specific to INAD since we also found the initial targeting of TRPL in late pupae may be defective (data not shown). Future studies will need to elucidate whether chaoptin indeed functions in the localization of some signaling proteins, or if retinal degeneration from loss of chaoptin affects the localization of proteins differently. The isolation of new trp alleles from our screen further verifies the reliability of our screen since trp has previously been shown to be required for anchoring INAD in the rhabdomere.18,21 The identification, however, of only two third-chromosome genes suggests that a very limited number of genes/proteins are needed for properly assembling and localizing INAD-signaling complexes in the rhabdomere. Although we anticipated the identification of many more mutants from this screen, one hypothesis is that formation and targeting of INAD-signaling complexes depends largely on components in the complex itself. Signaling proteins in the complex may function, for example, in a novel trafficking capacity. It will be interesting to see if the subcellular localization of other macromolecular complexes also relies primarily on new roles of signaling proteins within the complexes.
Another possibility is that components required for INAD-signaling complex assembly/localization are encoded by essential genes, rather than non-essential eye-specific genes. Although all the signaling components in INAD-complexes are eye-specific, or nearly so, it is possible that the trafficking of these eye-specific components is dependent on motors and chaperone proteins that function ubiquitously, or in processes vital for survival. The assay described here could be used, for example, to screen a collection of lethal mutants. Genetic manipulation described by Stowers et al.39 could be performed to generate heterozygous flies with homozygous mutant eyes. Candidate motor and chaperone proteins, for which there are existing mutants, could be screened similarly.
In this study, TRP has been confirmed again to play an essential role in the rhabdomeric localization of INAD. Two new alleles of trp are identified here, one affecting a conserved residue in the second N-terminal ankyrin domain (idl-B1 ; L(74)F), and one affecting a highly conserved residue in the S6 domain of TRP (idlB2 ; L(653)H). Each mutation appears to make the TRP protein unstable. Although this may be the result of defective targeting or anchoring of TRP, it is difficult to draw such conclusions since TRP could simply be misfolded with these alterations. Additional structure-function studies may help to determine how TRP anchors INAD in the rhabdomere. For example, no function has been identified for the ankyrin domains on the N-teminus of TRP channels, and one possibility is that these domains may bind components of the actin cytoskeleton, thereby stabilizing INAD-signaling complexes in the rhabdomere. Based on our results, we do not anticipate a lot of genes to be involved in INAD localization, but a similar screen of the X and 2nd chromosome may reveal the few remaining, and perhaps key, players involved in INAD-complex localization.
Methods and Materials
Fly stocks
Fly lines were maintained at 25°C on standard fly food. The following Drosophila stocks were used: w1118 or bw;st as wild type, trpP343,40 trp301,38 chp2,23 and P(inaD-GFP); bw; l29DTS4 D/TM6B (Tsunoda and Zuker, previously unpublished). For P(inaD-GFP) transformants, inaD cDNA was fused in-frame to a C-terminal GFP sequence, and subcloned into the pGMR transformation vector to drive expression in photoreceptors.41 Transgenic fly lines were generated by standard P-element mediated transformation techniques. Third chromosome mutant flies from the Zuker Collection24 were used for the genetic screen. The Zuker identification number is listed in parenthesis for all third chromosome mutants discussed in this manuscript.
Analysis of INAD-GFP in intact eyes
Live flies expressing INAD-GFP were immobilized, with one eye pointing upwards, on double-stick tape mounted to glass microscope slides. Heads of immobilized flies were overlaid with Halocarbon oil 700 (Sigma-Aldrich, St. Louis, MO) and examined by standard epi-fluorescence at low (10X) magnification. In a wild-type background, every ommatidium in the field displayed clear GFP signal in all seven rhabdomeres.
Light/dark exposure
For light-induced retinal degeneration assays, all flies were first dark-raised in a 25°C incubator. Light-exposed flies were placed seven inches from a 15W white fluorescent lamp for 3 days at a light intensity that ranged between 3,100–4,300 lux, unless otherwise stated.
Immunostaining of retinal tissue sections
Flies were either dark-raised, light-exposed (as described above), or reared in the lab on an approximate 12-hour light/dark cycle. Heads were fixed in 3% paraformaldehyde in PBS for 1 hour on ice, washed with PBS, and infiltrated with 2.3 M sucrose in PBS overnight at 4°C. Heads were then bisected, positioned on ultramicrotomy pins (Ted Pella, Redding, CA) and frozen in liquid nitrogen. 1-μm thick sections were cut using a Leica Ultracut UCT attached to an EM FCS cryo unit (Leica Microscopy and Scientific Instruments Group, Heerbrugg, Switzerland) at −81°C. Frozen tissue sections were picked up with a drop of cold 2.3 M sucrose and 1% calf serum in PBS, and then placed on gelatin coated glass slides. Gelatin coated slides were prepared by dipping microscope glass slides in a solution containing 0.5% gelatin (Fisher, Pittsburgh, PA) and 0.05% chromium potassium sulfate (Sigma Aldrich, St. Louis, MO).
For retinal degeneration studies, tissue sections were washed with PBS and mounted in 90% glycerol (Sigma Aldrich, St. Louis, MO). For immunostaining studies, the retinal sections were incubated in blocking solution (1% BSA and 0.1% saponin in PBS) for at least 1 hour at room temperature, followed by 2 hours at room temperature for anti-PKC (1:100), or overnight at 4°C for: anti-INAD (1:1,000), anti-TRPL (1:100), 4C5 (1:50) or 24B10 (1:100). Antibodies against INAD and eye-PKC were received as a gift from C.S. Zuker (University of California, San Diego, CA). Antibodies against Chaoptin (24B10) and Rh1 (4C5) were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). The anti-TRPL antibody was generated by our lab, as described previously.42 After primary antibody incubation, tissue sections were washed (0.1% saponin in PBS) four times, followed by rhodamine-conjugated goat-anti-rabbit (1:200) or FITC-conjugated-anti-mouse secondary antibody (1:500) (Jackson ImmunoReaserch, West Grove, PA) incubation for 1 hour at room temperature. Following several 5 minute washes, the slides were mounted with 90% glycerol and p-phenylenediamine (Sigma Aldrich, St. Louis, MO). Retinal sections were analyzed using an Olympus BX51 fluorescent microscope and Olympus Magna-Fire cooled digital camera, S99806 (Optical Analysis Corporation, Nashua, NH). Images were acquired with MagnaFire 2.1C and edited for presentation in Adobe Photoshop.
Electroretinogram recordings
Dark-raised flies were immobilized with myristic acid (USB Corporation, Cleveland, OH) and dark-adapted for at least 5 minutes before light stimulation. Electroretinogram (ERG) recordings were performed using two glass microelectrodes filled with 1 M NaCl. The recording electrode was positioned on the surface of the fly eye, while the ground electrode was placed into the thorax of the fly. White light stimulation was provided by a xenon arc lamp (Lamba LS 175W; Sutter Instruments, Novato, CA) using a 400–700 nm bandpass filter. Signals were amplified using a differential amplifier (DAM 50, World Precision Instruments, Sarasota, FL) and sampled at 1 kHz (Digidata1322A; Molecular Devices, Sunnyvale, CA). Data traces were acquired using pClamp 8.0 software (Clampfit; Axon Instruments/Molecular Devices, Sunnyvale, CA). Traces were subsequently filtered off-line using a boxcar smoothing filter, at a range between 11–27 smoothing points, depending on noise level. All experiments were conducted at room temperature, under dim red light.
Immunoblot analysis
Immunoblot analysis was conducted with samples obtained from adult fly heads sonicated in 20 μl of cold SDS sample buffer (0.5 M Tris pH 6.8, 10% SDS, glycerol, bromophenol blue and DTT). Protein samples were run on 10% polyacrylamide gels by standard SDS-PAGE techniques, transferred to nitrocellulose membrane and incubated in blocking solution (5% milk in 0.05% Tween in PBS) for at least 30 minutes at room temperature. The blots were then subjected to standard immunoblot analysis using antibodies against: INAD (1:1,000), MAb83F6 (anti-TRP) (1:500), Rh1 (1:100). All incubations were conducted overnight at room temperature. The blots were washed (0.05% Tween in PBS) and exposed to the peroxidase-conjugated AffiniPure goat-anti-rabbit IgG or goatanti-mouse (Jackson ImmunoReaserch, West Grove, PA) secondary antibody (1:5,000) for 1 hour at room temperature. After several 5 minute washes, signal was detected using SuperSignal West Pico luminol/Enhancer and Stable Peroxidase Solutions (Thermo Scientific, Rockford, IL).
DNA extraction, PCR and sequencing
To extract total genomic DNA, flies were frozen in liquid nitrogen in a microcentrifuge tube and homogenized with a pestle to a powder consistency. The sample was vortexed in cold buffer-C (Tris and 50 mM EDTA) containing 10 mg/ml proteinase K and 10% SDS, and incubated at 65°C for 1 hour. Phenol/chloroform extraction of the homogenate was performed to separate proteins from nucleic acids. 3 M sodium azide and cold ethanol were added to the DNA fraction, vortexed and spun for 10 min at 14,000 rpm to precipitate DNA. The DNA pellet was washed with cold 70% ethanol, dried, dissolved in TE with 0.1 mg/ml RNase and placed at 37°C for 30 min before being used for PCR.
The trp gene was isolated from total genomic DNA obtained from (1) bw idl-B1;st, (2) bw idl-B2;st, and the parental (3) bw;st fly strains. Primers designed using Vector NTI software (Life Technologies, Carlsbad, CA), and generated by Invitrogen Corporation (Carlsbad, CA), were used to amplify overlapping ~1 kb fragments of the trp gene. Amplified PCR products were separated on a 1% agarose gel and purified via gel extraction (UltraClean15, Mo Bio Laboratories Inc., Carlsbad, CA). Genomic DNA fragments were sequenced by GENEWIZ, Inc., (South Plainfield, NJ) with the same primers used for amplification. Overlapping sequences were assembled and aligned using Vector NTI software.
Acknowledgements
We thank Dr. Charles Zuker for third chromosome mutant lines, Alex Ramos for his help with the characterization with the idl-B2 mutant and the following individuals who assisted in various aspects of the genetic screen: Rebecca Zee, Shannon White, James Botros, Emily Doughty, Melanie Scully and Vincent DiGiacamo. This work is supported by the National Eye Institute (EYO13751).
Abbreviations
- INAD
inactivation-no-afterpotential-D
- idl
INAD localization
- chp
chaoptic
- PLC
phospholipase C
- TRP
transient-receptor-potential
- TRPL
TRP-like
- PKC
eye-specific protein kinase C
- GFP
green fluorescent protein
- Rh1
rhodopsin
References
- 1.Tsunoda S, Zuker CS. The organization of INAD-signaling complexes by a multivalent PDZ domain protein in Drosophila photoreceptor cells ensures sensitivity and speed of signaling. Cell Calcium. 1999;26:165–71. doi: 10.1054/ceca.1999.0070. [DOI] [PubMed] [Google Scholar]
- 2.Huber A. Scaffolding proteins organize multimolecular protein complexes for sensory signal transduction. The European journal of neuroscience. 2001;14:769–76. doi: 10.1046/j.0953-816x.2001.01704.x. [DOI] [PubMed] [Google Scholar]
- 3.Ranganathan R, Ross EM. PDZ domain proteins: scaffolds for signaling complexes. Curr Biol. 1997;7:770–3. doi: 10.1016/s0960-9822(06)00401-5. [DOI] [PubMed] [Google Scholar]
- 4.Tsunoda S, Sierralta J, Zuker CS. Specificity in signaling pathways: assembly into multimolecular signaling complexes. Curr Opin Genet Dev. 1998;8:419–22. doi: 10.1016/s0959-437x(98)80112-3. [DOI] [PubMed] [Google Scholar]
- 5.Hardie RC, Raghu P. Visual transduction in Drosophila. Nature. 2001;413:186–93. doi: 10.1038/35093002. [DOI] [PubMed] [Google Scholar]
- 6.Ranganathan R, Malicki DM, Zuker CS. Signal Transduction in Drosophila Photoreceptors. Ann Rev Neurosci. 1995;18:283–317. doi: 10.1146/annurev.ne.18.030195.001435. [DOI] [PubMed] [Google Scholar]
- 7.Ranganathan R, Harris GL, Stevens CF, Zuker CS. A Drosophila mutant defective in extracellular calcium dependent photoreceptor inactivation and rapid desensitization. Nature. 1991;354:230–5. doi: 10.1038/354230a0. [DOI] [PubMed] [Google Scholar]
- 8.Smith DP, Stamnes MA, Zuker CS. Signal transduction in the visual system of Drosophila. Ann Rev Cell Biol. 1991;7:161–90. doi: 10.1146/annurev.cb.07.110191.001113. [DOI] [PubMed] [Google Scholar]
- 9.Hardie RC, Peretz A, Suss-Toby E, Rom-Glas A, Bishop SA, Selinger Z, Minke B. Protein kinase C is required for light adaptation in Drosophila photoreceptors. Nature. 1993;363:634–7. doi: 10.1038/363634a0. [DOI] [PubMed] [Google Scholar]
- 10.Dolph PJ, Ranganathan R, Colley NJ, Hardy RW, Socolich M, Zuker CS. Arrestin function in inactivation of G protein-coupled receptor rhodopsin in vivo. Science. 1993;260:1910–6. doi: 10.1126/science.8316831. [DOI] [PubMed] [Google Scholar]
- 11.Scott K, Sun Y, Beckingham K, Zuker CS. Calmodulin regulation of Drosophila light-activated channels and receptor function mediates termination of the light response in vivo. Cell. 1997;91:375–83. doi: 10.1016/s0092-8674(00)80421-3. [DOI] [PubMed] [Google Scholar]
- 12.Scott K, Zuker CS. Assembly of the Drosophila phototransduction cascade into signalling complex shapes elementary responses. Nature. 1998;395:805–8. doi: 10.1038/27448. [DOI] [PubMed] [Google Scholar]
- 13.Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, et al. A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature. 1997;388:243–9. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- 14.van Huizen R, Miller K, Chen D-M, Li Y, Lai Z-C, Raab RW, et al. Two distantly positioned PDZ domains mediate multivalent INAD-phospholipase C interactions essential for G protein-coupled signaling. EMBO J. 1998;17:2285–97. doi: 10.1093/emboj/17.8.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shieh BH, Zhu MY, Lee JK, Kelly IM, Bahiraei F. Association of INAD with NORPA is essential for controlled activation and deactivation of Drosophila phototransduction in vivo. Proc Natl Acad Sci USA. 1997;11:12682–7. doi: 10.1073/pnas.94.23.12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adamski FM, Zhu M-Y, Bahiraei F, Shieh B-H. Interaction of Eye Protein Kinase C and INAD in Drosophila. J Biol Chem. 1998;273:17713–9. doi: 10.1074/jbc.273.28.17713. [DOI] [PubMed] [Google Scholar]
- 17.Shieh BH, Zhu MY. Regulation of the TRP Ca2+ channel by INAD in Drosophila photoreceptors. Neuron. 1996;16:991–8. doi: 10.1016/s0896-6273(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 18.Li HS, Montell C. TRP and the PDZ protein, INAD, form the core complex required for retention of the signalplex in Drosophila photoreceptor cells. J Cell Biol. 2000;150:1411–22. doi: 10.1083/jcb.150.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–31. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 20.Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. Domains, motifs and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu Rev Biochem. 2006;75:655–80. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]
- 21.Tsunoda S, Sun Y, Suzuki E, Zuker C. Independent anchoring and assembly mechanisms of INAD signaling complexes in Drosophila photoreceptors. J Neurosci. 2001;21:150–8. doi: 10.1523/JNEUROSCI.21-01-00150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reinke R, Krantz DE, Yen D, Zipursky SL. Chaoptin, a cell surface glycoprotein required for Drosophila photoreceptor cell morphogenesis, contains a repeat motif found in yeast and human. Cell. 1988;52:291–301. doi: 10.1016/0092-8674(88)90518-1. [DOI] [PubMed] [Google Scholar]
- 23.Van Vactor D, Jr, Krantz DE, Reinke R, Zipursky SL. Analysis of mutants in chaoptin, a photoreceptor cellspecific glycoprotein in Drosophila, reveals its role in cellular morphogenesis. Cell. 1988;52:281–90. doi: 10.1016/0092-8674(88)90517-x. [DOI] [PubMed] [Google Scholar]
- 24.Koundakjian EJ, Cowan DM, Hardy RW, Becker AH. The Zuker collection: a resource for the analysis of autosomal gene function in Drosophila melanogaster. Genetics. 2004;167:203–6. doi: 10.1534/genetics.167.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott K, Becker A, Sun Y, Hardy R, Zuker C. Gqalpha protein function in vivo: genetic dissection of its role in photoreceptor cell physiology. Neuron. 1995;15:919–27. doi: 10.1016/0896-6273(95)90182-5. [DOI] [PubMed] [Google Scholar]
- 26.Niemeyer BA, Suzuki E, Scott K, Jalink K, Zuker CS. The Drosophila Light-Activated Conductance is Composed of the Two Channels TRP and TRPL. Cell. 1996;85:651–9. doi: 10.1016/s0092-8674(00)81232-5. [DOI] [PubMed] [Google Scholar]
- 27.Wakimoto BT, Lindsley DL, Herrera C. Toward a comprehensive genetic analysis of male fertility in Drosophila melanogaster. Genetics. 2004;167:207–16. doi: 10.1534/genetics.167.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laurencon A, Orme CM, Peters HK, Boulton CL, Vladar EK, Langley SA, et al. A large-scale screen for mutagen-sensitive loci in Drosophila. Genetics. 2004;167:217–31. doi: 10.1534/genetics.167.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan WD, Trout WE., 3rd The behavior of four neurological mutants of Drosophila. Genetics. 1969;61:399–409. doi: 10.1093/genetics/61.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montell C, Rubin GM. The Drosophila ninaC locus encodes two photoreceptor cell specific proteins with domains homologous to protein kinases and the myosin heavy chain head. Cell. 1988;52:757–72. doi: 10.1016/0092-8674(88)90413-8. [DOI] [PubMed] [Google Scholar]
- 31.Leonard DS, Bowman VD, Ready DF, Pak WL. Degeneration of photoreceptors in rhodopsin mutants of Drosophila. J Neurobiol. 1992;23:605–26. doi: 10.1002/neu.480230602. [DOI] [PubMed] [Google Scholar]
- 32.O’Tousa JE, Leonard DS, Pak WL. Morphological defects in oraJK84 photoreceptors caused by mutation in R1-6 opsin gene of Drosophila. J Neurogenet. 1989;6:41–52. doi: 10.3109/01677068909107099. [DOI] [PubMed] [Google Scholar]
- 33.Wang T, Montell C. Phototransduction and retinal degeneration in Drosophila. Pflugers Arch. 2007;454:821–47. doi: 10.1007/s00424-007-0251-1. [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Jiao Y, Montell C. Dissecting independent channel and scaffolding roles of the Drosophila transient receptor potential channel. J Cell Biol. 2005;171:685–94. doi: 10.1083/jcb.200508030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004;13:1435–48. doi: 10.1110/ps.03554604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaudet R. A primer on ankyrin repeat function in TRP channels and beyond. Mol Biosyst. 2008;4:372–9. doi: 10.1039/b801481g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosavi LK, Minor DL, Jr, Peng ZY. Consensus-derived structural determinants of the ankyrin repeat motif. Proc Natl Acad Sci USA. 2002;99:16029–34. doi: 10.1073/pnas.252537899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reuss H, Mojet MH, Chyb S, Hardie RC. In vivo analysis of the drosophila light-sensitive channels, TRP and TRPL. Neuron. 1997;19:1249–59. doi: 10.1016/s0896-6273(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 39.Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–9. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott K, Sun YM, Beckingham K, Zuker CS. Calmodulin regulation of Drosophila light-activated channels and receptor function mediates termination of the light response in vivo. Cell. 1997;91:375–83. doi: 10.1016/s0092-8674(00)80421-3. [DOI] [PubMed] [Google Scholar]
- 41.Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–9. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- 42.Cronin MA, Lieu MH, Tsunoda S. Two stages of light-dependent TRPL-channel translocation in Drosophila photoreceptors. J Cell Sci. 2006;119:2935–44. doi: 10.1242/jcs.03049. [DOI] [PubMed] [Google Scholar]