

SUMMARY
Liver cancers are highly heterogeneous with poor prognosis and drug response. A better understanding between genetic alterations and drug responses would facilitate precision treatment for liver cancers. To characterize the landscape of pharmacogenomic interactions in liver cancers, we developed a protocol to establish human liver cancer cell models at a success rate around 50% and generated Liver Cancer Model Repository (LIMORE) with 81 cell models. LIMORE represented genomic and transcriptomic heterogeneity of primary cancers. Interrogation of the pharmacogenomic landscape of LIMORE discovered unexplored gene-drug associations, including synthetic lethalities to prevalent alterations in liver cancers. Moreover, predictive biomarker candidates were suggested for the selection of sorafenib-responding patients. LIMORE provides a rich resource facilitating drug discovery in liver cancers.
Keywords: Liver cancer, patient-derived cancer models, pharmacogenomics, sorafenib
Graphical Abstract

Qiu et al. establish Liver Cancer Model Repository, combining public and newly-generated cell lines, that represents genomic and transcriptomic heterogeneity of Eastern Asian hepatocellular carcinomas and use it to reveal gene-drug associations and potential biomarkers for selecting sorafenib-responding patients.
INTRODUCTION
Primary liver cancers, of which hepatocellular carcinoma (HCC) is the major type, are the second leading cause of cancer-related mortality worldwide (Zucman-Rossi et al., 2015). Limited progress has been made in systemic treatment for liver cancers over the past decade. Sorafenib is the first FDA approved drug for advanced HCC management, and regorafenib and lenvatinib are lately approved. However, due to their low drug response rates, additional improvement is required for their application in clinics (Bruix et al., 2017; Kudo et al., 2018; Llovet et al., 2008). Variable response rates may be partially attributed to different etiologies among countries. For example, most HCCs in China and southeastern Asia are caused by hepatitis B virus (HBV), whereas nonalcoholic-associated steatosis appears to be one of the main causes of HCCs in Western countries (Yang and Roberts, 2010). Large-scale genome sequencing also revealed large heterogeneity in liver cancers, which represents another major challenge in precision treatment of liver cancers (Zucman-Rossi et al., 2015). The use of predictive biomarkers has been proposed to select responsive patients (Holohan et al., 2013), which apparently requires systematic knowledge of pharmacogenomic landscape in liver cancers.
To materialize the precision treatment, it depends on properly modeling cancer heterogeneity using experimental systems. Recent years witnessed an increasing interest in using pan-cancer platforms of widely used cell lines to model cancers and study pharmacogenomics, including Cancer Cell Line Encyclopedia (CCLE), Cancer Therapeutics Response Portal (CTRP) and Genomics of Drug Sensitivity in Cancer (GDSC) (Barretina et al., 2012; Basu et al., 2013; Garnett et al., 2012; Iorio et al., 2016). Because mutational profiles and drug responses differ greatly across cancer types (Garnett and McDermott, 2014), several studies in breast, lung and melanoma cancers have also demonstrated the necessarity to use tissue-specific models (Lin et al., 2008; McMillan et al., 2018; Neve et al., 2006; Sos et al., 2009). However, most of these lines were generated decades ago, lacking proper control or clinical annotations. To better model cancer heterogeneity, great effforts have been made to create in vitro models for various types of cancers (Boj et al., 2015; Broutier et al., 2017; Gao et al., 2014; Lee et al., 2018; Pauli et al., 2017; Sachs et al., 2018; van de Wetering et al., 2015; Vlachogiannis et al., 2018), leading to international collaborations including Human Cancer Model Initiative (HCMI) and Cancer Cell Line Factory (CCLF). Most of these reports focused on generating cancer cell models as a first step, yet had analyzed limited pharmacogenomics (Boehm and Golub, 2015; Williams and McDermott, 2017). To bridge the precision medicine and cancer heterogeneity, it is important to perform a full spectrum of pharmacogenomic characterization of patient-derived cancer models at scale.
For the liver cancer, there are only around 30 cell lines available to the community, which are insufficient to capture the genomic and transcriptomic diversity of this disease (Goodspeed et al., 2016). Moreover, available HCC cell lines underrepresent HBV-associated HCCs, which accounts for more than half of HCCs worldwide. On the top of that, it has been recently reported that many of the widely used HCC cell lines were actually contaminated by HeLa cells (Rebouissou et al., 2017). Therefore, to systematically analyze genetic heterogeneity and drug responses, it is imperative to develop a large panel of patient-derived liver cancer cell models and, accordingly, discover gene-drug associations.
RESULTS
Establishment of Liver Cancer Model Repository (LIMORE)
We built LIMORE by collecting 31 public liver cancer cell lines and generating patient-derived models (Figures S1A and S1B). To generate liver cancer cell models, we optimized the primary culture protocol by adding the ROCK inhibitor Y-27632 and the TGF-β inhibitor A83-01, based on a previous study (Qiu et al., 2016). Y-27632 facilitates attachment of primary cells in vitro whereas A83-01 inhibits mesenchymal cells and supports epithelia cell growth (Katsuda et al., 2017; Liu et al., 2012b). The addition of Y-27632 and A83-01 promoted the success rate of primary culture to 46%, likely allowing long-term survival and proliferation of tumor epithelial cells (Figures S1C and S1D). These models were named as Chinese Liver Cancer (CLC) cell models. In total, 50 models were generated from 49 Chinese HCCs (CLC19 and CLC20 were subclones from the same HCC) with detailed clinicopathological information (Table S1). Among them, 8 were from Edmondson Grade II HCCs and 40 from Edmondson Grade III. These models were enriched in HBV infection (47/50) with other etiologies underrepresented. No significant correlation was found between clinicopathological parameters and the success of model establishment (Table S1). In line with previous findings (Qiu et al., 2016), comparison of cell models and primary cancers from 9 patients suggested that these generated models retained mutational and transcriptional landscapes of original primary cancers (Figures S1E–S1G).
LIMORE consisted of 81 authenticated liver cancer cell models, including 79 HCC models and 2 hepatoblastoma models (Table S1). Compared to CCLE and GDSC that collected 26 and 16 liver cancer models, respectively, LIMORE increased the number by more than 3 times (Figure 1A). LIMORE models represented specific epidemiological characteristics of primary liver cancers, such as the predominance of Chinese patients, the infection of HBV and HCV as the major etiologies, and the high incidence in the male and the aged (Figures 1B and S1H–S1J). Notably, after transplantation into immune-deficient mice, LIMORE cell model-derived cancers showed comparable histopathological features of matched primary HCCs (Figure 1C).
Figure 1. Comparison between LIMORE and primary liver cancers.
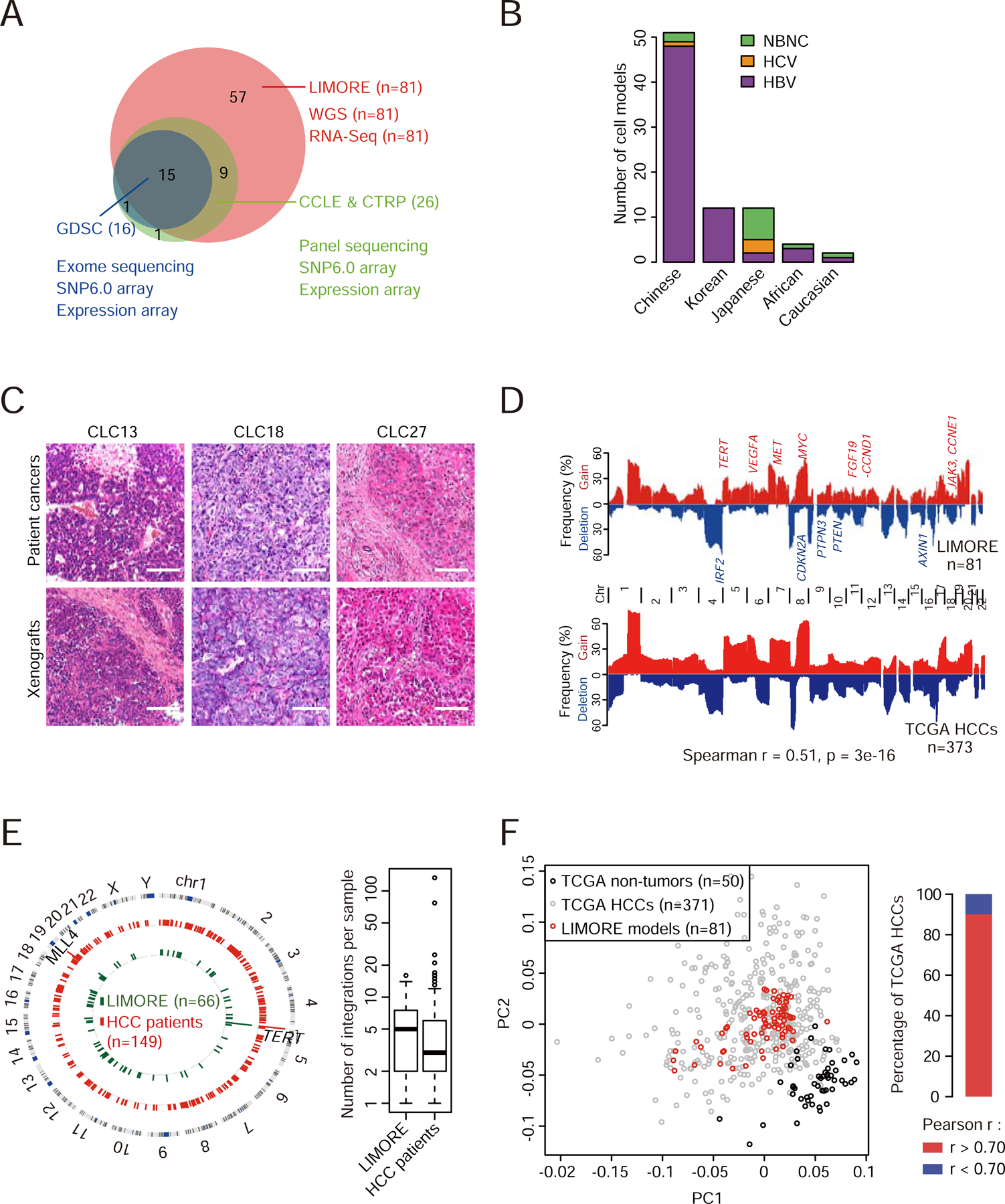
(A) Numbers of cell models in LIMORE and other panels.
(B) Population and virus status of patients whose tumors were used to generate LIMORE models. NBNC, non-HBV and non-HCV.
(C) Representative hematoxylin and eosin (H&E) stainings of subcutaneous tumors from LIMORE models and matched original cancers. Scale bars, 100 µm.
(D) CNA frequencies in LIMORE and TCGA HCCs. Spearman correlation of CNA frequencies is shown. Chr, chromosome.
(E) Circos plot shows HBV integration breakpoints in LIMORE and primary liver cancers (left) and boxplot shows the number of HBV integrations in each LIMORE model and patient sample (right). For box-and-whisker plot, the box indicates interquartile range (IQR), the line in the box indicates the median, the whiskers indicate points within Q3+1.5×IQR and Q1−1.5×IQR and the points beyond whiskers indicate outliers. Q1 and Q3, the first and third quartiles, respectively.
(F) Comparison of gene expressions between LIMORE and TCGA HCCs. Principle component analysis using top 3,000 variable genes (left) and barplot showing the percentage of TCGA HCCs highly correlated with at least 1 LIMORE model (right).
LIMORE Retains Heterogeneity of Primary Liver Cancers
To characterize the extent to which LIMORE represented genomic and transcriptomic landscapes of primary liver cancers, we identified copy number alterations (CNAs), somatic mutations and HBV integrations in 81 models using whole genome sequencing (WGS) (Table S2). Expression profiles were also determined by RNA-Seq. Genetic data were collected from The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017) and other published cohorts to represent primary human liver cancers (Table S2).
Typical CNAs in primary liver cancers were identified by WGS in LIMORE models, including arm-level gains (1q, 8q) and losses (4q, 17p), homozygous deletions of CDKN2A and AXIN1 and focal amplifications containing FGF19 and CCND1. The overall copy number profile of LIMORE models is very similar to that of primary liver cancers (Figure 1D). We further compared CNA profiles and exome somatic mutations between cell models and primary cancers from various types of human cancers. Notably, LIMORE showed the highest correlation with liver cancers (Figures S1K and S1L). Clustering of somatic mutations also showed that LIMORE models grouped closely with 80% of primary liver cancers (Figure S1M and Table S2). A total of 353 HBV integration breakpoints were detected in 60 of 66 HBV-positive models (Figure 1E and Table S2). Breakpoints in TERT, the most prevalent HBV integration site, occurred in 28.6% (16/60) of LIMORE models, a frequency comparable to that of primary liver cancers (Sung et al., 2012). Together, these data indicate that LIMORE models reflect the altered genomic landscape of primary liver cancers.
We next compared transcriptomes between LIMORE models and primary cancers. Principle component analysis showed that LIMORE models were grouped together with TCGA liver cancers (Figure 1F). Moreover, 90% of TCGA liver cancers showed expression profiles correlated with at least 1 LIMORE model (Pearson correlation r > 0.7), indicating that LIMORE retained transcriptomic features of a portion of primary liver cancers. We further determined whether LIMORE retained transcriptome-related functional heterogeneity. Primary liver cancers were reported to be grouped into three subclasses associated with invasion capabilities (Hoshida et al., 2009). LIMORE models were classified to Hoshida S1 (40%), S2 (30%), and S3 (30%) subclasses with slightly higher percentage of S1 (28%–32% in primary HCCs) and lower percentage of S3 (45%–57% in primary HCCs) (Figure S1N). Subclasses of LIMORE models showed good correlations with migration capabilities (Figures S1O and S1P), suggesting that LIMORE models retained some functional heterogeneity of primary liver cancers.
LIMORE Captures Oncogenic Alterations of Liver Cancers
We next characterized whether LIMORE captured major oncogenic alterations in primary liver cancers. Cancer genes were compiled from 6 published cohorts (Table S2). In total, 70 genes with recurrent mutations and 2 genes with CNAs were identified as cancer functional genes (CFGs) of liver cancers (Table S3). 29 CFGs were specific to liver cancers when compared to 27 other types of cancers (Rubio-Perez et al., 2015), including ALB, RPS6KA3 and HNF4A (Figure S2A). These unique alterations highlighted the necessity of liver cancer-specific models.
CFG alterations in primary liver cancers were captured in LIMORE models, including high-frequency alterations in TP53, TERT and FGF19 and low-frequency alterations in HNF4A and NFE2L2 (Figure 2A and Table S3). The overall profiles of CFG alterations were comparable between LIMORE and primary liver cancers (Spearman r = 0.70, p = 7.2e-12, Figure 2B). 10 CFGs, including TP53 and CTNNB1, showed different mutation rates between LIMORE and primary liver cancers (Figure 2B). In total, 61 CFGs (85%) were covered by at least 1 model, and 37 CFGs (51%) were covered by at least 3 models (Figure S2B). By contrast, only 10 (14%) CFGs were covered by at least 3 liver cancer models in previous panels. LIMORE increased the coverage of prevalent CFGs, such as TERT HBV integration (16 vs 3 models) and CTNNB1 activating mutation (8 vs 3 models) and captured CFGs that were not covered by previous panels, including PIK3CA and RPS6KA3 (Figure S2C). For CFGs not retrieved by LIMORE, alteration frequencies were all less than 5% in primary cancers (Figure S2D).
Figure 2. Coverage of oncogenic alterations in primary liver cancers by LIMORE.
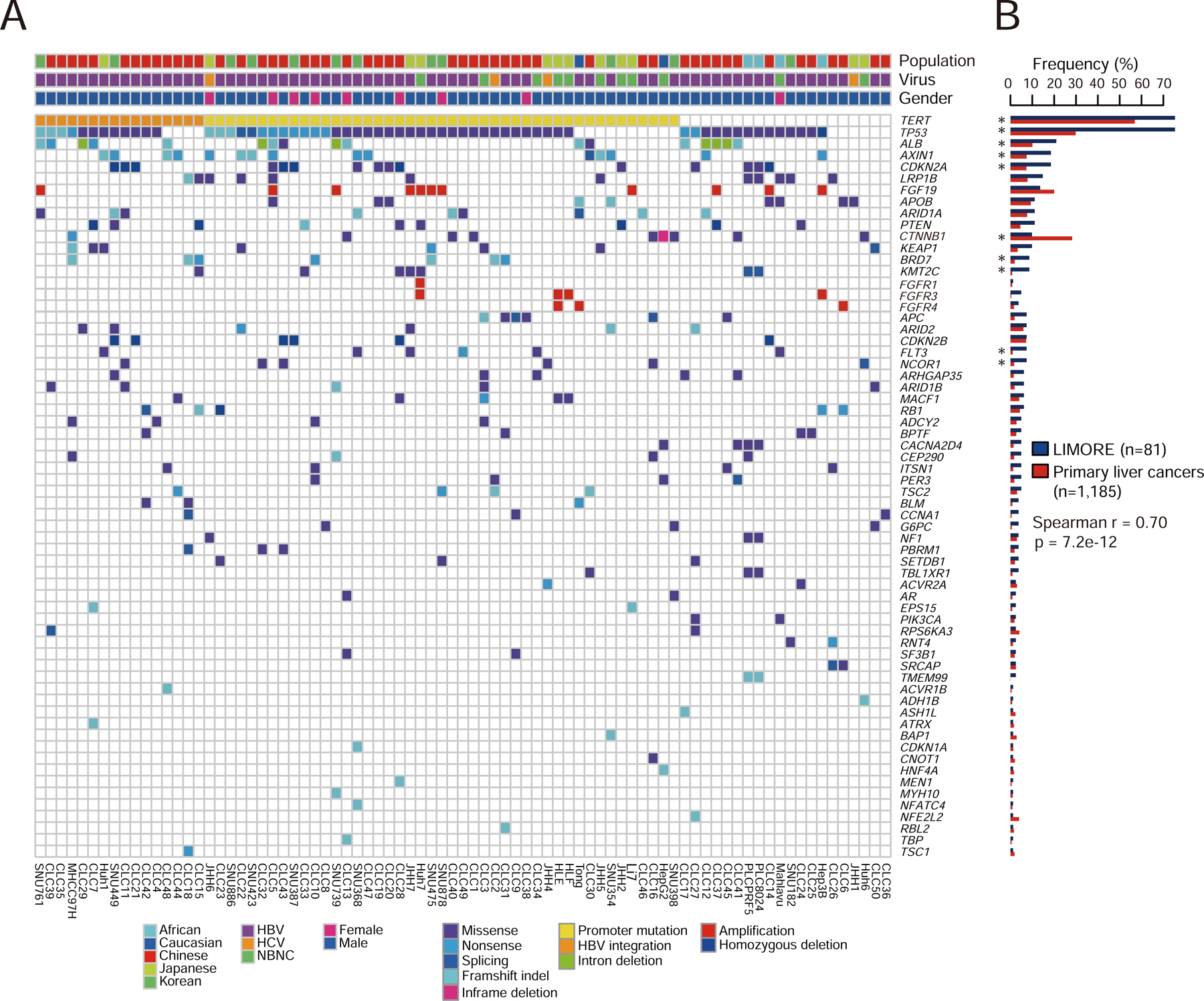
(A) Heatmap shows the alteration landscape of cancer functional genes (CFGs) in LIMORE. Amplifications of FGFR1, FGFR3, and FGFR4 were additionally shown in comparison with FGF19 amplification.
(B) Barplot shows alteration frequencies of CFGs in LIMORE and primary liver cancers. Spearman correlation of CFG frequencies was calculated. Significance of individual CFG frequency between LIMORE and primary liver cancers was determined by Fisher’s exact test. *FDR < 0.05.
Prevalent CFGs in primary liver cancers, such as TERT and Wnt signaling alterations, were observed in LIMORE. Common alterations of TERT in liver cancers were all identified in LIMORE (Figure S2E). In line with that seen in primary liver cancers (Totoki et al., 2014), TERT promoter mutations and HBV integrations were mutually exclusive in LIMORE. Wnt signaling genes (CTNNB1, AXIN1 and APC) were altered in a mutually exclusive manner in > 30% of LIMORE models and primary liver cancers (Figure S2F). Loss-of-function (LOF) alterations were enriched in AXIN1 (15/15) and APC (3/6), while 8 activating mutations were found in the 9 CTNNB1 alterations. MHCC-97H, harboring a heterozygous nonsense mutation at codon 500 of CTNNB1, showed no increase in the expression of β-catenin target genes (Abitbol et al., 2018). Moreover, alterations in potential therapeutic targets, such as FGF19 and MET, were identified in LIMORE models (Figure S2G).
Diversified Drug Responses in LIMORE Models
We next applied an in vitro drug screening in LIMORE (Figure S1A). In total, we compiled 90 anti-cancer drugs, including 15 chemotherapeutic and 75 molecularly targeted drugs against 9 cellular functions (Figure 3A and Table S4). Most of these drugs were approved for clinical use (n=43) or in clinical trials (n=32). All the 81 LIMORE models were screened against this drug panel with 7 or 10 doses for each drug, generating >218,000 measurements of cell-drug interactions. Of the 871 screened plates, the average Z-prime as a control for robustness was 0.84, and in 99% of the plates the Z-prime was greater than 0.5, indicating that the screening assay was experimentally robust (Figures S3A and S3B). Half maximal inhibitory concentration (IC50), maximal effect level (Emax), and activity area (AA) were calculated to reflect drug responses among LIMORE models (Table S4), which showed good correlation with each other (Figure S3C). Analysis of biological replications in 22 randomly selected LIMORE models showed high reproducibility between experiments (Pearson r = 0.91, p < 2.2e-16, Figure S3D). Moreover, a strong correlation of drug responses was observed when a panel of 18 drugs were tested in 6 estabilished models (> 20 passages) and paired early-passage cells (< 10 passages) (Figure S3E), which might reflect the retaination of genomic landscape in LIMORE models during the passage (Figures S1E–S1G) (Qiu et al., 2016). Notably, by comparing 52 drugs and 21 cell models that were also characterized by CTRP or GDSC, we found that response profiles of these drugs were comparable across studies (Figure S3F). We also analyzed the effect of Y-27632 and found that overall drug response profile was not changed by Y-27632 (Figure S3G and Table S4). No signaling pathways were enriched in Y-27632-cultured models (Figure S3H). Interestingly, the response to paclitaxel appeared to be slightly affected in some Y-27632-derived models (Figure S3I).
Figure 3. Diverse drug responses in LIMORE.
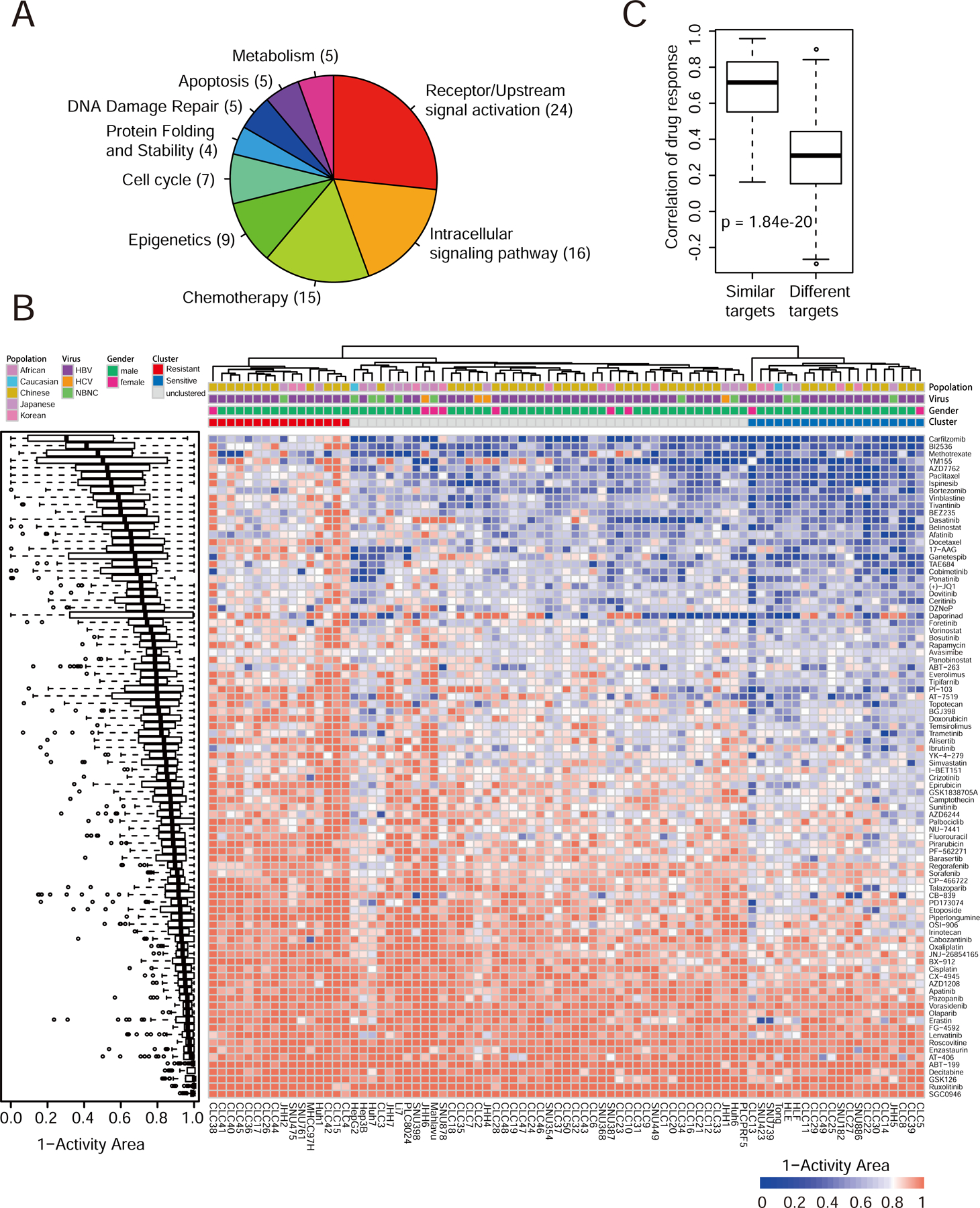
(A) Pieplot shows mechanism of action of 90 screened drugs.
(B) Boxplot shows drug response distributions (left) and heatmap shows drug responses in 81 LIMORE models (right). Red represents resistant cluster (Cluster R) and blue represents sensitive cluster (Cluster S). Drug response value is presented as the 1-Activity Area.
(C) Boxplot shows Spearman correlations of drug pairs with similar or different targets in LIMORE dataset.
For box-and-whisker plot, the box indicates IQR, the line in the box indicates the median, the whiskers indicate points within Q3+1.5×IQR and Q1−1.5×IQR and the points beyond whiskers indicate outliers. See also Figure S3 and Table S4.
Drug response profiles varied among different LIMORE models (Figure 3B). The large variations in drug responses (coefficient of variation range from 0.25 to 3.14) together with the genetic heterogeneity enabled us to discover genetic markers for drug responses. Clustering analysis identified two clusters of LIMORE models (Figure 3B). Cluster R was generally more resistant to drugs than cluster S. Although DNA repair-associated genes were enriched in a subset of cluster S models, common sensitive mechanisms were not obvious for cluster S. Genes involved in drug detoxification and transportation were enriched in cluster R, which was in line with their resistant phenotype (Figure S3J). These data suggest that a significant portion of liver cancers appeared to be resistant to multiple drugs intrinsically, likely due to their high drug-turnover capability. When models were separated according to their status of HBV infection, we found that HBV-positive cell models tended to be less sensitive to doxorubicin and epirubicin but more sensitive to ibrutinib than HBV-negative models (Figure S3K and Table S4).
Drugs with similar mechanism of action (MoA) showd correlated response profiles (Figures 3C and S3L), suggesting the robustness of drug response data. We identified a panel of 26 drugs showing strong inhibition effect with IC50 < 1 µM in at least 25% of LIMORE models. Among them, there were chemotherapeutic drugs, such as doxorubicin and topotecan, likely reflecting their general cytostatic effects. Interestingly, targeted drugs in clinical use for HCC treatment, including sorafenib, regorafenib and lenvatinib, were not in the list, which might be correlated with their relatively low response rates in HCC patients. Nevertheless, we identified other targeted drugs with strong potencies in LIMORE models, including dasatinib and cobimetinib (Figure S3M). Despite the fact that the molecular basis of these drugs is not well understood in liver cancers, this dataset provides drug candidates and their repurposing for liver cancer treatment. Together, these data suggest that high-throughput drug screening in LIMORE captures variable drug responses and provides an opportunity for pharmacogenomic analysis in liver cancers.
The Pharmacogenomic Analysis in LIMORE
To capture the diverse drug response patterns across LIMORE models, we defined the drug responding score (DRS), which was the normalized z-score of the most variable parameter from IC50, Emax and AA for each drug. To define the contributions of CFG and expression features, robust predictive features for drug responses were inferred from elastic net (EN) models with bootstrapping for 1,000 times (Barretina et al., 2012; Garnett et al., 2012). EN score threshold was set to 0.60, which means that the feature was selected in >60% of bootstrapping. A median of 54 mutation features (23 CFGs and 31 noncoding mutations) and 249 expression features were identified as predictive for each drug (Figures S4A and S4B and Table S5). Notably, predictive features included known gene-drug associations, such as associations of MRP1-Etoposide (Moitra et al., 2012) and SLC35F2-YM155 (Winter et al., 2014) (Figure S4C).
Overall, we identified 1,508 significant interactions of CFG-drug pairs, among which 56 pairs were associated with responses to approved liver cancer drugs like sorafenib, regorafenib and lenvatinib (Table S5). In total, 727 CFG-drug pairs were associated with drug resistance, while a cluster of 781 CFG-drug pairs predicted drug sensitivity (Figure 4A). Intriguingly, we found that HBV integration in TERT promoter was associated with drug resistance, whereas TERT promoter mutations were sensitive to a large group of drugs (Figure 4A). Because similar result was obtained when HBV-positive models were specificically analyzed, this finding unlikely resulted from a general etiology driven by HBV infection. Collectively, LIMORE supplied a large set of CFG-drug interactions, which were readily retrievable from the dataset and could be further pursued as biomarkers.
Figure 4. The pharmacogenomic landscape in LIMORE.
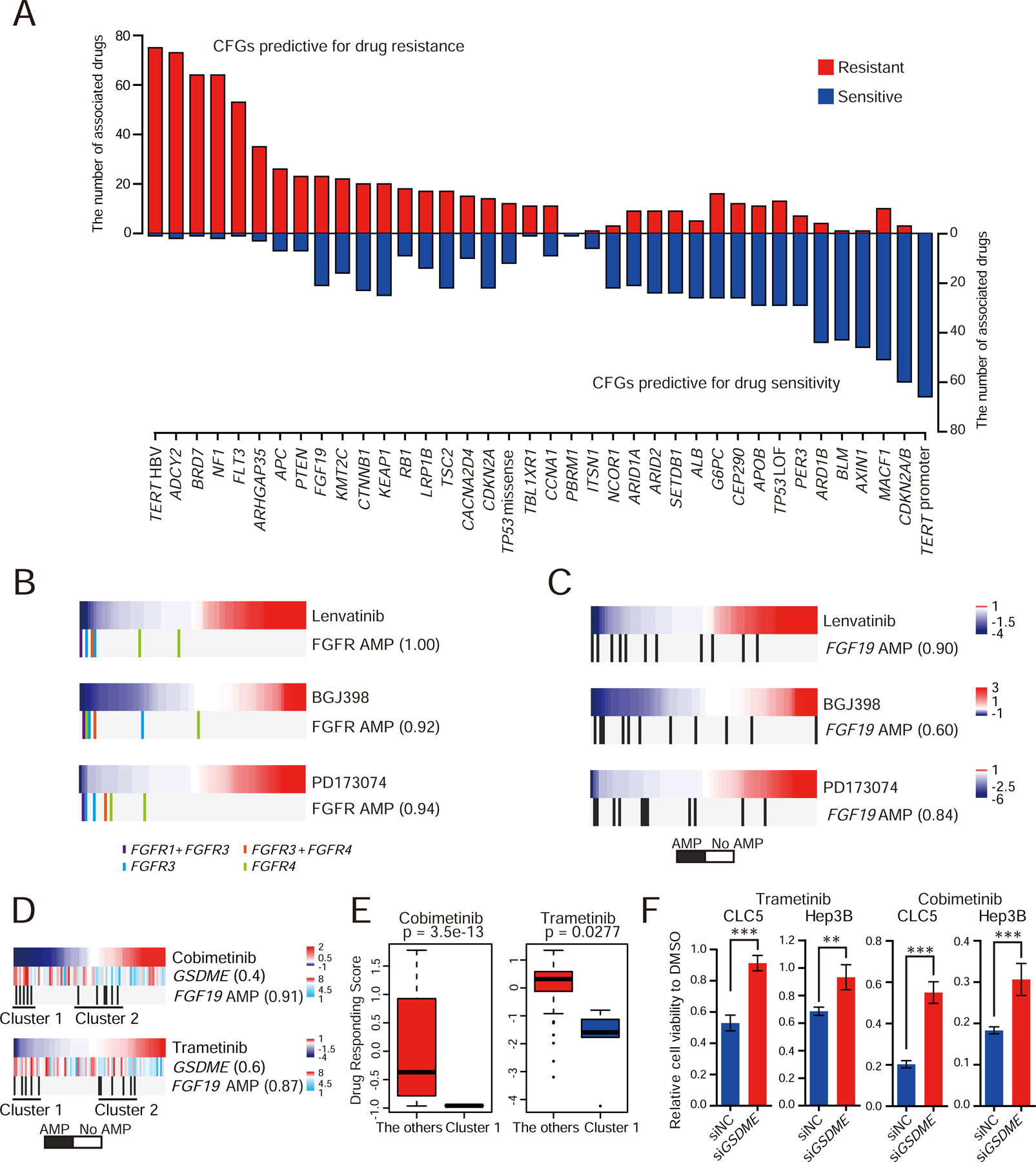
(A) Barplot shows the pharmacogenomic interactions for recurrent CFGs in liver cancers. Red, predicitive for drug resistant; blue, predicitive for drug sensitivity.
(B and C) FGFR inhibitor sensitivity and amplificaitons of FGFR genes (FGFR1, FGFR3, and FGFR4) (B) or FGF19 (C). Color bars indicate Drug Responding Score (DRS). Blue, sensitivity; red, resistance. Vertical bars represent cell models with indicated alterations, FGFR1+FGFR3 and FGFR3+FGFR4 indicate concurrent amplifications of two FGFR genes.The number in parentheses indicates EN score, which is the percentage of models where a feature was selected as predictive in bootstrapping. AMP, copy number amplification.
(D) Predictive biomarkers and sensitivity to MEK inhibitors. Rank-ordered drug responses are shown in upper panel. Blue, sensitivity; red, resistance. GSDME expression and FGF19 amplification in LIMORE models are shown below. Cluster 1 are models with both GSDME overexpression and FGF19 amplification. The number in parentheses indicates EN score.
(E) Boxplots show the DRSs for MEK inhibitors in Cluster 1 subgroup and the other LIMORE models. For box-and-whisker plot, the box indicates IQR, the line in the box indicates the median, the whiskers indicate points within Q3+1.5×IQR and Q1−1.5×IQR and the points beyond whiskers indicate outliers. Statistics, unpaired Student’s t-test.
(F) Relative cell viability of GSDME-knockdown CLC5 and Hep3B cells treated with Cobimetinib (0.625 µM) or Trametinib (0.5 µM) for 72 hr. Experiments were biologically repeated in triplicate and one representative result is shown. Data are presented as mean±SD.
**p < 0.01, ***p < 0.001 by unpaired Student’s t-test. See also Figure S4 and Table S5.
The interaction between FGF/FGFR and anti-cancer drugs was ranked top in CFG-drug interaction list. FGFR inhibitors, including lenvatinib, BGJ398 and PD173074, showed selective sensitivity to amplifications of both FGFR (including FGFR1, 3 and 4 with copy number >=4) (Figure 4B) and FGF19 (Figure 4C) in liver cancer cells. These data suggested that FGF19 and FGFR amplification may serve as biomarkers for lenvatinib. In concordance with the role of FGF19 amplification in the MAPK pathway activation (Zucman-Rossi et al., 2015), FGF19 amplification correlated with the sensitivity to MEK inhibitors (MEKi) cobimetinib and trametinib (Figure 4D). Notably, MEKi-induced cell death showed morphological change of large bubbles from membranes, a typical characteristic of pyroptosis (Figure S4D). Accordingly, the sensitivity to MEKi moderately correlated with high expression of GSDME, a key regulator of pyroptosis (Figure 4D). While FGF19 amplification or GSDME overexpression alone predicted MEKi sensitivity, cell models with both FGF19 amplification and high GSDME expression were extremely sensitive to MEKi (Figures 4D, 4E and S4E). Depletion of GSDME expression led to reduced MEKi sensitivity in FGF19-amplified models (Figures 4F and S4F). These data together indicated MEK inhibition as a selective vulnerability to liver cancers with FGF19 amplification and GSDME overexpression.
LIMORE Identifies Drugs Targeting Synthetic Lethal Interactions
Some CFGs, such as CTNNB1, were considered as undruggable, but could be exploited for therapies if proper synthetic lethal interactions were established (Naik et al., 2009). A cluster of drugs were found to preferentially eradicate LIMORE models with CTNNB1 activating mutations (Figure 5A). CTNNB1 activating mutations correlated with sensitivity to HDAC inhibitors, panobinostat, vorinostat and belinostat (Figures 5B and 5C, Cohen’s d 0.69, 0.67 and 0.43, respectively), which was in line with the finding that HDACs were required for the β-catenin signaling (Billin et al., 2000). HDACs may function redundantly in mediating drug sensitivity, because knockdown of individual HDACs did not apparently impair cell proliferation in β-catenin activated models (Figure S5A). Expression of a constitutively active form of β-catenin in β-catenin wide-type models increased their sensitivity to HDAC inhibitors (Figures 5D and S5B). When transplanted in vivo, panobinostat inhibited the growth of CTNNB1 mutant models but not CTNNB1 wild-type models (Figures 5E and S5C). Moreover, over-expression of activated β-catenin endowed panobinostat sensitivity to CTNNB1 wild-type JHH7 in vivo (Figures 5F, S5D and S5E). These data suggested HDAC inhibition as a potential strategy to target HCCs with CTNNB1 activating mutations.
Figure 5. Synthetic lethal interactions with Wnt and MYC activation.
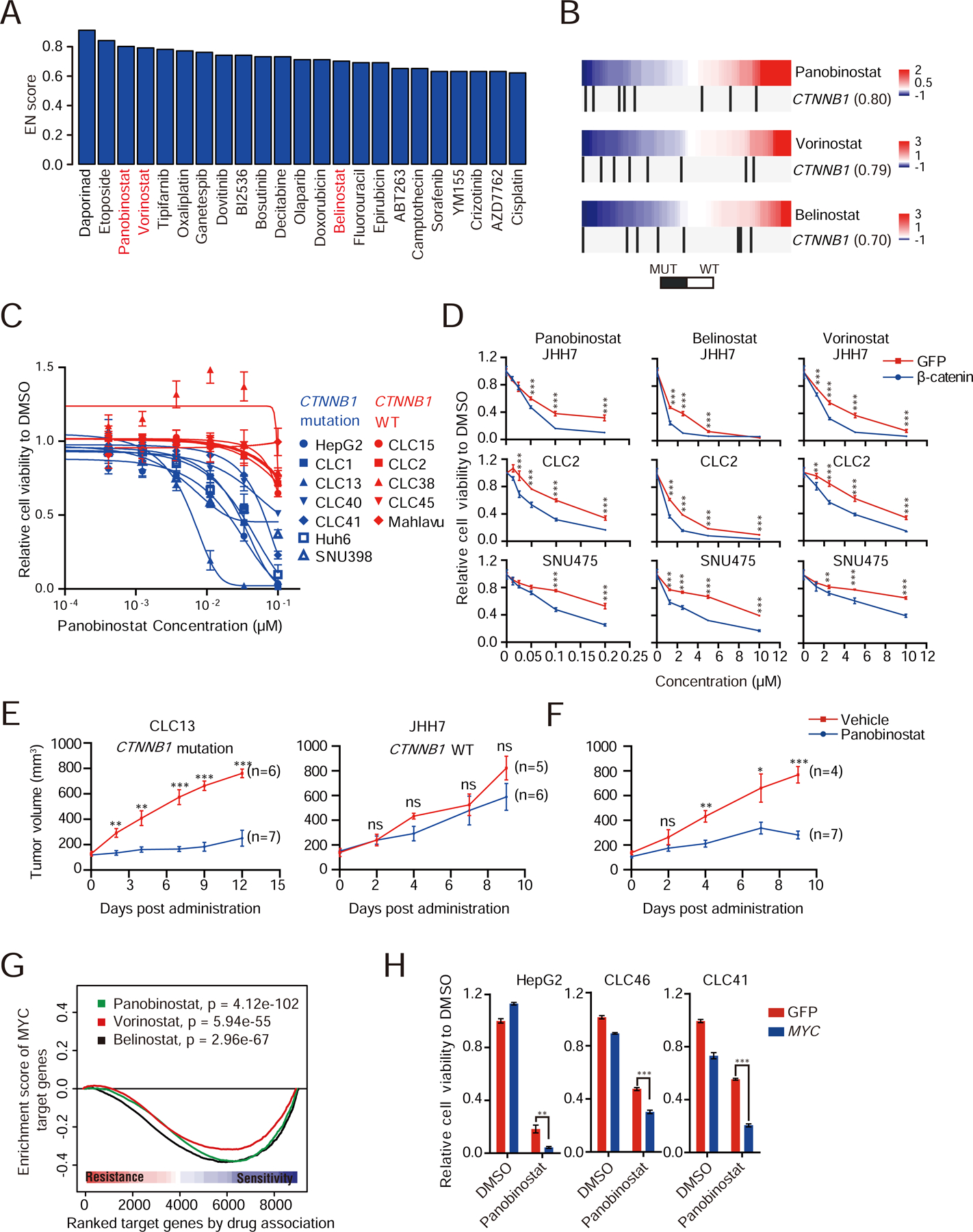
(A) Barplot shows the drugs targeting synthetic lethal interactions with CTNNB1 mutations.
(B) HDAC inhibitor sensitivity and CTNNB1 activating mutations. Color bars indicate DRS. Blue, sensitivity; red, resistance. Vertical black bars represent cell models with CTNNB1 activating mutations.
(C) Dose response curves for LIMORE models with (blue) or without (red) CTNNB1 activating mutations. Data are presented as mean±SD.
(D) Dose response curves of indicated models ectopically expressing ∆N90-β-catenin and treated with indicated drugs for 72 hr. Data are presented as mean±SD. Experiments were biologically repeated in triplicate and one representative result is shown.
(E) Tumor growth curves of CLC13 and JHH7 treated with panobinostat or vehicle. Data are presented as mean±SEM.
(F) Tumor growth curves of JHH7 after ∆N90-β-catenin overexpression treated with panobinostat or vehicle. Data are presented as mean±SEM.
(G) Enrichment plot of MYC-regulated transcription program and drug responses. The curve represents enrichment scores of MYC targets ranked by correlation with drug responses.
(H) Relative cell viabilities of HepG2, CLC46 and CLC41 cells overexpressing MYC and treated with 0.2 µM panobinostat for 72 hr. Data are presented as mean±SD. Experiments were biologically repeated in triplicate and one representative result is shown.
*p < 0.05, **p < 0.01, ***p < 0.001 by unpaired Student’s t-test. See also Figure S5.
MYC is another undruggable oncogenic protein in HCCs. Recent evidence suggested that the Wnt pathway crosstalks with MYC-mediated transcription in hepatoblastoma and colorectal cancer (Cairo et al., 2008; Sansom et al., 2007). Indeed, by analyzing published ChIP-Seq data of TCF4/7, key transcription factors of Wnt signalling, and MYC, we found that Wnt targets were significantly co-bound by both TCF4/7 and MYC (Figure S5F). Using a transcription factor-based analysis, we found that MYC-regulated transcription program correlated with sensitivity to HDAC inhibitors (Figures 5G and S5G, Cohen’s d 0.91, 0.60 and 1.18, respectively). The increased sensitivity to HDAC inhibitors was validated by MYC overexpression (Figures 5H and S5H). When the prediction power for HDAC inhibitors was assessed, MYC-regulated transcription program appeared to be comparable to or slightly stronger than CTNNB1 mutations (75%–81.8% vs 62.5%–75%). These data together showed that pharmacogenomics landscape in LIMORE could be interrogated to find potential synthetic lethal strategies for undruggable oncogenes in liver cancers.
Prediction Models and Biomarker Candidates for Sorafenib
Sorafenib-related CFGs and gene expressions were of great interest, as sorafenib is the widely-used standard of care for HCCs. 51 mutation features (18 CFGs and 33 noncoding mutations) and 77 expression features were identified predictive for sorafenib (Table S5). Among top predictive CFG features for sorafenib resistance was KEAP1 (Figure 6A). Mutations of KEAP1 correlate with activation of NRF2 signaling and sorafenib resistance (Sun et al., 2016). Indeed, downstream targets of NRF2 were highly expressed in KEAP1-mutated LIMORE models (Figure S6A). Moreover, NRF2 knockdown increased sensitivity of KEAP1-mutated models to sorafenib (Figures S6B and S6C), supporting the role of the KEAP1/NRF2 pathway in sorafenib resistance. We also identified expression features for sorafenib sensitivity (Figure 6A). As an example, EZH2 expression was highly associated with sensitivity to sorafenib (Figure 6A). This was confirmed by the finding that EZH2 knockdown increased resistance to sorafenib (Figures S6D and S6E). Pharmacologic inhibition of EZH2 by DZNep showed antagonistic effects with sorafenib in 33 of 46 LIMORE models (CDI>1), further suggesting that EZH2 overexpression might synergize with sorafenib (Figures S6F and S6G). Collectively, these data unveiled the molecular features associated with sorafenib sensitivity.
Figure 6. Prediction models and biomarkers for sorafenib.
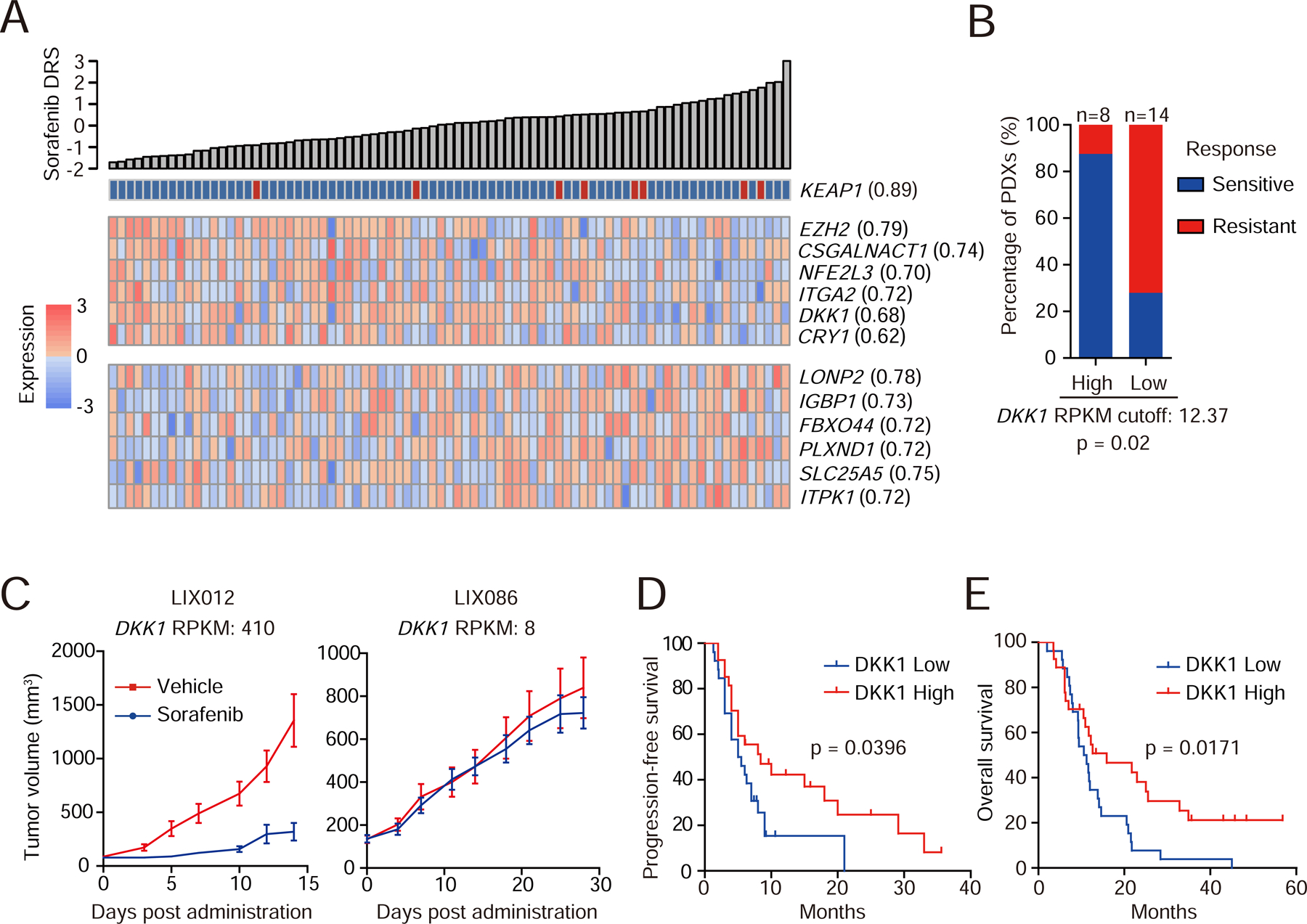
(A) Heatmap shows top predictive CFG and expression features associated with sorafenib response.
(B) The 22 PDXs were classified into DKK1-high or -low group using the optimal cutoff. PDXs with treatment-to-control ratio < 0.25 were considered sensitive to sorafenib, otherwise resistant. Gene expressions in RNA-Seq are presented as Reads Per Kilobase per Million mapped reads (RPKM). Statistics, Fisher’s exact test.
(C) Tumor growth curves of two representative PDXs (LIX012 and LIX086) with different DKK1 levels. Data are presented as mean±SEM. n=10 mice/group.
(D and E) Kaplan-Meier plots for progression-free (D) and overall (E) survival of HCC patients grouped by the median of serum DKK1 levels. Statistics, Log-rank test.
We next explored whether the pharmacogenomic landscape could be translated into predictive models or biomarkers for sorafenib. Elastic net regression models were developed based on response-related mutation and expression features in LIMORE models (Figures S6H and S6I). Prediction performance was assessed by Spearman correlation between predicted and detected responses in LIMORE (Iorio et al., 2016). To evaluate the prediction model in vivo, we analyzed the sorafenib prediction model using an independent dataset of 22 HCC PDXs with sorafenib treatment and found significant correlation between predicted and experimental responses (Figure S6J and Table S6).
We then searched biomarker candidates for potential clinic practice. DKK1 was of particular interest (Figure 6A), because it is involved in the Wnt signaling (Niida et al., 2004). We first characterized the prediction power of DKK1 in PDX models. The optimal cutoff to distinguish high and low DKK1 mRNA levels in PDXs was determined by ROC analysis (Figure S6K). Notably, PDXs with high DKK1 levels showed increased response rate to sorafenib (Figures 6B and 6C), suggesting that DKK1 expression might predict sorafenib response in vivo.
We then investigated whether DKK1 would be a possible biomarker to predict sorafenib response in patients. DKK1 is a secreted protein that can be measured in serum, we thus analyzed the correlation between patient’s serum DKK1 levels and their sorafenib response. Serum samples from 54 HCC patients either before or after sorafenib treatment were retrospectively collected, and DKK1 levels were measured (Table S6). Patients in the DKK1-high group had longer progression-free survival and overall survival (Figures 6D and 6E). When only patients whose serum samples were collected before sorafenib treatment were analyze, similar trends were observed (Figures S6L and S6M). Given that high DKK1 expression was associated with poor survival of HCC patients without sorafenib treatment (Tao et al., 2013; Tung et al., 2011), these data suggested that DKK1 might be a serum biomarker to select sorafenib-responding patients.
DISCUSSION
Liver cancer-specific pharmacogenomics analysis requires a large number of models. Yet liver cancer models are not prioritized in the international initiations HCMI or CCLF (Boehm and Golub, 2015; Williams and McDermott, 2017). Our study is in line with these efforts to generate representative cancer models and focuses specifically on liver cancer. Compared to 31 established cell lines, LIMORE models were characterized by well-annotated clinicopathological information. By increasing the number of models, LIMORE better captures the heterogeneity of primary liver cancers. Cancer organoids are recently developed as in vitro models. Compared to generating HCC orgnaoids (Broutier et al., 2017; Nuciforo et al., 2018), LIMORE provided a relatively higher efficiency (50% vs ~20%). It is also worth noting that 8 LIMORE models were established from Grade II HCCs, which were reported to fail to grow as organoids (Nuciforo et al., 2018). Organoids are superior in maintaining cancer tissue architecture, 2D-cultured cells nonetheless could form authentic cancer structures in vivo. Moreover, 2D-cultured cells are relatively easy to passage and expand in a large quantity, making LIMORE models amenable to large-scale pharmacogenomics analysis.
In this study, LIMORE models were developed from resected cancers, so that it was not possible to compare the exact in vivo response of these cancers with LIMORE models. In the future, to directly compare in vitro and in vivo drug responses, it would be necessary to generate models from biopsies or circulating liver cancer cells with annotated responses to drug treatment. Moreover, as most of LIMORE models were each established from one sub-clone of the primary culture, it is important to generate multiple models from the same HCC to study intratumoral heterogeneity. Addition of ROCK inhibitor Y-27632 significantly increased the success rate of primary culture of liver cancer cells. It remains unclear whether ROCK inhibitor affected the establishment of cell models in regarding to their drug response. Our data suggested that it may have limited effect on drug responses of established cell models. In addition, because LIMORE models could be maintained independent of Y-27632 and A83-01, it is possible to use LIMORE models in medium free of these compounds.
Because of the improved protocol, LIMORE models increase the coverage of liver cancer CFGs. For CFGs not covered by LIMORE models currently, additional representative models could be generated. However, given the low percentage of these remaining CFGs in liver cancers, it would require a large amount of investment. Alternatively, it is possible to introduce CFGs of interest specificially into functional hepatocytes generated by reprogramming technologies (Gao et al., 2017). It is notable that compared to TCGA primary HCCs, frequencies of several CFGs, such as TP53 and CTNNB1, were significantly different in LIMORE. Although concurrent mutations of TP53 and CTNNB1 were at a frequency (6%) concordant with that in TCGA HCCs (6%), TP53 mutation rate was higher and CTNNB1 was lower in LIMORE. It is possible that LIMORE were mainly derived from HBV-positive HCCs, which showed high TP53 and low CTNNB1 mutation rates (Hsu et al., 2000; Levrero and Zucman-Rossi, 2016; Qi et al., 2015). Moreover, TP53-mutated liver cancer cells may survive better during the culture (Caruso et al., 2019). For transcriptome, LIMORE models covered a subset of primary HCCs as shown by PCA analysis, and our pervious study has revealed that genes related to cell cycle and extracellular matrix were affected by in vitro culture (Qiu et al., 2016).
LIMORE provides sufficient cell models to evaluate drug potency and efficacy in consideration of intertumoral heterogeneity. It should be noted, however, that LIMORE models were mainly developed from HBV-positive HCCs and showed limited representation of other etiologies. Although HBV-positive cancer cells showed some specificity, such as the resistance to doxorubicin and epirubicin and the sensitivity to ibrutinib, a large part of drug responses were found to be shared between HBV-positive and HBV-negative models. Interrogation of drug screening matrix revealed a list of drugs with better potency than currently approved therapies for HCCs. Some of these drugs, such as dasatinib and cobimetinib, have been approved in other cancer types. By additional validation, more drugs and biomarkers might be repurposed and tested for liver cancers, thus accelerating drug development for HCCs.
A goal of cell platform is offering prediction of therapeutic response from genetic profiles (Goodspeed et al., 2016). In GDSC, genomic features performed better than gene expression in tissue-specific predictions (Iorio et al., 2016), which was not observed in LIMORE. This might be explained by the fact that, unlike other cancers, druggable alterations are rare in liver cancers. On the other hand, target alterations may not always predict drug sensitivity, e.g. some LIMORE models with FGF alterations did not respond to FGFR inhibitors. These complexities highlight the importance of a large collection of liver cancer models. LIMORE showed the power of pharmacogenomic analysis using WGS, RNA-Seq and drug sensitivity from a large collection of models and identified genetic alterations and expression markers for the selection of drug-repsonsive patients.
We retrospectively analyzed DKK1 as a predictive biomarker candidate for sorafenib. It was reported that serum DKK1 could complement AFP in HCC diagnosis and were associated with poor diagnosis of HCCs (Shen et al., 2012). We found that the correlation between DKK1 and sorafenib response was consistently observed in vitro, in PDXs, and in patients. To further confirm the predictive value of serum DKK1, it is important to perform a perspective clinical study and to trace DKK1 levels during the sorafenib treatment. We wish to emphasize that in addition to above examples, other gene-drug interactions, especially those related to regorafenib, lenvatinib and cabozantinib, were readily retrievable from supplementary data or the online website (www.picb.ac.cn/limore/ or http://limore.sibcb.ac.cn/).
In summary, our study defines a framework of using cell platform to improve drug response in human HCC. We built a knowledge base on gene-drug interactions in liver cancers, which, if properly validated, could help clinical design and accelerate precision medicine in liver cancers. For the community, LIMORE represents a rich resource to choose proper models and provide an opportunity to study pharmacogenomics of liver cancers. Recently, a study has characterized drug response using 34 public liver cancer cell lines (Caruso et al., 2019). Together, these efforts help to understand pharmacogenomics of liver cancers. With other techniques, such as gene editing and organoid culture (Gao et al., 2014; Sachs and Clevers, 2014), LIMORE could be further improved to model liver cancer heterogeneity in genomics and drug response.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lijian Hui (ljhui@sibcb.ac.cn). Liver cancer cell lines generated in this study are publicly available from the Center of Cell Resources, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, and will be distributed under Material Transfer Agreement (MTA).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Models
A total of 31 publicly available liver cancer cell lines were collected from cell line banks or as gifts from other labs. We have also generated 50 HCC cell models from Chinese HCC tissues as in vitro HCC models, 9 of which have been reported previously (Qiu et al., 2016). Culture condition, authentication and resources were provided in Table S1. All the cell models are authenticated by Short Tandem Repeat (STR) analysis using primers from Powerplex 1.1 kit (Promega) and compared to Database of Cross-contaminated or Misidentified Cell Lines (Yu et al., 2015) to avoid cell line contamination.
Patient Samples
Surgically resected tumor samples were collected from Chinese patients diagnosed with hepatocellular carcinoma (HCC). The tissue resource hospitals included Eastern Hepatobilliary Surgery Hospital, The First Affiliated Hospital of Nanjing Medical University, Zhongshan Hospital and the Affiliated Drum Tower Hospital of Medical School of Nanjing University. The pathologies of the tissues were confirmed by the department of pathology in the hospitals. This study was approved by the ethical committees of Eastern Hepatobilliary Surgery Hospital, The First Affiliated Hospital of Nanjing Medical University, Zhongshan Hospital and the Affiliated Drum Tower Hospital of Medical School of Nanjing University, and the informed consent was obtained from the patients involved in this study. Methods were carried out in accordance with the approved guidelines. Fresh tumor tissues were subjected to preparation of single cell suspensions and subsequent primary culture. The study of DKK1 was approved by the ethical committee of Fudan University Shanghai Cancer Center, and the informed consent was obtained from the patients. Peripheral serum samples from 54 Chinese HCC patients with sorafenib treatment in Fudan University Shanghai Cancer Center were collected and stored at −80 °C until use.
Animals
4 weeks-old immuno-deficient athymic BALB/c-nu/nu male mice were purchased from SLAC Laboratory Animal, China. 5–6 weeks-old NOD.CB17-Prkdcscid/scid/shjh (NOD/SCID) male mice were purchased from Shanghai Jihui Laboratory Animal Care, China. 6–8 weeks-old immuno-deficient athymic BALB/c-nu/nu female mice were purchased from Beijing Vital River Laboratory Animal Technology, China. The mouse experiments were approved by the Institutional Animal Care and Use Committees (IACUCs) of Shanghai Institute of Biochemistry and Cell Biology and Shanghai ChemPartner, respectively, and performed in accordance with the approved protocols.
METHOD DETAILS
Cell Model Generation
To generate cell models from clinical specimens, we have reported a protocol using primary culture medium (Qiu et al., 2016). The primary culture medium was RPMI1640 supplemented with 10% fetal bovine serum (FBS), 1 × ITS (Insulin, Transferrin, Selenium Solution) and 40 ng/mL EGF (epithelial growth factor). Currently, we have been generating liver cancer cell models using a modified protocol with the addition of ROCK (Rho-associated coiled-coil-containing kinase) inhibitor Y-27632 (10 µM) in primary culture medium (termed as ROCKi medium) or both Y-27632 (10 µM) and TGFβ inhibitor A83-01 (5 µM) in primary culture medium (termed as YA medium) (Table S1). The success rate of generating cancer cell models from Chinese HCCs is up to 50%. Briefly, the fresh tissues were surgically resected from Chinese HCC patients. Either apparently necrotic or normal tissues were discarded. The remainder was finely minced with scissors to small fragments (1 to 2 mm in diameter) and digested by 0.1% Collagenase Type IV in phosphate buffer saline (PBS) for 30–90 min at 37 °C. Cell suspensions were then filtered by 70 μm cell strainer and centrifuged consecutively at 1000 rpm, 800 rpm and 600 rpm for 5 min. Cancer cells were re-suspended in ROCKi and YA medium and transferred to rat collagen I-coated dishes for culture in a humidified incubator at 37 °C with 5% CO 2. ROCKi or YA medium were changed every three days. Frequent microscopic examination of the cell culture is required to check the proliferation and expanding of epithelial cell clones. Picking out epithelial clones was used to purify epithelial cells and avoid fibroblast contamination. Once confluent, epithelial cells were digested by 0.05% trypsin-EDTA for passage at a ratio of 1:3 (1:5 after passage 10 for most of the cell models). Collagen coating was withdrawn after 10 passages. So far, we have generated 50 liver cancer lines from 49 Chinese HCC patients. We usually kept one subclone from each cancer sample for long-term propagation, except that CLC19 and CLC20 were two subclones derived from the same HCC. Because the two clones show distinct morphologies and gene expression profiles of liver marker genes, we remained them in further analysis.
Collecting Public Liver Cancer Cell Lines
A total of 31 publicly available liver cancer cell lines were collected from cell line banks or as gifts from other labs. An estimation of around 30 liver cancer cell lines are publicly available in several cell line banks, which were reported to be derived from HCC, hepatoblastoma (HepG2 and Huh-6 clone 5) and hepatocellular adenomas (SK-HEP-1). However, SK-HEP-1 was reported to be of endothelial origin, and was excluded in our analysis. We collected 27 authenticated liver cancer cell lines from cell line banks, including American Type Culture Collection (ATCC, www.atcc.org), Japanese Collection of Research Bioresources (JCRB, http://cellbank.nibiohn.go.jp/english/), Korean Cell Line Bank (KCLB, https://cellbank.snu.ac.kr/english/) and Cell Bank of Chinese Academy of Sciences in Shanghai (www.cellbank.org.cn). In addition, 2 cell lines, MHCC-97H and PLC8024, are gifts from Terence Kin Wah Lee (The University of Hong Kong). PLC/PRF/5 and PLC8024 share the same STR profile, but are included due to the different morphologies. MHCC-97H was established from Chinese HCC in Liver Cancer Institute, Fudan University. 2 cell lines, Mahlavu and Tong, are gifts from Yuh-Shan Jou (Institute of Biomedical Sciences, Academia Sinica). In addition, HLE and HLF are subclones derived from the same patient, but show different morphologies.
Transwell assay
50,000 cells were suspended in 200 µL serum-free medium and added to the upper compartment of Transwell insert (Corning, 12-well plate, pore size 8 µm). The lower compartment was added 600 µL culture medium with 10% FBS. After culture for 14 hr, cells were fixed in ethanol for 10 min and stained by crystal violet. At least 5 fields were randomly selected and counted under light microscopy. Assays were performed in triplicate or twice.
Subcutaneous xenograft
Two million cells were harvested in 150 µL PBS and injected into the hind legs of 3 immuno-deficient athymic mice (BALB/c-nu/nu, 4-week-old, male). Tumor formation was examined for up to four months or until the tumors reached a volume of 2000 mm3.
To assess the efficacy of the HDAC inhibitor panobinostat in vivo, NOD/SCID mice (5–6 weeks old) were injected with 2–5 million tumor cells subcutaneously. Randomization into 2 groups was performed when tumors reached a volume of 100–350 mm3. Panobinostat or vehicle was injected intraperitoneally at 15 mg/kg daily for 5 days and subsequently 2 days off for the first week. Treatments were continued every other day for another week due to the toxicity of panobinostat (Lachenmayer et al., 2012). Animals were euthanized and tumors were collected when reaching around 1,000 mm3 in volume or body weight loss > 30%.
Whole Genome Sequencing
Genomic DNA was extracted using Qiagen DNeasy kit. All 81 LIMORE models have been subjected to whole genome sequencing. Whole-genome DNA from cell models was used to construct sequencing libraries and sequenced on the Illumina HiSeq X TEN platform with 2x150 bp paired-end reads. DNA sequencing was performed by WuXi NextCODE, Macrogene and Novogene. Raw sequencing reads were filtered to remove low-quality reads. The remaining reads were mapped to human reference genome (HG38) using BWA algorithm (Li and Durbin, 2009) with default parameters. Genome Analysis Toolkit (GATK) (DePristo et al., 2011) was applied to call variants from DNA sequencing data. The HaplotypeCaller in GATK was ran per sample to call variants and generate a corresponding gVCF file. Multiple-sample joint genotyping was performed on multiple gVCF files to obtain a combined VCF file. The called variants were scored using the recalibration model. Low-quality variants were removed. Functional effects of variants were annotated by ANNOVAR (Wang et al., 2010). Because most of existing liver cancer cell models are lacking paired normal tissues, we applied the commonly used pipeline to call putative somatic mutations in cancer cell model analysis (Iorio et al., 2016; Liu et al., 2012a; Mouradov et al., 2014). The variants were compared to the known germline variation databases, including dbSNP138, 1000 Genomes, ESP6500 and ExAC v0.3. We removed variations with frequencies higher than 0.1% in any of these databases, and variations which occurred in normal samples from our other sequencing projects. The variations would be retained if it was present in at least 10 cancer patients in COSMIC database. The remained variations were regarded as putative somatic mutations. Of note, lagre InDels have not been explored in this study due to limitations in sequencing and analytical methodology.
Control-FREEC (Boeva et al., 2011) was used to obtain copy number profiles as previously described (Qiu et al., 2016). Control-FREEC could calculate copy number (CN) profiles without paired normal. We used the default parameters with the genomic window size set to 50k bp. Whole genome gene copy number alteration (CNA) profiles were obtained using GISTIC 2.0 (Mermel et al., 2011). The copy numbers of cancer functional genes (CFGs) were manually curated in Integrative Genomic Viewer (IGV) tool (Robinson et al., 2011). The visualization of CNA was done in IGV. CNA status were defined as below in specific gene evaluation: Normal, CN=2; deletion, CN=1; homozygous deletion, CN=0; gain, CN=3 or 4; amplification, CN>4.
HBV integration breakpoints were identified and annotated by Virus-Clip profiles as previously described (Ho et al., 2015; Qiu et al., 2016). Sequencing reads were aligned to the HBV genome. Then soft-clipped reads were extracted to identify the breakpoint positions. We merged any two adjacent breakpoints, whose locations on human genome are within 200 bp and on HBV genome are within 50 bp. The breakpoints supported by only one soft-clipped read were removed.
RNA Sequencing
Total RNA was extracted using Trizol (Invitrogen) according to the manufacturer’s instructions. All 81 cell models were subjected to RNA sequencing (RNA-Seq). Single-end 100 bp read sequencing was performed on Illumina HiSeq 2000 sequencer for 60 cell models; the remaining 21 cell models were subjected to paired-end 150 bp sequencing on HiSeq 4500 sequencer or X ten sequencer respectively. One cell model was sequenced by different sequencers to confirm no obvious batch effect. Sequencing reads were mapped to HG38 genome by TopHat2 (Kim et al., 2013). Cufflinks algorithm (Trapnell et al., 2010) was used to calculate gene expression levels. The expression profiles were normalized by the “fragment per kilobase of exon per million fragments mapped” (FPKM) method and log2 transformed. Molecular classification of liver cancer cell models using RNA-Seq-derived gene expression data was performed using the gene signature and Nearest Template Prediction (NTP) algorithm from the report of this classification (Hoshida et al., 2009). The NTP module is implemented in GenePattern (https://genepattern.broadinstitute.org/). Gene Set Enrichment Analysis (GSEA) was performed on normalized RNA-Seq expression data using the Desktop Application (Subramanian et al., 2005).
Comparison of Cell Lines and Patients
We have collected genetic data, including next generation sequencing and microarray analysis, from a total of 1,856 liver cancer patients published in recent years. A summary of these published cohorts is listed in Table S2. This large collection of liver cancer patients should guarantee the representation of heterogeneity in liver cancer patients to a large extent. Mutational profiles were downloaded from publications or database for 5 large HCC cohorts with 1,185 HCC patients, including those from Chinese (Kan et al., 2013), French (Schulze et al., 2015), Korea (Ahn et al., 2014) and Japanese (Totoki et al., 2014) as well as TCGA (The Cancer Genome Atlas) liver cancers (Broad GDAC FIREHOSE, http://firebrowse.org/) (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017). The synonymous mutations were excluded. CNA data of TCGA liver cancers were downloaded from FIREHOSE. To identify cancer functional genes (CFGs) in liver cancers, we have analyzed whole exome or genome sequencing data from 6 studies (Ahn et al., 2014; Fujimoto et al., 2016; Kan et al., 2013; Rubio-Perez et al., 2015; Schulze et al., 2015; Totoki et al., 2014) (Table S2). These studies recruited HCCs from different populations, including Chinese, Japanese, Korea and Western countries. CFGs were identified as significantly mutated genes by MutSigCV (Lawrence et al., 2013) adjusting for background mutation rate with a threshold of FDR < 0.05. We also included 30 liver cancer driver genes reported in IntOGen database (Rubio-Perez et al., 2015). All the identified driver genes were summarized to a final list of 72 cancer functional genes (CFGs) for liver cancer. The list here included nearly all the driver genes reported in other liver cancer sequencing studies. HBV integration data from two HCC cohorts were obtained from the publications (Sung et al., 2012; Totoki et al., 2014). For transcriptome comparison, RNA-Seq data of TCGA liver cancers were downloaded from FIREHOSE. Microarray expression files for GSE9843 and GSE14520 were downloaded from GEO, and the clinical information was obtained from the publications (Chiang et al., 2008; Roessler et al., 2012).
The similarities in whole genome CNAs and somatic coding mutations between cancer cell models and patients were investigated in 7 types of cancers, including hepatocellular carcinoma (LIHC), breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), esophageal carcinoma (ESCA), lung adenocarcinoma (LUAD), ovarian serous cystadenocarcinoma (OV) and stomach adenocarcinoma (STAD). All of these primary cancers were characterized by TCGA. Processed files, including copy number segments and somatic mutation files, of these primary cancers were downloaded from FIREHOSE. For cancer cell models, the CNAs and somatic mutations were obtained from CCLE or GDSC, except for liver cancer cell models which were characterized by WGS/RNA-Seq in LIMORE. MutSig algorithm was used to identify significantly mutated genes in each dataset of primary cancers and cell models (Lawrence et al., 2013). Spearman correlations of mutation profiles between patients and cell models from different types of cancers were calculated to measure their similarities. GISTIC2.0 was used to identify significantly amplified or deleted regions in each dataset(Mermel et al., 2011) using default parameters. To compare CNA similarities between patients and cell models from different types of cancers, CNA regions were splitted into 1Mb windows, and Spearman correlation was used to evaluate the similarities of CNA frequencies. In the whole genome visualization of CNA frequencies between LIMORE and TCGA primary liver cancers, the default parameters were used in IGV.
High-throughput Drug Screening
The information of 90 screened drugs, including drug names, MoAs, clinical status and resources, is provided in Table S4. Regarding for the drug selection, drugs that are approved for liver cancer treatment or in clinical trials of liver cancers were prioritized. To cover different MoAs in cancer treatment, we retrieved the drug lists that were commonly used in published cancer cell screenings (Basu et al., 2013; Crystal et al., 2014; Iorio et al., 2016). Drugs showing potencies in at least a subset of liver cancer cell lines or under clinical development were in priority. In total, we collected 90 drugs which covered 9 MoAs and targeted different pathways in liver cancers. Drug stocks (10 mM in DMSO) were stored in −80 °C. Because DMSO would inactivate platinum complexes, cisplatin was dissolved in 0.9% NaCl and oxaliplatin in water, respectively. The optimal number of seeding cells for each cell model was determined to avoid over-confluency at the end of the drug treatment (around 90% confluency). The cells were cultured in their preferred medium at 37 °C during screening. The screening was performed in Chemical Biology Core Facility of our institute (http://www.sibcb.ac.cn/ep4-5.asp).
For drug screening, cells were seeded in 384-well plates at the pre-determined cell density at a volume of 50 µL by Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific). After overnight incubation, cells were treated with one drug of either 3-fold serial dilutions of 7 doses or 2-fold serial dilutions of 10 doses using Mosquito HTS (TTP labtech), and the plates were transferred to the incubator for 72 hr. At the end point of drug treatment, each well was added 25 µL CellTiter-Glo reagent (Promega), and after 10 min incubation in room temperature, the luminescent signals were measured by En1Vision Multilabel Reader (PerkinElmer) to determine the cell viabilities. There are three replicate wells for one drug dose. 22 cell models were screened at least twice as biological replicates to confirm the robustness of our screening system. Bortezomib at a high single-point concentration which achieved complete cell killing was used as positive control in all the screening plates. The Z-prime score comparing negative and positive control wells was calculated for all the screening plates.
Multi-parametric analysis of drug response curves using R package (GRmetrics) yielded values for IC50, Emax and Activity Area (AA). IC50 and Emax are measures of drug potency and efficacy (Fallahi-Sichani et al., 2013), and AA reflects magnitude of drug response (Barretina et al., 2012). IC50, Emax and AA can all be used to define the drug response and provide information from different aspects. To reflect the maximal drug response variation across LIMORE models for each drug, we calculated the Coefficient of Variation (CV) of IC50, Emax and AA. We then used the parameter with the largest CV for individual drug and defined the normalized Z-score of this parameter as Drug Responding Score (DRS) for this drug. DRS of a cell line for a drug is the normalized Z-score of the most variable parameter from IC50, Emax and AA. In addition, when Emax parameter was choosen for DRS, we manually checked Emax values in LIMORE models, and confirmed that more than 30% LIMORE models with Emax > 0.3 to avoid general cytotoxic effect at the maximal concentration screened.
We also applied the growth rate inhibition metrics (Hafner et al., 2016) to evaluate the potential effect of doubling time on drug response. Notably, the inferred GRAOC, a corrected AUC based on the growth rate inhibition metrics, is highly correlated with traditional AUC in LIMORE (Pearson r = 0.87, p < 2.2e-16), excluding the strong influcence of doubling time.
To examine the consistency of drug screening data, we compared LIMORE drug results with CTRP and GDSC data (Iorio et al., 2016; Seashore-Ludlow et al., 2015). A total of 38 drugs were shared among CTRP, GDSC and LIMORE. Drug data in LIMORE (area under curve, AUC) were overlapped with 22 liver cancer cell models in CTRP (area under curve, AUC) and with 13 models in GDSC (loge IC50). Twelve liver cancer cell models were shared between CTRP and GDSC. The Spearman correlation coefficient was calculated in each paired comparison of drug profile.
Pharmacogenomic Analysis in LIMORE
To find drug response associated features and build prediction models, the 81 LIMORE models were split into two datasets. The first batch of 54 cell models were used as the training dataset, and the other batch of 27 independent cell models were regarded as testing dataset.
Elastic net (EN) algorithm is powerful to create parsimonious models from a large number of features and a relatively small number of samples. It has been successfully used to find drug sensitivity related genes and build prediction models (Barretina et al., 2012; Garnett et al., 2012). We used a bootstrapping strategy in the analysis of gene-drug associations to control the stability of results. Therefore, elastic net regression algorithm combined with bootstrapping was applied to identify drug response associated features from the training dataset. Gene expression profiles from RNA-Seq were converted to a numeric matrix. Genomic alterations in CFGs and 166 genes/regions (non-coding mutations present in > 5 patients and > 5 LIMORE models) with recurrent non-coding mutations from ICGC liver cancer WGS data were converted to a gene-level binary matrix. A total of 1,000 resampled datasets were generated by sampling with replacement. For each resampling dataset, the elastic net regression model was built using R package “glmnet” to obtain the regression coefficients for all the input features. To evaluate the predictive ability of molecular features, results from 1,000 resampling datasets were summarized to an EN score (S) by the following formula:
where Npos and Nneg are the times of positive and negative regression coefficients.
Genes with larger EN scores were more robust for the prediction. We defined 0.6 as the EN score threshold, which means that the feature was selected in >60% of 1,000 EN models among resampling datasets.
For prediction models of drug responses, elastic net regression models were built for the drug based on the pre-selected features. These models were used to predict the drug response of the 27 independent cell models in the testing dataset. Genomic alterations in CFGs, non-coding mutations and gene expression features were used - in combination to assess their contributions to drug response. Prediction performance was evaluated by Spearman correlation between real drug responses and the predicted values. We also built prediction models using support vector machine and random forest algorithms, and the results were similar to EN in LIMORE.
Furthermore, we combined cell model and PDX datasets to optimize the prediction model for sorafenib. Leave-one-out technique was used to improve sorafenib associated features and assess prediction ability to PDX. Suppose n is the number of PDX models; Fc is the sorafenib associated feature set, which were selected from LIMORE by EN algorithm combined with bootstraping. PDXs’ responses to sorafenib were predicted as follows:
For i in [1:n], omit the i-th PDX, and used other PDXs to calculate the spearman correlation coefficients (cor) between sorafenib response and gene expression. Fm is the gene set with cor > 0.2. The improved feature set is defined as .
Use genes in F to build EN model, and then predict sorafenib response for the i-th PDX.
Repeat Step 1) and 2) n times, and obtain predicted sorafenib responses for all the PDX models.
Finally, we used Spearman correlation to assess the prediction performance. We also built prediction models only using PDX dataset. However, its performance was not as good as the improved prediction model combing both LIMORE cell models and PDXs.
Drug Response Associated Transcription Regulators
The pipeline to define the contribution of transcription regulators to drug responses consists of four steps (https://github.com/coexps/Rephine). We applied this pipeline to analyze MYC and drug responses in LIMORE. First, ChIP-Seq data for MYC in HepG2 and 5 other cell lines were downloaded from ENCODE database (Consortium, 2012). To quantitatively determine the targets of MYC, we implemented regulatory potential (RP) score by considering binding site’s distance to TSS (transcription start site) of a gene and the signal strength of ChIP-Seq peaks (Tang et al., 2011). Therefore, RP score indicates the regulatory strength of MYC on the specific gene. The higher the score is, the stronger the regulatory strength is. RP score of MYC was calculated for each gene. Next, in LIMORE, we used partial correlation to determine the correlation between each gene’s RNA-Seq expression levels and each drug’s response profile after adjusting for confounding factors, such as CNA and CFG mutations by adaptive lasso selection. For each drug, a list of genes ranked by their expression correlations with drug response profile was identified. Third, we determined the concordant enrichment of MYC-regulated target genes in drug response-related gene lists using elastic-net regression. The coefficient from elastic net model was calculated to indicate a positive or negative correlation with drug response values. The p value was calculated by the likelihood ratio test to indicate the statistical significance. Finally, to visualize the relationship between a transcription regulator and response profile of a drug, we adopted canonical GSEA for the continuous variable (RP score) in the calculation and plot of enrichment score (Subramanian et al., 2005). Additional processed bigWig files for EZH2 and TCF4/7 were downloaded from Cistrome database (http://cistrome.org/db) and visualized at the gene loci in UCSC web browser (https://genome.ucsc.edu/). The heatmap was plotted using SeqPlots software (http://przemol.github.io/seqplots/).
MYC target genes (from ChIP-Seq data) were selected based on their association with drug response, and MYC transcription program activity was measured by calculating the sum of Z-score normalized expression values of these target genes. The continuous variable of the activity of MYC transcription program was then converted to a binary variable by selecting a threshold to maximum the difference of drug response.
Cell models were classified into two groups by the status of CTNNB1 mutation or MYC transcription program activity. Cohen’s d considers the standardized mean difference between two populations. The difference is considered as large if d >= 0.8 and medium if d >= 0.5. Cohen’s d was calculated to measure the effect size of the two cell model groups with or without CTNNB1 mutation and with high or low MYC transcription program activity. In addition, cell models were separated to sensitive or resistant group based on the mean of IC50 of HDAC inhibitors. Cell models with mutated CTNNB1 or activated MYC were predicted to be sensitive to HDAC inhibitor. The percentage of correctly predicted sensitive cell models were calculated to evaluate the prediction power.
Experimental Validation of Gene Functions
The construction of the modified pWPI plasmids overexpressing the activating isoform of β-catenin (∆N90-β-catenin) or MYC was described previously (Li et al., 2016). For lentivirus packaging, the pWPI or pLKO.1 plasmids were introduced into 293FT cells with the plasmids psPAX2 and pMD2.G (Addgene). After 48 hr incubation, lentiviruses in the supernatant were collected and stored at −80 °C until use. For gene overexpression, lentiviruses (multiplicity of infection, MOI = 2) carrying ∆N90-β-catenin or MYC were added into the medium when we seeded the cells, and 4 days later cells were infected for the second time at MOI = 1–1.5. For siRNA transfection in 384-well format, 0.05 or 0.1 µL siRNA (20 µL ) in 10 µL serum-free opti-MEM medium (Thermo Fisher scientific) was mixed with either 0.075 µL or 0.1 µL of RNAimax (invitrogen) in 10 µL serum-free opti-MEM medium. Following a 20 min incubation, the siRNA-lipid mixture was transferred to a 384-well plate followed by seeding cells at a concentration ranging from 1000 cells/well to 3000 cells/well (depending on cellular growth rate) in 30 µL medium. In 96-well format, 0.2 or 0.4 µL siRNA in 40 µL serum-free opti-MEM medium was mixed with either 0.3 µL or 0.4 µL of RNAimax in 40 µL serum-free opti-MEM medium, cell number plated ranged from 3000 to 10000 cells/well in 120 µL medium. 48 hr later, transfected cells were treated with drugs for 72 hr at the indicated doses. Relative gene expressions were measured by Real-time qPCR and normalized to GAPDH or β-actin mRNA levels. The primers in qRT-PCR were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) or PrimerBank (https://pga.mgh.harvard.edu/primerbank/) online tools. The oligonucleotide sequences for human genes used in this study were listed below: MYC forward primer, gcgtcctgggaagggagatccggagc; MYC reverse primer, ttgaggggcatcgtcgcgggaggctg; β-catenin forward primer (mouse), cttccatcccttcctgctta; β-catenin reverse primer (mouse), aggtgctgtctgtctgctcta; two pairs of GSDME primers (forward primer 1, acatgcaggtcgaggagaagt; reverse primer 1, tcaatgacaccgtaggcaatg and forward primer 2, cccaggatggaccattaagtgt; reverse primer 2, ggttccaggaccatgagtagtt ); two pairs of EZH2 primers (forward primer 1, agtgtgaccctgacctctgt; reverse primer 1, agatggtgccagcaatagat and forward primer 2, ttgttggcggaagcgtgtaaaatc; reverse primer 2, tccctagtcccgcgcaatgagc); NRF2 forward primer, cacatccagtcagaaaccagtgg; NRF2 reverse primer, ggaatgtctgcgccaaaagctg (Sun et al., 2016). For siRNA mediated gene knockdown, a mixture of two specific siRNAs (5’-gcggtcctatttgatgatgaa-3’ and 5’-gatgatggagtatctgatctt-3’), reported in a previous study (Wang et al., 2017), was used to knock down GSDME expression. Two NRF2 specific siRNAs (5’- gagatgaacttagggcaaa-3’ and 5’-tggagtaagtcgagaagta-3’) and EZH2 specific siRNAs (5’- gactctgaatgcagttgct-3’ and 5’- gctgaagcctcaatgttta-3’) were transfected to knock down gene expression individually. siRNAs were transfected to knock down HDAC4 (5’-cgacaggcctcgtgtatga-3’, 5’-aaattacggtccaggctaa-3’, 5’-gagtgtcgacctcctataa-3’ and 5’-gaacggtggtcatgccgat-3’), HDAC5 (5’-gggcgtcgtccgtgtgtaa-3’, 5’-aaagtgcgttcaaggctaa-3’, 5’-ggactgttattagcacctt-3’ and 5’-tacgacacgttcatgctaa-3’), HDAC7 (5’-gacaagagcaagcgaagtg-3’, 5’-gcagataccctcggctgaa-3’, 5’-ggtgagggcttcaatgtca-3’ and 5’-tggctgctcttctgggtaa-3’) and HDAC11 (5’-cacacgaggcgctatctta-3’, 5’-cgacaagcgtgtatacatc-3’, 5’-gcaatgggcatgagcgaga-3’ and 5’-gcacagaactcagacacac-3’). Three siRNAs (5’-ugguuuacaugucgacuaa-3’, 5’-ugguuuacauguuuucuga-3’ and 5’-uucuccgaacgugucacgutt-3’) were used as nontargeting control. TOX (From Dharmacon), TOX transfection control was used to assess transfection efficiency.
HCC PDX Models
To examine sorafenib efficacies in 22 HCC PDX models, mice (6–8 weeks-old immuno-deficient athymic BALB/c-nu/nu female mice) were randomized in sorafenib treatment (n=10 mice) or control group (n=10 mice) for each PDX model. The durations of sorafenib treatment (40 mg/kg, daily, orally) for each model varied from 10 days to 28 days. The tumors were measured twice weekly using the caliper. Tumor volume was calculated by the following formula: V = (L × W2) / 2, where L and W are the long and short diameters of the tumors, respectively. Tumor growth inhibition, the ratio of sorafenib treatment group compared to control group (Treatment-to-control ratio, T/C ratio) was calculated at the end-point day to indicate the efficacy of sorafenib. Response categories were defined as previously reported (Schutte et al., 2017): T/C ratio < 0.25, moderate response; T/C ratio 0.26 – 0.50, minor response; T/C ratio > 0.50, resistance. PDXs with “moderate response” were considered as sensitive to sorafenib treatment, whereas PDXs with “minor response” or “resistance” were considered as resistant to sorafenib treatment. RNA-Seq raw data were obtained from Shanghai ChemPartner, and analyzed using the same pipeline of LIMORE. The DKK1 gene expression levels were retrieved from RNA-Seq data. In the DKK1 analysis, PDXs with moderate response were regarded as sensitive PDXs, while those with minor response or resistance to sorafenib were resistant PDXs. ROC (receiver operating characteristics) curve was performed using R package “pROC”.
Serum DKK1 Analysis in HCC Patients
To detect DKK1 levels in serum, the commercially available ELISA kit (R&D systems, Cat No. DKK100) was applied according to the manufacturer’s instructions. Briefly, a monoclonal antibody specific for human DKK1 has been pre-coated onto a microplate. Diluted samples or standards (100 µL) were incubated for 2 hr at room temperature. A horseradish peroxidase conjugated polyclonal antibody for human DKK1 was added and incubated for 2 hr at room temperature. After washing away any unbound antibody-enzyme reagent, 200 µL Substrate solution is added and color development was stopped by 50 µL Stop Solution containing sulfuric acid. The optimal density of each well was determined at 450 nm and 570 nm. DKK1 concentrations were calculated from the standard curve. The patients’ responses to sorafenib treatment within one year after sorafenib treatment initiation were evaluated by Response Evaluation Criteria In Solid Tumors (RECIST, www.irrecist.com).
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. Statistical significance tests, including unpaired Student’s t-test, Fisher’s exact test, Chi-square test, Kruskal-Wallis test, Pearson or Spearman correlation test, Kolmogorov-Smirnov normality test, and receiver operating characteristics (ROC) curve were performed using R or Graphpad Prism v5 software, as denoted in each analysis. Data in the barplot or curve are presented as mean±SD (technical or biological replicates from a single cell model) or mean±SEM (mean of means in different cell models or PDX mice). For box-and-whisker plot, the box indicates interquartile range (IQR), the line in the box indicates the median, the whiskers indicate points within Q3+1.5×IQR and Q1−1.5×IQR and the points beyond whiskers indicate outliers. Q1 and Q3, the first and third quartiles, respectively. All the statistical tests were two-tailed. p < 0.05 of the two-tail was taken to indicate statistical significance unless otherwise stated.
DATA AND SOFTWARE AVAILABILITY
Genome data have been deposited at the European Genome-phenome Archive (EGA) hosted at the EBI under accession number EGAS00001002237 and EGAS00001001678. RNA-Seq data have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE97098 and GSE78236.
Supplementary Material
Table S1. Information of 81 liver cancer cell models in LIMORE, Related to Figure 1.
Table S2. Summary of sequencing data and liver cancer patient data, list of somatic mutations and HBV integration sites used in this study, Related to Figure 1.
Table S3. List of cancer functional genes (CFGs) and alterations in liver cancer cell models, Related to Figure 2.
Table S4. Information of 90 drugs and drug response matrix in liver cancer cell models, Related to Figure 3.
Table S5. List of gene-drug associations in liver cancer cell models, Related to Figure 4.
Table S6. Information of PDXs and HCC patients with sorafenib treatment, Related to Figure 6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Glutamine Synthase | BD bioscience | Cat#610517; RRID:AB_397879 |
| Biological Samples | ||
| Surgically resected tumor samples from Chinese HCC patients | Eastern Hepatobilliary Surgery Hospital, Second Military Medical University; Zhongshan Hospital, Fudan University; The First Affiliated Hospital of Nanjing Medical University; the Affiliated Drum Tower Hospital of Medical School of Nanjing University | N/A |
| Serum samples from Chinese HCC patients | Fudan University Shanghai Cancer Center | N/A |
| Patient-derived xenografts (PDXs) from HCCs | ChemPartner | www.chempartner.com |
| Chemicals, Peptides, and Recombinant Proteins | ||
| (+)-JQ1 | Selleck Chemicals | Cat#S7110 |
| 17-AAG | Selleck Chemicals | Cat#S1141 |
| ABT-199 | Selleck Chemicals | Cat#S8048 |
| ABT-263 | Selleck Chemicals | Cat#S1001 |
| Afatinib | Selleck Chemicals | Cat#S1011 |
| Alisertib | Selleck Chemicals | Cat#S1133 |
| Apatinib | Selleck Chemicals | Cat#S2221 |
| AT-406 | Selleck Chemicals | Cat#S2754 |
| AT-7519 | Selleck Chemicals | Cat#S1524 |
| Avasimibe | Selleck Chemicals | Cat#S2187 |
| AZD1208 | Selleck Chemicals | Cat#S7104 |
| AZD6244 | Selleck Chemicals | Cat#S1008 |
| AZD7762 | Selleck Chemicals | Cat#S1532 |
| Barasertib | Selleck Chemicals | Cat#S1147 |
| Belinostat | Selleck Chemicals | Cat#S1085 |
| BEZ235 | Selleck Chemicals | Cat#S1009 |
| BGJ398 | Selleck Chemicals | Cat#S2183 |
| BI2536 | Selleck Chemicals | Cat#S1109 |
| Bortezomib | Selleck Chemicals | Cat#S1013 |
| Bosutinib | Selleck Chemicals | Cat#S1014 |
| BX-912 | Selleck Chemicals | Cat#S1275 |
| Cabozantinib | Selleck Chemicals | Cat#S1119 |
| Camptothecin | Selleck Chemicals | Cat#S1288 |
| Carfilzomib | Selleck Chemicals | Cat#S2853 |
| CB-839 | Selleck Chemicals | Cat#S7655 |
| Ceritinib | Selleck Chemicals | Cat#S7083 |
| Cisplatin | Selleck Chemicals | Cat#S1166 |
| Cobimetinib | Selleck Chemicals | Cat#S8041 |
| CP-466722 | Selleck Chemicals | Cat#S2245 |
| Crizotinib | Selleck Chemicals | Cat#S1068 |
| CX-4945 | Selleck Chemicals | Cat#S2248 |
| Daporinad | Selleck Chemicals | Cat#S2799 |
| Dasatinib | Selleck Chemicals | Cat#S1021 |
| Decitabine | Selleck Chemicals | Cat#S1200 |
| Docetaxel | Selleck Chemicals | Cat#S1148 |
| Dovitinib | Selleck Chemicals | Cat#S1018 |
| Doxorubicin | Selleck Chemicals | Cat#S1208 |
| DZNeP | Selleck Chemicals | Cat#S7120 |
| Enzastaurin | Selleck Chemicals | Cat#S1055 |
| Epirubicin | Selleck Chemicals | Cat#S1223 |
| Erastin | Selleck Chemicals | Cat#S7242 |
| Etoposide | Selleck Chemicals | Cat#S1225 |
| Everolimus | Selleck Chemicals | Cat#S1120 |
| FG-4592 | Selleck Chemicals | Cat#S1007 |
| Fluorouracil | Selleck Chemicals | Cat#S1209 |
| Foretinib | Selleck Chemicals | Cat#S1111 |
| Ganetespib | Selleck Chemicals | Cat#S1159 |
| GSK126 | Selleck Chemicals | Cat#S7061 |
| GSK1838705A | Selleck Chemicals | Cat#S2703 |
| I-BET151 | Selleck Chemicals | Cat#S2780 |
| Ibrutinib | Selleck Chemicals | Cat#S2680 |
| Irinotecan | Selleck Chemicals | Cat#S2217 |
| Ispinesib | Selleck Chemicals | Cat#S1452 |
| JNJ-26854165 | Selleck Chemicals | Cat#S1172 |
| Lenvatinib | Selleck Chemicals | Cat#S1164 |
| Methotrexate | Selleck Chemicals | Cat#S1210 |
| NU-7441 | Selleck Chemicals | Cat#S2638 |
| Olaparib | Selleck Chemicals | Cat#S1060 |
| OSI-906 | Selleck Chemicals | Cat#S1091 |
| Oxaliplatin | Selleck Chemicals | Cat#S1224 |
| Paclitaxel | Selleck Chemicals | Cat#S1150 |
| Palbociclib | Selleck Chemicals | Cat#S1116 |
| Panobinostat | Selleck Chemicals | Cat#S1030 |
| Pazopanib | Selleck Chemicals | Cat#S1035 |
| PD173074 | Selleck Chemicals | Cat#S1264 |
| PF-562271 | Selleck Chemicals | Cat#S2890 |
| PI-103 | Selleck Chemicals | Cat#S1038 |
| Piperlongumine | Selleck Chemicals | Cat#S7551 |
| Pirarubicin | Selleck Chemicals | Cat#S1393 |
| Ponatinib | Selleck Chemicals | Cat#S1490 |
| Rapamycin | Selleck Chemicals | Cat#S1039 |
| Regorafenib | Selleck Chemicals | Cat#S1178 |
| Roscovitine | Selleck Chemicals | Cat#S1153 |
| Ruxolitinib | Selleck Chemicals | Cat#S1378 |
| SGC0946 | Selleck Chemicals | Cat#S7079 |
| Simvastatin | Selleck Chemicals | Cat#S1792 |
| Sorafenib | Selleck Chemicals | Cat#S1040 |
| Sunitinib | Selleck Chemicals | Cat#S1042 |
| TAE684 | Selleck Chemicals | Cat#S1108 |
| Talazoparib | Selleck Chemicals | Cat#S7048 |
| Temsirolimus | Selleck Chemicals | Cat#S1044 |
| Tipifarnib | Selleck Chemicals | Cat#S1453 |
| Tivantinib | Selleck Chemicals | Cat#CS-0030 |
| Topotecan | Selleck Chemicals | Cat#S1231 |
| Trametinib | Selleck Chemicals | Cat#S2673 |
| Vinblastine | Selleck Chemicals | Cat#S4505 |
| Vorasidenib | Selleck Chemicals | Cat#S8611 |
| Vorinostat | Selleck Chemicals | Cat#S1047 |
| YK-4-279 | Selleck Chemicals | Cat#S7679 |
| YM155 | Selleck Chemicals | Cat#S1130 |
| Y-27632 | Selleck Chemicals | Cat#S1049 |
| A83-01 | Tocris Bioscience | Cat#2939 |
| Collagen type I | Corning | Cat#354236 |
| Recombinant human EGF | PeproTech | Cat#AF-100-15 |
| ITS-A | Gibco | Cat#51300-044 |
| Collagenase, Type IV, powder | Gibco | Cat#17104019 |
| Critical Commercial Assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat#G7573 |
| Human Dkk-1 Quantikine ELISA Kit | R&D systems | Cat#DKK100 |
| Transwell® Permeable Supports | Corning | Cat#3422 |
| DNeasy Blood and Tissue kit | QIAGEN | Cat#69504 |
| Deposited Data | ||
| DNA sequencing data for LIMORE | This paper | EGA: EGAS00001002237, EGAS00001001678 |
| RNA sequencing data for 72 LIMORE models | This paper | GEO: GSE97098 |
| RNA sequencing data for 9 LIMORE models | (Qiu et al., 2016) | GEO: GSE78236 |
| Drug screening data of LIMORE | This paper | www.picb.ac.cn/limore/ |
| Drug screen data of cell line (CCLE&CTRP) | CTRP | http://portals.broadinstitute.org/ctrp/ |
| Drug screen data of cell line (GDSC) | GDSC | www.cancerrxgene.org |
| Experimental Models: Cell Lines | ||
| See Table S1 | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: Immuno-deficient athymic mice (BALB/c-nu/nu) | SLAC Laboratory Animal (www.slaccas.com); Beijing Vital River Laboratory Animal Technology Co., Ltd | N/A |
| Mouse: NOD.CB17-Prkdcscid/scid/shjh (NOD/SCID) | Shanghai Jihui Laboratory Animal Care Co., Ltd | N/A |
| Oligonucleotides | ||
| siGSDME (gcggtcctatttgatgatgaa) | (Wang et al., 2017) | N/A |
| siGSDME (gatgatggagtatctgatctt) | (Wang et al., 2017) | N/A |
| siNRF2 (gagatgaacttagggcaaa) | (McMillan et al., 2018) | N/A |
| siNRF2 (tggagtaagtcgagaagta) | This paper | N/A |
| siEZH2 (gactctgaatgcagttgct) | This paper | N/A |
| siEZH2 (gctgaagcctcaatgttta) | This paper | N/A |
| siHDAC4 (cgacaggcctcgtgtatga) | Dharmacon | J-003497-07 |
| siHDAC4 (aaattacggtccaggctaa) | Dharmacon | J-003497-08 |
| siHDAC4 (gagtgtcgacctcctataa) | Dharmacon | J-003497-09 |
| siHDAC4 (gaacggtggtcatgccgat) | Dharmacon | J-003497-10 |
| siHDAC5 (gggcgtcgtccgtgtgtaa) | Dharmacon | J-003498-09 |
| siHDAC5 (aaagtgcgttcaaggctaa) | Dharmacon | J-003498-10 |
| siHDAC5 (ggactgttattagcacctt) | Dharmacon | J-003498-11 |
| siHDAC5 (tacgacacgttcatgctaa) | Dharmacon | J-003498-12 |
| siHDAC7 (gacaagagcaagcgaagtg) | Dharmacon | J-009330-07 |
| siHDAC7 (gcagataccctcggctgaa) | Dharmacon | J-009330-08 |
| siHDAC7 (ggtgagggcttcaatgtca) | Dharmacon | J-009330-09 |
| siHDAC7 (tggctgctcttctgggtaa) | Dharmacon | J-009330-10 |
| siHDAC11 (cacacgaggcgctatctta) | Dharmacon | J-004258-05 |
| siHDAC11 (cgacaagcgtgtatacatc) | Dharmacon | J-004258-06 |
| siHDAC11 (gcaatgggcatgagcgaga) | Dharmacon | J-004258-07 |
| siHDAC11 (gcacagaactcagacacac) | Dharmacon | J-004258-08 |
| siNC (ugguuuacaugucgacuaa) | Dharmacon | D-001810-01-05 |
| siNC (ugguuuacauguuuucuga) | Dharmacon | D-001810-03-05 |
| siNC (uucuccgaacgugucacgutt) | This paper | N/A |
| Primer: MYC Forward: gcgtcctgggaagggagatccggagc | This paper | N/A |
| Primer: MYC Reverse: ttgaggggcatcgtcgcgggaggctg | This paper | N/A |
| Primer: β-catenin Forward: cttccatcccttcctgctta | This paper | N/A |
| Primer: β-catenin Reverse: aggtgctgtctgtctgctcta | This paper | N/A |
| Primer: GSDME Forward #1: acatgcaggtcgaggagaagt | This paper | N/A |
| Primer: GSDME Reverse #1: tcaatgacaccgtaggcaatg | This paper | N/A |
| Primer: GSDME Forward #2: cccaggatggaccattaagtgt | This paper | N/A |
| Primer: GSDME Reverse #2: ggttccaggaccatgagtagtt | This paper | N/A |
| Primer: EZH2 Forward #1: agtgtgaccctgacctctgt | This paper | N/A |
| Primer: EZH2 Reverse #1: agatggtgccagcaatagat | This paper | N/A |
| Primer: EZH2 Forward #2: ttgttggcggaagcgtgtaaaatc | This paper | N/A |
| Primer: EZH2 Reverse #2: tccctagtcccgcgcaatgagc | This paper | N/A |
| Primer: NRF2 Forward: cacatccagtcagaaaccagtgg | (Sun et al., 2016) | N/A |
| Primer: NRF2 Reverse: ggaatgtctgcgccaaaagctg | (Sun et al., 2016) | N/A |
| Recombinant DNA | ||
| pWPI-mouse-ΔN90-β-catenin | (Li et al., 2016) | N/A |
| pWPI-human-c-Myc | (Li et al., 2016) | N/A |
| Software and Algorithms | ||
| Burrows-Wheeler Aligner (BWA) | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| Genome Analysis Toolkit (GATK) | (DePristo et al., 2011) | https://software.broadinstitute.org/gatk/ |
| ANNOVAR | (Wang et al., 2010) | http://annovar.openbioinformatics.org/en/latest/ |
| Control-FREEC | (Boeva et al., 2011) | http://boevalab.com/FREEC/ |
| Integrative Genomic Viewer (IGV) tool | (Robinson et al., 2011) | http://software.broadinstitute.org/software/igv/ |
| Virus-Clip | (Ho et al., 2015) | http://web.hku.hk/~dwhho/Virus-Clip.zip |
| TopHat2 | (Kim et al., 2013) | http://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | (Trapnell et al., 2010) | http://cole-trapnell-lab.github.io/cufflinks/ |
| MutSigCV | (Lawrence et al., 2013) | http://software.broadinstitute.org/cancer/software/genepattern/modules/docs/mutsigcv |
| GISTIC2.0 | (Mermel et al., 2011) | http://portals.broadinstitute.org/cgi-bin/cancer/publications/pub_paper.cgi?mode=view&paper_id=216&p=t |
| Growth rate inhibition metrics (GR metrics) | (Hafner et al., 2016) | http://www.grcalculator.org/grcalculator/ |
| RePhine | N/A | https://github.com/coexps/Rephine |
| R Version 3.4.1 | R | www.r-project.org |
| Prism Version 5 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Gene Set Enrichment Analysis (GSEA) | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/index.jsp |
| glmnet | R Bioconductor | https://bioconductor.org/ |
| pHeatmap | R Bioconductor | https://bioconductor.org/ |
| Other | ||
| Web portal for LIMORE models | This paper | www.picb.ac.cn/limore/ |
Liver Cancer Model Repository (LIMORE) consists of 81 liver cancer cell models
LIMORE recapitulated genetic heterogeneity of human liver cancers
Molecular and drug screenings provide a pharmacogenomic landscape in liver cancers
Interrogation of the landscape informs biomarkers for liver cancer treatment
SIGNIFICANCE.
International effort has been made in recent years to generate patient-derived cancer models. However, there have been insufficient experimental models to sufficiently represent the extensive heterogeneity of liver cancers, partially due to the difficulty to generate liver cancer cell models in vitro. The method reported here provides the opportunity to efficiently generate liver cancer models. Interrogation of these genomically validated models provides a pharmacogenomic dataset in liver cancers that allows the identification of gene-drug associations for potential therapies and biomarker candidates, such as CTNNB1 mutations-HDAC inhibitors and DKK1-sorafenib. Patient-derived liver cancer cell lines, together with cancer organoids and patient-derived xenografts, provide model systems with different levels of complexity and advantage.
ACKNOWLEDGMENTS
We thank Xiang Chen (St. Jude Children’s Research Hospital), Xiangsheng Ye (Eli Lilly and Company) and Hai Jiang (Shanghai Institute of Biochemistry and Cell Biology) for critical reading and suggestions. We thank Chemical Biology Core Facility at SIBCB for technical assistance in drug screening. This study is supported by Chinese Academy of Sciences (XDA16020201 and XDA12050104), the National Natural Science Foundation of China (31630044, 81703093, and 31801228), Science and Technology Commission of Shanghai Municipality (16JC1400202), National Major Science and Technology Projects of China (2018ZX09711002-009), and National Special Support Plan for Top Talents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
He Zhou, Wei Wang, Fubo Xie and Ying Gu are employees of Shanghai ChemPartner Co., Ltd.. The remaining authors declare no competing financial interests.
ADDITIONAL RESOURCES
A website for LIMORE (www.picb.ac.cn/limore/ and http://limore.sibcb.ac.cn/) is built and updated regularly. The website contains the information of liver cancer cell models in this study in order to support the community to use these models.
REFERENCES
- Abitbol S, Dahmani R, Coulouarn C, Ragazzon B, Mlecnik B, Senni N, Savall M, Bossard P, Sohier P, Drouet V, et al. (2018). AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of beta-catenin activation. J Hepatol 68, 1203–1213. [DOI] [PubMed] [Google Scholar]
- Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, Baek D, Haq F, Ansari AA, Lee SY, et al. (2014). Genomic Portrait of Resectable Hepatocellular Carcinomas: Implications of RB1 and FGF19 Aberrations for Patient Stratification. Hepatology 60, 1972–1982. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al. (2013). An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billin AN, Thirlwell H, and Ayer DE (2000). Beta-catenin-histone deacetylase interactions regulate the transition of LEF1 from a transcriptional repressor to an activator. Mol Cell Biol 20, 6882–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm JS, and Golub TR (2015). An ecosystem of cancer cell line factories to support a cancer dependency map. Nat Rev Genet 16, 373–374. [DOI] [PubMed] [Google Scholar]
- Boeva V, Zinovyev A, Bleakley K, Vert JP, Janoueix-Lerosey I, Delattre O, and Barillot E (2011). Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics 27, 268–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarro LM, Bradshaw CR, Allen GE, Arnes-Benito R, Sidorova O, Gaspersz MP, et al. (2017). Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med 23, 1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruix J, Qin S, Merle P, Granito A, Huang Y-H, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al. (2017). Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet 389, 56–66. [DOI] [PubMed] [Google Scholar]
- Cairo S, Armengol C, De Reynies A, Wei Y, Thomas E, Renard CA, Goga A, Balakrishnan A, Semeraro M, Gresh L, et al. (2008). Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 14, 471–484. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Electronic address, w.b.e., and Cancer Genome Atlas Research, N. (2017). Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169, 1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso S, Calatayud AL, Pilet J, La Bella T, Rekik S, Imbeaud S, Letouze E, Meunier L, Bayard Q, Rohr-Udilova N, et al. (2019). Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology. [DOI] [PubMed] [Google Scholar]
- Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, LeBlanc AC, Donovan DJ, Thung SN, Sole M, et al. (2008). Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer research 68, 6779–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, et al. (2014). Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346, 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallahi-Sichani M, Honarnejad S, Heiser LM, Gray JW, and Sorger PK (2013). Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat Chem Biol 9, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, et al. (2016). Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nature genetics 48, 500–509. [DOI] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. (2014). Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhang X, Zhang L, Cen J, Ni X, Liao X, Yang C, Li Y, Chen X, Zhang Z, et al. (2017). Distinct Gene Expression and Epigenetic Signatures in Hepatocyte-like Cells Produced by Different Strategies from the Same Donor. Stem Cell Reports 9, 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. (2012). Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, and McDermott U (2014). The evolving role of cancer cell line-based screens to define the impact of cancer genomes on drug response. Curr Opin Genet Dev 24, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodspeed A, Heiser LM, Gray JW, and Costello JC (2016). Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Molecular cancer research : MCR 14, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Niepel M, Chung M, and Sorger PK (2016). Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods 13, 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DW, Sze KM, and Ng IO (2015). Virus-Clip: a fast and memory-efficient viral integration site detection tool at single-base resolution with annotation capability. Oncotarget 6, 20959–20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, and Johnston PG (2013). Cancer drug resistance: an evolving paradigm. Nature reviews Cancer 13, 714–726. [DOI] [PubMed] [Google Scholar]
- Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, et al. (2009). Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 69, 7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Jeng YM, Mao TL, Chu JS, Lai PL, and Peng SY (2000). Beta-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am J Pathol 157, 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, et al. (2016). A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, et al. (2013). Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res 23, 1422–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuda T, Kawamata M, Hagiwara K, Takahashi RU, Yamamoto Y, Camargo FD, and Ochiya T (2017). Conversion of Terminally Committed Hepatocytes to Culturable Bipotent Progenitor Cells with Regenerative Capacity. Cell Stem Cell 20, 41–55. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo M, Finn RS, Qin S, Han K-H, Ikeda K, Piscaglia F, Baron A, Park J-W, Han G, Jassem J, et al. (2018). Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. The Lancet 391, 1163–1173. [DOI] [PubMed] [Google Scholar]
- Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, Minguez B, Tsai HW, Ward SC, Thung S, et al. (2012). Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol 56, 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, Chua CW, Barlow LJ, Kandoth C, Williams AB, et al. (2018). Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 173, 515–528 e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levrero M, and Zucman-Rossi J (2016). Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol 64, S84–S101. [DOI] [PubMed] [Google Scholar]
- Li D, Fu J, Du M, Zhang H, Li L, Cen J, Li W, Chen X, Lin Y, Conway EM, et al. (2016). Hepatocellular carcinoma repression by TNFalpha-mediated synergistic lethal effect of mitosis defect-induced senescence and cell death sensitization. Hepatology 64, 1105–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WM, Baker AC, Beroukhim R, Winckler W, Feng W, Marmion JM, Laine E, Greulich H, Tseng H, Gates C, et al. (2008). Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res 68, 664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lee W, Jiang Z, Chen Z, Jhunjhunwala S, Haverty PM, Gnad F, Guan Y, Gilbert HN, Stinson J, et al. (2012a). Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res 22, 2315–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, et al. (2012b). ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 180, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. (2008). Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359, 378–390. [DOI] [PubMed] [Google Scholar]
- McMillan EA, Ryu MJ, Diep CH, Mendiratta S, Clemenceau JR, Vaden RM, Kim JH, Motoyaji T, Covington KR, Peyton M, et al. (2018). Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell 173, 864–878 e829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitra K, Im K, Limpert K, Borsa A, Sawitzke J, Robey R, Yuhki N, Savan R, Huang da W, Lempicki RA, et al. (2012). Differential gene and microRNA expression between etoposide resistant and etoposide sensitive MCF7 breast cancer cell lines. PLoS One 7, e45268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, Arango D, Strausberg RL, Buchanan D, Wormald S, et al. (2014). Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res 74, 3238–3247. [DOI] [PubMed] [Google Scholar]
- Naik S, Dothager RS, Marasa J, Lewis CL, and Piwnica-Worms D (2009). Vascular Endothelial Growth Factor Receptor-1 Is Synthetic Lethal to Aberrant {beta}-Catenin Activation in Colon Cancer. Clin Cancer Res 15, 7529–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. (2006). A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, Sugano S, and Akiyama T (2004). DKK1, a negative regulator of Wnt signaling, is a target of the beta-catenin/TCF pathway. Oncogene 23, 8520–8526. [DOI] [PubMed] [Google Scholar]
- Nuciforo S, Fofana I, Matter MS, Blumer T, Calabrese D, Boldanova T, Piscuoglio S, Wieland S, Ringnalda F, Schwank G, et al. (2018). Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep 24, 1363–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, Sailer V, Augello M, Puca L, Rosati R, et al. (2017). Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov 7, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LN, Bai T, Chen ZS, Wu FX, Chen YY, De Xiang B, Peng T, Han ZG, and Li LQ (2015). The p53 mutation spectrum in hepatocellular carcinoma from Guangxi, China : role of chronic hepatitis B virus infection and aflatoxin B1 exposure. Liver Int 35, 999–1009. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Zou K, Zhuang L, Qin J, Li H, Li C, Zhang Z, Chen X, Cen J, Meng Z, et al. (2016). Hepatocellular carcinoma cell lines retain the genomic and transcriptomic landscapes of primary human cancers. Sci Rep 6, 27411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebouissou S, Zucman-Rossi J, Moreau R, Qiu Z, and Hui L (2017). Note of caution: Contaminations of hepatocellular cell lines. J Hepatol 67, 896–897. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler S, Long EL, Budhu A, Chen Y, Zhao X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, et al. (2012). Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology 142, 957–966 e912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C, Mestres J, Gonzalez-Perez A, and Lopez-Bigas N (2015). In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 27, 382–396. [DOI] [PubMed] [Google Scholar]
- Sachs N, and Clevers H (2014). Organoid cultures for the analysis of cancer phenotypes. Curr Opin Genet Dev 24, 68–73. [DOI] [PubMed] [Google Scholar]
- Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H, et al. (2018). A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 172, 373–386 e310. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, and Clarke AR (2007). Myc deletion rescues Apc deficiency in the small intestine. Nature 446, 676–679. [DOI] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. (2015). Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nature genetics 47, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte M, Risch T, Abdavi-Azar N, Boehnke K, Schumacher D, Keil M, Yildiriman R, Jandrasits C, Borodina T, Amstislavskiy V, et al. (2017). Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nature communications 8, 14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, et al. (2015). Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov 5, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Fan J, Yang XR, Tan Y, Zhao W, Xu Y, Wang N, Niu Y, Wu Z, Zhou J, et al. (2012). Serum DKK1 as a protein biomarker for the diagnosis of hepatocellular carcinoma: a large-scale, multicentre study. The Lancet Oncology 13, 817–826. [DOI] [PubMed] [Google Scholar]
- Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, Li D, Ullrich R, Koker M, Fischer F, et al. (2009). Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest 119, 1727–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al. (2012). Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nature genetics 44, 765–769. [DOI] [PubMed] [Google Scholar]
- Tang Q, Chen Y, Meyer C, Geistlinger T, Lupien M, Wang Q, Liu T, Zhang Y, Brown M, and Liu XS (2011). A comprehensive view of nuclear receptor cancer cistromes. Cancer research 71, 6940–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YM, Liu Z, and Liu HL (2013). Dickkopf-1 (DKK1) promotes invasion and metastasis of hepatocellular carcinoma. Dig Liver Dis 45, 251–257. [DOI] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. (2014). Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nature genetics 46, 1267–1273. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung EK, Mak CK, Fatima S, Lo RC, Zhao H, Zhang C, Dai H, Poon RT, Yuen MF, Lai CL, et al. (2011). Clinicopathological and prognostic significance of serum and tissue Dickkopf-1 levels in human hepatocellular carcinoma. Liver Int 31, 1494–1504. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, et al. (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, and Shao F (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. [DOI] [PubMed] [Google Scholar]
- Williams SP, and McDermott U (2017). The Pursuit of Therapeutic Biomarkers with High-Throughput Cancer Cell Drug Screens. Cell Chem Biol 24, 1066–1074. [DOI] [PubMed] [Google Scholar]
- Winter GE, Radic B, Mayor-Ruiz C, Blomen VA, Trefzer C, Kandasamy RK, Huber KVM, Gridling M, Chen D, Klampfl T, et al. (2014). The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nature chemical biology 10, 768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JD, and Roberts LR (2010). Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol 7, 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Selvaraj SK, Liang-Chu MM, Aghajani S, Busse M, Yuan J, Lee G, Peale F, Klijn C, Bourgon R, et al. (2015). A resource for cell line authentication, annotation and quality control. Nature 520, 307–311. [DOI] [PubMed] [Google Scholar]
- Zucman-Rossi J, Villanueva A, Nault JC, and Llovet JM (2015). Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 149, 1226–1239 e1224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Information of 81 liver cancer cell models in LIMORE, Related to Figure 1.
Table S2. Summary of sequencing data and liver cancer patient data, list of somatic mutations and HBV integration sites used in this study, Related to Figure 1.
Table S3. List of cancer functional genes (CFGs) and alterations in liver cancer cell models, Related to Figure 2.
Table S4. Information of 90 drugs and drug response matrix in liver cancer cell models, Related to Figure 3.
Table S5. List of gene-drug associations in liver cancer cell models, Related to Figure 4.
Table S6. Information of PDXs and HCC patients with sorafenib treatment, Related to Figure 6.
Data Availability Statement
Genome data have been deposited at the European Genome-phenome Archive (EGA) hosted at the EBI under accession number EGAS00001002237 and EGAS00001001678. RNA-Seq data have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE97098 and GSE78236.