

Abstract
Constructing efficient cellular factories often requires integration of heterologous pathways for synthesis of novel compounds and improved cellular productivity. Few genomic sites are routinely used, however, for efficient integration and expression of heterologous genes, especially in nonmodel hosts. Here, a data-guided framework for informing suitable integration sites for heterologous genes based on ATAC-seq was developed in the nonmodel yeast Komagataella phaffii. Single-copy GFP constructs were integrated using CRISPR/Cas9 into 38 intergenic regions (IGRs) to evaluate the effects of IGR size, intensity of ATAC-seq peaks, and orientation and expression of adjacent genes. Only the intensity of accessibility peaks was observed to have a significant effect, with higher expression observed from IGRs with low- to moderate-intensity peaks than from high-intensity peaks. This effect diminished for tandem, multicopy integrations, suggesting that the additional copies of exogenous sequence buffered the transcriptional unit of the transgene against effects from endogenous sequence context. The approach developed from these results should provide a basis for nominating suitable IGRs in other eukaryotic hosts from an annotated genome and ATAC-seq data.
Keywords: ATAC-seq, heterologous gene, RNA-seq, locus, genome engineering
Significant genomic engineering of cellular hosts is often required for efficient production of complex or novel molecules1,2 Genome-scale screens can now rapidly identify genes and associated pathways that enhance the production of desired chemicals, fuels, or biologics.3−5 While modern gene editing tools such as CRISPR/Cas9 enable precise genomic editing,6 deciding where to integrate new genes and pathways remains challenging, especially in nonmodel organisms.
Currently, integration sites for heterologous cassettes are chosen semirandomly or by gene replacement, and promising sites are reused in future applications or hosts where similar sites exist. Integration mediated by retroviruses is performed routinely in mammalian cells.7 The resultant integration is semirandom, however, and creates heterogeneity among transformants, requiring significant post-transformational screening. Targeted integration is preferred, but identifying “safe harbor” sites that promote stable transgene expression without harmful off-target effects remains challenging. Current sites employed for targeted integration of recombinant cassettes are often uncovered through empiricism, and a limited number of validated sites are recycled in subsequent uses for a given organism.1,8
In yeast, integration into, and disruption of, native genes such as auxotrophic markers can achieve stable expression and native gene knockout in a single step.8,9 Strategies using gene replacement were essential prior to the development of whole-genome sequencing, which enabled mapping of nontranscribed regions. Knockout screens could potentially identify nonessential genes for integration sites.10 This approach, however, is complex: essentiality is often specific to the tested condition(s), and harmful disruptions to multigene interactions may be initially overlooked. In higher order eukaryotes such as humans, finding genomic safe harbors has been especially elusive, and requires extensive screening to validate a particular site for use.11,12 The number of trusted integration sites is often insufficient, especially in nonmodel organisms, for complex cellular engineering, which may require introduction of several heterologous pathways that may comprise several genes each.
Methods have been developed to engineer an organism in cases where few genomic safe harbors are known. Integration of large constructs containing multiple genes reduces the number of integration sites needed, but this approach is often limited by the transformation and recombination efficiency of the organism.7,13 An artificial chromosome can obviate the need for genomic safe harbors, but often is challenging to synthesize and stably integrate; these constructs are also not yet available for many organisms.13 Screens to identify and validate positional effects of various integration sites have thus far required intensive upfront screening and have not resulted in an organized framework for use by synthetic biologists.7,14,15 A simple, genome-wide assay to inform optimal sites for integration of heterologous constructs would facilitate construction of cellular factories, even in nonmodel organisms.
ATAC-seq measures genome-wide chromatin accessibility by using a hyperactive Tn5 transposase to add sequencing adaptors to accessible regions of the genome.16,17 ATAC-seq is simple and highly scalable, enabling genome-wide evaluation of several strains, conditions, or time points for accessibility and nucleosome positioning in a single assay.18 Identifying sites for gene integration using ATAC-seq is especially appealing because it only requires knowledge of the genome sequence and annotated coding sequences; detailed functional annotations, transcription factor binding sites, or promoter annotations are useful but not necessary. Recently, ATAC-seq has been used to investigate varied performance of limited integration sites,19 but the properties of a desirable integration site are still poorly understood. To enable construction of complex cell factories, a framework is needed that can predict optimal sites for integration of heterologous genes in almost any host using a simple, scalable, assay such as ATAC-seq.
Here, we collected paired ATAC-seq and RNA-seq data to characterize the properties that might inform the suitability of a particular intergenic region (IGR) for integration of heterologous genes. We performed this characterization in the nonmodel, biotechnological yeast, Komagataella phaffii (Pichia pastoris), for which only a few sites for integration are routinely used and functional annotations are not well developed. Heterologous constructs expressing a single copy of a fluorescent reporter were integrated into 38 diverse intergenic regions (IGRs) using CRISPR/Cas9 to test the effects of IGR size, expression and orientation of adjacent genes, and intensity of ATAC-seq peaks, among other factors. We further evaluated these effects for expression of secreted recombinant proteins from tandem, multicopy integrations. From this analysis, we present a framework for leveraging ATAC-seq to inform integration sites that should facilitate genomic engineering of many other eukaryotic cell factories.
Results and Discussion
Analysis of Transcriptomic and Epigenetic Features in K. phaffii
We first characterized genomic, epigenetic, and transcriptomic features that might influence the suitability of a given locus for heterologous gene integration. We focused our methods for characterization on simple, genome-wide assays RNA-seq and ATAC-seq, which require only a genome sequence with annotated coding sequences (CDSs). To this end, we cultivated wild-type K. phaffii under relevant conditions for this host: biomass accumulation in a glycerol-containing medium followed by simulated induction of transgene expression in a methanol-containing medium. We collected samples after each stage of the cultivation for RNA-seq and ATAC-seq, which enables mapping of epigenetically accessible regions and nucleosomes (Figure 1). We sought to identify integration sites that would permit strong expression across both of these typical growth conditions. (The same methodology, however, should also allow identification of sites that otherwise use epigenetic change across conditions to modulate the expression of a gene.)
Figure 1.

Genome-wide analysis of gene expression (RNA-seq) and chromatin accessibility (ATAC-seq). (A) Workflow for cultivation and sampling for RNA-seq and ATAC-seq after 24 h growth on glycerol and an additional 24 h growth on methanol. (B) Relative frequency of mapped fragment sizes recovered in ATAC-seq libraries. (C) Log2(fold-change) in gene expression and accessibility score relative to genome-wide averages for 7.5 kbp intervals across each chromosome. Approximate positions of centromeres are depicted for each chromosome. (D) Nucleosome positioning around translation start sites in K. phaffii as determined by NucleoATAC.
Starting with an annotated genome,20 we reduced the design space of potential integration sites to intergenic regions (IGRs) located away from chromosome ends and centromeres. Chromosomal ends are not ideal regions for integration of heterologous constructs due to the presence of telomeres, repetitive sequences, and susceptibility to accumulation of polymorphisms.21 For these reasons, we avoided regions of 5–10 kb at the distal ends of chromosomes where there were no annotated CDSs with high-quality mRNA transcript. We excluded centromeres, which we confirmed to be generally inaccessible as expected (Figure S2).22 Finally, we excluded regions in the extrachromosomal plasmids maintained in variants of K. phaffii,21 since the function and stability of these plasmids are not well characterized.
As with most nonmodel organisms, functional annotation of genes in K. phaffii is incomplete, so we did not consider intragenic sites to prevent unwanted deleterious effects. Nearly all the genes in K. phaffii were expressed under both conditions, further supporting the choice of IGRs as potential integration sites (Figure S2).
We first investigated the impact of global dynamics of chromatin on IGRs by searching for large regions in any chromosome in which gene expression or chromatin accessibility was universally high or low. We hypothesized accessible, strongly transcribed regions may contain the most suitable IGRs, while closed chromatin areas are undesirable. We divided each chromosome into equal intervals about 7.5 kb in length, and visualized the average gene expression and accessibility for each interval across the genome (Figure S1). Both accessibility and gene expression varied considerably from one interval to the next, suggesting that gene-specific regulation, and not global changes in chromatin, strongly influenced accessibility and expression. We similarly compared gene expression and accessibility under growth on methanol relative to glycerol. These environmental conditions altered expression of many genes (Figure 1C, Figure S2).20 There were no large hotspots of enhanced expression or closed chromatin on the global scale (>20 kb) across these conditions. Given these findings, we considered each IGR in the genome independently, and next hypothesized which features might affect the suitability of a particular IGR for use as an integration locus.
Nomination of Potential Features That Affect Suitability for Integration
We examined the size of an IGR for its influence on integration sites. IGRs should have less disruptive effects on cellular functions, but these regions do contain many functional elements such as transcription factor binding sites, enhancer-like sequences, small RNAs, and transcription terminators that could be unintentionally disrupted. To our knowledge, these features have not been annotated comprehensively in K. phaffii, as is the case in most nonmodel hosts. ATAC-seq can partially inform the location of potential sites of transcriptional regulation through the mapping of accessibility peaks in putative promoter regions and the nucleosomes that typically flank these sites.18 For example, we mapped the relative frequency of nucleosomes around the start codon in K. phaffii and found that two nucleosomes were often centered 200 bp upstream of the start codon as well as just after the start codon, suggesting the importance of the contained region for transcription (Figure 1D). The genome of K. phaffii is extremely compact, however, with a median IGR length of only 300 bp (Figure 2A). Larger IGRs may be desirable, therefore, to provide sufficient distancing between the integration sites for heterologous genes and regions of the genome that are essential for transcription of neighboring genes or other essential functions.
Figure 2.

Characterization of genomic properties of IGRs that potentially influence suitability for integration of a heterologous gene. (A) Distribution of intergenic region (IGR) length for each chromosome. (B) Distribution of IGR length for each orientation of adjacent genes. (C,D) Overall accessibility score in the promoter region (defined as 600 bp upstream of translation start site) for low (bottom 25%), medium (middle 50%), and high (top 25%) expression of genes under growth on (C) glycerol or (D) methanol.
In addition to size, we considered the combination of the expression of genes adjacent to, and intensities of accessibility peaks contained within, a given IGR. Previous studies have demonstrated a correlation between the intensity of accessibility peaks in a promoter region and transcriptional activity of that gene in S. cerevisiae, particularly when expression is induced in response to a stimulus.18 We attempted to detect a similar response in K. phaffii using ATAC-seq by analyzing changes in methanol utilization genes between the glycerol and methanol conditions. As expected, the change in the expression of these genes across conditions was correlated with the change in the overall accessibility scores of the respective promoter regions (Figure S3). We extended this analysis to all methanol- and glycerol-specific genes, and observed a similar relationship primarily for methanol-specific genes (Figure S3).
To characterize the relationship between gene expression and accessibility of the promoter region, we divided genes by expression level into low (bottom 25%), medium (middle 50%), and high (top 25%) groups, and compared the overall score for peak intensity within promoters (Figure 2C,D). Expression levels and accessibility peaks were obtained for >92% of genes across conditions. Under both conditions, the promoter regions of highly expressed genes had higher accessibility scores (p < 2.2 × 10–16) than those of medium expressed genes, and the trend continued from medium to lowly expressed genes (p < 6.4 × 10–06). These results supported our hypothesis that the combination of accessibility of an IGR and the expression of adjacent genes were important factors for determining the suitability of sites for integration of heterologous constructs.
The final feature of an IGR that we considered was the orientation of the adjacent genes (codirectional, convergent, or divergent). Proper DNA supercoiling is a requirement for transcription: it influences the strength of expression, and it can be affected by the neighboring genes.23 IGRs bordered by convergent genes tend to accumulate more positive supercoiling, which can impede transcription.23 Convergent gene pairs can also form mRNA–mRNA interactions that regulate expression post-transcriptionally.24 Nearly 90% of the 1500 IGRs in K. phaffii for which there were no recovered accessibility peaks were convergent with a median size less than 50 bp and might form such interactions. Disruption of these interactions by integration of a heterologous construct may disrupt cellular functions since gene pairs that form these interactions are often conserved and associated with stress response.24 Codirectional genes may be coregulated by a single promoter, while expression of divergent genes may be driven by a bidirectional promoter. Additionally, the orientation of an IGR is correlated with its size, with convergent IGRs being significantly smaller than divergent or codirectional IGRs (Figure 2B).
Construction and Characterization of an IGR Library
To assess the importance of these four genomic and transcriptomic features, we selected 72 candidate IGRs that spanned different sizes, orientations, accessibility peak scores, and expression strengths of adjacent genes. For this demonstration, we sought IGRs that would permit strong expression both under growth on glycerol and on methanol. We, therefore, identified approximately 1300 of the 3500 IGRs with detected accessibility peaks as constitutive, defined here as a change in expression of adjacent genes by less than 4-fold and in accessibility by less than 30%. For these 1300 IGRs, we classified the size of IGRs as small (<450 bp) or large (>550 bp), and average expression of adjacent genes as low, medium, or high using the same criteria as previously. Finally, we calculated an overall accessibility score by summing the intensity scores of all peaks within each IGR, and classified these scores as low (<250), medium (300–850), or high (>1000). Categorization of expression-accessibility into four groups (low-low, low-high, med-med, and high-high) covered the design space observed in the genome (Figure S4). Our selection of 72 IGRs comprised three IGRs for each of the 24 combinations of sizes, orientations, and expression-accessibility categories. Because many of these combinations were at the extremes of the design space, three IGRs per combination corresponded to a median representation of roughly 20% of possible constitutive IGRs meeting each set of criteria (Figure S5). We reasoned, therefore, that the selected subset of IGRs adequately represented the genome-wide design space for size, expression, accessibility, and orientation.
We constructed a library of clones expressing eGFP for heterologous insertion into each of the selected IGRs (Figure 3). Expression of eGFP was controlled by the strong, constitutive TEF1 promoter and 450 bp of IGR-specific homology arms on either end of the donor DNA mediated homologous recombination. To ensure targeted integration, a plasmid containing Cas9 and a sgRNA targeting the desired IGR was cotransformed with the eGFP linear DNA into a strain previously modified to express mCherry. Successful integrants were obtained for 38 of the 72 selected IGRs. (The unsuitability of particular IGRs for integration by CRISPR/Cas9 may have affected our ability to obtain successful integrants. Several confounding factors exist, however, such as the efficiency of the sgRNA or the suitability of the local nucleotide sequence chosen for each IGR, which prevent us from drawing conclusions about these unsuccessful integrants.) The 38 successful integrants included representatives for all categories (size, orientation, expression-accessibility), and were characterized for gene expression and growth (Figure S5).
Figure 3.

Evaluation of the impact of IGR properties on transgene expression and cell growth. (A) Workflow diagram for creation and analysis of an IGR targeting library. (B) Normalized GFP fluorescence versus IGR size. (C) Fluorescence versus IGR orientation. (D) Fluorescence versus category of adjacent gene expression and overall IGR accessibility. (E) Normalized max growth rate versus IGR size. (F) Growth versus IGR orientation. (G) Growth versus expression-accessibility category. Adjusted p-values computed using a Wilcoxon signed-rank test with the Benjamini–Hochberg correction for multiple hypotheses.
Effects of IGR Size and Orientation on Growth and Expression
We performed flow cytometry to evaluate relative expression from each IGR in the library, and independently monitored the growth rates of each strain. We measured the expression of eGFP relative to mCherry to correct for intrinsic heterogeneity in expression among single cells. Expression was well correlated (R2 > 0.9) between glycerol and methanol conditions, confirming that integration sites permitted constitutive expression as intended.
We did not observe significant differences in expression of GFP or growth between large and small IGRs (Figure 3B, Figure 3E). Given the compact structure of the K. phaffii genome, it was unsurprising that robust expression was observed from a heterologous construct despite a small distance between it and neighboring genes.
Similarly, the orientation of genes neighboring an IGR did not significantly affect expression levels or growth (Figure 3C, Figure 3F). Even when considering the pairwise orientations of the transgene with each adjacent gene, expression did not differ dramatically (Figure S6). This result might suggest that the local sequence context of the heterologous construct determined and regulated the proper supercoiling necessary for robust expression of the transgene.23 Interference from adjacent genes, therefore, appears insignificant. Previous studies have focused primarily on the influence of orientation within synthetic gene clusters.25,26 The influence of orientation between an integrated heterologous gene and adjacent endogenous genes, especially in eukaryotes has been less characterized, however. Between endogenous genes, a relatively weaker promoter increases susceptibility to transcriptional interference.27,28 This relationship is consistent with our observations: the strong TEF1 promoter was robust to interference from weaker promoters adjacent to the integration site. While adjacent genes did not appear to influence expression of the transgene, the reverse may not have been the case. Interestingly, the maximum growth rate was higher for strains with a convergent orientation of the transgene and downstream gene versus when this orientation was codirectional (Figure S6). Integration of a strongly expressed transgene might therefore interfere with the promoter of the downstream gene. Nearly half of the IGRs in the library are located upstream of genes involved in central functions such as metabolism, cell cycle, or stress response, which, if disrupted, might inhibit cell growth.
While we did not observe local effects from gene orientation, expression of eGFP did correlate with the chromosome on which a particular IGR was located. The highest expression was observed for IGRs located on chromosome 1, then chromosome 2, 3, and 4 in that order (Figure S6). This ordering follows the decreasing length of the chromosomes. The mechanism underlying this observation is unclear, and further study on the topology or structure of the chromosomes could yield additional insights.29
Effects of IGR Accessibility and Expression Levels of Adjacent Genes
Unexpectedly, normalized expression of GFP was significantly higher upon integration into IGRs with low to moderate expression and accessibility scores (low-low and med-med categories) than those with high accessibility scores (low-high and high-high categories, Figure 3D). Three-fold differences in GFP expression were observed between IGRs with similarly low levels of expression of adjacent genes but large differences in accessibility scores (low-high versus high-high categories). This result suggests that the intensity of accessibility peaks dominated the observed locus effects, rather than the expression levels of adjacent genes. Transcriptional interference depends on the relative expression level of adjacent genes in concert with the orientation and proximity of the respective promoters.30 It is unsurprising, therefore, that the expression levels of adjacent genes did not significantly affect expression of the transgene, given that we also observed a minimal impact from orientation and proximity (IGR size).
The observed trend—that integration into IGRs with high overall accessibility scores led to lower transgene expression—may seem counterintuitive. An important distinction exists, however, between a high overall accessibility score and an open chromatin state. Open chromatin is required for robust expression, especially in multicellular organisms, where gene expression is more tightly controlled by epigenetics.19,31 In K. phaffii, we detected accessibility peaks or expression in almost every transcriptional unit under both conditions tested (Figures S1, S2, S5A), making it likely that nearly all chromosomal IGRs in this organism are always in an open state. This result suggests that even IGRs with relatively low peak intensities are still accessible to recombination and expression machinery, and may explain why integration into all IGRs resulted in GFP-positive cells by flow cytometry.
In contrast to chromatin state, peak intensity (as measured by ATAC-seq) corresponds to accessibility to the transposase, which is correlated with chromatin activity by chromatin remodeling and transcription factors.17 The differences in overall accessibility score among IGRs in our library therefore corresponded to differences in the activity of factors that immediately surround the transcriptional unit of the transgene, not whether the chromatin is open. Our results suggest that a high amount of this activity nearby the transgene is correlated with lower expression of that transgene. This correlation was most prominent in IGRs with divergent or codirectional orientations, which contain promoter regions and transcription factor binding sites unlike IGRs with convergent orientations.
Extension to Multicopy Integrations
After uncovering a locus effect related to the intensity of accessibility peaks for single-copy integration, we next sought to see if there was a similar effect for multicopy integrations. In K. phaffii, expression of the recombinant gene of interest is typically driven by several tandem copies of the heterologous cassette, integrated via homologous recombination into a single locus just upstream of a promoter.32 To evaluate potential locus effects, we constructed strains to secrete human growth hormone (hGH) or granulocyte-colony stimulating factor (G-CSF) under control of three different promoters (PAOX1, PDAS2, POLE1) integrated into the genome upstream of various promoters (AOX1, DAS2, TDH3, OLE1, or PIF1). These IGRs spanned a range of overall accessibility and expression levels of adjacent genes (Table S1). We did not observe the integration locus to have a significant effect on expression for any of the promoters or genes tested, as measured by RNA-seq (Figure 4). The copy numbers of transgenes spanned a similar range among loci, ruling out the possibility of bias in copy number. Expression was positively correlated with copy number for each promoter tested (PAOX1: r = 0.62, PDAS2: r = 0.38, POLE1: r = 0.84). In these multicopy insertions, the transgene is separated from the endogenous locus by multiple copies of heterologous contextual elements, which can total 30 kb in length for high-copy integrations. It is therefore likely that the epigenetic state of heterologous elements, and not that of the endogenous locus, most influences expression of the transgene.
Figure 4.
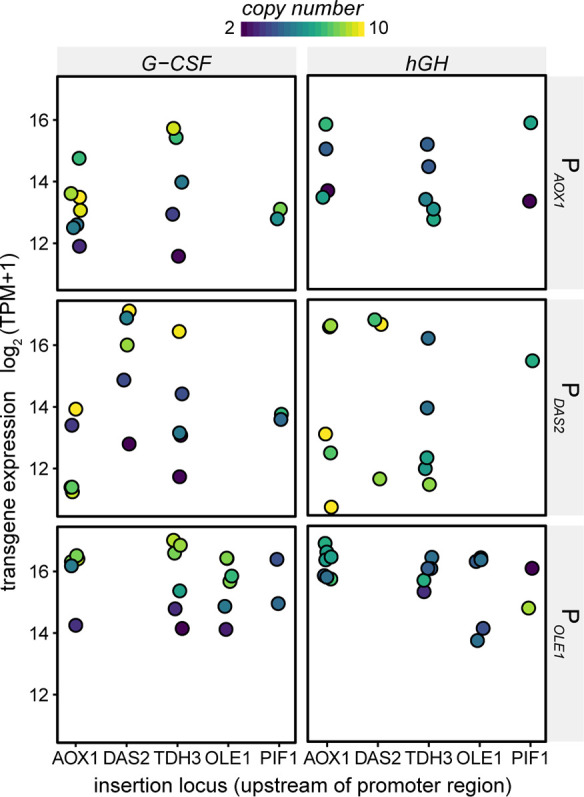
Comparison of transgene expression among insertion loci across three promoters (PAOX1, PDAS2, and POLE1) and two heterologous genes (G-CSF and hGH). Transgene expression was measured by RNA-seq as log2(TPM + 1). Each point is a unique clone and represents the average of triplicate samples analyzed by RNA-seq. The transgene copy number is indicated for each clone.
To characterize the epigenetic state of these heterologous elements, we performed ATAC-seq on three hGH-expressing strains with multicopy insertions upstream of the native AOX1, DAS2, or OLE1 promoters (Figure S8). Sequence elements of these constructs occurring natively in the genome (such as the AOX1 transcription terminator) adopted similar peak position, peak intensity, and nucleosome occupancy as the native copy. Interestingly, aligned reads from ATAC-seq formed a much stronger, but flat (ill-defined peaks), signal across most of the portion of the cassette that contained the bacterial origin of replication and the transcriptional unit for the selectable marker—all sequence elements originating in other yeast or bacteria. Within this portion of the cassette, only the promoter region of the selectable marker appeared to have a defined peak, and no nucleosomes were predicted with high confidence (>80% occupancy). These results may suggest that the non-native elements of each construct adopted an especially accessible chromatin state, but without strong evidence of much chromatin activity such as by chromatin remodeling or transcription factors. This accessible and largely inactive context surrounds the transcriptional unit of the transgene in the case of multicopy insertions.
Our collective findings uncovered an effect of the locus on transgene expression, but our previous library results suggested a potential impact of transgene expression on the downstream native gene. To confirm this downstream impact, we compared the expression of TDH3 and PIF1 among strains integrated upstream of the TDH3 promoter, the PIF1 promoter, or another promoter elsewhere in the genome (Figure 5). The strong TDH3 promoter is the most common constitutive promoter used in K. phaffii and PIF1 is adjacent to TDH3 in the genome. We chose to drive transgene expression in this case by one of three other promoters to isolate the effect of the integration site on the adjacent genes.
Figure 5.
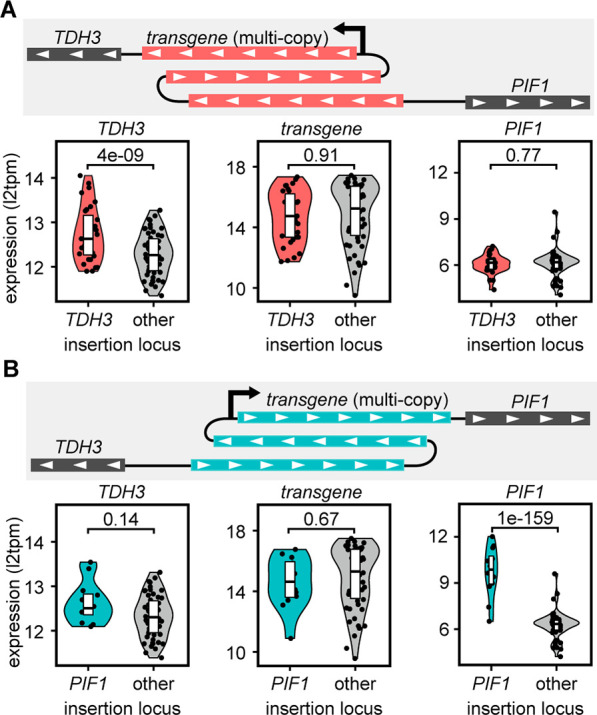
Influence of expression of heterologous genes on the downstream native gene. (A) Expression of TDH3, the transgene (hGH or G-CSF), and PIF1 as a function of integration either upstream of the TDH3 promoter or elsewhere in the genome. Each point represents the average of triplicate RNA-seq measurements of expression in log2(TPM + 1). Adjusted p-values were calculated using DESeq2. (B) Expression of TDH3, the transgene (hGH or G-CSF), and PIF1 as a function of integration either upstream of the PIF1 promoter or elsewhere in the genome.
Surprisingly, integration upstream of the PIF1 promoter led to an 8-fold increase in expression of PIF1 (adjusted p-value = 2.2 × 10–163) relative to integration elsewhere in the genome or even at the same IGR but oriented in the direction of TDH3. We observed no significant effect on expression of the transgene or TDH3. We observed the same effect upon integration upstream of the TDH3 promoter, though to a lesser degree, with a near 2-fold increase in expression of TDH3 (adjusted p-value = 4.4 × 10–09) relative to the other integration groups. These results confirmed the impact of transgene expression on the downstream gene observed with single-copy integrants and may suggest that the effect was even greater with multicopy integrants, perhaps due to positive feedback with each additional copy.
Reconciling Single-Copy and Multicopy Locus Effects
Together, our results suggest a significant locus effect on transgene expression for single-copy integrations that diminished for multicopy, tandem integrations. Each subsequent copy of the heterologous construct further increases the average distance between the transgene and the original genomic context. The introduction of multiple copies is thus likely to surround the transgene in a new epigenetic context, which contains multiple copies of the promoter driving expression of the transgene. In yeast, DNA looping can bring together enhancer-like elements with promoters and even join whole clusters of transcription units.23,33 It is possible, therefore, that tandem copies of a heterologous construct encourage close association of repetitive sequence elements in three-dimensional space, forming a new microenvironment. Such an association of the same binding sites within promoters may lead to cooperativity and further encourage the binding of factors needed to promote transcription.34
Selecting Integration Sites from ATAC-seq
Our data and observations here provide guidance on the selection of novel integration sites for genome engineering from a simple, one-time, genome-wide assay such as ATAC-seq. Our evaluation of the relative impacts of tested features may reduce the need for performing similar screens of IGRs in many other hosts. We envision that only an annotated genome and an ATAC-seq data set collected under the desired conditions may be sufficient to enable selection of suitable integration sites in many eukaryotic hosts.
Our results suggest ideal sites for integration are IGRs that are (1) sufficiently far from telomeres or centromeres, (2) in an open region of chromatin but with only low to moderate intensities of accessibility peaks nearby, and (3) in a convergent orientation with the native, downstream gene to prevent unwanted interference. In cases where weaker promoters drive expression of the transgene, it may be useful to avoid placing these constructs near highly transcribed genes to minimize any transcriptional interference. These criteria may be especially important for single-copy integration of heterologous genes such as by CRISPR/Cas9. For tandem, multicopy integrations to drive extremely strong expression, a convergent orientation with the downstream gene may be most important.
We believe that the framework here using a host’s epigenetic landscape should guide selection of heterologous insertion sites in many other eukaryotes. In developing this framework, we have also uncovered surprising biological relationships between transgene expression and the locus of integration that warrant further study in yeasts and higher eukaryotes. With a greater understanding of the impactful features of an integration site, novel engineering solutions can be developed to modify these interactions and promote robust expression. Such solutions might include the development of improved landing pads with enhancer-like elements or the optimization of synthetic gene clusters for pathway engineering. Our work underscores the importance of genome-wide assays such as ATAC-seq to better understand biological mechanisms and inform engineering decisions for the construction of improved cell factories.
Materials and Methods
Strains and Cultivations
K. phaffii Y-11430 was obtained from the USDA and modified for subsequent CRISPR/Cas9-based integration by frameshift knockout of KU70.35 This strain was further modified to express mCherry by cotransformation of a donor cassette and a circular plasmid containing Cas9 and guide RNA sequences, described previously.36 IGR libraries were similarly created by cotransformation of a donor cassette for eGFP expression and a circular plasmid for CRISPR/Cas9-based targeting to the desired IGR. Donor cassettes contained the following (5′ to 3′): a 450 bp 5′ homology arm, the TEF1 promoter, the mCherry or eGFP gene, a transcription terminator, and a 450 bp 3′ homology arm. The eGFP cassette contained a phleomycin D1 (Thermo Fisher Scientific) selection marker to facilitate screening for positive transformants. Guide RNA sequences were designed using the Broad Institute’s Genetic Perturbation Platform sgRNA tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design),6,10 constraining GC content to between 20% and 80% and requiring no homopolymers >4 bp. Successful on-target integration was confirmed by PCR amplification of the target locus.
Strains were generated to express either human growth (hGH) or granulocyte-colony stimulating factor (G-CSF) under control of multiple native promoters (AOX1, DAS2, OLE1) by modifying the commercial vector pPICZ A (Thermo Fisher Scientific). A single homology sequence was inserted upstream of the promoter to direct integration into a locus other than that of the promoter (AOX1, DAS2, GAPDH, OLE1, PIF1). Plasmids were linearized in the middle of the homology sequence or native promoter for multicopy insertion into that particular locus.
For collection of samples for ATAC-seq and RNA-seq, Y-11430 wild-type cells or multicopy hGH and GCSF cells were grown in 24-well deep well plates (25 °C, 600 rpm) using glycerol-containing media (BMGY-Buffered Glycerol Complex Medium, Teknova) supplemented to 4% (v/v) glycerol. After 24 h of biomass accumulation, cells were pelleted and resuspended in BMMY (Buffered Methanol Complex Medium, Teknova) containing 3% (v/v) methanol. Samples were collected for ATAC-seq and RNA-seq from triplicate cultures after 24 h initial growth in BMGY and after an additional 24 h growth in BMMY for wild-type cells and only the BMMY phase for protein-expressing cells.
ATAC-seq Analysis
Two million cells per sample were prepared for transposition as described previously.18 Spheroplasts were washed once with transposition buffer37 prior to transposition and PCR amplification for sequencing, which are described elsewhere.16 Amplified libraries were size selected using 0.4× followed by 0.8× Ampure XP beads according to manufacturer instructions (Beckman Coulter). Size-selected libraries were sequenced on an Illumina NextSeq to generate 40-nt paired-end reads.
Alignments were performed using Burrows-Wheeler Aligner (BWA-MEM) v0.7.5a,38 sorted, duplicates were marked, and .bam files were indexed using Picard v1.94 and samtools v0.1.19. Accessibility peaks were called and scored using MACS2, while nucleosome position and occupancy were called using NucleoATAC, both as described previously.18 The −log10(q-value) score from the narrowPeak output of MACS2 was defined to be the accessibility score of a peak. Overall scores for promoter regions or IGRs were calculated as the sum of scores of peaks contained within these regions.
RNA-seq Analysis
RNA was extracted and purified according to the Qiagen RNeasy kit and RNA quality was analyzed to ensure RNA Quality Number >7. RNA libraries were prepared using the 3′ DGE method39 and sequenced on an Illumina HiSeq2500 to generate paired reads of 17 bp (read 1) + 46 bp (read 2). Sequenced mRNA transcripts were quantified with Salmon v0.9.1,40 using a transcript database consisting of a single K. phaffii transcript per gene, each with a 100-nt extension on the 3′ end, as well as rhGH and rhG-CSF transgenes. Expression was visualized using log2(Transcripts per Million +1) values.
Cell Growth and Fluorescence Characterization
Strains with confirmed integration of the eGFP cassette into the proper IGR were grown to saturation in YPD in a 96-well plate at ambient temperature and 1000 rpm shaking for 2–3 days. From these cultures, complex media containing either 4% (v/v) glycerol or 3% (v/v) methanol was inoculated to an initial OD600 of 0.1 in a 96-well plate. Cultures were subsequently grown in a microplate reader (Tecan), with OD600 measurements every hour for 24 h. Growth measurements were performed on biological triplicates of each library member for each carbon source. Growth data for each well was fit to a Baranyi model to obtain a maximum growth rate. Maximum growth rates were scaled within each plate to account for heterogeneity between plates. In parallel, cultures were similarly started at an initial OD600 of 0.1, grown for 4 h, and then analyzed by flow cytometry for GFP and mCherry fluorescence, gating for horizontal and vertical singlets. Cells were further gated for positive mCherry fluorescence to eliminate nonviable or transiently nonexpressing cells. Flow cytometry was performed on four biological replicates of each library member for each carbon source.
Analytical Assays for Strain Characterization
For hGH- and G-CSF-secreting strains, copy number analysis by qPCR and determination of protein concentration by sandwich ELISA were both performed as described previously.21
Data Availability
Raw and processed ATAC-seq data used in this study can be obtained from the NCBI Gene Expression Omnibus (accession number: GSE154330).
Acknowledgments
We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the Bioinformatics & Computing, Flow Cytometry, and Genomics core facilities. This work was supported by the Defense Advanced Research Projects Agency (DARPA) and SPAWAR Systems Center Pacific (SSC Pacific) (contract no. N66001-13-C-4025), by the Bill & Melinda Gates Foundation, and by the AltHost Consortium. This work was also supported in part by the Koch Institute Support (core) grant P30-CA14051 from the National Cancer Institute. J.R.B. and N.C.D. were partially supported by a NIGMS/MIT Biotechnology Training Program Fellowship (NIH contract no. 2T32GM008334-26). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCI, NIH, DARPA, SSC Pacific, the Bill & Melinda Gates Foundation, or the AltHost Consortium.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00299.
Figure S1, Gene expression and accessibility score across the K. phaffii genome; Figure S2, Gene expression across carbon sources; Figure S3, Effect of changes in chromatin accessibility on changes in gene expression; Figure S4, Selection of IGRs with varied adjacent gene expression and accessibility scores; Figure S5, Demographics of IGRs in K. phaffii; Figure S6, Effect of integration site on expression for additional genomic properties; Figure S7, Copy number distributions for multicopy strains; Figure S8, ATAC-seq profiles across heterologous cassettes for three multicopy strains; Table S1, Expression and accessibility of loci used for multicopy integration (PDF)
Author Contributions
J.R.B., K.R.L., and J.C.L. developed the concepts and designed the study. J.R.B., M.C.T., and N.A.C. performed the experiments. J.R.B., K.R.L., and J.C.L. wrote the manuscript. J.R.B. and C.A.W. performed bioinformatics analyses. N.D. developed constructs subsequently used for construction of the library.
The authors declare no competing financial interest.
Supplementary Material
References
- Li Y.; Li S.; Thodey K.; Trenchard I.; Cravens A.; Smolke C. D. (2018) Complete Biosynthesis of Noscapine and Halogenated Alkaloids in Yeast. Proc. Natl. Acad. Sci. U. S. A. 115 (17), E3922–E3931. 10.1073/pnas.1721469115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton S. R.; Davidson R. C.; Sethuraman N.; Nett J. H.; Jiang Y.; Rios S.; Bobrowicz P.; Stadheim T. a; Li H.; Choi B.; et al. (2006) Humanization of Yeast to Produce Complex Terminally Sialylated Glycoproteins. Science (Washington, DC, U. S.) 313 (5792), 1441–1443. 10.1126/science.1130256. [DOI] [PubMed] [Google Scholar]
- Deaner M.; Alper H. S. (2017) Systematic Testing of Enzyme Perturbation Sensitivities via Graded DCas9Modulation in Saccharomyces Cerevisiae. Metab. Eng. 40, 14–22. 10.1016/j.ymben.2017.01.012. [DOI] [PubMed] [Google Scholar]
- Löbs A. K.; Schwartz C.; Thorwall S.; Wheeldon I. (2018) Highly Multiplexed CRISPRi Repression of Respiratory Functions Enhances Mitochondrial Localized Ethyl Acetate Biosynthesis in Kluyveromyces Marxianus. ACS Synth. Biol. 7 (11), 2647–2655. 10.1021/acssynbio.8b00331. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Wang S.; Halim A.; Schulz M. A.; Frodin M.; Rahman S. H.; Vester-Christensen M. B.; Behrens C.; Kristensen C.; Vakhrushev S. Y.; et al. (2015) Engineered CHO Cells for Production of Diverse, Homogeneous Glycoproteins. Nat. Biotechnol. 33 (8), 842–844. 10.1038/nbt.3280. [DOI] [PubMed] [Google Scholar]
- Doench J. G.; Fusi N.; Sullender M.; Hegde M.; Vaimberg E. W.; Donovan K. F.; Smith I.; Tothova Z.; Wilen C.; Orchard R.; et al. (2016) Optimized SgRNA Design to Maximize Activity and Minimize Off-Target Effects of CRISPR-Cas9. Nat. Biotechnol. 34 (2), 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidukov L.; Wroblewska L.; Teague B.; Nelson T.; Zhang X.; Liu Y.; Jagtap K.; Mamo S.; Tseng W. A.; Lowe A.; et al. (2018) A Multi-Landing Pad DNA Integration Platform for Mammalian Cell Engineering. Nucleic Acids Res. 46 (8), 4072–4086. 10.1093/nar/gky216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nett J. H.; Hodel N.; Rausch S.; Wildt S. (2005) Cloning and Disruption of the Pichia Pastoris ARG1, ARG2, ARG3, HIS1, HIS2, HIS5, HIS6 Genes and Their Use as Auxotrophic Markers. Yeast 22 (4), 295–304. 10.1002/yea.1202. [DOI] [PubMed] [Google Scholar]
- Gassler T.; Sauer M.; Gasser B.; Egermeier M.; Troyer C.; Causon T.; Hann S.; Mattanovich D.; Steiger M. G. (2020) The Industrial Yeast Pichia Pastoris Is Converted from a Heterotroph into an Autotroph Capable of Growth on CO2. Nat. Biotechnol. 38, 210. 10.1038/s41587-019-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson K. R.; Hanna R. E.; Hegde M.; Donovan K. F.; Strand C.; Sullender M. E.; Vaimberg E. W.; Goodale A.; Root D. E.; Piccioni F.; et al. (2018) Optimized Libraries for CRISPR-Cas9 Genetic Screens with Multiple Modalities. Nat. Commun. 9 (1), 1–15. 10.1038/s41467-018-07901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M.; Papapetrou E. P.; Bushman F. D. (2012) Safe Harbours for the Integration of New DNA in the Human Genome. Nat. Rev. Cancer 12 (1), 51–58. 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- Papapetrou E. P.; Schambach A. (2016) Gene Insertion into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 24 (4), 678–684. 10.1038/mt.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceroni F.; Ellis T. (2018) The Challenges Facing Synthetic Biology in Eukaryotes. Nat. Rev. Mol. Cell Biol. 19 (8), 481–482. 10.1038/s41580-018-0013-2. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zhang J. (2016) The Genomic Landscape of Position Effects on Protein Expression Level and Noise in Yeast. Cell Syst. 2 (5), 347–354. 10.1016/j.cels.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Licon K.; Otsuka R.; Pillus L.; Ideker T. (2013) Decoupling Epigenetic and Genetic Effects through Systematic Analysis of Gene Position. Cell Rep. 3 (1), 128–137. 10.1016/j.celrep.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.; Wu B.; Chang H.; Greenleaf W. (2015) ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J. D.; Giresi P. G.; Zaba L. C.; Chang H. Y.; Greenleaf W. J. (2013) Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 10 (12), 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep A. N.; Buenrostro J. D.; Denny S. K.; Schwartz K.; Sherlock G.; Greenleaf W. J. (2015) Structured Nucleosome Fingerprints Enable High-Resolution Mapping of Chromatin Architecture within Regulatory Regions. Genome Res. 25 (11), 1757–1770. 10.1101/gr.192294.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien S. A.; Lee K.; Fu H. Y.; Lee Z.; Le T. S.; Stach C. S.; McCann M. G.; Zhang A. Q.; Smanski M. J.; Somia N. V.; et al. (2018) Single Copy Transgene Integration in a Transcriptionally Active Site for Recombinant Protein Synthesis. Biotechnol. J. 13 (10), 1800226. 10.1002/biot.201800226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love K. R.; Shah K. A.; Whittaker C. A.; Wu J.; Bartlett M. C.; Ma D.; Leeson R. L.; Priest M.; Borowsky J.; Young S. K.; et al. (2016) Comparative Genomics and Transcriptomics of Pichia Pastoris. BMC Genomics 17, 550. 10.1186/s12864-016-2876-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady J. R.; Whittaker C. A.; Tan M. C.; Kristensen D. L.; Ma D.; Dalvie N. C.; Love K. R.; Love J. C. (2020) Comparative Genome-Scale Analysis of Pichia Pastoris Variants Informs Selection of an Optimal Base Strain. Biotechnol. Bioeng. 117 (2), 543–555. 10.1002/bit.27209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturmberger L.; Chappell T.; Geier M.; Krainer F.; Day K. J.; Vide U.; Trstenjak S.; Schiefer A.; Richardson T.; Soriaga L.; et al. (2016) Refined Pichia Pastoris Reference Genome Sequence. J. Biotechnol. 235, 121–131. 10.1016/j.jbiotec.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achar Y. J.; Adhil M.; Choudhary R.; Gilbert N.; Foiani M. (2020) Negative Supercoil at Gene Boundaries Modulates Gene Topology Topological Context of Pol II Genes. Nature 577, 701. 10.1038/s41586-020-1934-4. [DOI] [PubMed] [Google Scholar]
- Gilet J.; Conte R.; Torchet C.; Benard L.; Lafontaine I. (2020) Additional Layer of Regulation via Convergent Gene Orientation in Yeasts. Mol. Biol. Evol. 37 (2), 365–378. 10.1093/molbev/msz221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L. W.; Hussein R.; Block D. H. S.; Lim H. N. (2013) Minimal Effect of Gene Clustering on Expression in Escherichia Coli. Genetics 193 (2), 453–465. 10.1534/genetics.112.147199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung E.; Dy A. J.; Martin K. B.; Ng A. H.; Del Vecchio D.; Beck J. L.; Collins J. J.; Murray R. M. (2017) Biophysical Constraints Arising from Compositional Context in Synthetic Gene Networks. Cell Syst. 5 (1), 11–24. 10.1016/j.cels.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Shearwin K. E.; Callen B. P.; Egan J. B. (2005) Transcriptional Interference - A Crash Course. Trends Genet. 21 (6), 339–345. 10.1016/j.tig.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneppen K.; Dodd I. B.; Shearwin K. E.; Palmer A. C.; Schubert R. A.; Callen B. P.; Egan J. B. (2005) A Mathematical Model for Transcriptional Interference by RNA Polymerase Traffic in Escherichia Coli. J. Mol. Biol. 346 (2), 399–409. 10.1016/j.jmb.2004.11.075. [DOI] [PubMed] [Google Scholar]
- Hildebrand E. M.; Dekker J. (2020) Mechanisms and Functions of Chromosome Compartmentalization. Trends Biochem. Sci. 45 (5), 385–396. 10.1016/j.tibs.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N.; Palmer A. C.; Dodd I. B.; Shearwin K. E. (2017) Directing Traffic on DNA—How Transcription Factors Relieve or Induce Transcriptional Interference. Transcription 8 (2), 120–125. 10.1080/21541264.2017.1285851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal S. I. S.; Moazed D. (2003) Heterochromatin and Epigenetic Control of Gene Expression. Science (Washington, DC, U. S.) 301 (5634), 798–802. 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- Life Technologies (2014) Pichia Expression Kit User Guide. [Google Scholar]
- Petrascheck M.; Escher D.; Mahmoudi T.; Verrijzer C. P.; Schaffner W.; Barberis A. (2005) DNA Looping Induced by a Transcriptional Enhancer in Vivo. Nucleic Acids Res. 33 (12), 3743–3750. 10.1093/nar/gki689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D.; Shrinivas K.; Young R. A.; Chakraborty A. K.; Sharp P. A. (2017) A Phase Separation Model for Transcriptional Control. Cell 169 (1), 13–23. 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näätsaari L.; Mistlberger B.; Ruth C.; Hajek T.; Hartner F. S.; Glieder A. (2012) Deletion of the Pichia Pastoris Ku70 Homologue Facilitates Platform Strain Generation for Gene Expression and Synthetic Biology. PLoS One 7 (6), e39720. 10.1371/journal.pone.0039720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvie N. C.; Leal J.; Whittaker C. A.; Yang Y.; Brady J. R.; Love K. R.; Love J. C. (2020) Host-Informed Expression of CRISPR Guide RNA for Genomic Engineering in Komagataella Phaffii. ACS Synth. Biol. 9, 26–35. 10.1021/acssynbio.9b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Gu L.; Adey A.; Radlwimmer B.; Wang W.; Hovestadt V.; Bähr M.; Wolf S.; Shendure J.; Eils R.; et al. (2013) Tagmentation-Based Whole-Genome Bisulfite Sequencing. Nat. Protoc. 8 (10), 2022–2032. 10.1038/nprot.2013.118. [DOI] [PubMed] [Google Scholar]
- Li H.; Durbin R. (2010) Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 26 (5), 589–595. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M., Cacchiarelli D., Semrau S., van Oudenaarden A., and Mikkelsen T. S. (2014) Characterization of Directed Differentiation by High-Throughput Single-Cell RNA-Seq. bioRxiv, Mar 5, 2014, 003236. 10.1101/003236, accessed 2020-06-07. [DOI]
- Patro R.; Duggal G.; Love M. I.; Irizarry R. A.; Kingsford C. (2017) Salmon: Fast and Bias-Aware Quantification of Transcript Expression Using Dual-Phase Inference. Nat. Methods 14 (4), 417. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed ATAC-seq data used in this study can be obtained from the NCBI Gene Expression Omnibus (accession number: GSE154330).