

Abstract
Background
Fabry disease (FD), the second most prevalent lysosomal storage disorder, is classified as a rare disease. It often leads to significant quality of life impairments and premature death. Many cases remain undiagnosed due to the rarity and heterogeneity. Further, costs related to treatment often constitute a substantial financial burden for patients and health systems. While its epidemiology is still unclear, newborn screenings suggest that its actual prevalence rate is significantly higher than previously suspected.
Methods
Based on well‐established methodologies, this study gives an overview about the background of the development of FD‐related research and provides a critical view of future needs.
Results
On the grounds of benchmarking findings, an increasing research activity on FD can be observed. Most publishing countries are the USA, some European countries, Japan, Taiwan, and South Korea. In general, high‐income countries publish comparably more on FD than low‐ or middle‐income economies. The countries' financial and infrastructural background are unveiled as crucial factors for the FD research activity.
Conclusions
Overall, there is a need to foster FD research infrastructure in developing and emerging countries with focus on cost‐intensive genetic research that is independent from economic interests of big pharmaceutical companies.
Keywords: alpha‐galactosidase A deficiency, Anderson–Fabry, angiokeratoma diffuse, bibliometrics, GLA deficiency, hereditary dystopic lipidosis
In general, high‐income countries publish comparably more on Fabry disease (FD) than low‐ or middle‐income economies. The countries' financial‐ and infrastructural background as well as the presence of pharmaceutical companies are unveiled as crucial factors for the FD research activity. Therefore, our findings stress that collaborative efforts should be strengthened worldwide to facilitate a better understanding of FD in terms of epidemiological background, diagnosis, and treatment and to also develop standardized guidelines to improve patient outcomes.
1. INTRODUCTION
There are currently more than 7,000 known rare diseases, which are affecting more than 300 million people worldwide. It is estimated that the majority (~80%) is of genetic origin (GlobalGenes, 2018). Rare diseases are defined to affect less than 5:10,000 in the European Union (EU; EU, 1999) and fewer than 200,000 people in the USA (Orphan‐Drug‐Act 1983). The prevalence rate of classic Fabry disease (FD), an X‐linked lysosomal storage disorder, is estimated to range widely from 1:8,454 to 1:117,000 males (Houge & Skarbovik, 2005; Meikle, Hopwood, Clague, & Carey, 1999). More than 800 disease‐associated mutations have been described until today (Benjamin et al., 2017).
Fabry disease is the second most prevalent genetic metabolic storage disease worldwide (Shin et al., 2008) and often leads to progressive organ failure and premature death (Hoffmann, 2009). Fabry disease is caused by a mutation in the galactosidase alpha gene, which leads to absent or deficient function of the lysosomal enzyme α‐galactosidase A (GALA). Consequently, this results in an impairment of the metabolization of globotriaosylceramide (Gb3) and related glycosphingolipids, and subsequently, in a progressive lysosomal accumulation of Gb3 and glycosphingolipids. Those accumulations are thought to have cytotoxic, proinflammatory, and profibrotic effects (Germain, 2010; Oliveira & Ferreira, 2019). Cells particularly compromised by the glycosphingolipid accumulations include vascular cells, cells of the heart, kidney, brain, eye, skin, and the nervous system. Two different types of FD can be differentiated: the early onset variant, occurring predominantly in males with little or absent GALA activity, and the late‐onset variant with residual GALA activity (Germain, 2010). The clinical symptoms of the early onset form of FD usually begin between childhood and adolescence and include acroparesthesias, angiokeratomas and telangiectasias, renal and gastrointestinal problems, corneal opacity, microalbuminuria, and other nonspecific manifestations such as tinnitus and hypohidrosis (Oliveira & Ferreira, 2019). In addition to a significant quality of life impairment, early onset FD may lead to progressive renal, cardiac, and cerebrovascular consequences, which often result in premature death (Oliveira & Ferreira, 2019).
Patients with late‐onset FD are mostly seen later in life and their symptoms usually center around one organ system, most often the heart. However, diagnosis of FD can be challenging due to its rarity, multifaceted symptoms, and often controversial diagnostic criteria (Germain, 2010; Zarate & Hopkin, 2008), which often result in a delay of diagnosis. Its nature as a rare disease is linked to a low awareness among physicians worldwide. This often results in a significant delay in diagnosis or patients remaining undiagnosed, which is in turn oftentimes associated with tremendous psychosocial consequences among others. Thus, it is assumed that the prevalence rates are largely underestimated given their only fragmentary ascertainment (Zarate & Hopkin, 2008).
Screenings for FD among newborn males have revealed frequencies as high as ≈1:3,000 in Italy and Japan, even reaching ≈1:1,500 in Taiwan (Germain, 2010; Hwu et al., 2009; Spada et al., 2006), but the inclusion of nonpathogenic DNA variants may impact these rates (Ortiz et al., 2018; Schiffmann et al., 2017).
The diagnosis of FD, the determination of the global prevalence, and incidence rates of FD remain challenging. The opinions are varying about the effectiveness of the expansion of screenings to enable early diagnosis and initiation of treatment before disease onset and progression. Some society and government‐sponsored guidelines from selected countries and regions around the world exist, but there have to be standardized approaches all over the world.
Although FD research evolved over the years, it still represents an enormous burden for clinicians and health systems worldwide due to its difficult diagnosis, high costs related to treatment, and the multidisciplinary approach required for an effective management. Consequently, further scientific efforts have to be directed to collaborative efforts to facilitate a better understanding of the underlying pathophysiology and to develop standardized guidelines for the recognition, assessment, and monitoring of disease‐associated morbidities, as well as therapeutic concepts, including enzyme replacement and other adjunctive approaches, to improve patient outcomes (Eng et al., 2006). Robust prognosis of future trends and necessities requires the profound knowledge of the past development and the current status of the global FD research infrastructure. Therefore, here we provide the first in‐depth analysis of the global FD research activity for scientists, funders, and decision makers interested in FD to create a productive and promising scientific environment for times to come.
2. RESULTS
2.1. Publication development over time
The first entry on FD listed in the Web of Science (WoS), a publisher‐independent global citation database, can be traced back to an article published in 1916 by G. Stümpke in the journal “Archiv für Dermatologie und Syphilis”. Stümpke's article reports a case study of angiokeratoma corporis diffusum in Germany (Stumpke, 1916). Interestingly, in his article, he refers to a report in the same journal describing a similar occurrence, which was already published in 1898. This was also the year of the first clinical description of FD, respectively, by Johannes Fabry (Fabry, 1898), in Germany, and William Anderson (Anderson, 1898), in England. Based on this first presentation of the disease, the name Fabry–Anderson disease was coined. In the following years until the 1960s, an only sporadic publication activity on FD was seen. The few publications during this time episode were mainly composed of case descriptions of angiokeratomas.
In terms of the qualitative aspects of FD‐related articles, we observed a major peak of the citation rate in 1962, which can be referred back to a highly cited study (180 citations) of eight affected families (Wise, Wallace, & Jellinek, 1962). The third most cited article was published in 1967 by geneticist and biochemist Roscoe O. Brady, an early pioneer of lysosomal storage diseases. In his work, he discovered the underlying enzyme deficiency of FD (Brady et al., 1967).
Figure 1a shows the development of the publication output, the number of citations, and the annual average citation rates and clearly unveils the impact of the highly cited publication of Brady in 1967 (Brady et al., 1967). In the 1960s, Hashimoto et al. characterized firstly lysosomes and suggested the genetic reason for an abnormality (Hashimoto, Gross, & Lever, 1965). Afterward, studies from Opitz et al. (1965) and Brady et al. (1967) specified the genetic background. These new insights prepared the ground for novel approaches to evaluate the underlying pathophysiology of FD, clearly superior to the previously predominantly descriptive analyses (Fabry, 2001). During these years, the publication output per year further increased to a peak of n = 23 articles in 1978. After a decline in the following years, the publication activity rose again to 23 articles/year in 1990, shortly after the description of the structural gene organization in 1988 (Bishop, Kornreich, & Desnick, 1988) and detection of the mutations in the responsible gene in 1989 (Bernstein et al., 1989). The first significant increase in the publication output to n = 71 was observed during the time period from 2000 to 2002. This rise in the publication activity can be linked to the introduction of intravenous enzyme replacement therapy (ERT) in 2001, which led to a new era of FD research in terms of a better understanding of the underlying pathophysiology of the disease, the clinical progression, treatment, and prognosis (Hoffmann, 2009). Not surprisingly, the highest citation counts in FD research were observed for the two articles that, in 2001, reported the results of the seminal clinical trials of ERT with Agalsidase alfa and Agalsidase beta, which also led to the maximum of annual citation numbers (Eng et al., 2001; Schiffmann et al., 2001; Table 1). In 2001, another highly cited article determining the baseline parameters, such as prevalence, mortality, and frequency of FD manifestations, in a cohort of 98 hemizygous males using the UK/Eire register was published. Further, this study was the first to also evaluate the psychosexual impacts of FD, such as, for example, impotence (MacDermot, Holmes, & Miners, 2001). In 2007, another high impact article authored by Banikazemi et al. showed evidence for a delay in disease progression and premature death for patients under Agalsidase‐beta therapy (Banikazemi et al., 2007). After the approval of Agalsidase‐beta ERT in the EU and USA, another decline in the publication numbers from the maximum of n = 134 in 2010 to only 67 articles in 2014 followed. A novel approach of FD treatment, pharmacological chaperone therapy, has been developed for the treatment of patients with amenable mutations. Pharmacological chaperones are chemical compounds that can facilitate the proper folding of proteins and hereby improve its stability and trafficking to lysosomes (Hollak, Vedder, Linthorst, & Aerts, 2007). To date, the only approved therapeutic chaperone for FD in the USA is migalastat (Germain et al., 2016). It received regulatory approval as oral monotherapy for adults and adolescents over 16 years of age in the EU in 2016 (Markham, 2016). Maybe this might explain a new increase in publication numbers, resulting in n = 126 articles in 2017, the year with the second highest publication output on FD in this study. From these articles, 29 articles could be directly assigned to chaperone, meaning approximately 23%. In total, we could determine a minimum number of 176 articles on FD that are directly linked to chaperone.
Figure 1.
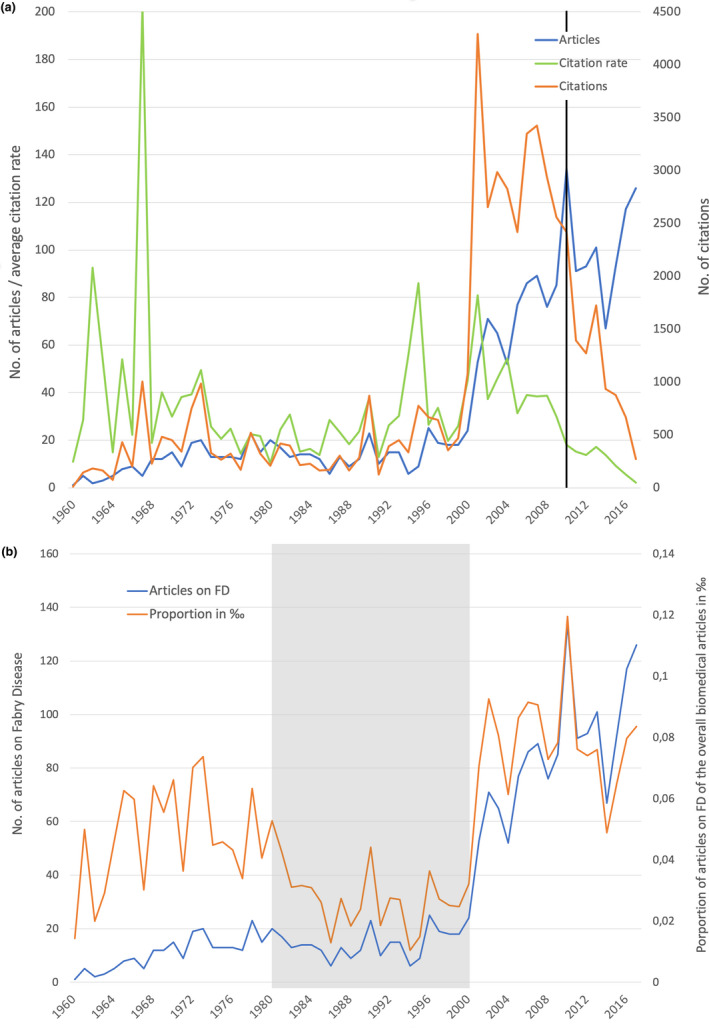
Development of publication parameters (original articles and reviews) from 1960 to 2017. (a) Number of articles, number of citations, average citation rate per year, black line: cited half‐life. (b) Number of articles on Fabry disease (FD) and their proportion of the overall biomedical articles in ‰, gray area: relatively low proportion of articles on FD
Table 1.
The most cited articles on Fabry disease
| Country | Authors | Year | Citations | Title |
|---|---|---|---|---|
| USA, France, UK, the Netherlands | Eng et al. (2001) | 2001 | 976 | Safety and efficacy of recombinant human alpha‐galactosidase a replacement therapy in Fabry's disease |
| USA | Schiffmann et al. (2001) | 2001 | 841 | Enzyme replacement therapy in Fabry disease – a randomized controlled trial |
| USA | Brady et al. (1967) | 1967 | 832 | Enzymatic defect in Fabry's disease – ceramidetrihexosidase deficiency |
| USA, Italy, Japan | Spada et al. (2006) | 2006 | 490 | High incidence of later‐onset Fabry disease revealed by newborn screening |
| Japan | Nakao et al. (1995) | 1995 | 484 | An atypical variant of Fabry's disease in men with left ventricular hypertrophy |
| UK, Italy, Switzerland, Belgium, Spain, Germany, Czech Republic, Austria | Mehta et al. (2004) | 2004 | 432 | Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey |
| UK | MacDermot et al. (2001) | 2001 | 425 | Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males |
| USA, France | Desnick et al. (2003) | 2003 | 389 | Fabry disease, an under‐recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy |
| USA, Czech Republic, UK, Canada | Banikazemi et al. (2007) | 2007 | 369 | Agalsidase‐beta therapy for advanced Fabry disease – a randomized trial |
| UK, Japan | Sachdev et al. (2002) | 2002 | 338 | Prevalence of Anderson–Fabry disease in male patients with late onset hypertrophic cardiomyopathy |
In terms of the number of citations, a steady decline since 2007 was observed. This is not due to a lower recognition of these studies. It is merely related to the shorter time span in which these articles had to reach their maximum number of citations. This effect is called cited half‐life, the time an article needs to reach 50% of its citations. It is assumed that it can last up to 7–8 years for biomedical publications (Magri & Solari, 1996). Future studies will show if the downswing remains.
The development of articles on FD in relation to the overall biomedical research output showed a relatively low FD activity between 1980 and 2000 (Figure 1b). The share of FD articles reached its lowest value in 1994 with only 0.01 per mil, clearly indicating a time period with low interest probably due to few new insights in this rare disease.
2.2. Countries' performance
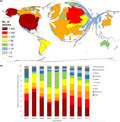
Since only the FD research‐related entries after 1973 included sufficient information to evaluate the underlying countries of origin, the geographical evaluations were only based on the research activity between 1973 and 2018. Our study unveiled a total of n = 1,975 articles during this period with the USA in the leading position (n = 500), followed by Germany (n = 325 articles), Italy (n = 240), UK (n = 238), France (n = 212), and Japan (n = 202). Brazil and Argentina were the only South American countries with more than 50 FD‐related articles, most Asian countries showed an only marginal contribution, and the research activity of African countries was close to zero.
In terms of the countries' relative contribution to the overall FD research output over time, a relatively balanced involvement of the 10 most publishing countries during the first (1973–1977) and last investigation interval (2013–2017) was revealed, whereas varying shares were found for the years between 1978 and 2013 (Figure 2b). As for the USA, an increase in their contribution from 31.34% (1973–1977) to 48.85% (1978–1982) with a subsequent decrease to 17.16% in the last interval was observed. Germany exhibited an extremely low involvement in FD research of only 1.49% of the total FD output between 1978 and 1997, but demonstrated an increase in its peak of 17.86% during the period 2008 to 2012. Italy's share increased over time from 1.49% to 15.86%, whereas the share of France decreased from 18.75% in the second period to 5.6% in the last period.
Figure 2.
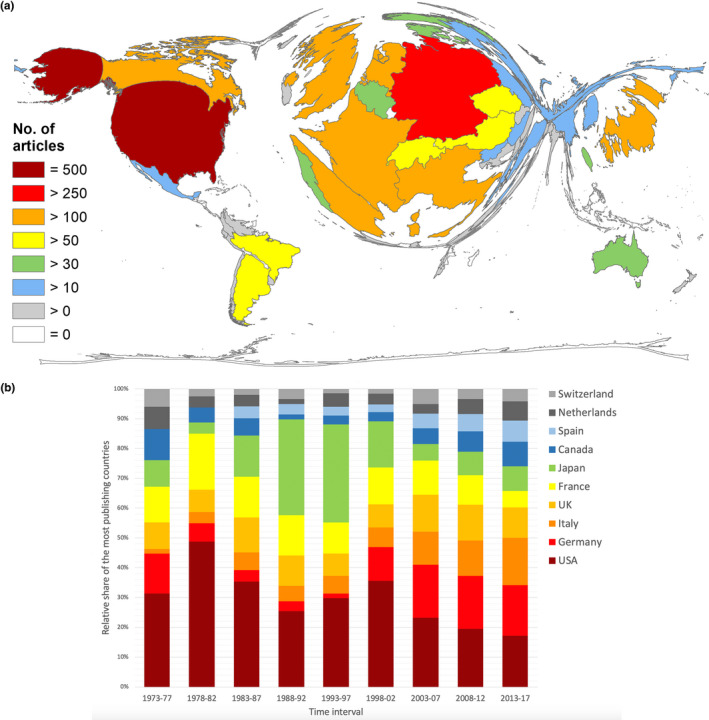
Global publication output on Fabry disease. (a) Number of articles per country. (b) Development of the relative proportion of the most publishing countries
Although the time between 1980 and 2000 was characterized by a relatively low FD research activity (Figure 1b) with a low contribution of some of the main protagonists, Japan on the contrary exhibited an increasing publication performance throughout this whole interval (Figure 2b).
As an indicator for the scientific resonance of the countries' FD research performance, we determined the country‐specific citation parameters. In terms of the overall number of received citations (c), the USA was again the clear leader with the c = 23,183 and was followed by the UK (c = 11,558), who here ranked second as opposed to fourth in terms of the overall publication output. The UK was followed by Germany (c = 9,631), France (c = 6,468), and Italy (c = 5,557; Figure 3a).
Figure 3.
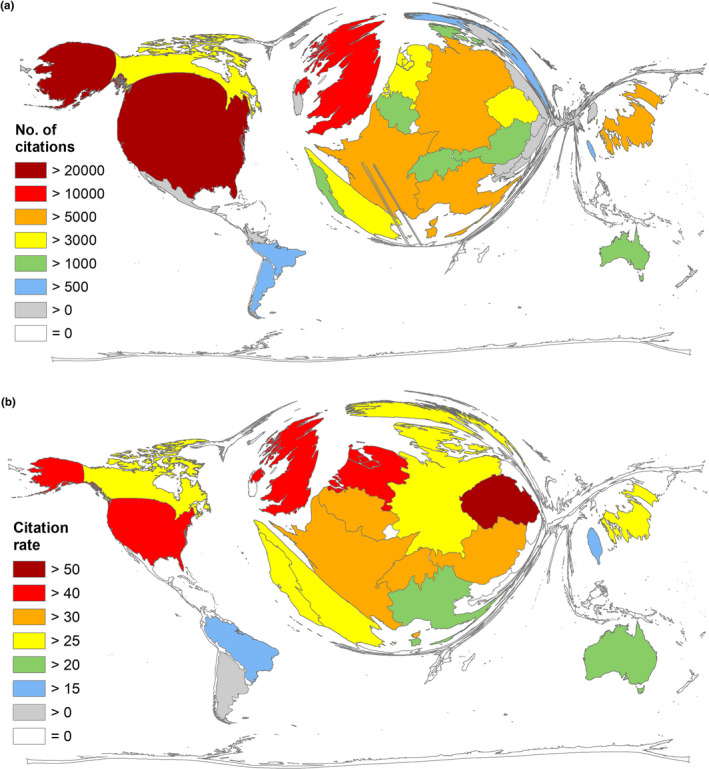
Global citation parameters. (a) Number of citations per country. (b) Citation rate (threshold ≥30 articles per country)
Strikingly, the analysis of the country‐specific citation rate (cr = number of citations per country/number of articles per country; only countries with ≥30 articles) was led by the Czech Republic with cr = 50.98. The UK ranked second (cr = 48.56) and was followed by the USA (cr = 46.36), the Netherlands (cr = 46.15), and Belgium (cr = 38.68; Figure 3b).
To enable the assessment and comparison of the countries' FD research performances in relation to their respective population and economic status, we determined the country‐specific ratios, R POP = articles/population in mill. and R GDP = articles/GDP in 1,000 bn US dollar for countries with ≥30 articles (threshold). The best FD‐related performance in relation to the countries' population was found in Switzerland (R POP = 9.78) and in Austria (R POP = 7.12) (Figure 4a). The best performance relative to the economic status (Figure 4b) was seen in the Czech Republic (R GDP = 176.69), Switzerland (R GDP = 161.84), and Austria (R GDP = 149.07). The USA stood out with a relatively poor performance and ranked only second to last before Brazil (Table 2; Figure 5).
Figure 4.
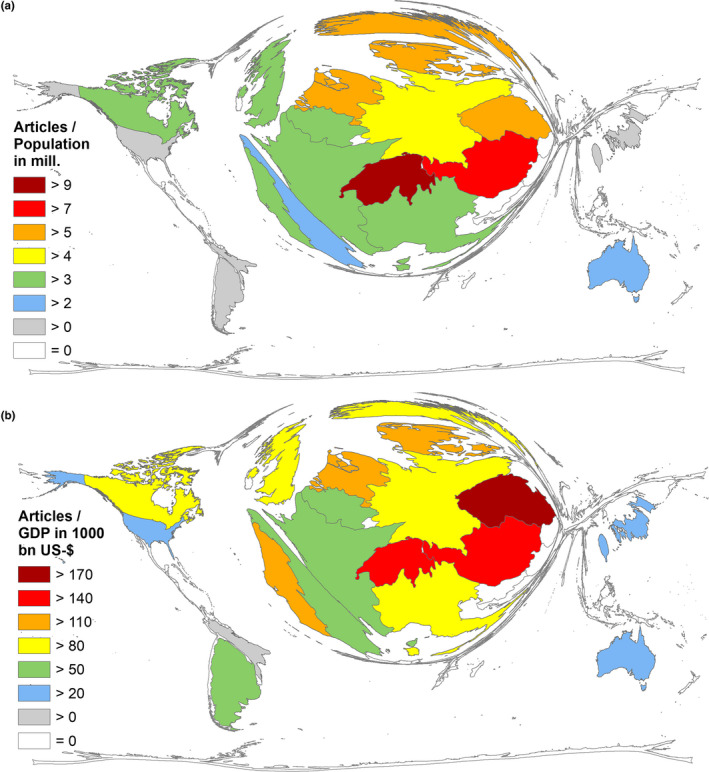
Socioeconomic parameters of the publishing countries with ≥30 articles. (a) Ratio of number of articles and population (in mill.). (b) Ratio of number of articles and gross domestic product in 1,000 bn USD
Table 2.
Countries' ranking according to the socioeconomic parameters (WorldBank, 2019)
| Country | R GDP | Rank 1 | R POP | Rank 2 |
|---|---|---|---|---|
| Switzerland | 161.85 | HI 2 | 9.78 | HI 1 |
| Austria | 149.07 | HI 3 | 7.12 | HI 2 |
| Denmark | 139.73 | HI 4 | 6.61 | HI 3 |
| Norway | 90.49 | HI 8 | 6.27 | HI 4 |
| The Netherlands | 118.95 | HI 6 | 6.05 | HI 5 |
| Czech Republic | 176.69 | HI 1 | 5.82 | HI 6 |
| Germany | 81.68 | HI 10 | 4.03 | HI 7 |
| Italy | 108.06 | HI 7 | 3.87 | HI 8 |
| Canada | 80.65 | HI 11 | 3.82 | HI 9 |
| United Kingdom | 85.37 | HI 9 | 3.69 | HI 10 |
| Portugal | 127.90 | HI 5 | 3.51 | HI 11 |
| France | 77.46 | HI 12 | 3.17 | HI 12 |
| Belgium | 68.82 | HI 13 | 3.07 | HI 13 |
| Spain | 63.91 | HI 14 | 2.22 | HI 14 |
| Australia | 39.53 | HI 16 | 2.04 | HI 15 |
| Japan | 40.96 | HI 15 | 1.59 | HI 16 |
| Argentina | 77.33 | UMI 1 | 1.55 | UMI 1 |
| United States | 26.94 | HI 18 | 1.54 | HI 17 |
| Taiwan | 27.56 | HI 17 | 1.32 | HI 18 |
| Brazil | 17.22 | UMI 2 | 0.26 | UMI 2 |
Abbreviations: HI, high‐income countries; R GDP, articles/GDP in 1,000 bn US dollars; R POP = articles/population in mill. Inhabitants; UMI, upper middle income countries.
Figure 5.
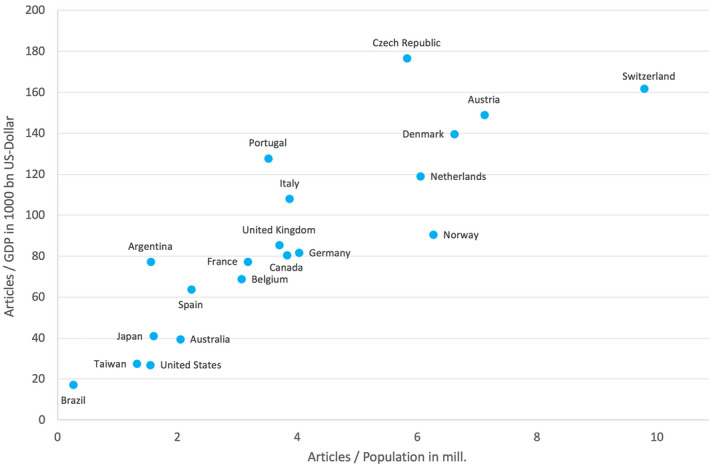
Countries' positioning regarding the Ratio of number of articles and population (in mill.) and the ratio of number of articles and gross domestic product (GDP) in 1,000 bn USD
2.3. Networking
From 1900 to 2018, an increasing number of FD articles were authored in international collaborations forming the basis of a, to date, well‐established global FD research network (Figure 6a). Overall, 443 articles (22.43%) originated from international collaborations with the majority 62.75% (n = 278) being authored in bilateral collaboration. In contrast, one article published in 2015 (Biegstraaten et al., 2015) originated from the joint work of FD experts from 15 different European countries, under the self‐designation of European Fabry Working Group. In this article, the group presents recommendations for indications for initiation and termination of ERT. The collaboration between Germany and the UK scientists was unveiled as the most productive collaboration on a bilateral level with n = 73 articles, followed by USA/Canada (n = 63), USA/Germany (n = 57), and Germany/Italy (n = 51).
Figure 6.

International collaborations, the thickness of lines corresponds to the publication numbers. (a) Global network. Number in brackets: number of articles/number of collaboration articles. (b) Collaborations of institutions with ≥8 publications. Numbers: numbers of common articles/numbers of the citations
In terms of collaborations on an institutional level, a complex network of European institutions was identified. The universities of London (UK), Mainz (Germany), Vienna (Austria), Zurich (Switzerland), and Cattolica Sacro Cuore (Italy) have been major contributors (Figure 6b).
Since 2001, two major pharma‐sponsored, multicenter, observational FD registries have provided solid platforms for formal and informal international networking among participant institutions and investigators, respectively, the “Fabry Outcome Survey” (FOS; NIH, 2019b) sponsored by Shire—since 2019 acquired by the Japan company Takeda—and the “Fabry Disease Registry” (NIH, 2019a) sponsored by Genzyme (a Sanofi Company).
2.4. Funding
Overall, our analysis was able to identify the funding sources of 661 FD‐related articles (31.55% of all publications). Funding for FD‐related research was provided by a total of 199 different organizations. In total, 1141 research grants (g) on FD were bestowed, half privately (48.47%) and half publicly (51.53%) financed. Governmental funding provided the highest number of FD grants (g = 485) and was followed by industrial sponsors (g = 458), universities (g = 76), hospitals (g = 8), foundations (g = 64), nonprofit organizations (g = 23), or societies (g = 12).
On an individual basis, the most influential funding source with a yield of 222 FD‐related grants was the US government, mainly represented by the National Institutes of Health (NIH) as part of the U.S. Department of Health and Human Services (Table 2).
Further crucial funding sources of FD research were identified as pharmaceutical companies specialized on rare diseases: Genzyme Corp. (USA) gave 220 grants, followed by Shire HGT (Human Genetic Therapies/USA) with a contribution of 133 grants, and Amicus Therapeutics (USA) with an amount of 29 grants. The Fabry registry is supported by Genzyme. Shire HGT, as already mentioned above, initiated the FOS originally to gain a better understanding of patients eligible for or under Agalsidase‐alfa therapy, but evolved to a full registry over the years.
Other important financial driving forces behind FD research were governmental organizations of Japan, South Korea, Germany, Canada, China, Brazil, EU, Italy, and Spain. A more recent stakeholder in the field of FD‐related research is Protalix Biotherapeutics, an Israeli company developing Pegunigalsidase alfa as a possible advantageous alternative for ERT (Table 3).
Table 3.
Most funding organizations, governmental institutions
| Funder | Grants | Country | Funding type |
|---|---|---|---|
| Genzyme Corp., USA | 220 | USA | Private, economic |
| NIH, USA | 212 | USA | Public, government |
| Shire HGT, USA | 133 | USA | Private, economic |
| Amicus Therapeutics, USA | 29 | USA | Private, economic |
| Japan Government | 29 | Japan | Public, government |
| South Korea Government | 24 | South Korea | Public, government |
| Canada Government | 18 | Canada | Public, government |
| China Government | 17 | China | Public, government |
| Brazil Government | 16 | Brazil | Public, government |
| EU | 16 | International | Public, government |
| Protalix Biotherapeutics, Israel | 16 | Israel | Private, economic |
| Telethon Foundation, Italy | 15 | Italy | Private, foundation |
| Italy Government | 14 | Italy | Public, government |
| Spain Government | 14 | Spain | Public, government |
The Italian charity foundation Telethon plays another crucial role in terms of financial support of FD research. It provides financial aid for three institutes conducting FD‐related research: the TIGEM (Telethon Institute of Genetics and Medicine), TIGET (Telethon Institute for Gene Therapy at the San Raffaele Hospital), and DTI (Dulbecco Telethon Institute; Telethon, 2018).
3. DISCUSSION
3.1. Chronological development
In accordance with studies analyzing other biomedical topics (Bruggmann et al., 2018; Gotting, Schwarzer, Gerber, Klingelhofer, & Groneberg, 2017; Van Noorden, 2014), we observed a significant increase in the number of articles and received citations over time, too. Milestones of FD research as well as the introduction of new therapeutic approaches paved the way for an increasing FD research activity over the years. However, the findings show that in relation to the general biomedical research output, the FD research activity between 1980 and 2000 was characterized by a relative stagnation. The only country with a constantly rising publication output throughout the whole investigation period was Japan. Before discovery of new therapeutic approaches and better epidemiological characterizations, the involvement in FD research did not yield much success. According to our findings, it can be assumed that the introduction of novel treatment approaches and the realization of a significantly higher prevalence through large genetic screenings started a new era of FD research with a rising global interest. As a consequence, many countries intensified comprehensive screenings and are connected in international collaborations shown by the increase of international publications over time. Nevertheless, it is crucial to expand this network to elucidate the many remaining uncertainties of regional differences in terms of management and treatment of FD. Especially low‐income countries should be supported due to the high costs of enzyme replacement therapies as well as the pharmacological chaperone.
3.2. Global publication output and funding
The most publishing countries on FD are the USA, some European countries, Japan, Taiwan, and South Korea. In line with previous studies evaluating other biomedical topics, the USA was also unveiled as the leading country in terms of FD research activity. This can mainly be explained by its high scientific standards, well‐established scientific infrastructure, and its attraction to world renowned scientists (Groneberg et al., 2015; Lammer et al., 2018). It is mainly involved in the Fabry Disease Registry. The EU nations Germany, Italy, UK, and France are identified as the main protagonists involved in multinational programs and studies, like the FOS or Fabry Disease Registry. As also shown by some other studies (Lammer et al., 2018; Trost et al., 2018), the leading countries afford high expenditures for Research and Development, so that their FD rankings are not astonishing. The results of the present study reveal that high‐income countries publish much more on FD than low‐ or middle‐income economies.
Here it seems safe to assume that genetic screenings influenced the publication numbers by providing incentives and data reflected by the high publication outputs of these countries.
Further critical influences of the countries' FD activity are shown to be the amount of financial support by the respective governments and also the presence of pharmaceutical companies in the respective country. In recent years, governmental measures legalizing the promotion of orphan drugs as well as regulations that led to significant improvements in the management of rare diseases were strengthened in the USA, EU countries, Japan, South Korea, and Taiwan (Thomas & Caplan, 2019). However, our findings also revealed private pharmaceutical organizations as pioneers in terms of financial support of FD research. This is at least partly surprising, since due to the nature of rare diseases, low patient numbers did not promise high profits for the pharmaceutical industry. Yet, in turn, to remain profitable, this often results in high costs of the developed medications, which is passed on to patients and health systems. Consequently, low‐income countries often struggle with the financial burden linked to effective treatment for FD resulting in many patients not having access to quality of life‐improving drugs (Pinto, Dennis, & Parra, 2009).
Noteworthy in terms of the scientific resonance of the countries' FD performances is the outstanding position of the Czech Republic regarding the average citation rate and economic analysis of this study. Their leading position can be explained by their involvement in the FOS with the FOS Investigators and the European FOS Investigator Group as well as in the Fabry Disease Registry and the Fabry Registry European Board. Additionally, their participation in these author groups yielded highly cited articles that are ranking among the 10 most cited publications on FD (Banikazemi et al., 2007; Mehta, Ricci, Widmer, Dehout, de Lorenzo, et al., 2004).
Similarly, also Switzerland's and Austria's high ranking in terms of the citation rate can also be referred to their participation in the FOS (Mehta et al., 2004, 2009).
However, China was here unveiled as an outlier with a relatively poor FD output indicating a low interest in FD research. Looking at China's contribution over time, we observed a poor performance throughout the whole investigation period. In contrast to their otherwise massively expanding research efforts in the recent years (Klingelhofer et al., 2018), China only published 11 FD‐related articles from 2013 to 2017. However, rare diseases in China are increasingly recognized as an important public health issue and it is assumed that China is going to expand its investment in rare diseases through the National Natural Science Foundation of China in the future (Chen, Yao, & Liu, 2015), albeit occidental pharma industries do not penetrate easily in Asia until now (Floether, 2010).
Overall, our analyses show the urgent need to enhance the infrastructure for FD research. In the light of the low participation of low‐income economies, this is particularly important for these countries (Forero et al., 2016) with regard to the ethical questions of distributive justice raised by the prohibitively high costs of FD treatment, whether through enzyme replacement or pharmacological accompaniment. Here, the market monopoly of the pharmaceutical manufacturers allows to charge high prices that are argued by the companies to be justified because they are adapted to market level. Since each company generates the market by itself alone, this argument should be questioned (Roos, Hyry, & Cox, 2010).
4. CONCLUSIONS
The present study supplies scientists, funders, and decision makers with information about the incentives and benchmarks as well as about the demands of FD research measured as publication output. The finding emphasizes the contributions of US‐American and European scientists and the important function of governmental support.
Although FD research evolved over time, to date, only very few effective treatment options are available and these options are further linked to a substantial financial burden for patients and health systems. Many patients have no access to treatment, especially in low‐income nations. Consequently, some governments are increasingly recognizing the need to foster basic research on FD independently of economic interests of big pharmaceutical companies. It is therefore internationally important to support low‐income economies with know‐how and financial resources to improve their scientific infrastructure and their participation in international networks. Collaborative scientific efforts, resulting in an increasing publication output should be strengthened to facilitate a better understanding of FD in terms of epidemiological background, diagnosis, and treatment and to also develop standardized guidelines to improve patient outcomes. Countries worldwide should be made aware of the enormous burden of FD for patients and health systems.
5. METHODS
This study is embedded in the bibliometric platform NewQIS (New Quality and Quantity in Science) (Groneberg‐Kloft, Fischer, Quarcoo, & Scutaru, 2009), that was established to elucidate in‐depth the research activity of a multitude of biomedical areas and topics with focus on the chronological and geographical aspects of the respective issues (Lammer et al., 2018; Quarcoo, Bruggmann, Klingelhofer, & Groneberg, 2015). NewQIS was created in 2009 and has since analyzed the research activity of various areas of biomedical science and other public health issues of international relevance. The platform is evolving constantly and novel indices enable new insights into the global scientific activity.
The data for the bibliometric part of this study were obtained from the online database WoS. In order to produce the most representative dataset of the global FD research activity, the entry terms of the MeSH (Medical Subject Heading) database of the US National Library of Medicine were used to create a unique search term. This search term was then utilized for a “Title” search for all entries on FD between 1900 and 2018. Only original articles and reviews on FD were included. The bibliometric metadata were then downloaded and transferred to a Microsoft Access database. Additional socioeconomic data of countries publishing on FD were retrieved from the World Factbook (CIA, 2017).
Based on these data, we evaluated the overall publication output between 1900 and 2018, elucidated the development of the FD research activity over time, depicted the global contribution as well as the international network, and also analyzed the qualitative aspects (citation parameters) of FD research. Furthermore, socioeconomic influences of the countries' contribution to FD research were assessed (CIA, 2017).
The visualization of the geographical findings was realized by means of cartograms by Gastner and Newman. These cartograms, also called Density Equalizing Map Projections, are based on the physical principle of the density equilibrium and re‐sizes the countries according to an evaluation parameter (Gastner & Newman, 2004).
5.1. Methodological limitations and strengths
This study, embedded in the NewQIS platform, was established to provide in‐depth insights into the global research activity of a wide range of relevant biomedical and public health issues. Using the WoS as source for the bibliometric data implies a persistent bias toward English literature and does not reflect the entirety of scientific articles on FD. However, here we need to emphasize that due to its strict selection processes, the utilization of the WoS is of clear advantage in terms of the quality of the underlying data of this study, especially against the background of the steadily increasing flood of low quality and predatory journals (Clark & Smith, 2015). Another advantageous feature of the WoS is the possibility to access the FD research‐related citation parameters via the Journal Citation Report, which enables the qualitative assessment of FD articles. Objective of the decision to search the “Title” for FD‐related articles was to minimize the entries not associated with FD research, although this might have resulted in the omission of some relevant publications (Li et al., 2004). In summary, we aimed to create a database containing a representative dataset of high‐quality work on FD.
Further, since the assessment of the funding organizations behind FD research was limited to the entries included in our database, some funding sources may not have been included.
CONFLICT OF INTERESTS
None declared.
AUTHOR CONTRIBUTIONS
DK, DAG contributed to the development of the methodological platform. DK concepted, designed, drafted the initial manuscript, and carried out the literature search and the analyses. DAG contributed to conception, design, and analyses. MB, RKS, DQ, DB contributed to the literature search, interpretation, and design of results. MB, RKS corrected the draft. RKS, DQ, MB contributed with important expert content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
ACKNOWLEDGMENTS
Not applicable.
Klingelhöfer D, Braun M, Seeger‐Zybok RK, Quarcoo D, Brüggmann D, Groneberg DA. Global research on Fabry's disease: Demands for a rare disease. Mol Genet Genomic Med. 2020;8:e1163 10.1002/mgg3.1163
DATA AVAILABILITY STATEMENT
The bibliometric data is owned by and have been obtained from the WoS database. Any researcher with access to the WoS database can obtain the data using the methods described in the paper. Readers who do not have access to WoS should contact Clarivate Analytics to obtain a license.
REFERENCES
- Anderson, W. (1898). A case of Angeio‐Keratoma. Brit J Dermatol, 113–117. 10.1111/j.1365-2133.1898.tb16317.x [DOI] [Google Scholar]
- Banikazemi, M. , Bultas, J. , Waldek, S. , Wilcox, W. R. , Whitley, C. B. , McDonald, M. , … Desnick R. J.; Fabry Disease Clinical Trial Study Group . (2007). Agalsidase‐beta therapy for advanced Fabry disease – A randomized trial. Annals of Internal Medicine, 146(2), 77–86. 10.7326/0003-4819-146-2-200701160-00148 [DOI] [PubMed] [Google Scholar]
- Benjamin, E. R. , Della Valle, M. C. , Wu, X. , Katz, E. , Pruthi, F. , Bond, S. , … Lockhart, D. J. (2017). The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genetics in Medicine, 19(4), 430–438. 10.1038/gim.2016.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, H. S. , Bishop, D. F. , Astrin, K. H. , Kornreich, R. , Eng, C. M. , Sakuraba, H. , & Desnick, R. J. (1989). Fabry disease – 6 Gene rearrangements and an exonic point mutation in the alpha‐galactosidase gene. Journal of Clinical Investigation, 83(4), 1390–1399. 10.1172/Jci114027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegstraaten, M. , Arngrímsson, R. , Barbey, F. , Boks, L. , Cecchi, F. , Deegan, P. B. , … Hollak, C. E. M. (2015). Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: The European Fabry Working Group consensus document. Orphanet Journal of Rare Diseases, 10 10.1186/s13023-015-0253-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, D. F. , Kornreich, R. , & Desnick, R. J. (1988). Structural organization of the human alpha‐galactosidase A gene: Further evidence for the absence of a 3' untranslated region. Proceedings of the National Academy of Sciences of the United States of America, 85(11), 3903–3907. 10.1073/pnas.85.11.3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady, R. O. , Gal, A. E. , Bradley, R. M. , Martensson, E. , Warshaw, A. L. , & Laster, L. (1967). Enzymatic defect in Fabrys disease – Ceramidetrihexosidase deficiency. New England Journal of Medicine, 276(21), 1163–2000. 10.1056/Nejm196705252762101 [DOI] [PubMed] [Google Scholar]
- Brüggmann, D. , Alafi, A. , Jaque, J. , Klingelhöfer, D. , Bendels, M. H. , Ohlendorf, D. , … Groneberg, D. A. (2018). World‐wide research architecture of vitamin D research: Density‐equalizing mapping studies and socio‐economic analysis. Nutrition Journal, 17(1), 3 10.1186/s12937-018-0313-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, K. , Yao, L. , & Liu, Z. Y. (2015). Towards government‐funded special biomedical research programs to combat rare diseases in China. Bioscience Trends, 9(2), 88–90. 10.5582/bst.2015.01048 [DOI] [PubMed] [Google Scholar]
- CIA . (2017). CIA, the world factbook, 2017. Retrieved from https://www.cia.gov/library/publications/the-world-factbook/ [Google Scholar]
- Clark, J. , & Smith, R. (2015). Firm action needed on predatory journals. British Medical Journal, 350, h210–h210. 10.1136/bmj.h210 [DOI] [PubMed] [Google Scholar]
- Desnick, R. J. , Brady, R. , Barranger, J. , Collins, A. J. , Germain, D. P. , Goldman, M. , … Wilcox, W. R. (2003). Fabry disease, an under‐recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Annals of Internal Medicine, 138(4), 338–346. 10.7326/0003-4819-138-4-200302180-00014 [DOI] [PubMed] [Google Scholar]
- Eng, C. M. , Germain, D. P. , Banikazemi, M. , Warnock, D. G. , Wanner, C. , Hopkin, R. J. , … Wilcox, W. R. (2006). Fabry disease: Guidelines for the evaluation and management of multi‐organ system involvement. Genetics in Medicine, 8(9), 539–548. https://doi.org/10.109701.gim.0000237866.70357.c6 [DOI] [PubMed] [Google Scholar]
- Eng, C. M. , Guffon, N. , Wilcox, W. R. , Germain, D. P. , Lee, P. , Waldek, S. , … Desnick, R. J. International Collaborative Fabry Disease Study Group . (2001). Safety and efficacy of recombinant human alpha‐galactosidase a replacement therapy in Fabry's disease. New England Journal of Medicine, 345(1), 9–16. 10.1056/Nejm200107053450102 [DOI] [PubMed] [Google Scholar]
- EU . (1999). Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products. Retrieved from http://eur-lex.europa.eu/legal-content/CS/ALL/?uri=URISERV:l21167 [Google Scholar]
- Fabry, H. (2001). An historical overview of Fabry disease. Journal of Inherited Metabolic Disease, 24(Suppl 2), 3–7. 10.1023/a:1012443001449 [DOI] [PubMed] [Google Scholar]
- Fabry, J. (1898). Ein Beitrag zur Kenntnis der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). Arch Dermatol Syph, XL111, 187‐200. [Google Scholar]
- Floether, F. U. (2010). Emerging Asian biotech‐pharma industry – Comparative perspectives In Strategic alliances in biotechnology and pharmaceuticals. Biotechnology in agriculture industry and medicine (pp. 107–136). Hauppauge, NY: Nova Science Publishers. [Google Scholar]
- Forero, D. A. , Wonkam, A. , Wang, W. , Laissue, P. , Lopez‐Correa, C. , Fernandez‐Lopez, J. C. , … Perry, G. (2016). Current needs for human and medical genomics research infrastructure in low and middle income countries. Journal of Medical Genetics, 53(7), 438–440. 10.1136/jmedgenet-2015-103631 [DOI] [PubMed] [Google Scholar]
- Gastner, M. T. , & Newman, M. E. (2004). Diffusion‐based method for producing density‐equalizing maps. Proceedings of the National Academy of Sciences of the United States of America, 101(20), 7499–7504. 10.1073/pnas.0400280101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain, D. P. (2010). Fabry disease. Orphanet Journal of Rare Diseases, 5, 30 10.1186/1750-1172-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain, D. P. , Hughes, D. A. , Nicholls, K. , Bichet, D. G. , Giugliani, R. , Wilcox, W. R. , … Schiffmann, R. (2016). Treatment of Fabry's disease with the pharmacologic chaperone migalastat. New England Journal of Medicine, 375(6), 545–555. 10.1056/NEJMoa1510198 [DOI] [PubMed] [Google Scholar]
- GlobalGenes . (2018). Global genes, rare disease facts and figures. Retrieved from https://globalgenes.org/raredaily/rare-disease-facts-and-figures/ [Google Scholar]
- Gotting, M. , Schwarzer, M. , Gerber, A. , Klingelhofer, D. , & Groneberg, D. A. (2017). Pulmonary hypertension: Scientometric analysis and density‐equalizing mapping. PLoS ONE, 12(1), e0169238 10.1371/journal.pone.0169238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groneberg, D. A. , Weber, E. , Gerber, A. , Fischer, A. , Klingelhoefer, D. , & Brueggmann, D. (2015). Density equalizing mapping of the global tuberculosis research architecture. Tuberculosis (Edinb), 95(4), 515–522. 10.1016/j.tube.2015.05.003 [DOI] [PubMed] [Google Scholar]
- Groneberg‐Kloft, B. , Fischer, T. C. , Quarcoo, D. , & Scutaru, C. (2009). New quality and quantity indices in science (NewQIS): The study protocol of an international project. Journal of Occupational Medicine and Toxicology, 4, 16 10.1186/1745-6673-4-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, K. , Gross, B. G. , & Lever, W. F. (1965). Angiokeratoma corporis diffusum (Fabry) – Histochemical and electron microscopic studies of skin. Journal of Investigative Dermatology, 44(2), 119–128. 10.1038/jid.1965.22 [DOI] [PubMed] [Google Scholar]
- Hoffmann, B. (2009). Fabry disease: Recent advances in pathology, diagnosis, treatment and monitoring. Orphanet Journal of Rare Diseases, 4 10.1186/1750-1172-4-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollak, C. E. M. , Vedder, A. C. , Linthorst, G. E. , & Aerts, J. M. F. G. (2007). Novel therapeutic targets for the treatment of Fabry disease. Expert Opinion on Therapeutic Targets, 11(6), 821–833. 10.1517/14728222.11.6.821 [DOI] [PubMed] [Google Scholar]
- Houge, G. , & Skarbovik, A. J. (2005). Fabry disease – A diagnostic and therapeutic challenge. Tidsskrift for den Norske Laegeforening, 125(8), 1004–1006. [PubMed] [Google Scholar]
- Hwu, W. L. , Chien, Y. H. , Lee, N. C. , Chiang, S. C. , Dobrovolny, R. , Huang, A. C. , … Hsu, L. W. (2009). Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later‐onset GLA mutation c.936+919G>A (IVS4+919G>A). Human Mutation, 30(10), 1397–1405. 10.1002/humu.21074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingelhofer, D. , Zhu, Y. , Braun, M. , Bendels, M. H. K. , Bruggmann, D. , & Groneberg, D. A. (2018). Aflatoxin – Publication analysis of a global health threat. Food Control, 89, 280–290. 10.1016/j.foodcont.2018.02.017 [DOI] [Google Scholar]
- Lammer, E. , Klingelhofer, D. , Bendels, M. H. K. , Ohlendorf, D. , Spallek, M. , & Groneberg, D. A. (2018). Development of the global schizophrenia research under epidemiological and socio‐economic influences. Schizophrenia Research, 199, 458–460. 10.1016/j.schres.2018.04.001 [DOI] [PubMed] [Google Scholar]
- Li, Y. J. , Scott, C. R. , Chamoles, N. A. , Ghavami, A. , Pinto, B. M. , Turecek, F. , & Gelb, M. H. (2004). Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clinical Chemistry, 50(10), 1785–1796. 10.1373/clinchem.2004.035907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermot, K. D. , Holmes, A. , & Miners, A. H. (2001). Anderson‐Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males. Journal of Medical Genetics, 38(11), 750–760. 10.1136/jmg.38.11.750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri, M. , & Solari, A. (1996). The SCI Journal citation reports: A potential tool for studying journals? Scientometrics, 35(1), 93–117. 10.1007/BF02018235 [DOI] [Google Scholar]
- Markham, A. (2016). Migalastat: First global approval. Drugs, 76(11), 1147–1152. 10.1007/s40265-016-0607-y [DOI] [PubMed] [Google Scholar]
- Mehta, A. , Beck, M. , Elliott, P. , Giugliani, R. , Linhart, A. , Sunder‐Plassmann, G. , … Clarke, J. (2009). Enzyme replacement therapy with agalsidase alfa in patients with Fabry's disease: An analysis of registry data. The Lancet, 374(9706), 1986–1996. 10.1016/S0140-6736(09)61493-8 [DOI] [PubMed] [Google Scholar]
- Mehta, A. , Ricci, R. , Widmer, U. , Dehout, F. , Garcia de Lorenzo, A. , Kampmann, C. , … Beck, M. (2004). Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. European Journal of Clinical Investigation, 34(3), 236–242. 10.1111/j.1365-2362.2004.01309.x [DOI] [PubMed] [Google Scholar]
- Meikle, P. J. , Hopwood, J. J. , Clague, A. E. , & Carey, W. F. (1999). Prevalence of lysosomal storage disorders. Journal of the American Medical Association, 281(3), 249–254. 10.1001/jama.281.3.249 [DOI] [PubMed] [Google Scholar]
- Nakao, S. , Takenaka, T. , Maeda, M. , Kodama, C. , Tanaka, A. , Tahara, M. , … Tanaka, H. (1995). An atypical variant of Fabrys‐disease in men with left‐ventricular hypertrophy. New England Journal of Medicine, 333(5), 288–293. 10.1056/Nejm199508033330504 [DOI] [PubMed] [Google Scholar]
- NIH . (2019a). U.S. National Library of Medicine, ClinicalTrials.gov. Retrieved from https://clinicaltrials.gov/ct2/show/NCT00196742 [Google Scholar]
- NIH . (2019b). U.S. National Library of Medicine, ClinicalTrials.gov. Retrieved from https://clinicaltrials.gov/ct2/show/NCT03289065 [Google Scholar]
- Oliveira, J. P. , & Ferreira, S. (2019). Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype‐phenotype correlations. Application of Clinical Genetics, 12, 35–50. 10.2147/Tacg.S146022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz, J. M. , Stiles, F. C. , Wise, D. , Race, R. R. , Sanger, R. , Von Gemmingen, G. R. , … De Groot, W. P. (1965). Genetics of Angiokeratoma corporis diffusum (Fabrys disease) and its linkage relations with Xg locus. American Journal of Human Genetics, 17(4), 325. [PMC free article] [PubMed] [Google Scholar]
- Orphan‐Drug‐Act . (1983). Public Law 97-414, 97th Congress . Retrieved from https://www.fda.gov/media/99546/download
- Ortiz, A. , Germain, D. P. , Desnick, R. J. , Politei, J. , Mauer, M. , Burlina, A. , … Wilcox, W. R. (2018). Fabry disease revisited: Management and treatment recommendations for adult patients. Molecular Genetics and Metabolism, 123(4), 416–427. 10.1016/j.ymgme.2018.02.014 [DOI] [PubMed] [Google Scholar]
- Pinto, D. M. , Dennis, R. , & Parra, O. (2009). The economic burden of Gaucher and Fabry's disease in Colombia. Implications for National Health Insurance. Value in Health, 12(7), A487–A487. 10.1016/S1098-3015(10)75301-8 [DOI] [Google Scholar]
- Quarcoo, D. , Bruggmann, D. , Klingelhofer, D. , & Groneberg, D. A. (2015). Ebola and its global research architecture‐need for an improvement. PLoS Neglected Tropical Diseases, 9(9), e0004083 10.1371/journal.pntd.0004083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos, J. C. P. , Hyry, H. I. , & Cox, T. M. (2010). Orphan drug pricing may warrant a competition law investigation. British Medical Journal, 341, c6471–c6471. 10.1136/bmj.c6471 [DOI] [PubMed] [Google Scholar]
- Sachdev, B. , Takenaka, T. , Teraguchi, H. , Tei, C. , Lee, P. , McKenna, W. J. , & Elliott, P. M. (2002). Prevalence of Anderson‐Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation, 105(12), 1407–1411. 10.1161/01.CIR.0000012626.81324.38 [DOI] [PubMed] [Google Scholar]
- Schiffmann, R. , Hughes, D. A. , Linthorst, G. E. , Ortiz, A. , Svarstad, E. , Warnock, D. G. , … Walter, J. (2017). Screening, diagnosis, and management of patients with Fabry disease: Conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney International, 91(2), 284–293. 10.1016/j.kint.2016.10.004 [DOI] [PubMed] [Google Scholar]
- Schiffmann, R. , Kopp, J. B. , Austin, H. A. III , Sabnis, S. , Moore, D. F. , Weibel, T. , … Brady, R. O. (2001). Enzyme replacement therapy in Fabry disease – A randomized controlled trial. Journal of the American Medical Association, 285(21), 2743–2749. 10.1001/jama.285.21.2743 [DOI] [PubMed] [Google Scholar]
- Shin, S. H. , Kluepfel‐Stahl, S. , Cooney, A. M. , Kaneski, C. R. , Quirk, J. M. , Schiffmann, R. , … Murray, G. J. (2008). Prediction of response of mutated alpha‐galactosidase A to a pharmacological chaperone. Pharmacogenetics and Genomics, 18(9), 773–780. 10.1097/FPC.0b013e32830500f4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spada, M. , Pagliardini, S. , Yasuda, M. , Tukel, T. , Thiagarajan, G. , Sakuraba, H. , … Desnick, R. J. (2006). High incidence of later‐onset fabry disease revealed by newborn screening. American Journal of Human Genetics, 79(1), 31–40. 10.1086/504601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpke, G. (1916). A case of angiokeratoma corporis diffusum. Archiv fur Dermatologie und Syphilis, 121, 291–295. [Google Scholar]
- Telethon . (2018). The Telethon Foundation. Retrieved from http://www.tigem.it/the-institute/the-telethon-foundation [Google Scholar]
- Thomas, S. , & Caplan, A. (2019). The Orphan Drug Act revisited. Journal of the American Medical Association, 321(9), 833–834. 10.1001/jama.2019.0290 [DOI] [PubMed] [Google Scholar]
- Trost, M. , Wanke, E. M. , Ohlendorf, D. , Klingelhöfer, D. , Braun, M. , Bauer, J. , … Brüggmann, D. (2018). Immigration: Analysis, trends and outlook on the global research activity. Journal of Global Health, 8(1), 010414 10.7189/jogh.08.010414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Noorden, R. (2014). Nature News blog. Retrieved from http://blogs.nature.com/news/2014/05/global-scientific-output-doubles-every-nine-years.html [Google Scholar]
- Wise, D. , Wallace, H. J. , & Jellinek, E. H. (1962). Angiokeratoma corporis diffusum. A clinical study of eight affected families. Quarterly Journal of Medicine, 31, 177–206. [PubMed] [Google Scholar]
- WorldBank . (2019). World bank country and lending groups. Retrieved from https://datahelpdesk.worldbank.org/knowledgebase/articles/906519#High_income [Google Scholar]
- Zarate, Y. A. , & Hopkin, R. J. (2008). Lysosomal storage disease 3 – Fabry's disease. Lancet, 372(9647), 1427–1435. 10.1016/S0140-6736(08)61589-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The bibliometric data is owned by and have been obtained from the WoS database. Any researcher with access to the WoS database can obtain the data using the methods described in the paper. Readers who do not have access to WoS should contact Clarivate Analytics to obtain a license.