

Abstract
Monitoring host cell proteins (HCPs) is one of the most important analytical requirements in production of recombinant biopharmaceuticals to ensure product purity and patient safety. Enzyme‐linked immunosorbent assay (ELISA) is the standard method for monitoring HCP clearance. It is important to validate that the critical reagent of an ELISA, the HCP antibody, covers a broad spectrum of the HCPs potentially present in the purified drug substance. Current coverage methods for assessing HCP antibody coverage are based on 2D‐Western blot or immunoaffinity‐purification combined with 2D gel electrophoresis and have several limitations. In the present study, we present a novel coverage method combining ELISA‐based immunocapture with protein identification by liquid chromatography–tandem mass spectrometry (LC–MS/MS): ELISA‐MS. ELISA‐MS is used to accurately determine HCP coverage of an early process sample by three commercially available anti‐Escherichia coli HCP antibodies, evading the limitations of current methods for coverage analysis, and taking advantage of the benefits of MS analysis. The results obtained comprise a list of individual HCPs covered by each HCP antibody. The novel method shows high sensitivity, high reproducibility, and enables tight control of nonspecific binding through inclusion of a species‐specific isotype control antibody. We propose that ELISA‐MS will be a valuable supplement to existing coverage methods or even a replacement. ELISA‐MS will increase the possibility of selecting the best HCP ELISA, thus improving HCP surveillance and resulting in a final HCP profile with the lowest achievable risk. Overall, this will be beneficial to both the pharmaceutical industry and patient safety.
Keywords: biopharmaceuticals, coverage analysis, ELISA‐MS, host cell protein, reagent characterization
1. INTRODUCTION
Characterization of host cell proteins (HCPs) is required in production of recombinant biopharmaceuticals to ensure product purity and patient safety.1 The composition and abundance of HCPs depends on a number of factors: the host cell expression system (bacterial cells, mammalian cell, fungi, etc.); the manner of expression, for example, intracellular expression in the cytoplasm or in inclusion bodies in Escherichia coli or secretion into the culture medium by Chinese hamster ovary cells; the bioprocess itself with a variation of purification steps; and the physiochemical properties of the recombinant protein being expressed.2 Some HCPs co‐purify with the drug substance due to similar physiochemical properties whereas others co‐purify as “hitchhiker” HCPs due to direct interaction with the drug.3, 4, 5, 6 The HCP profile of a bioprocess, where a specific recombinant protein is expressed in a certain host cell, is therefore very difficult to predict and needs to be monitored closely during process development.
Characterization and quantification of HCPs are important as they impose a potential risk for the patient safety or may effect drug substance degradation.7 HCPs can have direct biological activity, for example, protease activity impacting product stability8; immunomodulatory effects, for example, by triggering Toll‐like receptor mediated responses9; or immunogenic effects by inducing direct antibody responses or functioning as adjuvants increasing immune response against the drug substance.7, 10 Enzyme‐linked immunosorbent assay (ELISA) is the standard method for monitoring HCP clearance during a purification process due to its availability, high throughput, sensitivity, and selectivity.11 The ELISA gives a semi‐quantitative measure of the total HCP content of a sample determined as immuno‐equivalent nanogram HCP per milligram drug substance (ng/mg or ppm). It is important to ensure that the critical reagent of an ELISA, the HCP antibody, covers a broad spectrum of the HCPs that potentially could end up in the purified drug substance.1 A limitation of ELISA is that nonimmunoreactive or weakly immunoreactive HCPs are not detected by the HCP antibody, as antibodies are only generated against immunocompetent proteins, and that some HCPs may not be measured by the assay due to poor antibody/antigen binding conditions, antibody affinity/avidity, or availability of HCP epitopes. To detect such proteins, orthogonal methods, such as mass spectrometry (MS), should be used.
Biopharmaceutical companies usually develop process‐specific ELISAs for late‐stage clinical trials, whereas commercial ELISA kits are available for early process development. The ELISA should cover the majority of HCPs potentially found in the product and have sufficient sensitivity, enabling the assay to detect appearance of new HCPs in case of process change or process failure.12, 13 ELISA HCP antibody coverage of HCPs is usually assessed by techniques, such as two‐dimensional (2D)‐Western blot or immunoaffinity‐purification combined with 2D gel electrophoresis. When using these techniques, the number of gel spots, observed after immunostaining or after immunoaffinity‐purification, are compared to the number of spots observed after a total protein stain. There are several limitations to these techniques resulting in high variability in HCP coverage‐percentages, such as over‐transfer through the membrane or incomplete transfer of HCPs to the blotting membrane; unreliable spot counting with one HCP accounting for several spots; overloading or comigration where one spot may overlay another spot; difficulties comparing spots in blots and gels because of unlike staining methods with different sensitivity; and denaturing conditions during 2D gel electrophoresis destroying native epitopes.13, 14 In addition, the methods relying on immunoaffinity purification require large amounts of HCP antibody, which might be a limiting factor as the amount of HCP antibody should last the anticipated lifetime of the product.13 An alternative coverage method, combining immunoaffinity‐purification on magnetic beads and MS, has recently been described.15 The method shows high sensitivity with identification and quantification of individual HCPs by MS, avoiding the limitations of 2D‐Western blot or immunoaffinity‐purification combined with 2D gel electrophoresis.15
Monitoring HCPs by ELISA is an important tool in process development and as a release test of the purified drug substance. However, ELISA does not enable quantification and identification of individual HCPs of potential risk for product stability or patient safety. The use of MS to identify and quantify HCPs of interest in the downstream purification steps and in the purified drug substance is increasing.16, 17, 18, 19, 20, 21 MS is an important tool in process development to identify HCPs with a high‐risk assessment enabling development of a strategy for their removal. This warrants a final HCP profile with the lowest achievable risk.22
The present study describes a new experimental method to determine ELISA HCP antibody coverage, evading the limitations of coverage analysis by 2D‐Western blot or immunoaffinity‐purification, and taking advantage of the benefit of identification and quantification of individual HCPs by MS. This novel method combines ELISA‐based immunocapture with protein identification by liquid chromatography–tandem mass spectrometry (LC–MS/MS) to accurately determine HCP coverage. The method is named and referred to as ELISA‐MS. To calculate the HCP coverage percentages, HCPs were identified by LC–MS/MS after immunocapture and compared to the total number of HCPs identified in an early process sample. A species‐specific isotype control antibody was analyzed in parallel to assess nonspecific binding during immunocapture. The resulting list of HCPs covered by the HCP antibody was compared to the HCPs identified in the purified drug substance to calculate the specific coverage of the final product. This novel workflow can be applied to both process‐specific HCP antibodies and to generic commercially available HCP antibodies to find the most suitable ELISA antibody for the specific process and purified drug substance.
2. MATERIALS AND METHODS
2.1. Cell culture
Recombinant granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) was expressed in E. coli BL21 (DE3) essentially as described by Schwanke and colleagues.23 In short, cells were transfected with an IPTG inducible vector including the gene encoding human GM‐CSF. The cells were propagated in minimal media and induced with IPTG at high cell density for GM‐CSF expression. After induction for 3–4 hr, the cells were harvested, washed, and lysed by cavitation technology and inclusion bodies were recovered by centrifugation. The inclusion bodies were frozen and stored until further processing. The denatured product (GM‐CSF) was recovered by inclusion body solubilization under reducing and denaturing condition followed by renaturation by controlled oxidation and folding of the product. The resolubilized inclusion body sample without further processing (referred to as early process sample) was used as antigen for coverage analysis of three generic anti‐E. coli HCP polyclonal antibodies (referred to as HCP antibodies). The final drug substance was purified by standard chromatographic conditions including three preparative ion chromatography and reversed phase chromatography unit operations. The purified drug substance was subjected to MS‐based HCP analysis and the specific coverage of remaining HCPs was evaluated.
2.2. Antibodies for HCP coverage analysis
The coverage of HCPs in the early process sample containing recombinant GM‐CSF expressed in E. coli was evaluated using three commercially available HCP antibodies: rabbit anti‐E. coli HCP pAb A (Vendor A), goat anti‐E. coli HCP pAb B (Vendor B), and goat anti‐E. coli HCP pAb C (Vendor C). The specific HCP antibodies are referred to as Ab A, Ab B, and Ab C, respectively. Species‐specific isotype controls (referred to as control antibodies) were included in all experiments to monitor nonspecific binding of antigens: normal rabbit IgG (R&D Systems), normal rabbit IgG (Invitrogen), and normal goat IgG (R&D Systems).
2.3. Coverage analysis by ELISA‐MS
Prior to analysis, ELISA conditions were optimized to the antigen (early process sample) using the three different HCP antibodies, the control antibodies, and their biotinylated counterparts. The purpose of the optimization was to capture as much antigen as possible with minimal nonspecific binding. For coverage experiments, the goat HCP antibodies (Ab B and Ab C) and their corresponding control antibodies were coated in 96‐well micro‐titer plates at a concentration of 5 μg/ml (100 μl/well), whereas the rabbit HCP antibody (Ab A) was used at a concentration of 2.5 μg/ml (100 μl/well) due to high nonspecific binding of antigen to the rabbit control antibody. The plates (Thermo Scientific) were coated overnight at 4°C with HCP antibodies or control antibodies. For comparison, equal amounts of antigen (9 μg/ml, 100 μl/well) from the early process sample containing E. coli HCPs were added to each plate. Plates were washed extensively with phosphate‐buffered saline (PBS), pH 7.4 (Gibco), and 0.01% Tween‐20 (Thermo Scientific) (PBS‐T) before and after each step to remove excess antibody and antigen. The bound antigens were digested in the plate using trypsin. An aliquot of the early process sample was digested under similar conditions in parallel without antibodies and served as a measurement of total protein content to identify all HCPs before immunocapture (referred to as total protein sample). Reduced and alkylated peptides were analyzed by LC–MS/MS.
2.4. HCP analysis of purified drug substance
The HCP content of the purified drug substance was analyzed by LC–MS/MS. Two hundred micrograms of drug substance was diluted in 50 mM NH4·HCO3 (Sigma‐Aldrich) and proteins were precipitated in four volumes acetone (Sigma‐Aldrich) overnight at −20°C. The precipitated proteins were dissolved in 8 M urea (Sigma‐Aldrich), reduced, and alkylated. The proteins were digested by lysyl endopeptidase (1:50 enzyme‐to‐protein ratio, Wako) followed by 1:10 dilution in 50 mM NH4·HCO3 and digestion using trypsin (1:50 enzyme‐to‐protein ratio, Promega). Reduced (using dithiotreiol, final concentration of 6 mM) and alkylated (using iodoaceamide, final concentration of 12 mM) peptides were analyzed by LC–MS/MS.
2.5. Mass spectrometry
2.5.1. Coverage analysis
Protein identification by information‐dependent acquisition‐mediated LC–MS/MS (IDA‐LC–MS/MS).
Samples were analyzed on an Exigent nanoLC connected online to a TripleTOF 6600 mass spectrometer (AB Sciex). Peptides were separated on a reversed phase C18 column (nanoEase M/Z Peptide CSH C18, 130 Å, 1.7 μM, 300 μM × 150 mm, Waters) using an 80 min gradient at a flow of 5 μL/min. The data were acquired in data‐dependent mode where a survey spectrum (m/z range 350–1,700) is followed by MS/MS (m/z range 130–2,000) of the most intense multiply charged ions using collision induced dissociation. The total protein sample was analyzed in a twofold dilution starting at 8 μg on column.
The IDA‐LC–MS/MS data, converted to mgf‐files using ProteinPilot (AB Sciex), were searched against a protein sequence database by Mascot (Matrix Sciences)24 to create protein identification lists. The database contained: protein sequences from the UniProt E. coli proteome strain BL21 (DE3) (UP000002032, last updated February 27, 2018, n = 4,156); the T7 bacteriophage (UP000000840, last updated October 22, 2018, n = 57); goat‐ and rabbit‐related antibodies; common contaminants such as human hair/skin keratins; bovine serum albumin (used for instrument calibration); the drug substance; and trypsin. The mgf‐files from each set of HCP antibody, control antibody, and total protein sample were merged. The following settings were used: fixed modifications: carbamidomethyl (C), variable modifications: oxidation (M), enzymes: trypsin, one missed cleavage, peptide charge 1+ to 3+, peptide tolerance: 20 ppm, MS/MS tolerance: 0.05 Da. A decoy database search was performed to estimate false discovery rate. All protein and peptide data were extracted and processed in Skyline25 to match peptide identifications between runs and reintegration of peaks/peptides. Criteria for identification were identification by minimum two unique peptides, 5% false discovery rate threshold, and a q value below 0.01 for quantification (“identified proteins”). The number of covered proteins is defined as the sum of the protein identifications unique to the HCP antibody and the proteins enriched more than twofold by the HCP antibody compared to the control antibody. A protein is considered enriched by the HCP antibody compared to the control antibody if the SumAll peptide area (sum of MS peak areas for all peptides assigned to a specific protein) for the protein after immunocapture with the HCP antibody is at least twofold higher than the SumAll peptide area for the protein after immunocapture with the control antibody. The total number of HCPs in the early process sample is the sum of proteins identified in the total protein sample and the proteins only identified after immunocapture with the HCP antibody or control antibody.
2.5.2. HCP analysis of purified drug substance
IDA‐LC–MS/MS: samples were analyzed in triplicate on an Exigent nanoLC connected online to a TripleTOF 6600 mass spectrometer (AB Sciex). The peptides (8 μg) were separated on a reversed phase C18 column using a 40 min gradient at a flow of 5 μL/min. The data were acquired in data‐dependent mode where a survey spectrum (m/z range 350–1,700) is followed by MS/MS (m/z range 130–2,000) of the most intense multiple charged ions using collision induced dissociation.
Sequential Window Acquisition of all Theoretical fragment ion spectra‐LC–MS/MS (SWATH‐LC–MS/MS): Peptides were separated using the same chromatography as the IDA analysis. The SWATH analyses consist of a parent ion spectrum, followed by 52 MS/MS acquisitions that transmit and fragment all ions within variable window sizes from m/z 130–2,000. All samples were analyzed in triplicate by SWATH‐LC–MS/MS.
The IDA‐LC–MS/MS data were searched against the protein sequence database described above (with addition of the protein sequence of lysyl endopeptidase) using ProteinPilot (AB Sciex) to create sample‐specific protein libraries. The following settings were used: sample type: Identification, Cys Alkylation: Iodoacetamide, digestion: Trypsin, Instrument: TripleTOF 6600, no special factors, species or ID focus, search effort: rapid ID (tryptic peptides only), results quality: detected protein, threshold: 2.0 (99.0%), and a false discovery rate analysis was performed. Criteria for identification: identified by minimum two unique peptides.
2.6. Statistical analysis
GraphPad Prism version 8 was used for statistical analysis of data presented in Figures 3, 6, and S2. Three independent experiments were performed (n = 3). Data are presented as mean + SEM with individual data points shown as dots (Figure 3) or mean ± SEM (Figure 6) or +SEM (Figure S2). Data in Figures 2 and 5 are one representative of three independent experiments (n = 1).
Figure 3.
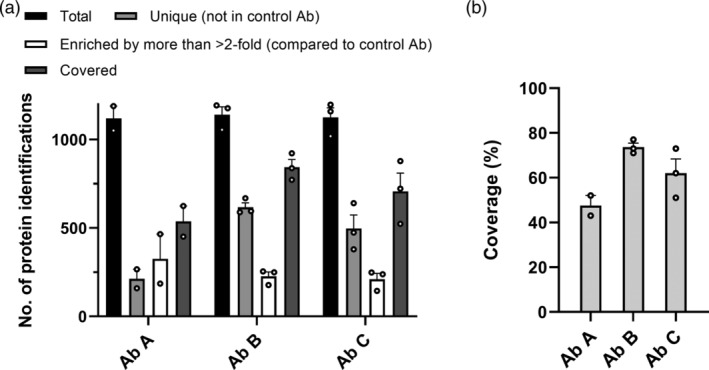
HCP antibody coverage of E. coli HCPs assessed by ELISA‐MS. Data are presented as mean + SEM with individual data points shown as dots (Ab A [n = 2], Ab B, and C [n = 3])
Figure 6.
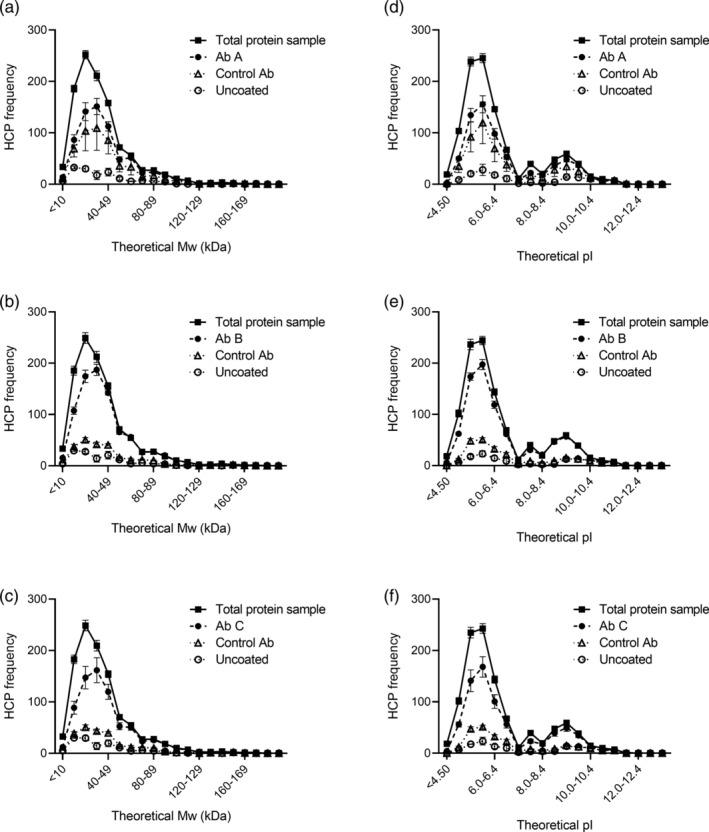
Frequency of HCPs identified in the different samples across the range of theoretical MW (a–c) and pI (d–f). Data are presented as mean ± SEM (n = 3. For the rabbit control antibody, n = 2)
Figure 2.
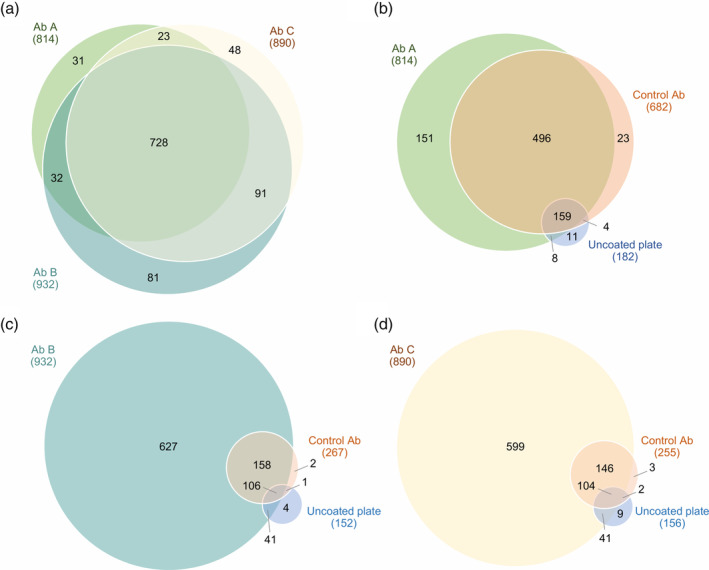
Comparison of proteins recognized by the three HCP antibodies. (a) Comparison of E. coli HCPs recognized by Ab A, B, or C. (b‐d) Comparison of E. coli HCPs recognized by the HCP antibody, control antibody, or bound to uncoated plates. The negative control antibodies are species‐specific isotype controls (rabbit IgG [Ab A] or goat IgG [Ab B and C]). Data shown are one representative of three independent experiments (n = 1)
Figure 5.
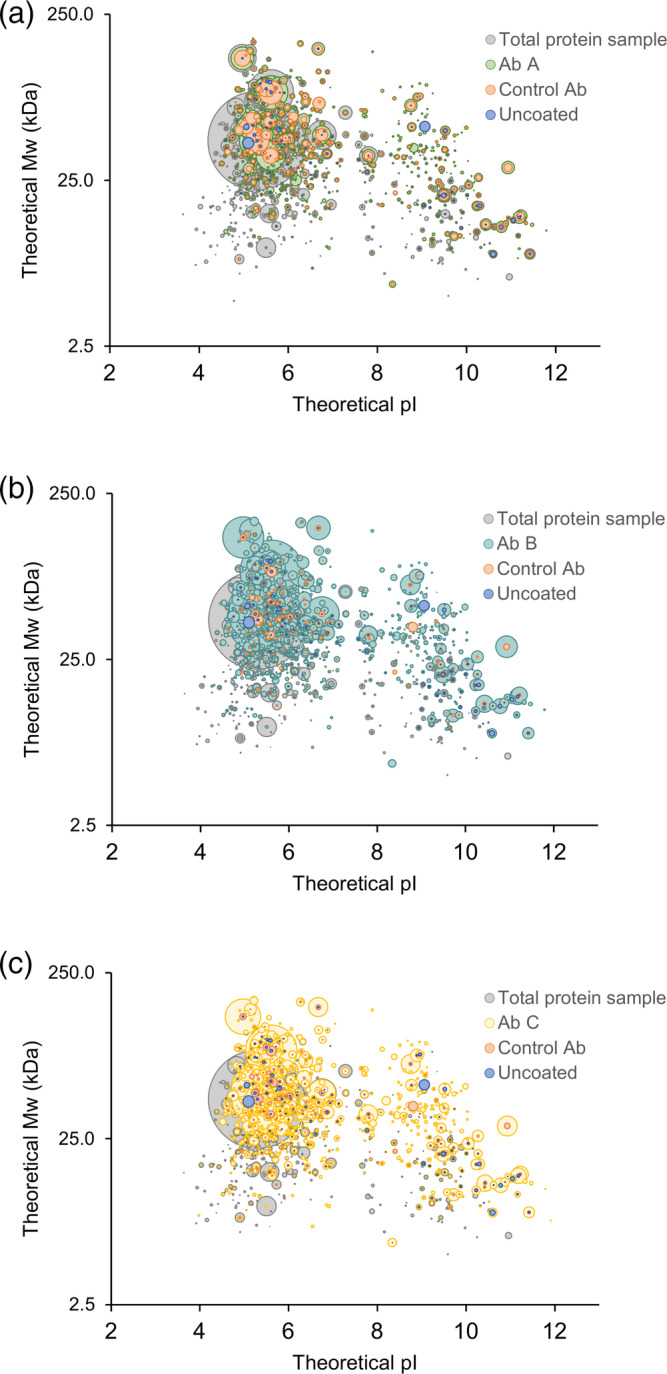
HCP antibody coverage across the theoretical molecular weight and isoelectric point (pI) range of the HCPs identified in the early process sample. 3D‐plots comparing HCPs identified in total protein sample to HCPs recognized by the three different HCP antibodies (Ab A, Ab B, and Ab C), species‐specific control antibodies, or bound to uncoated plates. Each spot represents a unique HCP and the spot size is scaled relative to the protein quantifications. Data shown are one representative of three independent experiments (n = 1)
3. RESULTS AND DISCUSSION
Sandwich ELISA is the standard method for monitoring HCP clearance during process development and release testing. It is important to determine ELISA HCP antibody coverage to select an appropriate ELISA for a specific bioprocess. Here, we describe a novel coverage method: ELISA‐MS, combining ELISA‐based immunocapture, mimicking the experimental conditions found in the sandwich ELISA setup, and protein identification by LC–MS/MS. The experimental setup is depicted in Figure 1. The results described in the following sections provide information about the HCP coverage of an early process sample and the purified drug substance by three commercially available HCP antibodies.
Figure 1.
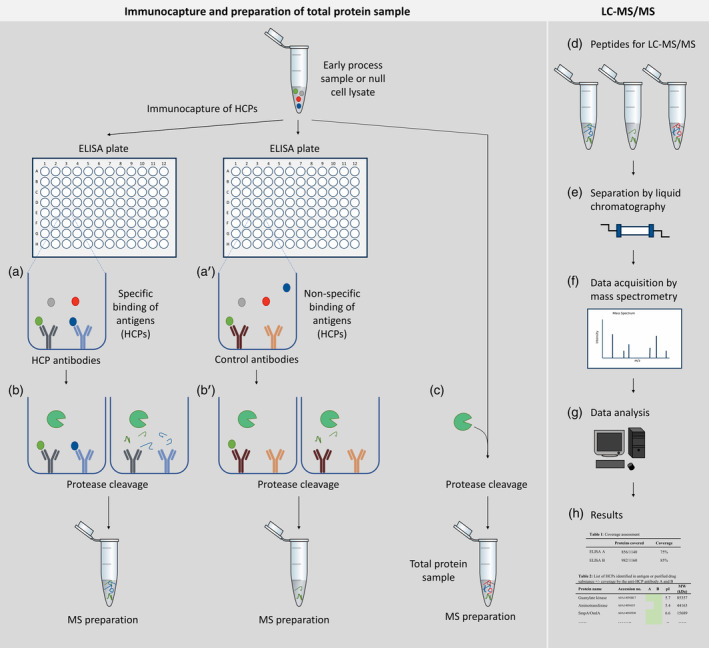
Experimental setup ELISA‐based immunocapture followed by protein identification by LC–MS/MS (ELISA‐MS). Left panel: immunocapture and preparation of total protein sample. For immunocapture, ELISA plates are coated with specific HCP antibodies or control antibodies. Antigen, an early process sample or a null cell lysate, containing HCPs are added to each plate and unbound HCPs are removed during washing steps (a, a'). Bound HCPs are digested in the plate using trypsin (b, b′). An aliquot of the early process sample or null cell lysate is digested under similar conditions in parallel without antibodies (c) and serves as a measurement of total protein content to identify all HCPs (referred to as total protein sample). Right panel: peptides from the immunocapture and the total protein sample are analyzed by LC–MS/MS (d). During LC–MS/MS, peptides are separated by liquid chromatography (e) and data are acquired by mass spectrometry (f). The LC–MS/MS data are searched against a protein sequence database (g) to create resulting protein identification lists for each sample (h). HCPs identified by LC–MS/MS after immunocapture is compared to the total number of HCPs identified in the early process sample or null cell lysate to calculate HCP coverage (h)
3.1. Comparison of protein identifications after immunocapture
Three commercially available HCP antibodies were used for coverage analysis of the early process sample: Ab A, Ab B, and Ab C. The proteins recognized by each HCP antibody are compared in Figure 2a. Protein identifications were highly similar between the three different HCP antibodies: 70.4% of the proteins were recognized by all three HCP antibodies (728 out of 1,034), and 84.5% of the proteins were recognized by at least two out of the three HCP antibodies (874 out of 1,034) (Figure 2a).
HCPs interacting nonspecifically (nonepitope‐driven) with the HCP antibodies would result in false positive coverage. This is highly unfortunate as it could result in a decision to monitor HCP clearance using an ELISA antibody that lacks true HCP coverage. To investigate the level of nonspecific binding, negative control experiments were performed to identify proteins captured on plates coated with control antibodies or uncoated plates. The proteins identified after immunocapture on plates coated with control antibodies or uncoated ELISA plates were compared to the proteins recognized by the HCP antibodies. Approximately, 28% of the immunocaptured HCPs were identified after immunocapture using the goat control antibody: 267 out of 939 unique proteins in total for the experiment using Ab B (Figure 2c) and 255 out of 904 unique proteins in total for the experiment using Ab C (Figure 2d). Compared to the goat control antibody, a high number of proteins were identified after immunocapture with the rabbit control antibody: 682 out of 852 unique proteins in total for the experiment, corresponding to 80% (Figure 2b). To examine if the high background from the immunocapture was due to this specific rabbit control antibody, we conducted the same experiment using a different rabbit antibody, but no notable difference was observed (Figure S1).
Few proteins were identified from the uncoated plates (experiment using Ab A: 182 out of 852 immunocaptured HCPs [21.4%], experiment using Ab B: 152 out of 939 immunocaptured HCPs [16.2%], and experiment using Ab C: 156 out of 904 immunocaptured HCPs [17.3%]; Figure 2b–d). Of these, 94–97% were also recognized by the HCP antibodies or control antibodies (Figure 2b–d). The low nonspecific binding of antigen to the uncoated plates is due to efficient blocking of nonspecific interactions with the plate surface by washing in the method. This saturates the unoccupied binding sites on the plate, before the addition of antigen. The remaining nonspecific binding to the control antibodies can be nonspecific “sticky” adhesion not blocked by the washing steps. This emphasizes the importance of including a control antibody to account for nonspecific binding of antigens.
3.2. HCP coverage
To determine the coverage of HCPs by the HCP antibodies, the number of E. coli proteins found in the early process sample was determined. Since proteins in low abundance in the early process sample may only be detectable after enrichment by immunocapture, the total number of proteins used to calculate the coverage for each HCP antibody was defined as the sum of the proteins identified in the total protein sample and the proteins only identified after immunocapture with the HCP antibody or control antibody. The number of proteins identified in the early process sample varied between 1,019 and 1,197 individual proteins among individual experiments (n = 8) (Figure 3a). Covered proteins were defined as the sum of the protein identifications unique to the HCP antibody and the proteins enriched more than twofold by the HCP antibody compared to the control antibody. The number of covered proteins was used to calculate the percentage of HCPs identified in the early process sample covered by the HCP antibody (Figure 3). The number of covered proteins varied between 451 and 922 individual HCPs for the three HCP antibodies in the experiments (n = 8) (Figure 3a). Ab B had the highest coverage percent with a mean coverage of 73.7% (n = 3) compared to a mean coverage of 47.5% for Ab A (n = 2) and 62.0% for Ab C (n = 3) (Figure 3b).
These data show that ELISA‐MS is a sensitive method that can be used to asses coverage of HCP antibodies, with a tight control of nonspecific binding through implementation of control antibodies. Currently used coverage methods, 2D gel electrophoresis in combination with 2D‐Western blot or immunoaffinity‐purification, have several technical limitations resulting in high variability in HCP coverage‐percentages and often do not include a control experiment with a negative control antibody.13 An alternative coverage method, combining immunoaffinity‐purification on magnetic beads and MS, has recently been described by Henry and colleagues.15 Our method has similar advantages as the bead‐based immunocapture, such as high sensitivity and identification and quantification of individual HCPs by MS. In addition, the immunocapture using our method resembles the actual experimental conditions in the sandwich ELISA, as the immunocapture is performed in an ELISA plate.
Opposed to coverage methods relying on spot counting, the results obtained using the method described here not only provide a coverage assessment, but also include a list of HCPs providing valuable information regarding individual proteins, their relative quantities, biological function, and physiochemical properties. Furthermore, the identification of individual protein species enables a more accurate risk assessment, a major requirement for evaluation of product safety for biological applications. An example of a list with 30 selected HCPs identified in the early process sample and the coverage by the three HCP antibodies is shown in Table 1. The HCP was considered covered/identified in this table if it was covered/identified by the HCP antibody in at least one out of three experiments.
Table 1.
Coverage of HCPs in the early process sample
| Protein name | Accession no. | Total protein sample | Ab A | Ab B | Ab C | pI | MW (kDa) |
|---|---|---|---|---|---|---|---|
| Guanylate kinase | A0A140NBE7 |

|

|
|
|
5.7 | 85,357 |
| Aminotransferase | A0A140N655 |
|
|
|
|
5.4 | 44,163 |
| SmpA/OmlA domain protein | A0A140NFD8 |
|
|
|
|
6.6 | 15,689 |
| DNA‐directed RNA polymerase subunit beta | A0A140NGD5 |
|
|
|
|
5.2 | 34,775 |
| Thioredoxin | A0A140NCS9 |
|
|
|
|
5.5 | 36,649 |
| Beta‐galactosidase | A0A140NDN8 |
|
|
|
|
6.5 | 37,999 |
| DNA polymerase III, delta prime subunit | A0A140NAC4 |
|
|
6.2 | 40,655 | ||
| Ubiquinone/menaquinone biosynthesis C‐methyltransferase UbiE | A0A140NBU4 |
|
|
|
|
6.2 | 45,679 |
| TonB‐dependent siderophore receptor | A0A140NFD2 |
|
|
|
|
5.1 | 29,418 |
| Periplasmic serine endoprotease DegP‐like | A0A140N6M4 |
|
|
|
|
4.8 | 34,893 |
| Isochorismatase | A0A140NCD4 |
|
6.0 | 15,935 | |||
| S‐ribosylhomocysteine lyase | A0A140NCE4 |
|
|
|
|
6.3 | 96,127 |
| UPF0250 protein YbeD | A0A140N8X2 |
|
|
|
|
5.1 | 25,535 |
| Chromosomal replication initiator protein DnaA | A0A140NI72 |
|
|
|
|
5.2 | 54,069 |
| Muramoyltetrapeptide carboxypeptidase | A0A140NCB5 |
|

|
|
|
7.7 | 17,229 |
| UPF0227 protein YcfP | A0A140N6A8 |
|
|
|
|
5.2 | 51,455 |
| Chaperone protein DnaK | A0A140NBA5 |
|
|
|
|
4.9 | 61,158 |
| Alanine–tRNA ligase | A0A140N5K8 |
|
|
|
|
9.7 | 22,087 |
| Peptidylprolyl isomerase | A0A140N8V9 |
|
|
|
|
5.9 | 22,321 |
| Threonine–tRNA ligase | A0A140N4K1 |
|
|
|
|
10.3 | 25,983 |
| Acetyl‐coenzyme A carboxylase carboxyl transferase subunit beta | A0A140N7A3 |
|
|
|
|
5.6 | 26,801 |
| Alkyl hydroperoxide reductase C | A0A140N3G7 |
|
|
|
|
9.9 | 22,244 |
| Type I secretion outer membrane protein | A0A140N9D7 |
|
|
|
|
7.8 | 33,402 |
| Alcohol dehydrogenase zinc‐binding domain protein | A0A140NDZ9 |
|
|
|
|
6.8 | 26,726 |
| ABC transporter related | A0A140N5X9 |
|
|
|
|
9.4 | 23,335 |
| DNA polymerase I | A0A140N583 |
|
|
|
|
5.3 | 51,051 |
| Uncharacterized protein | A0A140N7G4 |
|
11.0 | 6,542 | |||
| Aldo/keto reductase | A0A140NEE0 |
|
|
|
7.9 | 46,633 | |
| DNA gyrase subunit B | A0A140NH65 |
|
|
|
|
4.9 | 57,329 |
| Histone family protein DNA‐binding protein | A0A140N7E6 |
|
|
|
|
5.8 | 74,014 |
Note: List includes 30 proteins out of 1,524 identified HCPs. Diagonal shading indicates identified in total protein sample without immunocapture. Gray fill indicates coverage by Ab A, B, or C. Dark gray fill indicates that the protein was identified in the sample after immunocapture with the HCP antibody, but not considered covered as the protein was also found in similar or higher amounts in the sample after immunocapture using the control antibody (i.e., a false positive). The HCP was considered covered/identified in this table if it was covered/identified by the HCP antibody in at least one out of three experiments.
3.3. Theoretical MW and pI range of the HCPs identified in the early process sample
The early process sample used in the present study contained HCPs derived from inclusion bodies. In total, 1,524 unique proteins were identified in the early process sample during all experiments with or without immunocapture. This is a high number of proteins compared to currently used coverage methods: 2D gel electrophoresis in combination with 2D‐Western blot or immunoaffinity‐purification, which relies on spot counting. This high resolution, demonstrated by the high number of identified and named HCPs using ELISA‐MS, is a considerable advantage of the method compared to currently used methods. After 2D gel electrophoresis of E. coli BL21 cell lysates, between 250 and 900 unidentified spots are often observed, depending on staining method and fractionation of samples before the analysis.26 Furthermore, the number of spots in a gel or blot does not convert directly into a number of proteins, as some proteins may account for several spots due to protein isoforms, post‐translational modification, and degradation forms. For instance, it has previously been shown that 1,185 protein spots identified after 2D gel electrophoresis of an E. coli MG1655 cell lysate corresponded to around 723 unique proteins.27 The theoretical molecular weight (MW) and isoelectric point (pI) of the HCPs identified by this method in the early process sample are shown in Figure 4. The HCPs identified are within a theoretical MW of 2.8 to 179.3 kDa (mean MW of 38.5 kDa) and a theoretical pI of 3.6 to 11.9 (mean pI 6.5). This corresponds well to the range of MW (2.5–248.6 kDa, mean MW of 34.3 kDa) and pI (3.6–13, mean pI 6.9) of the E. coli BL21 (DE3) proteome (UniProt UP000002032).
Figure 4.
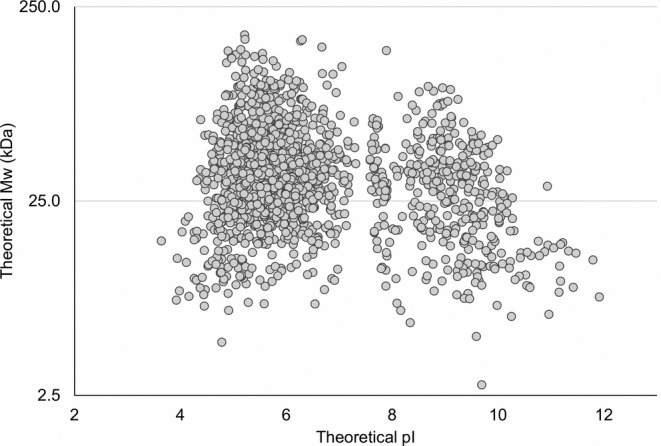
The theoretical molecular weight and isoelectric point (pI) range of the HCPs identified in the early process sample
3.4. HCP antibody coverage across the theoretical MW and pI range of the early process sample
To ensure coverage of a wide range of MW and pI, the distribution of HCP coverage across the range of the individual HCPs found in the early process sample was investigated (Figure 5). The HCP antibodies recognize proteins across the entire range of MW and pI of the early process sample (proteins in every quadrant of the plot). As expected, some HCPs are enriched by the immunocapture and some are underestimated after immunocapture relative to the amount in the total protein sample (Figure 5).
To assess if the immunoreactivity of the HCP antibodies is biased against HCPs with certain MWs or pIs, we compared the frequency of HCPs identified in the different samples across the range of theoretical MW and pI (Figure 6). A complementary analysis of the proportion (percentage) of HCPs identified in the total protein sample covered by the HCP antibody, the control antibody, or bound to uncoated plates is found in Figure S2. The HCPs recognized by the HCP antibody, the control antibody, or bound to uncoated plates followed the tendencies of the HCPs identified in the total protein sample across the entire range of MW and pI (Figure 6). Collectively, these results show that the HCP antibodies cover HCPs across the entire range of MW and pI of the HCPs found in the early process sample, which also corresponds well to the proteins in the E. coli proteome. Thus, in general, the ELISA‐MS method is not biased against a certain MW or pI range.
3.5. Specific coverage of HCPs in drug substance
The list of proteins covered by the HCP antibodies can be compared to a list of HCPs identified in a late process sample or in the purified drug substance. Table 2 shows the specific coverage of five HCPs identified in the drug substance purified from the early process sample used as antigen for the coverage analysis. The five HCPs were identified in the purified drug substance by LC–MS/MS. Ab B had the highest specific coverage of the HCPs found in the purified drug substance with a coverage of four out of five HCPs (Table 2). Ab A and C had a low coverage of only one out of five HCPs and two out of five HCPs, respectively. This illustrates the value of assessing the specific coverage of HCPs in the purified drug substance in combination with the overall coverage of upstream HCPs. Assessment of specific coverage might change the conclusion regarding the suitability of the different ELISA reagents. In this case, the overall coverage of the HCP population found in the early process sample was quite similar between Ab B (73.7%) and Ab C (62.0%). However, the specific coverage of HCPs in the purified drug substance was considerably higher for Ab B (four out of five HCPs) compared to Ab C (two out of five HCPs). This shows that the commercially available Ab B covers the majority of upstream HCPs present in inclusion bodies and HCPs in the purified drug substance. Thus, Ab B is suitable for monitoring HCP clearance of this given bioprocess.
Table 2.
Specific coverage of five HCPs identified in purified drug substance
| Protein name | Accession no. | Ab A | Ab B | Ab C |
|---|---|---|---|---|
| Ferric uptake regulation protein | A0A140NE13 |
|
||
| PTS system, glucose subfamily | A0A140N9A4 |
|
|
|
| UPF0250 protein YbeD | A0A140NE54 |
|
|
|
| Uncharacterized protein | A0A140NFF9 | |||
| ATP synthase subunit delta | A0A140NFT6 |
|
||
| Specific coverage | 1 out of 5 | 4 out of 5 | 2 out of 5 |
Note: Gray fill indicates coverage by Ab A, B, or C.
4. CONCLUSIONS
The study describes a novel coverage method, ELISA‐MS, allowing detailed analysis of HCP coverage by combining ELISA‐based immunocapture with protein identification by LC–MS/MS. Coverage analysis using this method provides a list of HCPs covered by an HCP antibody. This list is compared to a list of HCPs found in an early process sample, to calculate the HCP coverage percentage. Furthermore, the list of covered proteins can be compared to HCPs identified in the purified drug substance. This provides a measure of how well the HCP ELISA detects impurities in the final product that may impose a potential risk for patients or effecting drug substance stability. This novel method reassures data‐driven decisions to guide process development and monitoring, ensuring adequate coverage of the chosen ELISA(s).
ELISA is a valuable tool to monitor process performance during drug development and in product release testing. However, currently used methods to evaluate ELISA reagent coverage have several limitations as they rely on spot counting in a gel. We propose that ELISA‐MS, as presented here, will be a valuable supplement to existing methods or even a replacement. Coverage analysis by ELISA‐MS mirrors the actual conditions found in an ELISA, as the HCPs are bound to the HCP antibodies in an ELISA plate under native conditions as in the immunoassay. Furthermore, ELISA‐MS does not involve immunoblotting, thus avoiding problems with protein transfer from gel to blotting membrane; requires a low amount of HCP antibody for the analysis; and does not require spot counting, thus avoiding problems with lack of standardization of spot counting. However, ELISA‐MS requires sophisticated and expensive mass spectrometry instrumentation, compared to the more commonly used gel and blotting equipment, and may underrepresent some HCPs if they are not digested by trypsin. In combination with HCP analysis by MS, coverage analysis by ELISA‐MS will contribute to improved product characterization and increased understanding of HCP contamination of biopharmaceutical products and process clearance. The experimental setup can be tailored to assess HCP coverage of process‐specific and commercial ELISAs using antigen from different host cells.
The method can be used to evaluate several ELISA antibodies simultaneously, screening for a suitable generic ELISA before deciding to develop a process‐specific ELISA or use a platform method. The results demonstrate the suitability of commercially available ELISA reagents for HCP measurements. For the tested biopharmaceutical (GM‐CSF), Ab B covered the majority of the HCPs identified in the early process sample (73.7%), as well as four out of the five HCPs identified in the purified drug substance, making it acceptable for HCP surveillance for this specific bioprocess.
Our method enables tight control of nonspecific binding through experiments using control antibodies and provides identification and relative quantification of individual HCPs. Compared to currently used coverage methods, ELISA‐MS has a high resolution demonstrated by the high number of identified HCPs in the E. coli sample compared to for example, the number of spots in a 2D‐PAGE. An additional advantage is that the method can use an early process sample, containing the drug substance, as well as a null cell lysate as antigen. The HCP profile of a null cell lysate may differ from the HCPs found in an early process sample. Some HCPs are co‐expressed and co‐purified with the drug substance, which is why a coverage analysis using an early process sample is preferable. This saves time and investments by evading the production of an expensive null cell line not expressing the drug substance.
In conclusion, ELISA‐based immunocapture followed by LC–MS/MS combines the information provided by ELISA and MS‐based HCP analysis, allowing evaluation of protein‐specific coverage, and provides detailed information about the reagent suitability. Potential gaps in the coverage can be identified. This can improve HCP surveillance and result in a final HCP profile with the lowest achievable risk, which is beneficial to both the pharmaceutical industry and patient safety.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Figure S1 Comparison of E. coli HCP identifications after immunocapture with rabbit control antibody from two different vendors. The Rabbit IgG from R&D Systems was used in the experiments presented in the results and discussion section.
Figure S2 Proportion (percentage) of HCPs identified in the total protein sample covered by the HCP antibodies, the control antibodies, or bound to uncoated plates across the range of MW (A‐C) and pI (D‐F). Data are presented as mean + SEM (n = 3. For the rabbit IgG, n = 2). The coverage of HCPs was consistent for HCPs with a theoretical MW of 40–160 kDa varying between a mean of 70–105% of the number of HCPs identified in the total protein sample (A‐C). The coverage of HCPs below 40 kDa was slightly decreased (mean of 63–78%) and only around 50% (45–56%) of the HCPs identified in the total protein sample with a theoretical MW below 20 kDa was covered by the HCP antibodies (A‐C). This might reflect the known difficulties of eliciting effective antibody responses against low MW HCPs (Hanly, Artwohl, & Bennett, 1995). The coverage of HCPs with a MW above 160 kDa was slightly increased (>130%), but only four proteins were identified in this group. The coverage of HCPs by the HCP antibodies was consistent for HCPs with a theoretical pI between 5–10 with a coverage varying between a mean of 59–101% (D‐F). The coverage of HCPs with a pI below 5 was slightly decreased with only 50% (43–57%) of the HCPs identified in the total protein sample covered by the HCP antibodies (D‐F). Eleven HCPs identified in the early process sample with or without immunocapture have a pI above 11. The coverage of these was slightly increased (mean of 96–110%). The HCPs identified after immunocapture with the control antibody or bound to uncoated plates followed the coverage tendency of the HCP antibodies across the entire range of MW (A‐C). For the HCPs with a pI from below 5 to 10, the share of covered HCPs identified after immunocapture with the isotype control or on uncoated plates followed the coverage tendency of the HCP antibodies (D‐F). However, for the HCPs with a pI above 10 (50 protein identifications in the early process sample with or without immunocapture), the share of HCPs identified after immunocapture with the control antibodies or bound to the uncoated plate was highly increased (D‐F).
ACKNOWLEDGMENTS
The authors gratefully acknowledge the financial support of Eurostars [E10980 SaferBIOPHARMA].
Pilely K, Nielsen SB, Draborg A, et al. A novel approach to evaluate ELISA antibody coverage of host cell proteins—combining ELISA‐based immunocapture and mass spectrometry. Biotechnol Progress. 2020;36:e2983 10.1002/btpr.2983
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1002/btpr.2983.
Funding information Eurostars, Grant/Award Number: E10980 SaferBIOPHARMA
REFERENCES
- 1. Champion K, Madden H, Dougherty J, Shacter E. Defining your product profile and maintaining control over it, part 2. Bioprocess Int. 2005;3:52‐57.29899681 [Google Scholar]
- 2. Wang X, Hunter AK, Mozier NM. Host cell proteins in biologics development: identification, quantitation and risk assessment. Biotechnol Bioeng. 2009;103(3):446‐458. [DOI] [PubMed] [Google Scholar]
- 3. Shukla AA, Hinckley P. Host cell protein clearance during protein a chromatography: development of an improved column wash step. Biotechnol Prog. 2008;24(5):1115‐1121. [DOI] [PubMed] [Google Scholar]
- 4. Hunter AK, Wang X, Suda EJ, et al. Separation of product associating E. coli host cell proteins OppA and DppA from recombinant apolipoprotein A‐I Milano in an industrial HIC unit operation. Biotechnol Prog. 2009;25(2):446‐453. [DOI] [PubMed] [Google Scholar]
- 5. Levy NE, Valente KN, Choe LH, Lee KH, Lenhoff AM. Identification and characterization of host cell protein product‐associated impurities in monoclonal antibody bioprocessing. Biotechnol Bioeng. 2014;111(5):904‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aboulaich N, Chung WK, Thompson JH, Larkin C, Robbins D, Zhu M. A novel approach to monitor clearance of host cell proteins associated with monoclonal antibodies. Biotechnol Prog. 2014;30(5):1114‐1124. 10.1002/btpr.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vanderlaan M, Zhu‐Shimoni J, Lin S, Gunawan F, Waerner T, Van Cott KE. Experience with host cell protein impurities in biopharmaceuticals. Biotechnol Prog. 2018;34(4):828‐837. [DOI] [PubMed] [Google Scholar]
- 8. Gao SX, Zhang Y, Stansberry‐Perkins K, et al. Fragmentation of a highly purified monoclonal antibody attributed to residual CHO cell protease activity. Biotechnol Bioeng. 2011;108(4):977‐982. [DOI] [PubMed] [Google Scholar]
- 9. Huang L‐Y, Dumontelle JL, Zolodz M, Deora A, Mozier NM, Golding B. Use of toll‐like receptor assays to detect and identify microbial contaminants in biological products. J Clin Microbiol. 2009;47(11):3427‐3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bierich JR. Treatment of pituitary dwarfism with biosynthetic growth hormone. Acta Paediatr Scand Suppl. 1986;325:13‐18. [DOI] [PubMed] [Google Scholar]
- 11. Zhu‐Shimoni J, Yu C, Nishihara J, et al. Host cell protein testing by ELISAs and the use of orthogonal methods. Biotechnol Bioeng. 2014;111(12):2367‐2379. [DOI] [PubMed] [Google Scholar]
- 12. Khrenov A. Control of host cell proteins ‐ one Reviewer's perspective BioPharmaceutical Emerging Best Practices Association (BEBPA) HCP Conference. OTAT/CBER/FDA: San Pedro, CA; 2019. [Google Scholar]
- 13. U.S. Pharmacopeia Formulary N. Residual Host Cell Protein Measurement in Biopharmaceuticals; Rockville, MD: The United States Pharmacopeial Convention. 2016. [Google Scholar]
- 14. Shahrokh Z, Schmalzing D, Rawat R, et al. Science, risks, and regulations: current perspectives on host cell protein analysis and control. BioProcess Tech. 2016;14(8):40‐51. [Google Scholar]
- 15. Henry SM, Sutlief E, Salas‐Solano O, Valliere‐Douglass J. ELISA reagent coverage evaluation by affinity purification tandem mass spectrometry. MAbs. 2017;9(7):1065‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walker DE, Yang F, Carver J, Joe K, Michels DA, Yu XC. A modular and adaptive mass spectrometry‐based platform for support of bioprocess development toward optimal host cell protein clearance. MAbs. 2017;9(4):654‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doneanu CE, Anderson M, Williams BJ, Lauber MA, Chakraborty A, Chen W. Enhanced detection of low‐abundance host cell protein impurities in high‐purity monoclonal antibodies down to 1 ppm using ion mobility mass spectrometry coupled with multidimensional liquid chromatography. Anal Chem. 2015;87(20):10283‐10291. [DOI] [PubMed] [Google Scholar]
- 18. Doneanu CE, Xenopoulos A, Fadgen K, et al. Analysis of host‐cell proteins in biotherapeutic proteins by comprehensive online two‐dimensional liquid chromatography/mass spectrometry. MAbs. 2012;4(1):24‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Q, Goetze AM, Cui H, et al. Comprehensive tracking of host cell proteins during monoclonal antibody purifications using mass spectrometry. MAbs. 2014;6(3):659‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reisinger V, Toll H, Mayer RE, Visser J, Wolschin F. A mass spectrometry‐based approach to host cell protein identification and its application in a comparability exercise. Anal Biochem. 2014;463:1‐6. [DOI] [PubMed] [Google Scholar]
- 21. Schenauer MR, Flynn GC, Goetze AM. Identification and quantification of host cell protein impurities in biotherapeutics using mass spectrometry. Anal Biochem. 2012;428(2):150‐157. [DOI] [PubMed] [Google Scholar]
- 22. Bracewell DG, Francis R, Smales CM. The future of host cell protein (HCP) identification during process development and manufacturing linked to a risk‐based management for their control. Biotechnol Bioeng. 2015;112(9):1727‐1737. 10.1002/bit.25628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwanke RC, Renard G, Chies JM, et al. Molecular cloning, expression in Escherichia coli and production of bioactive homogeneous recombinant human granulocyte and macrophage colony stimulating factor. Int J Biol Macromol. 2009;45(2):97‐102. [DOI] [PubMed] [Google Scholar]
- 24. Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability‐based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551‐3567. [DOI] [PubMed] [Google Scholar]
- 25. MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966‐968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sigdel TK, Cilliers R, Gursahaney PR, Crowder MW. Fractionation of soluble proteins in Escherichia coli using DEAE‐, SP‐, and phenyl sepharose chromatographies. J Biomol Tech. 2004;15(3):199‐207. [PMC free article] [PubMed] [Google Scholar]
- 27. Vijayendran C, Burgemeister S, Friehs K, Niehaus K, Flaschel E. 2DBase: 2D‐PAGE database of Escherichia coli . Biochem Biophys Res Commun. 2007;363(3):822‐827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Comparison of E. coli HCP identifications after immunocapture with rabbit control antibody from two different vendors. The Rabbit IgG from R&D Systems was used in the experiments presented in the results and discussion section.
Figure S2 Proportion (percentage) of HCPs identified in the total protein sample covered by the HCP antibodies, the control antibodies, or bound to uncoated plates across the range of MW (A‐C) and pI (D‐F). Data are presented as mean + SEM (n = 3. For the rabbit IgG, n = 2). The coverage of HCPs was consistent for HCPs with a theoretical MW of 40–160 kDa varying between a mean of 70–105% of the number of HCPs identified in the total protein sample (A‐C). The coverage of HCPs below 40 kDa was slightly decreased (mean of 63–78%) and only around 50% (45–56%) of the HCPs identified in the total protein sample with a theoretical MW below 20 kDa was covered by the HCP antibodies (A‐C). This might reflect the known difficulties of eliciting effective antibody responses against low MW HCPs (Hanly, Artwohl, & Bennett, 1995). The coverage of HCPs with a MW above 160 kDa was slightly increased (>130%), but only four proteins were identified in this group. The coverage of HCPs by the HCP antibodies was consistent for HCPs with a theoretical pI between 5–10 with a coverage varying between a mean of 59–101% (D‐F). The coverage of HCPs with a pI below 5 was slightly decreased with only 50% (43–57%) of the HCPs identified in the total protein sample covered by the HCP antibodies (D‐F). Eleven HCPs identified in the early process sample with or without immunocapture have a pI above 11. The coverage of these was slightly increased (mean of 96–110%). The HCPs identified after immunocapture with the control antibody or bound to uncoated plates followed the coverage tendency of the HCP antibodies across the entire range of MW (A‐C). For the HCPs with a pI from below 5 to 10, the share of covered HCPs identified after immunocapture with the isotype control or on uncoated plates followed the coverage tendency of the HCP antibodies (D‐F). However, for the HCPs with a pI above 10 (50 protein identifications in the early process sample with or without immunocapture), the share of HCPs identified after immunocapture with the control antibodies or bound to the uncoated plate was highly increased (D‐F).