

Abstract
Background
Splicing is crucial for proper gene expression, and is predominately executed by the major spliceosome. Conversely, 722 introns in 699 human minor intron‐containing genes (MIGs) are spliced by the minor spliceosome. Splicing of these minor introns is disrupted in diseases caused by pathogenic variants in the minor spliceosome, ultimately leading to the aberrant expression of a subset of these MIGs. However, the effect of variants in minor introns and MIGs on diseases remains unexplored.
Methods
Variants in MIGs and associated clinical manifestations were identified using ClinVar. The HPO database was then used to curate the related symptoms and affected organ systems. Results: We found pathogenic variants in 211 MIGs, which commonly resulted in intellectual disability, seizures and microcephaly. This revealed a subset of MIGs whose aberrant splicing may contribute to the pathogenesis of minor spliceosome‐related diseases. Moreover, we identified 51 pathogenic variants in minor intron splice sites that reduce the splice site strength and can induce alternative splicing.
Conclusion
These findings highlight that disrupted minor intron splicing has a broader impact on human diseases than previously appreciated. The hope is that this knowledge will aid in the development of therapeutic strategies that incorporate the minor intron splicing pathway.
Keywords: disease, minor spliceosome, nervous system, splice site, variant
Pathogenic variants in minor spliceosome components result in disrupted splicing of minor introns and a wide range of symptoms affecting many organ systems. Here, we identified a subset of minor intron‐containing genes that may play a role in the pathogenesis of these minor spliceosome‐related diseases. Moreover, we report the presence of 51 pathogenic variants in minor intron splice sites that result in a reduced splice site strength.

1. INTRODUCTION
The human genome contains two types of introns, major (99.5%) and minor (<0.5%), that diverge in the consensus sequences at the 5′ splice site (5′SS), branch point sequence (BPS) and 3′SS (Alioto, 2007; Olthof, Hyatt, & Kanadia, 2019). The splicing of these two classes of introns therefore requires two separate splicing machineries, the major and minor spliceosome, which are made up of five small nuclear RNAs (snRNAs). Specifically, the major spliceosome consists of U1, U2, U4, U5 and U6 snRNAs, whereas the minor spliceosome contains the analogous snRNAs U11, U12, U4atac, U6atac and shares U5 with the major spliceosome (Tarn & Steitz 1996a,1996b). Moreover, the minor spliceosome contains seven unique proteins associated with U11 and U12 snRNA (Will et al., 2004). To date, the only known diseases associated with germline variants in snRNAs affect the minor spliceosome. Specifically, pathogenic variants in RNU4ATAC (OMIM: 601428) are linked to microcephalic osteodysplastic primordial dwarfism type 1 (MOPD1), Roifman syndrome and Lowry‐Wood syndrome (Edery et al., 2011; Farach et al., 2018; He et al., 2011; Merico et al., 2015). Moreover, pathogenic variants in RNU12, which encodes the U12 snRNA, have been linked to early onset cerebellar ataxia (Elsaid et al., 2017). Finally, variants in RNPC3 (OMIM: 618016), a minor spliceosome‐specific protein that is part of the U12 small nuclear ribonucleoprotein (snRNP), have been associated with isolated growth hormone deficiency (IGHD) (Argente et al., 2014). Finally, somatic mutations in ZRSR2, a protein component of both the major and minor spliceosome, have been shown to predominately affect minor intron splicing, resulting in myelodysplastic syndrome (Madan et al., 2015). Moreover, somatic mutations in U11 snRNA were recently identified in sonic hedgehog medulloblastomas (Suzuki et al., 2019). Despite these examples, overall the Mendelian minor spliceosome‐related diseases are rare, which might explain why the minor spliceosome is somewhat understudied.
The autosomal recessive inheritance pattern of minor spliceosome‐related diseases suggests that these pathogenic variants result in a loss‐of‐function (Argente et al., 2014; Edery et al., 2011; Elsaid et al., 2017; Farach et al., 2018; He et al., 2011; Merico et al., 2015). Consequently, this would result in the aberrant splicing and expression of minor intron‐containing genes (MIGs). Indeed, these molecular defects were observed in peripheral blood mononuclear cells (PBMCs) from individuals with Roifman syndrome and cerebellar ataxia (Elsaid et al., 2017; Merico et al., 2015). However, not all MIGs were affected in these individuals, suggesting that aberrant splicing of only a subset of MIGs underlies the symptoms associated with these diseases. Here we analyzed previous reports to determine shared symptoms among individuals with minor spliceosome‐related diseases. We then identify a subset of MIGs that, when disrupted, result in similar phenotypes and therefore may underlie the pathogenesis of minor spliceosome‐related diseases. Finally, we identify pathogenic variants in minor introns that are predicted to result in disrupted minor intron splicing and result in diseases commonly affecting the nervous system.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
This study does not contain any studies with human participants or animals performed by the authors, and therefore no ethical compliance is required.
2.2. Curation of variants and associated clinical manifestations in MIGs
All human MIGs from the Minor Intron DataBase (MIDB) (hg38, Ensembl version 95) were queried in ClinVar to identify associated variants listed as “pathogenic,” “likely pathogenic,” “uncertain significance,” and “conflicting results of pathogenicity.” Variants labeled as “uncertain significance” or “conflicting results of pathogenicity” were cross‐referenced with gnomAD to ensure the allelic frequency was < 0.01% (Karczewski et al., 2019). Clinical manifestations associated with each variant were then curated, and filtered to create a list of unique clinical manifestations associated with pathogenic variants in each MIG (Landrum et al., 2014; Olthof et al., 2019). Clinical manifestations linked to variants found in MIGs and overlapping non‐MIGs were excluded. The Human Phenotype Ontology (HPO) database was then used to curate symptoms associated with each clinical manifestation (Kohler et al., 2019). Here, the ORPHA code was used to extract symptoms listed as very frequent (>80%). If no ORPHA code was available, an OMIM code was used to extract the symptoms for the corresponding disease. Since these codes are not associated with a frequency, all phenotypes listed on HPO were included, regardless of their prevalence. Few variants were associated with diseases and ORPHA codes that were “in progress” or not recognized at all. These were excluded from further analysis.
2.3. Curation of symptoms
The Human Phenotype Ontology (HPO) database was used to curate symptoms associated with minor spliceosome‐related diseases (Kohler et al., 2019). Here, both the symptoms associated with the OMIM and ORPHA code were extracted. The frequency of these symptoms in minor spliceosome‐related disorders was then determined by manual curation of the primary literature. Only primary literature describing individuals for which the pathogenic variants were known, were included. Symptoms reported in at least two individuals were included in Figure 1. Some of the isolated symptoms were part of the same root but indicated different degrees of severity (e.g. Intellectual disability, mild (HP: 0001256) and Intellectual disability, moderate (HP: 0002342)). For these terms, we condensed the symptoms to the shared root (e.g. Intellectual disability (HP: 0001249)) in Figure 1.
Figure 1.
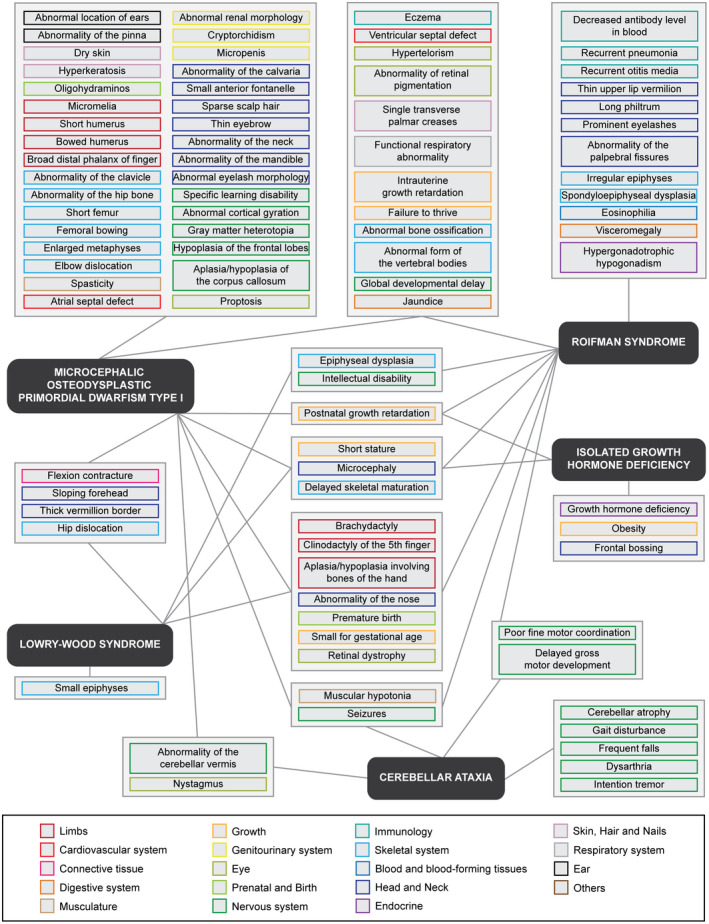
Pathogenic variants in disparate components of the minor spliceosome result in overlapping symptoms affecting many different tissues. Network of phenotypes associated with minor spliceosome‐related diseases according to HPO. Color of symptom outline corresponds to the affected organ system. Only symptoms reported in at least two individuals were linked to a disease
2.4. Identification of SNVs in minor introns
Minor intron coordinates from MIDB (hg38, Ensembl version 95) were cross‐referenced with all variants reported by ClinVar (both pathogenic and benign) (Landrum et al., 2014; Olthof et al., 2019). Variants within the first nine nucleotides of the intron were considered part of the 5′SS and variants within the last three nucleotides of the intron were classified as 3′SS. Finally, variants located within the BPS coordinates listed at MIDB were recorded as BPS, whereas the remainder was classified as “elsewhere in intron.” Three variants absent in ClinVar, but previously published in peer‐reviewed journals have also been included in Table S4. The allelic frequency and homozygosity of variants was determined by cross‐referencing of the coordinates with gnomAD (Karczewski et al., 2019). The associated clinical manifestations and symptoms were curated the same way as was done for MIGs.
2.5. Impact of variant on splice site strength
The strength of minor intron splice sites was based on their score against minor intron‐type PWMs and previously reported on MIDB (Olthof et al., 2019; Sheth et al., 2006). The splice sites including the variant were rescored against the same AT‐AC type PWM or GT‐AG type PWM as listed in (Sheth et al., 2006). LOD scores were then rescaled to be on a scale from 0 to 100 as described previously (Olthof et al., 2019).
3. RESULTS
3.1. Pathogenic variants in disparate components of the minor spliceosome result in overlapping symptoms affecting many different tissues
Pathogenic variants in different minor spliceosome components have all been shown to inhibit minor spliceosome activity and affect the splicing and/or expression of its targets, the MIGs. Since both the minor spliceosome and MIGs are ubiquitously expressed, these pathogenic variants were predicted to affect all tissues. To determine the footprint of disrupted minor intron splicing on tissue development, we curated all symptoms that have been associated with MOPD1, Roifman syndrome, Lowry‐Wood syndrome, early onset cerebellar ataxia and IGHD, using the HPO database (Kohler et al., 2019). The use of generalized nomenclature to describe all symptoms allowed us to compare the phenotypes associated with each disease, which is usually complicated by the varying terminology used in primary literature. The frequency of each symptom was then determined by manual curation of the primary literature to ensure it was reported in at least two individuals (Table S1). This resulted in a list of 64 symptoms associated with MOPD1, 41 symptoms for Roifman syndrome, and 17 for Lowry‐Wood syndrome (Figure 1). Moreover, we identified 11 symptoms in cerebellar ataxia, and 7 symptoms characteristic of individuals with IGHD (Figure 1). No symptoms were shared between all five diseases. However, microcephaly (HP: 0000252), short stature (HP: 0004322) and delayed skeletal maturation (HP: 0002750) were observed in four of the five minor spliceosome‐related diseases (Figure 1). Additionally, seven symptoms were shared in all three of the RNU4ATAC‐related disorders, but not reported for individuals with IGHD or cerebellar ataxia. These included brachydactyly (HP: 0001156), aplasia/hypoplasia involving bones of the hand (HP: 0005927), clinodactyly of the 5th finger (HP: 0004209), a premature birth (HP: 0001622), and retinal dystrophy (HP: 0000556). Moreover, muscular hypotonia (HP: 0001252) and seizures (HP: 0001250) were reported in individuals with MOPD1, Roifman syndrome and cerebellar ataxia, but not in individuals with Lowry‐Wood syndrome (Figure 1). Relating these commonly observed symptoms to the affected organ system revealed the pleiotropic nature of the minor spliceosome‐related diseases. For example, the aforementioned phenotypes were indicative of defects in limbs, head and neck, eye, musculature, growth, nervous system, as well as the skeletal system.
Given that MOPD1, Roifman syndrome and Lowry‐Wood syndrome all result from pathogenic variants in RNU4ATAC, we expected the most overlap of phenotypes between these diseases. Indeed, several symptoms were shared between individuals with MOPD1 and Roifman syndrome, such as abnormal bone ossification (HP: 0011849), intrauterine growth retardation (HP: 0001511), and an abnormal form of the vertebral bodies (HP: 0003312) (Figure 1). Moreover, symptoms such as flexion contracture (HP: 0001371) and a sloping forehead (HP: 0000340) were commonly observed in MOPD1 as well as Lowry‐Wood patients. However, the majority of the phenotypes were unique to one condition. Specifically, symptoms associated with the integumentary system like dry skin (HP: 0000958), hyperkeratosis (HP: 0000962), and sparse hair (HP: 0008070), were prevalent in individuals with MOPD1 (Figure 1). Similarly, several facial dysmorphisms were characteristic of individuals with MOPD1, such as abnormalities of the pinna (HP: 0000377) and the mandible (HP: 0000277) and proptosis (HP: 0000520). On the other hand, immunological defects like a decreased antibody level in the blood (HP: 0004313) and recurrent infections (HP: 0002719) were exclusively reported for individuals with Roifman syndrome (Figure 1). Interestingly, pathogenic variants in RNU12 and RNPC3 did not result in any overlapping symptoms, even though they are both predicted to disrupt U12 snRNP function. To date, relatively few individuals have been diagnosed with minor spliceosome‐related diseases, especially with early onset cerebellar ataxia and IGHD. Regardless, data from these individuals reveal that variants in the same minor spliceosome components can result in diseases with disparate phenotypes.
3.2. Disruption of MIG function predominantly affects the nervous system
Loss‐of function variants in the minor spliceosome components result in the aberrant splicing/expression of MIGs, which ultimately cause the disease (Cologne et al., 2019; Elsaid et al., 2017; Merico et al., 2015). To identify a subset of MIGs whose aberrant splicing might underlie the pathogenesis of minor spliceosome‐related diseases, we curated diseases resulting from independent pathogenic variants in all known MIGs. The rationale here was that shared symptomology between these diseases could provide insight into the effect of aberrant splicing/expression of each MIG. We identified variants in 225 MIGs, of which 211 were associated with a total of 753 clinical manifestations recorded in the HPO database (Table S2). These ranged from rare multisystem diseases like Yunis–Varón syndrome (ORPHA: 3472) and PHACE syndrome (ORPHA: 42775), to more prevalent clinical manifestations like attention‐deficit hyperactivity disorder (HP: 0007018) or neoplasms (Table S2). Several of the identified clinical manifestations were associated with variants in not just one, but multiple MIGs. Therefore, we curated all known genes linked to each disorder and determined whether MIGs were significantly enriched in this list. We found that there was a significant enrichment of protein‐coding MIGs among all protein‐coding genes that have been correlated with the cardiac diseases Brugada syndrome (ORPHA: 130), Long QT syndrome (OPRHA: 101016), and Wolff‐Parkinson‐White Pattern (OMIM: 194200) (Table 1). Moreover, a significant enrichment of MIGs was observed among all known causes of Jeune syndrome (ORPHA: 474) (Table 1). Finally, several MIGs have been linked to neurological disorders: we identified significant enrichments of MIGs for epileptic encephalopathy (ORPHA: 1934), Joubert syndrome (ORPHA: 2754), Charcot‐Marie‐Tooth disease (ORPHA: 166), and congenital disorder of glycosylation (ORPHA: 137) (Table 1). These findings underscore the importance of proper MIG expression for the functioning of the nervous system.
Table 1.
Overview of conditions that are associated with genes that contain a significant enrichment of minor introns
| Condition | Primary affected organ system | # of associated MIGs | # of total associated genes | Fisher exact p‐value | Affected MIGs |
|---|---|---|---|---|---|
| Brugada syndrome | Cardiovascular system | 4 | 28 | p = .0073 | CACNA1C; CACNA2D1; SCN10A; SCN5A |
| Long QT syndrome | Cardiovascular system | 8 | 112 | p = .0134 | CACNA1C; CACNA2D1; DNA2; HNRNPM; INTS8; RALGAPA1; SCN5A; TCTN3 |
| Wolff‐Parkinson‐White pattern | Cardiovascular system | 4 | 46 | p = .0391 | CACNA1C; CACNA1E; MYH11; SCN5A |
| Jeune syndrome | Skeletal system | 4 | 25 | p = .0048 | C2CD3; IFT74; IFT80; IFT88 |
| Epileptic encephalopathy | Nervous system | 14 | 105 | p < .00001 | ACTL6B; ATP6V1A; CACNA1A; CACNA1E; CACNA2D2; CAD; MAPK10 ; PLCB1; SCN1A; SCN2A; SCN3A; SCN8A; SLC12A5; TRIT1 |
| Joubert syndrome | Nervous system | 9 | 47 | p < .00001 | ARMC9; C2CD3; CEP41; KIAA0556; PDE6D; TCTN1; TCTN3; TMEM107; TMEM231 |
| Charcot‐Marie‐Tooth disease | Nervous system | 6 | 87 | p = .035 | FIG4 ; GARS; HARS; SCN9A; SLC12A6; SPG11 |
| Congenital disorder of glycosylation | Nervous system & Digestive system | 8 | 49 | p = .00001 | ALG12; ALG3; ALG6; ALG8; COG6; HMBS; MAGT1; TUSC3 |
Next, we established the organ systems that were most commonly affected upon pathogenic variants in MIGs. Of all 211 MIGs with known symptoms, disruption of the function of 119 MIGs would result in neurological defects (Figure 2). This enrichment was driven by global developmental delay (62 MIGs), intellectual disability (49 MIGs), and seizures (40 MIGs) (Figure 2). Many of these symptoms were due to variants in any of the voltage‐gated ion channels or solute carrier transporters that contain a minor intron. The second most affected organ system was the head and neck (80 MIGs), where the symptom microcephaly was prevalent (39 MIGs) (Figure 2). Finally, variants in 79 MIGs have been linked to eye‐related symptoms such as nystagmus and strabismus (Figure 2). It is of note that symptoms like microcephaly, macrocephaly, spasticity, hypotonia, and strabismus, can also be viewed as a defect in the development of the nervous system, thereby increasing the number of phenotypes associated with that system, even though they are classified as a symptom of another organ system by HPO. In contrast to these commonly affected organ systems, the respiratory system (29 MIGs) and immune system (26 MIGs) were only affected in conditions associated with few MIGs.
Figure 2.
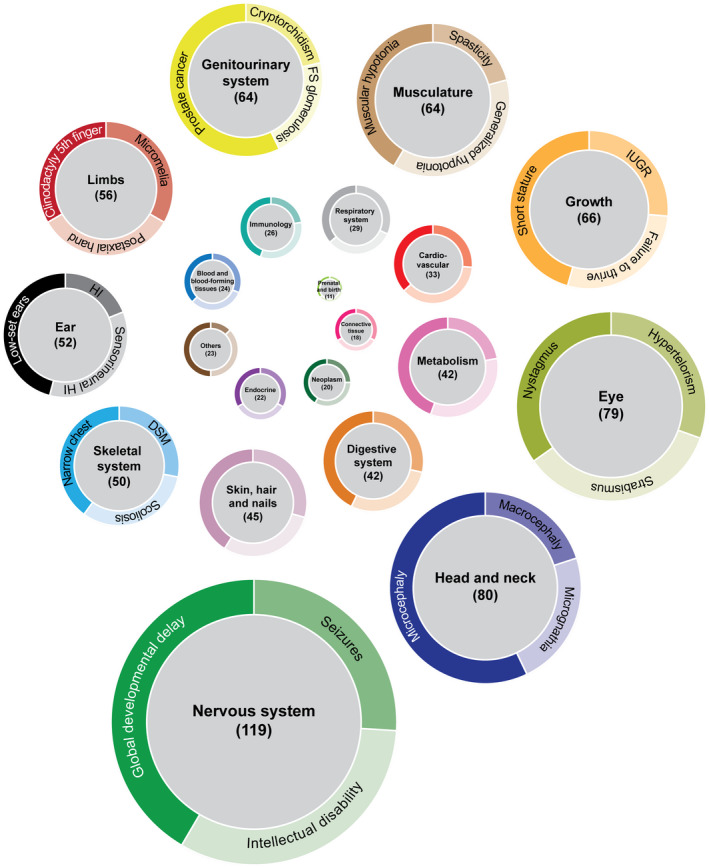
Pathogenic variants in MIGs predominately affect the nervous system. Doughnut charts of the affected organ systems due to variants in MIGs. Size of doughnut charts represents the number of MIGs that can result in a symptom related to the respective organ system when affected. If variants in MIGs were associated with several diseases that contained overlapping symptoms, then these symptoms were only counted once. Moreover, if different variants within the same gene were associated with the same symptoms, this symptom was also only included once. The three most prevalent phenotypes are listed in the ring based on their relative occurrence. Color‐coding of the organ systems is the same as in Figure 1. IUGR, intrauterine growth retardation; FS, focal segmental; HI, hearing impairment; DSM, delayed skeletal maturation
To test whether the low prevalence of immunological and respiratory symptoms was specific to MIGs, or whether these systems are generally less affected in case of pathogenic variants, we determined the percentage of all genotype–phenotype relationships listed in HPO that would involve each organ system. For example, of the 31,039 genotype–phenotype relationships listed in the HPO database, 2,903 involved genes that affected the nervous system when mutated, which is 9.4% of all relationships documented. Using a Fisher Exact test, we then compared this percentage to the percentage of MIGs that would result in a neurological phenotype when disrupted. This showed that a significantly higher proportion of MIG‐phenotype relationships affect the nervous system compared to all known genotype–phenotype relationships (Table 2). Besides the nervous system, we found no other enrichment of MIGs in genotype–phenotype relationships. However, we did observe a significantly lower number of MIG‐phenotype relationships that affected the digestive system, cardiovascular system and connective tissue (Table 2). Thus, this analysis underscores the importance of proper MIG functioning for nervous system development and function.
Table 2.
Distribution of affected organ systems in case of MIG‐ and total gene‐related pathogenic variants
| Organ system | Associated MIGs | Associated total genes | Fisher exact p‐value | ||
|---|---|---|---|---|---|
| Nervous system | 119 | 11.6% | 2,903 | 9.4% | p = .0168 |
| Head and neck | 80 | 7.8% | 2,274 | 7.3% | p = .543 |
| Eye | 79 | 7.7% | 2,238 | 7.2% | p = .5397 |
| Growth | 66 | 6.4% | 1964 | 6.3% | p = .8451 |
| Musculature | 64 | 6.2% | 2031 | 6.5% | p = .7482 |
| Genitourinary system | 64 | 6.2% | 1,600 | 5.2% | p = .1322 |
| Limbs | 56 | 5.5% | 1,361 | 4.4% | p = .1043 |
| Ear | 52 | 5.1% | 1,600 | 5.2% | p = 1.000 |
| Skeletal system | 50 | 4.9% | 1652 | 5.3% | p = .5715 |
| Skin, hair, and nails | 45 | 4.4% | 1,481 | 4.8% | p = .6543 |
| Digestive system | 42 | 4.1% | 1827 | 5.9% | p = .0146 |
| Metabolism | 42 | 4.1% | 1,422 | 4.6% | p = .5424 |
| Cardiovascular system | 33 | 3.2% | 1662 | 5.4% | p = .0017 |
| Respiratory system | 29 | 2.8% | 1,111 | 3.6% | p = .2296 |
| Immunology | 26 | 2.5% | 882 | 2.8% | p = .6324 |
| Blood and blood‐forming tissues | 24 | 2.3% | 863 | 2.8% | p = .497 |
| Others | 23 | 2.2% | 1,257 | 4.0% | p = .0026 |
| Endocrine | 22 | 2.1% | 874 | 2.8% | p = .2465 |
| Neoplasm | 20 | 2.0% | 613 | 2.0% | p = 1.000 |
| Connective tissue | 18 | 1.8% | 878 | 2.8% | p = .0423 |
| Prenatal and birth | 11 | 1.1% | 546 | 1.8% | p = .1133 |
3.3. Identification of candidate MIGs involved in the pathogenesis of minor spliceosome‐related diseases
Several of the commonly identified symptoms in MIG‐associated diseases were also observed in minor spliceosome‐related diseases, such as microcephaly and seizures (Figures 1 and 2). This suggests that it is indeed the aberrant expression of a subset of MIGs, that underlies the symptoms observed in the minor spliceosome‐associated diseases. To determine which MIGs are most likely playing a role in the pathogenesis of minor spliceosome‐related diseases, we focused on nine symptoms that were shared between at least three of the minor spliceosome‐related disorders (Figure 1). We found that variants in 51 MIGs resulted in at least two of these nine phenotypes (Figure 3). Functional annotation of these MIGs using DAVID revealed that they significantly enrich for only one GO‐term: cilium morphogenesis (p = 1.5E‐03), which was driven by CEP164 (OMIM: 614848) (Nephronophthisis), IFT122 (OMIM: 606045) (Cranioectodermal dysplasia), IFT80 (OMIM: 611177) (Short rib polydactyly syndrome), TCTN1 (OMIM: 609863) (Joubert syndrome and others), TCTN3 (OMIM: 613847) (Orofaciodigital syndrome), TMEM107 (OMIM: 616183) (Joubert syndrome and others), TMEM231 (OMIM: 614949) (Joubert syndrome and others). Cilia have been shown to play a role in cortical and cerebellar morphogenesis, and have been hypothesized to participate in the pathogenesis of Roifman syndrome (Gray, Sillence, & Kakakios, 2011; Guemez‐Gamboa, Coufal, & Gleeson, 2014). Thus, disrupted ciliogenesis may underlie some of the phenotypes observed in minor spliceosome‐related diseases. Variants in 23 of the 51 MIGs resulted in at least three of the phenotypes also commonly observed in minor spliceosome‐diseases, due to the association with a single disease (Figure 3).
Figure 3.
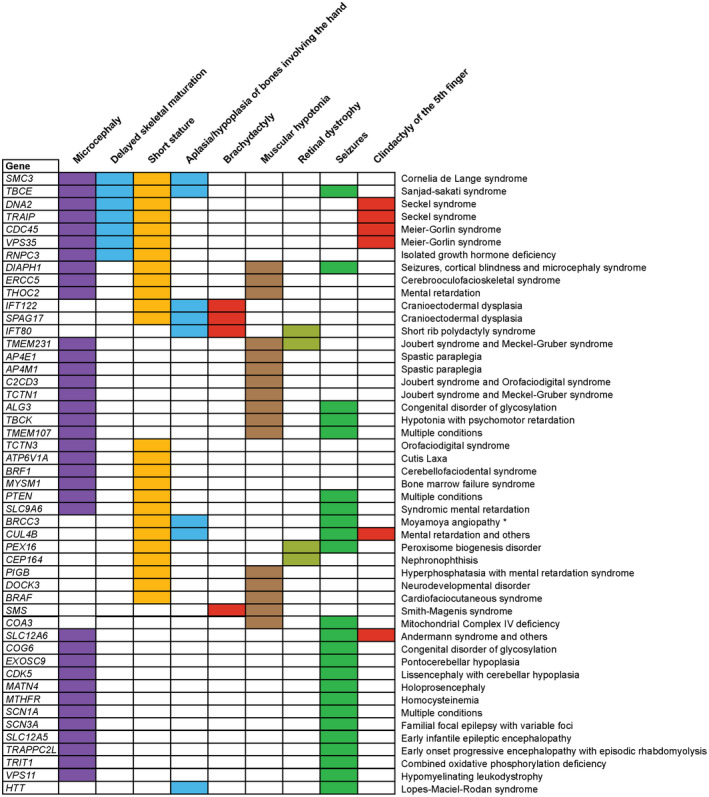
Pathogenic variants in MIGs result in similar phenotypes as minor spliceosome‐related diseases. Chart with MIGs that, when mutated, result in at least two of the commonly shared minor spliceosome‐related disease phenotypes. Diseases that are associated with the listed phenotypes are shown on the right. * = short stature‐facial dysmorphism‐hypergonadotropic hypogonadism syndrome
Finally, variants in eight MIGs resulted in four or five of the nine symptoms also observed in minor spliceosome‐related diseases (Figure 3). These MIGs include DNA2 (OMIM: 601810) (DNA replication helicase/nuclease 2) and TRAIP (OMIM: 605958) (TRAF interacting protein), which both have been linked to Seckel syndrome (Harley et al., 2016; Shaheen et al., 2014), SMC3 (OMIM: 606062) (Structural maintenance of chromosomes 3) which is linked to Cornelia de Lange syndrome and TBCE (OMIM: 604934) (Tubulin folding cofactor E), which has been linked to Sanjad‐sakati syndrome (Figure 3) (Deardorff et al., 2007; Parvari et al., 1998). While variants in most of these MIGs result in different diseases, they all share the common symptoms of microcephaly, short stature, and delayed skeletal maturation (Figure 3). Interestingly, Seckel syndrome, together with Meier‐Gorlin syndrome, MOPDI and MOPDII, make up a spectrum of microcephalic primordial dwarfism disorders (Tarnauskaite et al., 2019). As such, MIGs related to these diseases are prime candidates to be affected in minor spliceosome‐related disorders.
3.4. Pathogenic variants in minor intron splice sites reduce the splice site strength
Our analysis revealed a subset of MIGs whose disruption could contribute to the symptoms observed in the minor spliceosome‐related diseases. However, the MIG‐associated diseases we identified do not necessarily have to be due to disrupted minor intron splicing, but could also have been caused by variants in the coding regions of the MIG. Therefore, we next focused on the diseases that specifically resulted from variants in the minor introns of MIGs. ClinVar revealed 186 variants in 63 minor introns, found among 58 MIGs (Figure 4a; Table S3). Of these, 68 variants were found in one of the three consensus sequences of the minor intron. Specifically, we identified 33 variants in the 5′SS (+1 to +9 position), 19 variants in the BPS, and 16 variants in the 3′SS (−1 to −3 position) (Figure 4a). While the remaining variants were located outside of the splice sites, the majority was within close proximity (<20 nucleotides) of the 5′ and 3′ ends of the minor intron. For instance, we detected 12 variants at the −5 position of the 3′ end of the intron, which were identified in nine different MIGs (Figure 4b). We found that the most frequently mutated nucleotide within the 5′SS was the +1 position (Figure 4b). The nucleotide change at this position was relatively consistent, as all nine identified variants were either a G > A or A > G conversion (Figure 4b). Additionally, the variants at the +3 position of the 5′SS and the −2 position of the 3′SS were all A > G transitions (Figure 4b).
Figure 4.

Identification of 51 pathogenic variants in minor intron splice sites. (a) Bar graph with the number of variants observed in each of the splice site, as well as the number of minor introns and MIGs affected. (b) Frequency logo of 5′SS and 3′SS of all minor introns, generated using WebLogo. Below is a bar graph of the number of variants observed at each position and the type of variant. (c) Scatterplot with PWM score for splice sites with associated pathogenic variants. (d) Wordcloud with symptoms resulting from diseases associated with variants in splice sites of minor introns
Of the 68 minor intron splice site‐associated variants listed in ClinVar, 28 were considered pathogenic, 17 were considered benign, whereas the pathogenicity for the remaining 23 was uncertain. Since pathogenic variants tend to have a lower allelic frequency in the population due to natural selection, we cross‐referenced the 68 variants with all variants listed in gnomAD, a database describing allelic frequencies in control populations (Karczewski, 2019). Most of the variants classified as pathogenic by ClinVar were indeed not observed in gnomAD, whereas several benign variants were observed at a frequency > 0.1% (Table S4). Finally, for the 23 variants with uncertain pathogenicity, the allelic frequency was either lower than 0.05%, or they were not listed at all in gnomAD, suggesting that these may also be pathogenic (Table S4). Based on these results, we focused on the 51 variants that were likely pathogenic, of which 39 were associated with a clinical manifestation (Table S4). Since several variants were found within the same MIG (e.g., PTEN (OMIM: 601728), STK11 (OMIM: 602216), SCN1A (OMIM: 182389)), the overall number of affected MIGs was 25 (Table S4). These included several well‐characterized ion channels and transporters such as members from the CACNA, SCN, and SLC gene family (Table S4). Next, we assessed the effect of the 51 pathogenic variants on the splice site strength by scoring the sequence against a minor intron‐type PWM (resulting in a score ranging from 0 to 100) (Sheth et al., 2006). We found that 96% of pathogenic variants resulted in a reduction of the PWM score (Figure 4c; Table S4). On average, pathogenic variants in the 5′SS decreased the PWM score by 25.4 points, variants in the BPS lowered the PWM score by 17.5 points, and variants in the 3′SS reduced the PWM score by 29.5 points. Importantly, 41% of the likely benign variants in the consensus sequences actually enhanced the PWM score, suggesting an increased splice site strength and potentially enhanced splicing (Figure 4c). Thus, almost all variants classified as pathogenic or with uncertain significance by ClinVar reduced the splice site strength, which would be expected to affect the recognition of these minor introns and their splicing.
Finally, we wanted to determine the specific phenotypes that were observed in diseases associated with variants in these consensus sequences. We found that the most prominent symptoms included a global developmental delay (7 MIGs), intellectual disability (7 MIGs), seizures (6 MIGs), and microcephaly (3 MIGs) (Figure 4d). Thus, variants specifically identified in the splice sites of minor introns also heavily affect the nervous system.
4. DISCUSSION
The moniker, “minor spliceosome” refers to the relatively few (<0.5%) introns spliced by this machinery and not its biological significance. Most of the 699 MIGs in the human genome contain one or two minor introns, whereas the other introns are major introns (Olthof et al., 2019). Since minor introns cannot be spliced by the major spliceosome, minor intron splicing has a disproportionately large regulatory impact on the expression of MIGs. As such, pathogenic variants in minor spliceosome components that inhibit the function of the minor spliceosome, wield a large effect on the many biological processes MIGs execute. This is reflected by the multisystemic diseases such as MOPD1, Roifman syndrome, and Lowry‐Wood syndrome (Edery et al., 2011; Farach et al., 2018; He et al., 2011; Merico et al., 2015; Shelihan et al., 2018). Given that all of the pathogenic variants in these diseases ultimately inhibit the minor spliceosome function, it is not surprising that they result in overlapping symptoms such as short stature and microcephaly. However, these symptoms are found along a spectrum of severity in patients, such that the most severe microcephaly and growth retardation is associated with individuals with MOPD1 and the least severe with individuals with Lowry‐Wood syndrome (Shelihan et al., 2018). This gradation may be explained by the level of minor intron mis‐splicing that resulted from the individual variants in RNU4ATAC. It was reported that MOPD1‐associated variants resulted in a 90% loss of minor spliceosome activity as 100% loss would likely result in embryonic lethality (He et al., 2011). Moreover, individuals with Roifman syndrome and Lowry‐Wood syndrome experience fewer symptoms, as well as less severe growth retardation and microcephaly than individuals with MOPD1. Therefore, we hypothesize that minor spliceosome activity in Roifman syndrome and Lowry‐Wood syndrome might be affected to a lesser extent (Figure 1). Interestingly, individuals with Roifman syndrome are also characterized by several immunological phenotypes that are absent in individuals with MOPD1 and Lowry‐Wood syndrome, which suggests that different MIGs may be affected upon the different variants in RNU4ATAC (Figure 1). This is likely not the complete explanation, as the same g.51G > A pathogenic variant has been discovered in individuals with Lowry‐Wood syndrome, as well as individuals with Roifman syndrome and MOPD1 (Farach et al., 2018; He et al., 2011; Merico et al., 2015). These findings suggest that there are modifiers in the genome to these RNU4ATAC variants that can reduce the severity and/or penetrance of the phenotype in a tissue‐specific manner. Since most patients are compound heterozygotes, another explanation may be that the severity of disease is influenced by the pathogenic variant in the other RNU4ATAC allele. Indeed, variants uniquely found in individuals with MOPD1 were found to be more likely deleterious than those in individuals with Roifman syndrome and Lowry‐Wood syndrome (Shelihan et al., 2018).
Even though the majority of minor spliceosome‐related diseases result in pleiotropic phenotypes, most of the symptoms are indicative of defects in the nervous system or skeletal system (Figure 1). The source of these tissue‐enriched symptoms might be the tissue‐specific expression, splicing, and/or alternative splicing of MIGs that we reported previously (Olthof et al., 2019). The tissue‐specific regulation of MIGs is underscored by our findings that variants in MIGs commonly affect the nervous system (Figure 2). The most prevalent symptoms resulting from pathogenic variants in MIGs included a global developmental delay, intellectual disability and microcephaly (Figure 2). This is consistent with some of the cardinal sympotms observed in minor spliceosome‐related diseases. Together, the overlap of symptoms resulting from disrupted MIG function and minor spliceosome‐related diseases might shed light onto the molecular mechanism of pathogenesis underlying these syndromes. However, further investigation is required to understand how exactly the regulation of minor intron splicing contributes to the pathogenesis of minor spliceosome‐related diseases.
Since all minor spliceosome‐related diseases are inherited in an autosomal recessive manner, it is likely that the pathogenic variants result in a loss‐of‐function of the minor spliceosome. This was modeled in two different mouse models, through knockout of Rnpc3 and Rnu11 (Baumgartner et al., 2018; Doggett et al., 2018). Germline ablation of both Rnpc3 and Rnu11 resulted in embryonic lethality, again suggesting that the variants observed in minor spliceosome‐related diseases such as IGHD are hypomorphic (Argente et al., 2014; Baumgartner et al., 2018; Doggett et al., 2018). Ablation of Rnpc3 in adult mice resulted in severe gastrointestinal defects, as well as a reduction of peripheral lymphocytes and platelets (Doggett et al., 2018). While the latter phenotype resembles some of the immunological symptoms that individuals with Roifman syndrome experience, gastrointestinal defects are generally not found in individuals with IGHD (Argente et al., 2014). This may be due to a change in the expression pattern of Rnpc3 from embryonic development to adulthood. Therefore, it would be interesting to determine whether spatially restricted loss of Rnpc3 during embryonic development would recapitulate more minor spliceosome‐related phenotypes. Finally, loss of Rnu11 in the dorsal telencephalon resulted in severe microcephaly at birth, through disrupted splicing of a subset of MIGs (Baumgartner et al., 2018). As such, this mouse can be used to provide more insight into the molecular etiology of the microcephaly phenotype observed in individuals with minor spliceosome‐related diseases.
Of particular interest for the identification of targets affected in minor spliceosome‐related diseases are pathogenic variants in splice sites of minor introns, which are thought to compromise the splicing process. Variants in splice sites of major introns that reduce the strength of that splice site generally abolish splicing and are deemed pathogenic (Caminsky, Mucaki, & Rogan, 2014). These pathogenic variants are especially concentrated in the terminal dinucleotides, which are the only highly conserved nucleotides of these splice sites (Lord et al., 2019). In agreement with this, we found that variants classified as pathogenic generally result in a reduction in the minor intron splice site strength, whereas benign variants either do not affect or strengthen the splice site of minor introns (Figure 4c). However, since sequences at minor intron splice sites are much more conserved than those of major introns, we also identified many pathogenic variants outside the terminal dinucleotides of the minor intron. Thus, these data suggest a potential correlation between the effect of a variant on the splice site strength of minor introns and its pathogenicity.
The pathogenic variants in minor intron splice sites reveal the regulatory importance of minor intron splicing in proper MIG expression. Moreover, symptoms observed in diseases associated with splice site variants in minor introns can reveal the tissue types in which these MIGs perform nonredundant functions. These diseases are often being investigated by researchers who might not be aware of the fact that the primary defect underlying the disease is aberrant minor intron splicing. Moreover, variants in splice sites have been shown to result in alternative splicing of flanking exons and the use of cryptic splice sites, which means that variants in minor introns may also result in alternative splicing (Anna & Monika, 2018). Therefore, the aberrant expression of alternatively spliced isoforms of MIGs might contribute to the pathogenesis of these disease through a gain‐of‐function. To date, only the minor introns of STK11 and TRAPPC2 (OMIM: 300202) were known to contain pathogenic variants resulting in disease (Hastings et al., 2005; Shaw et al., 2003). Here, we report that at least another 28 minor introns are also associated with pathogenic variants in their splice sites (Figure S3). The number of aberrant minor intron splicing events that can result in disease might be much larger than this, as most variant detection is done through exome sequencing and advanced bioinformatics tools are required to detect variants in noncanonical splice sites (Lewandowska, 2013). Moreover, variants deeper in the intron, outside of the splice sites of minor introns, can also induce alternative splicing, thereby resulting in the production of aberrant isoforms (Vaz‐Drago, Custodio, & Carmo‐Fonseca, 2017). We hope that further studies on the intronic variants that we report here can reveal the role of aberrant minor intron splicing in disease pathogenesis. Together these findings expand the impact of minor intron splicing beyond pathogenic variants in the minor spliceosome components.
We found that the most common variant in the 5′SS of minor introns was located at the +1 position and involved a G > A or A > G change (Figure 4b). Minor introns can be classified based on their terminal dinucleotides as AT‐AC introns or GT‐AG introns and either contain an A or G at the +1 position, whereas the rest of their splice site sequence is the same. The presence of these two nucleotides normally does not hinder the recognition and splicing capacity of the minor spliceosome. Instead, the pathogenicity of the G > A and A > G conversion may come from the fact that the −1 position at the 3′SS is not mutated alongside these variants, as this combination of terminal dinucleotides has been shown to reduce canonical splicing using an in vitro splicing assay (Dietrich, Incorvaia, & Padgett, 1997).
Several pathogenic variants in splice sites of minor introns have been shown to result in alternative splicing. In the case of STK11, the variants in the 5′SS resulted in the occasional use of a cryptic 5′SS in conjunction with several cryptic 3′SS (Hastings et al., 2005). Moreover, pathogenic variants in the 5′SS of TRAPPC2 were shown to result in the utilization of a cryptic 5′SS and skipping of the downstream exon (Shaw et al., 2003). Several of the 5′SS variants listed in ClinVar also had associated manuscripts that described the effect on splicing. For instance, a + 1G > T variant in the 5′SS of the minor intron in AP4M1 (OMIM: 602296) has been shown to result in skipping of the upstream exon and the minor intron (Verkerk et al., 2009). Moreover, a + 6delC variant in the 5′SS of the minor intron in DNA2 resulted in the use of a cryptic 5′SS in combination with several cryptic 3′SS (Shaheen et al., 2014). Finally, a + 1G > A variant in the 5′SS of the minor intron in TMEM107 was shown to result in the inclusion of a single base‐pair, thereby resulting in a frameshift (Shaheen et al., 2015). These results highlight the potential of minor intron splice site variants to affect alternative splicing and regulate gene expression and/or protein production. While most of these alternative splicing events are predicted to be executed by the major spliceosome, the pathogenic variants in STK11 demonstrate the ability of the minor spliceosome to utilize cryptic splice sites. Since all evidence has currently resulted from variants in the 5′SS, it would be important to determine the effect of variants in the BPS and 3′SS on alternative splicing around minor introns. This would require experimental validation using patient samples and/or in vitro splicing constructs mimicking the human pathogenic variants. These results will be relevant for the identification of therapeutic strategies. Recently, antisense oligonucleotides have been a commonly used method to target‐specific splicing events and have been shown effective at treating splicing diseases (Daguenet, Dujardin, & Valcarcel, 2015; Finkel et al., 2016; Rigo, Hua, Krainer, & Bennett, 2012). Moreover, expression of U1 snRNA with compensatory mutations has been shown to rescue splicing defects in mutant mice (Balestra et al., 2014; Lee, Lee, Chen, Byrne, & Hwu, 2016). The rise in splicing‐targeted therapeutics therefore increases the importance of being cognizant of the splicing pathway that is affected by the specific splice site variants.
In all, our study was designed to interrogate the true impact of minor intron splicing defects on human diseases from both perspectives, that is, the minor spliceosome dysfunction and its targets, the MIGs. Here we report that the combined effect of minor spliceosome‐related diseases as well as MIG‐related diseases is significantly larger than has been appreciated. The field of minor spliceosome research has remained small because most diseases caused by loss of minor spliceosome function are rare. However, if one considers the MIGs and the associated diseases, the field of minor intron splicing expands. This study was aimed to create awareness among other scientists, that inherent to studying MIG‐related diseases is the understanding of a crucial form of gene regulation, that is, minor intron splicing.
CONFLICT OF INTEREST
The authors report no conflict of interest.
AUTHOR CONTRIBUTIONS
AMO and JSR curated the variant data, and associated diseases and symptoms. AMO analyzed the results. AMO, PMC, and RNK designed the study and edited the paper. AMO and RNK wrote the first draft of the paper.
Supporting information
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGMENTS
We thank members of the Kanadia laboratory for helpful discussions on this manuscript. This work was supported by grants R01NS102538 and R21NS101616 from the National Institute of Health, awarded to RNK.
Olthof AM, Rasmussen JS, Campeau PM, Kanadia RN. Disrupted minor intron splicing is prevalent in Mendelian disorders. Mol Genet Genomic Med. 2020;8:e1374 10.1002/mgg3.1374
Funding information
This study was supported by National Institute of Health (grant number R01NS102538 and R21NS101616), awarded to RNK.
DATA AVAILABILITY STATEMENT
The variant data reported in this manuscript are publicly available at NCBI Clinvar. The diseases associated with each MIG have been reported on the Minor Intron DataBase (MIDB), which can be accessed at midb.pnb.uconn.edu.
REFERENCES
- Alioto, T. S. (2007). U12DB: A database of orthologous U12‐type spliceosomal introns. Nucleic Acids Research, 35, D110–115. 10.1093/nar/gkl796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anna, A. , & Monika, G. (2018). Splicing mutations in human genetic disorders: Examples, detection, and confirmation. Journal of Applied Genetics, 59, 253–268. 10.1007/s13353-018-0444-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argente, J. , Flores, R. , Gutierrez‐Arumi, A. , Verma, B. , Martos‐Moreno, G. A. , Cusco, I. , … Perez‐Jurado, L. A. (2014). Defective minor spliceosome mRNA processing results in isolated familial growth hormone deficiency. EMBO Molecular Medicine, 6, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestra, D. , Faella, A. , Margaritis, P. , Cavallari, N. , Pagani, F. , Bernardi, F. , … Pinotti, M. (2014). An engineered U1 small nuclear RNA rescues splicing defective coagulation F7 gene expression in mice. Journal of Thrombosis and Haemostasis : JTH, 12, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner, M. , Olthof, A. M. , Aquino, G. S. , Hyatt, K. C. , Lemoine, C. , Drake, K. , … Kanadia, R. N. (2018). Minor spliceosome inactivation causes microcephaly, owing to cell cycle defects and death of self‐amplifying radial glial cells. Development, 145 dev166322 10.1242/dev.166322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caminsky, N. G. , Mucaki, E. J. , & Rogan, P. K. (2014). Interpretation of mRNA splicing mutations in genetic disease: review of the literature and guidelines for information‐theoretical analysis. F1000Research, 3, 282 10.12688/f1000research.5654.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cologne, A. , Benoit‐Pilven, C. , Besson, A. , Putoux, A. , Campan‐Fournier, A. , Bober, M. B. , … Lacroix, V. (2019). New insights into minor splicing‐a transcriptomic analysis of cells derived from TALS patients. RNA, 25, 1130–1149. 10.1261/rna.071423.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daguenet, E. , Dujardin, G. , & Valcarcel, J. (2015). The pathogenicity of splicing defects: Mechanistic insights into pre‐mRNA processing inform novel therapeutic approaches. EMBO Reports, 16, 1640–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff, M. A. , Kaur, M. , Yaeger, D. , Rampuria, A. , Korolev, S. , Pie, J. , … Krantz, I. D. (2007). Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. American Journal of Human Genetics, 80, 485–494. 10.1086/511888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, R. C. , Incorvaia, R. , & Padgett, R. A. (1997). Terminal intron dinucleotide sequences do not distinguish between U2‐ and U12‐dependent introns. Molecular Cell, 1, 151–160. 10.1016/S1097-2765(00)80016-7 [DOI] [PubMed] [Google Scholar]
- Doggett, K. , Williams, B. B. , Markmiller, S. , Geng, F. S. , Coates, J. , Mieruszynski, S. , … Heath, J. K. (2018). Early developmental arrest and impaired gastrointestinal homeostasis in U12‐dependent splicing‐defective Rnpc3‐deficient mice. RNA, 24, 1856–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edery, P. , Marcaillou, C. , Sahbatou, M. , Labalme, A. , Chastang, J. , Touraine, R. , … Leutenegger, A.‐L. (2011). Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science, 332, 240–243. 10.1126/science.1202205 [DOI] [PubMed] [Google Scholar]
- Elsaid, M. F. , Chalhoub, N. , Ben‐Omran, T. , Kumar, P. , Kamel, H. , Ibrahim, K. , … Aleem, A. A. (2017). Mutation in noncoding RNA RNU12 causes early onset cerebellar ataxia. Annals of Neurology, 81, 68–78. 10.1002/ana.24826 [DOI] [PubMed] [Google Scholar]
- Farach, L. S. , Little, M. E. , Duker, A. L. , Logan, C. V. , Jackson, A. , Hecht, J. T. , & Bober, M. (2018). The expanding phenotype of RNU4ATAC pathogenic variants to Lowry Wood syndrome. American Journal of Medical Genetics Part A, 176, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel, R. S. , Chiriboga, C. A. , Vajsar, J. , Day, J. W. , Montes, J. , De Vivo, D. C. , … Bishop, K. M. (2016). Treatment of infantile‐onset spinal muscular atrophy with nusinersen: A phase 2, open‐label, dose‐escalation study. Lancet, 388, 3017–3026. 10.1016/S0140-6736(16)31408-8 [DOI] [PubMed] [Google Scholar]
- Gray, P. E. , Sillence, D. , & Kakakios, A. (2011). Is Roifman syndrome an X‐linked ciliopathy with humoral immunodeficiency? Evidence from 2 new cases. International Journal of Immunogenetics, 38, 501–505. 10.1111/j.1744-313X.2011.01041.x [DOI] [PubMed] [Google Scholar]
- Guemez‐Gamboa, A. , Coufal, N. G. , & Gleeson, J. G. (2014). Primary cilia in the developing and mature brain. Neuron, 82, 511–521. 10.1016/j.neuron.2014.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley, M. E. , Murina, O. , Leitch, A. , Higgs, M. R. , Bicknell, L. S. , Yigit, G. , … Jackson, A. P. (2016). TRAIP promotes DNA damage response during genome replication and is mutated in primordial dwarfism. Nature Genetics, 48, 36–43. 10.1038/ng.3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings, M. L. , Resta, N. , Traum, D. , Stella, A. , Guanti, G. , & Krainer, A. R. (2005). An LKB1 AT‐AC intron mutation causes Peutz‐Jeghers syndrome via splicing at noncanonical cryptic splice sites. Nature Structural & Molecular Biology, 12, 54–59. 10.1038/nsmb873 [DOI] [PubMed] [Google Scholar]
- He, H. , Liyanarachchi, S. , Akagi, K. , Nagy, R. , Li, J. , Dietrich, R. C. , … de la Chapelle, A. (2011). Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science, 332, 238–240. 10.1126/science.1200587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , … Gauthier, L. D. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. BioRxiv, 531210. [Google Scholar]
- Köhler, S. , Carmody, L. , Vasilevsky, N. , Jacobsen, J. O. B. , Danis, D. , Gourdine, J.‐P. , … Robinson, P. N. (2019). Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Research, 47, D1018–D1027. 10.1093/nar/gky1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Riley, G. R. , Jang, W. , Rubinstein, W. S. , Church, D. M. , & Maglott, D. R. (2014). ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research, 42, D980–985. 10.1093/nar/gkt1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, N. C. , Lee, Y. M. , Chen, P. W. , Byrne, B. J. , & Hwu, W. L. (2016). Mutation‐adapted U1 snRNA corrects a splicing error of the dopa decarboxylase gene. Human Molecular Genetics, 25, 5142–5147. 10.1093/hmg/ddw323 [DOI] [PubMed] [Google Scholar]
- Lewandowska, M. A. (2013). The missing puzzle piece: Splicing mutations. International Journal of Clinical and Experimental Pathology, 6, 2675–2682. [PMC free article] [PubMed] [Google Scholar]
- Lord, J. , Gallone, G. , Short, P. J. , McRae, J. F. , Ironfield, H. , Wynn, E. H. , … Hurles, M. E. (2019). Pathogenicity and selective constraint on variation near splice sites. Genome Research, 29, 159–170. 10.1101/gr.238444.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan, V. , Kanojia, D. , Li, J. , Okamoto, R. , Sato‐Otsubo, A. , Kohlmann, A. , … Koeffler, H. P. (2015). Aberrant splicing of U12‐type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nature Communications, 6, 6042 10.1038/ncomms7042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico, D. , Roifman, M. , Braunschweig, U. , Yuen, R. K. C. , Alexandrova, R. , Bates, A. , … Scherer, S. W. (2015). Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nature Communications, 6, 8718 10.1038/ncomms9718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olthof, A. M. , Hyatt, K. C. , & Kanadia, R. N. (2019). Minor intron splicing revisited: Identification of new minor intron‐containing genes and tissue‐dependent retention and alternative splicing of minor introns. BMC Genomics, 20, 686 10.1186/s12864-019-6046-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvari, R. , Hershkovitz, E. , Kanis, A. , Gorodischer, R. , Shalitin, S. , Sheffield, V. C. , & Carmi, R. (1998). Homozygosity and linkage‐disequilibrium mapping of the syndrome of congenital hypoparathyroidism, growth and mental retardation, and dysmorphism to a 1‐cM interval on chromosome 1q42‐43. American Journal of Human Genetics, 63, 163–169. 10.1086/301915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo, F. , Hua, Y. , Krainer, A. R. , & Bennett, C. F. (2012). Antisense‐based therapy for the treatment of spinal muscular atrophy. The Journal of Cell Biology, 199, 21–25. 10.1083/jcb.201207087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen, R. , Almoisheer, A. , Faqeih, E. , Babay, Z. , Monies, D. , Tassan, N. , … Khalil, M. M. et al (2015). Identification of a novel MKS locus defined by TMEM107 mutation. Human Molecular Genetics, 24, 5211–5218. [DOI] [PubMed] [Google Scholar]
- Shaheen, R. , Faqeih, E. , Ansari, S. , Abdel‐Salam, G. , Al‐Hassnan, Z. N. , Al‐Shidi, T. , … Alkuraya, F. S. (2014). Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Research, 24, 291–299. 10.1101/gr.160572.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, M. A. , Brunetti‐Pierri, N. , Kadasi, L. , Kovacova, V. , Van Maldergem, L. , De Brasi, D. , … Gecz, J. (2003). Identification of three novel SEDL mutations, including mutation in the rare, non‐canonical splice site of exon 4. Clinical Genetics, 64, 235–242. 10.1034/j.1399-0004.2003.00132.x [DOI] [PubMed] [Google Scholar]
- Shelihan, I. , Ehresmann, S. , Magnani, C. , Forzano, F. , Baldo, C. , Brunetti‐Pierri, N. , & Campeau, P. M. (2018). Lowry‐Wood syndrome: Further evidence of association with RNU4ATAC, and correlation between genotype and phenotype. Human Genetics, 137, 905–909. 10.1007/s00439-018-1950-8 [DOI] [PubMed] [Google Scholar]
- Sheth, N. , Roca, X. , Hastings, M. L. , Roeder, T. , Krainer, A. R. , & Sachidanandam, R. (2006). Comprehensive splice‐site analysis using comparative genomics. Nucleic Acids Research, 34, 3955–3967. 10.1093/nar/gkl556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, H. , Kumar, S. A. , Shuai, S. , Diaz‐Navarro, A. , Gutierrez‐Fernandez, A. , De Antonellis, P. , … Vladoiu, M. C. (2019). Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature, 574, 707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn, W. Y. , & Steitz, J. A. (1996a). Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT‐AC introns. Science, 273, 1824–1832. 10.1126/science.273.5283.1824 [DOI] [PubMed] [Google Scholar]
- Tarn, W. Y. , & Steitz, J. A. (1996b). A novel spliceosome containing U11, U12, and U5 snRNPs excises a minor class (AT‐AC) intron in vitro. Cell, 84, 801–811. 10.1016/S0092-8674(00)81057-0 [DOI] [PubMed] [Google Scholar]
- Tarnauskaite, Z. , Bicknell, L. S. , Marsh, J. A. , Murray, J. E. , Parry, D. A. , Logan, C. V. , … Wise, C. et al (2019). Biallelic variants in DNA2 cause microcephalic primordial dwarfism. Human Mutation, 40, 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz‐Drago, R. , Custodio, N. , & Carmo‐Fonseca, M. (2017). Deep intronic mutations and human disease. Human Genetics, 136, 1093–1111. 10.1007/s00439-017-1809-4 [DOI] [PubMed] [Google Scholar]
- Verkerk, A. J. M. H. , Schot, R. , Dumee, B. , Schellekens, K. , Swagemakers, S. , Bertoli‐Avella, A. M. , … Mancini, G. M. S. (2009). Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. American Journal of Human Genetics, 85, 40–52. 10.1016/j.ajhg.2009.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will, C. L. , Schneider, C. , Hossbach, M. , Urlaub, H. , Rauhut, R. , Elbashir, S. , … Luhrmann, R. (2004). The human 18S U11/U12 snRNP contains a set of novel proteins not found in the U2‐dependent spliceosome. RNA, 10, 929–941. 10.1261/rna.7320604 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4
Data Availability Statement
The variant data reported in this manuscript are publicly available at NCBI Clinvar. The diseases associated with each MIG have been reported on the Minor Intron DataBase (MIDB), which can be accessed at midb.pnb.uconn.edu.