

Abstract
SIRT1 is a NAD+‐dependent deacetylase that controls key metabolic and signaling pathways, including inactivating the p53 tumor suppressor. However, the mechanisms controlling SIRT1 enzymatic activity in the context of cancer are unclear. Here, we show that the previously undescribed CSAG2 protein is a direct activator of SIRT1. CSAG2 is normally restricted to expression in the male germline but is frequently re‐activated in cancers. CSAG2 is necessary for cancer cell proliferation and promotes tumorigenesis in vivo. Biochemical studies revealed that CSAG2 directly binds to and stimulates SIRT1 activity toward multiple substrates. Importantly, CSAG2 enhances SIRT1‐mediated deacetylation of p53, inhibits p53 transcriptional activity, and improves cell survival in response to genotoxic stress. Mechanistically, CSAG2 binds SIRT1 catalytic domain and promotes activity independent of altering substrate affinity. Together, our results identify a previously undescribed mechanism for SIRT1 activation in cancer cells and highlight unanticipated approaches to therapeutically modulate SIRT1.
Keywords: apoptosis, cancer–testis antigen, genotoxic stress, NAD+‐dependent deacetylase, oncogene
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
CSAG2 is a cancer‐testis antigen that is transcriptionally activated in cancer cells and promotes tumorigenesis. CSAG2 binds to SIRT1 and enhances its catalytic activity to reprogram cellular key pathways, including the downregulation of p53 activity.

Introduction
SIRT1 is a NAD+‐dependent deacetylase that belongs to the class III histone/protein deacetylases (Vaziri et al, 2001; Lin & Fang, 2013). SIRT1 deacetylates diverse substrates, including p53, PGC‐1α, forkhead transcription factor (FOXO), and histones (Motta et al, 2004; Vaquero et al, 2004; Nemoto et al, 2005; Lee & Gu, 2013). SIRT1 influences gene silencing, apoptosis, stress resistance, senescence, and fat and glucose metabolism (Chalkiadaki & Guarente, 2015). The combination of these cellular functions has been suggested to contribute to anti‐aging in mammals. On the other hand, the pro‐survival functions of SIRT1 can be usurped in cancer to drive tumorigenesis (Kwon & Ott, 2008). SIRT1 knockout mice have increased p53 acetylation and radiation‐induced apoptosis (Cheng et al, 2003). Furthermore, inhibition of SIRT1 by Sirtinol induces cell growth arrest with decreased MAPK signaling (Ota et al, 2006). Additionally, DBC1 (deleted in breast cancer 1) acts as an inhibitor of SIRT1 in human cells. Repression of SIRT1 by DBC1 increases p53 acetylation levels and upregulates p53 function (Kim et al, 2008; Zhao et al, 2008). Importantly, SIRT1 participates in the silencing of tumor suppressor genes and SIRT1 overexpression has been observed in tumors (Saunders & Verdin, 2007). This includes several cancer types, such as prostate, melanoma, colon, and leukemia (Bradbury et al, 2005; Hida et al, 2007; Huffman et al, 2007; Stunkel et al, 2007). Therefore, elucidating the mechanisms regulating SIRT1 could have important implications.
p53 is a master regulator of tumor suppression and plays a critical role in cellular response to genotoxic stress through transcriptional activation of key cell cycle and apoptosis genes, including p53, Puma, and Bax (Beckerman & Prives, 2010). As such, p53 is subjected to a number of regulatory mechanisms, including acetylation and deacetylation. After DNA damage, the histone acetyltransferases CBP and p300 promote p53 acetylation. This includes acetylation of K382 that enhances p53 DNA binding and transcriptional activation of target genes (Reed & Quelle, 2014). Conversely, p53 is repressed through the action of SIRT1 that deacetylates p53 K382, thereby negatively regulating p53‐mediated transcriptional activation. Deacetylation of p53 by SIRT1 prevents cellular senescence and apoptosis and can be a mechanism employed by cancer cells to sustain cell growth (Yi & Luo, 2010).
Despite the extensive studies of SIRT1 cellular function, the mechanisms regulating SIRT1 in the context of disease are less understood. Cellular NAD+ concentration is a major determinant of SIRT1 activity, and AMP‐activated protein kinase (AMPK) signaling plays an important role in driving SIRT1 activity through upregulation of NAD+ (Canto et al, 2009). In addition to metabolic control of SIRT1, it is also regulated by post‐translational modifications, including the β‐adrenergic‐cAMP‐PKA pathway that controls SIRT1 activity in an NAD+‐independent and highly dynamic fashion (Gerhart‐Hines et al, 2011). In addition, SIRT1 activity is highly regulated by intramolecular interaction between its three regions: N‐terminal domain (NTD) that plays an important role in substrate binding and enzyme activity, catalytic domain (CD) required for catalysis, and C‐terminal regulatory (CTR) segment that contains a key ESA motif required for enzymatic activity (Pan et al, 2012). Intramolecular interactions between the NTD and CD, as well as the ESA and CD, are important for optimal SIRT1 activity. SIRT1 can also be regulated through protein–protein interactions occurring at the N‐ and C‐terminal regulatory regions or directly at the catalytic domain (Davenport et al, 2014; Dai et al, 2015; Ghisays et al, 2015). DBC1 (deleted in breast cancer‐1) protein has been implicated as a suppressor of SIRT1 activity through binding with SIRT1 catalytic domain (Kim et al, 2008). AROS (active regulator of SIRT1) has been reported to bind to SIRT1 and increases its activity, but its mechanism of action is unknown (Kim et al, 2007). Deletion analysis showed that amino acids 114–217 of SIRT1 are sufficient for the interaction between SIRT1 and AROS. Interestingly, this N‐terminal portion of SIRT1 plays an important role in promoting SIRT1 activity (Dai et al, 2015; Ghisays et al, 2015). However, subsequent studies failed to observe activation of SIRT1 by AROS in vitro or to regulate acetylation of SIRT1 targets, including p53 ac‐K382, in cells (Knight et al, 2013; Lakshminarasimhan et al, 2013; Kokkola et al, 2014). Thus, identification of new regulators of SIRT1 will provide better understanding of the molecular pathways that regulate cancer and other conditions, such as aging and diabetes, which may provide previously unrecognized therapeutic approaches.
Chondrosarcoma‐associated genes (CSAGs) are primate‐specific genes that encode for small proteins (78–127 amino acids). CSAGs are categorized as cancer–testis antigens (CTAs) because they are physiologically restricted to the testis but are aberrantly expressed in cancers (Duan et al, 1999; Materna et al, 2007; Karam et al, 2011). Accumulating evidence suggests that many CTAs make significant contributions to tumorigenesis (Pineda et al, 2015; Salmaninejad et al, 2016). However, CSAGs have no recognizable domains and the molecular mechanisms and oncogenic potential of CSAGs are unknown.
Here, we identify SIRT1 as a prominent binding partner of CSAG2 by unbiased, tandem affinity purification and mass spectrometry. CSAG2 directly interacts with SIRT1 catalytic domain and promotes SIRT1 activity in cells and in vitro without altering SIRT1 substrate binding affinity. Importantly, through enhancing SIRT1 activity, CSAG2 inhibits p53‐mediated transcriptional activity and promotes resistance of cells to genotoxic stress. Our findings suggest that CSAG2 is a novel cancer‐specific regulator of SIRT1 that provides insights into how SIRT1 is modulated in cancer and provides clues to chemoresistance mechanisms.
Results
CSAG genes are aberrantly expressed in cancer and correlate with poor patient prognosis
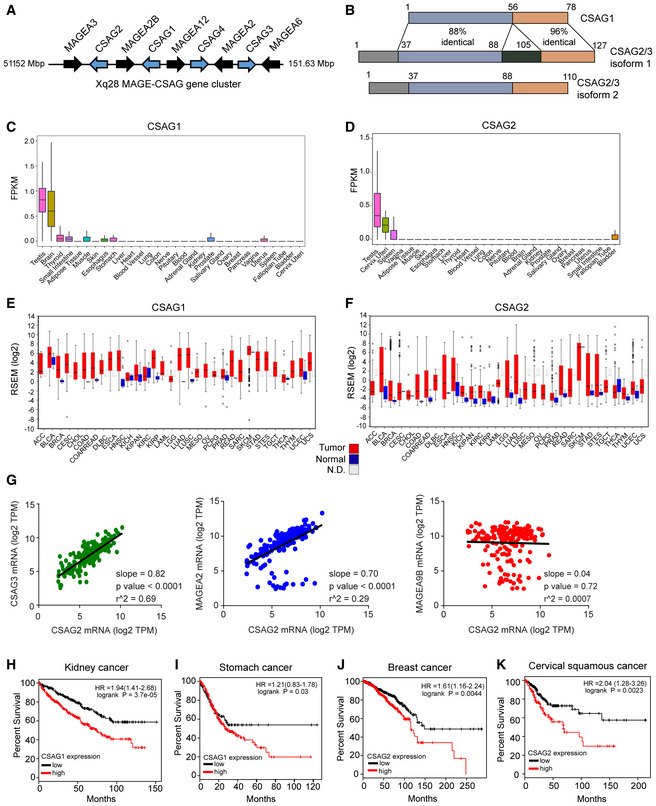
The CSAG family has four genes (CSAG1‐4), with CSAG4 being a pseudogene. Intriguingly, CSAG genes are interspersed in a periodic fashion with the previously described MAGE‐A CTAs (MAGE‐A2, ‐A3, ‐A6, and ‐A12) on chromosomal region Xq28 (Fig 1A; Bredenbeck et al, 2008). CSAG2/3 are virtually identical in sequences (126 of 127 identical residues) and are thus herein simply denoted as CSAG2. In comparison with CSAG2, CSAG1 lacks N‐terminal 37 amino acids, but otherwise is 73% identical to CSAG2 C‐terminus (Fig 1B). There are no previously identified domains or molecular functions for CSAGs. CSAGs show restricted expression in normal tissues, with CSAG2 being more restricted than CSAG1 and showing primarily expression in testis, cervix, and spleen (Fig 1C and D). Like MAGE‐As, CSAGs are frequently re‐activated in tumors with a number of cancer types showing higher expression of CSAGs in tumor tissue compared to normal tissue (Fig 1E and F). Given the unique chromosomal architecture of the MAGE‐CSAG Xq28 locus, we investigated whether these genes are co‐expressed in human lung squamous carcinomas. Indeed, gene expression analysis of the cancer genome atlas (TCGA) revealed strong correlated expression levels of the Xq28 MAGE‐CSAG genes, including CSAG2 with CSAG3 and MAGE‐A2, but not with non‐Xq28 MAGE‐A9B (Fig 1G). Furthermore, expression of CSAGs correlated with poor patient overall survival in several cancer types (Figs 1H–K and EV1A). This includes a median survival of breast cancer patients with CSAG2 low tumors of 12.4 years compared to 9.6 years for patients with CSAG2 high tumors (logrank P = 8.2e‐05; hazard ratio = 1.91). Together, these results suggest CSAGs are physiologically restricted to expression in select tissues, including reproductive tissues, but are aberrantly expressed in many cancer types where their expression correlates with poor patient prognosis.
Figure 1. CSAGs are aberrantly expressed in cancer and correlate with poor patient prognosis.
-
ACSAG genes cluster with MAGE‐As on chromosome Xq28.
-
BSequence identity between CSAG genes. Note isoforms of CSAG2/3 are splice variants.
-
C, DRNA‐Seq data from GTEx of the normalized expression of human CSAG1 (C) and CSAG2 (D) in the indicated tissues. Number of biological replicates are as follows: testis 361, brain 255, small intestine 187, white adipose 663, muscle 803, skin 604, esophagus 555, stomach 359, liver 226, heart 429. blood vessel 663, lung 578, colon 373, nerve 619, pituitary 283, blood 755, adrenal gland 258, kidney 85, prostate 245, salivary gland 162, ovary 180, breast 459, pancreas 328, vagina 156, uterus 142, spleen 241, fallopian tube 9, bladder 21, cervix 10. Central band indicates median, boxes define 25 and 75 percentiles, and whiskers define 5 and 95 percentiles.
-
E, FExpression of CSAG1 (E) and CSAG2 (F) in normal adjacent (blue) or tumor tissue (red) from TCGA RNA‐Seq data. N.D. represents normal adjacent is not available. Data visualized by firebrowse. Number of biological replicates are as follows: ACC 41, BLCA 307, BRCA 524, CESC 175, CHOL 11, COAD 153, COADREAD 232, DLBC 36, ESCA 121, HNSC 416, KICH 46, KIPAN 515, KIRC 411, KIRP 58, LAML 7, LGG 528, LUAD 301, LUSC 400, MESO 31, OV 183, PCPG 70, PRAD 204, READ 79, SARC 156, SKCM 422, STAD 288, STES 409, TGCT 126, THCA 408, THYM 75, UCEC 306, and UCS 37. Central band indicates median, boxes define 25 and 75 percentiles, and whiskers define 5 and 95 percentiles.
-
GXq28 MAGE and CSAG genes are frequently co‐expressed in lung squamous carcinoma tumors (n = 184). RNA‐Seq gene expression data analyzed from TCGA. Statistics shown were calculated by linear regression analysis.
-
H–KExpression of CSAG1 (H‐I) or CSAG2 (J‐K) in patients from indicated cancer types correlates with poor overall survival. Data are from TCGA and visualized by kmplotter.
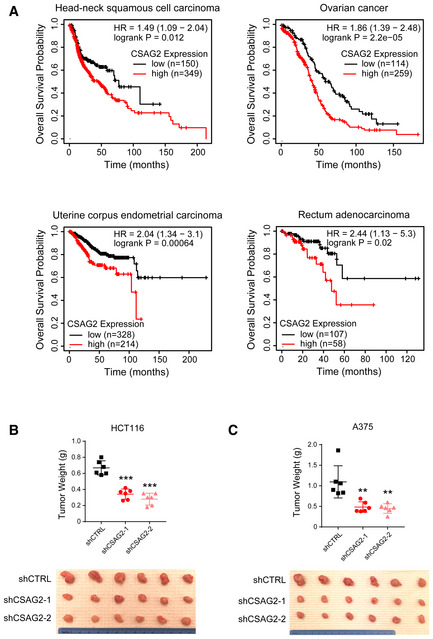
Figure EV1. Knockdown of CSAG2 impairs xenograft tumor growth.
-
AExpression of CSAG2 in patients from indicated cancer types correlates with poor overall survival. Data are from TCGA and visualized by kmplotter.
-
B, CKnockdown of CSAG2 in HCT116 (B) or A375 (C) cells decreases xenograft tumor growth in mice. Tumor weights (n = 6 for each group) are shown. **P < 0.01; ***P < 0.0001; unpaired, two‐tailed Student's t‐test.
CSAG2 is necessary and sufficient to drive cell and tumor growth
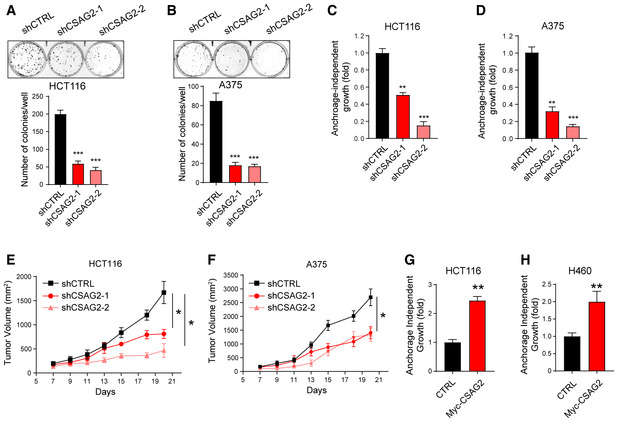
CSAG1 has previously been suggested to play a role in mitotic progression (Sapkota et al, 2020). Therefore, we focused our study on CSAG2 that has not been previously characterized. To determine whether the aberrant expression of CSAG2 in tumor cells is simply a passenger event due to global genomic dysregulation or whether CSAG2 has a more active role in driving tumorigenesis, we performed a series of gain and loss of function studies to elucidate the role of CSAG2 in cancer cell growth. First, we examined whether multiple cancer cells require the expression of CSAG2 for growth. Stable knockdown of CSAG2 with two independent shRNAs in HCT116 colorectal carcinoma cells or A375 malignant melanoma cells (both p53 wild type) resulted in decreased cell clonogenic growth on plastic (Fig 2A and B). Furthermore, knockdown of CSAG2 significantly decreased anchorage‐independent growth in soft agar (Fig 2C and D). To test the importance of CSAG2 in vivo, we performed inducible CSAG2 knockdown in mouse xenograft tumor formation assays. In both HCT116 and A375 cell models, depletion of CSAG2 with independent shRNAs significantly blunted xenograft tumor growth (Figs 2E and F, and EV1B and C). To determine whether overexpression of CSAG2 is sufficient to drive tumorigenic phenotypes, we stably expressed CSAG2 in H460 lung cancer cells that do not naturally express CSAG2 and overexpressed CSAG2 in HCT116 cells. Strikingly, expression of CSAG2 accelerated anchorage‐independent growth in both cellular models (Fig 2G and H). Together, these results suggest that CSAG2 functions as an oncogene to drive cancer cell growth and tumor formation. Additionally, cancer cells become addicted to CSAG2 upon its expression, suggesting that CSAG2 is a unique cancer cell‐specific vulnerability gene.
Figure 2. CSAG2 is necessary and sufficient to promote tumor cell growth in vitro and in vivo .
-
A, BThe effect of stable knockdown CSAG2 in HCT116 (A) or A375 (B) was assayed for clonogenic growth on plastic for 7 days. The number of colonies was quantified and is shown. Data are represented as the mean ± SD, n = 3 biological replicates.
-
C, DThe effect of stable knockdown CSAG2 in HCT116 (C) or A375 (D) was determined for anchorage‐independent growth in soft agar colony formation assays for 14 days. The number of colonies was quantified and is shown. Data are represented as the mean ± SD, n = 3 biological replicates.
-
E, FKnockdown of CSAG2 in A375 (F) or HCT116 (E) cells decreases xenograft tumor growth in mice. Cells with doxycycline‐inducible shRNAs were implanted into NOD SCID gamma mice. Doxycycline (2 mg/ml) was administered via drinking water. Tumor growth was monitored over time. Data are represented as the mean ± SD, n = 6 mice for each group.
-
G, HStable expression of CSAG2 in HCT116 (G) and CSAG2‐negative H460 (H) cells increases anchorage‐independent growth in soft agar colony formation assays, n = 3 biological replicates. Data are represented as the mean ± SD.
CSAG2 interacts with SIRT1 and promotes p53 deacetylation
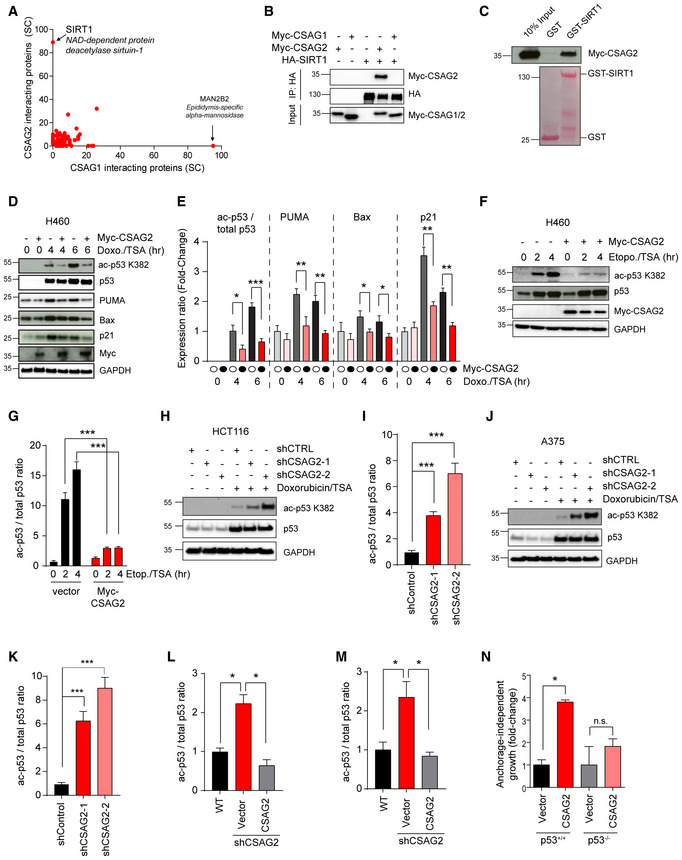
To elucidate the molecular mechanisms of CSAG2 oncogenic activity, we performed unbiased analysis of CSAG2 interacting proteins by tandem affinity purification (TAP) coupled to liquid chromatography‐tandem mass spectrometry (LC‐MS/MS). Vector only or CSAG1 were used as controls. Our data revealed SIRT1 as the most robust binding partner of CSAG2 (Fig 3A and Dataset EV1). Notably, SIRT1 was not detected in either vector control or CSAG1 pull downs, suggesting SIRT1 interaction is specific to CSAG2 (Fig 3A and Dataset EV1). Additionally, none of the other six SIRT family members were detected, suggesting specific interaction of CSAG2 with SIRT1 (Dataset EV1). Immunoprecipitation (IP) experiments confirmed the interaction between CSAG2 and SIRT1, with no interaction detected between CSAG1 and SIRT1 (Fig 3B). To determine whether CSAG2 directly bound SIRT1, we performed in vitro GST‐pull down experiments with recombinant GST‐SIRT1. Consistent with our IP and TAP‐MS results, CSAG2, but not CSAG1, bound to recombinant GST‐SIRT1 in vitro (Figs 3C and EV2A). Together, these results suggest that SIRT1 is a direct binding partner of CSAG2.
Figure 3. CSAG2 interacts with SIRT1 and promotes p53 deacetylation.
-
AHEK293 cells stably expressing TAP‐vector, TAP‐CSAG1, or TAP‐CSAG2 were subjected to pull down followed by SDS–PAGE and LC‐MS/MS. Spectral counts (SC) of the identified proteins in TAP‐CSAG1 and TAP‐CSAG2 pull downs, but absence in TAP‐vector control, are shown.
-
BHEK293FT cells stably expressing HA‐SIRT1 were transfected with Myc‐CSAG1 or Myc‐CSAG2 for 48 h before IP with anti‐HA followed by SDS–PAGE and immunoblotting for indicated proteins.
-
CRecombinant GST‐SIRT1, but not GST, binds in vitro translated Myc‐CSAG2. Myc‐tagged CSAG2 was in vitro translated, and GST‐pull down experiments were performed with recombinant GST or GST‐SIRT1 followed by SDS–PAGE and immunoblotting for anti‐Myc.
-
D–GExpression of CSAG2 decreased p53 ac‐K382 levels and downregulated the expression of downstream p53 target genes in H460 cells. Cells were treated by either 1 μM doxorubicin (D‐E) or 20 μM etoposide (F‐G) with 0.4 μM TSA for the indicated times. Cells were then harvested and blotted for the indicated proteins. Quantitation of expression levels is shown (E and G) as the mean ± SD, n = 3 biological replicates. Ac‐p53K382 levels normalized to total p53, whereas all other proteins were normalized to GAPDH.
-
H–KKnockdown of endogenous CSAG2 in HCT116 (H‐I) and A375 (J‐K) cells increased p53 ac‐K382 levels. CSAG2‐knockdown stable cells were treated by 1 μM doxorubicin/0.4 μM TSA for 6 h. Cells were then harvested and blotted for the indicated proteins. Quantitation of ac‐p53K382 levels relative to total p53 is shown (I and K) as the mean ± SD, n = 3 biological replicates.
-
L, MRe‐expression of CSAG2 rescued p53 ac‐K382 levels in CSAG2 knockdown stable cell lines. Myc‐CSAG2 was transfected in HCT116 (L) or A375 (M) CSAG2 knockdown stable cell lines. Cells were treated with 1 μM doxorubicin/0.4 μM TSA for 6 h and then blotted for the indicated proteins. Quantitation of ac‐p53K382 levels relative to total p53 is shown as the mean ± SD, n = 3 biological replicates.
-
NCSAG2 does not enhance anchorage‐independent growth of HCT116 p53‐null cells. CSAG2 was stably expressed in isogenic HCT116 p53‐wild‐type or p53‐null cells. Anchorage‐independent growth in soft agar colony formation assays was determined after 14 days. The number of colonies was quantified, normalized to each vector control cell line, and is shown as the mean ± SD, n = 3 biological replicates.
Figure EV2. CSAG2 promotes p53 and H3/4 deacetylation.
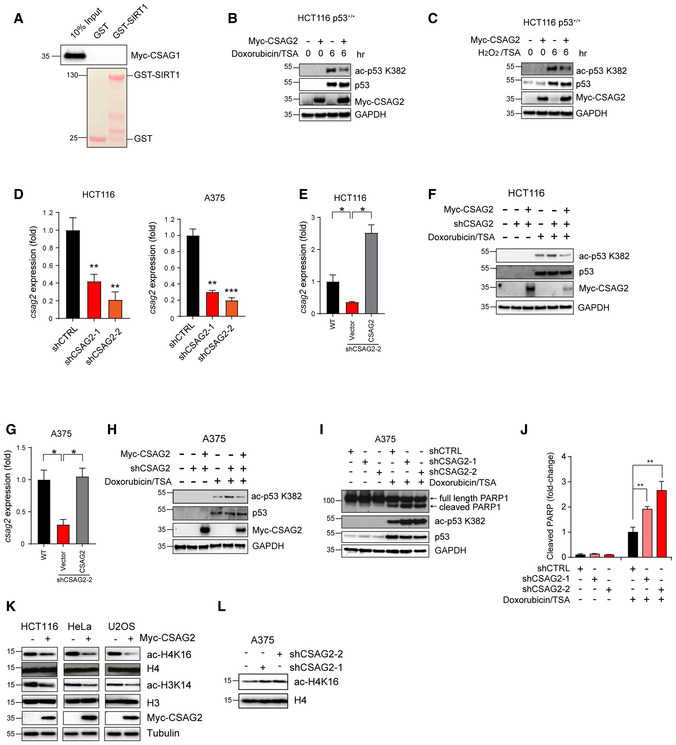
-
ACSAG1 does not bind SIRT1 in vitro. Myc‐tagged CSAG1 was in vitro translated followed by in vitro binding assays with recombinant GST or GST‐SIRT1, SDS–PAGE, and immunoblotting for anti‐Myc.
-
B, CExpression of CSAG2 decreased p53 ac‐K382 levels in HCT116 cells. Cells were treated with either 1 μM Doxorubicin (B) or 1 mM H2O2 (C) and 0.4 μM TSA for indicated times. Cells were then harvested and blotted for the indicated proteins.
-
DCSAG2 mRNA levels were determined by qRT–PCR (n = 3 biological replicates) in the indicated cell types.
-
E–HRe‐expression of CSAG2 rescued p53 ac‐K382 levels in CSAG2 knockdown stable cell lines. Myc‐CSAG2 was transfected in HCT116 (E‐F) or A375 (G‐H) CSAG2 knockdown stable cell lines. CSAG2 mRNA levels were determined by qRT–PCR (n = 3 biological replicates) (E and G). Cells were treated with 1 μM doxorubicin/0.4 μM TSA for 6 h and then blotted for the indicated proteins (F and H).
-
I, JA375 cells were transiently transfected with the indicated shRNAs for 48 h before treatment with 1 μM doxorubicin/0.4 μM TSA for 6 h. Cell lysates were collected and blotted for the indicated proteins (I). Quantitation (n = 3 biological replicates) of normalized cleaved PARP levels is shown (J).
-
KHeLa, U2OS, or HCT116 cells were transfected with Myc‐Vector (−) or Myc‐CSAG2 (+) for 48 h. Cell lysates were blotted for the indicated proteins.
-
LKnockdown of endogenous CSAG2 in A375 cells increased ac‐H4K16 levels. Cell lysates were blotted for the indicated proteins.
Next, we asked whether CSAG2 affected SIRT1 functions. Because p53 is one of the most well‐defined targets of SIRT1 and its importance as a tumor suppressor (Lee & Gu, 2013), we measured the effect of CSAG2 on p53 acetylation. Cells were treated with doxorubicin, etoposide, or H2O2 to induce genotoxic stress and p53 K382 acetylation. To ensure alterations in p53 K382 acetylation were primarily due to SIRT1, but not other HDAC deacetylases, cells were treated with the HDAC inhibitor trichostatin A (TSA). As expected, p53 ac‐K382 levels increased in a time‐dependent manner in response to genotoxic stress (Fig 3D–G). Surprisingly, expression of CSAG2 in CSAG2‐negative H460 cells significantly decreased p53 ac‐K382 levels in response to doxorubicin and etoposide (Fig 3D–G). On the contrary, knockdown of endogenous CSAG2 in HCT116 and A375 cells increased p53 ac‐K382 levels (Figs 3H–K and EV2D). This effect was specific to CSAG2 depletion as re‐expression of CSAG2 rescued p53 ac‐K382 levels in CSAG2 knockdown cell lines (Figs 3L and M, and EV2E–H).
p53 ac‐K382 promotes p53 transcriptional activation of target genes, including Bax, Puma, and p21 (Reed & Quelle, 2014). Therefore, we investigated whether CSAG2‐induced deacetylation of p53 K382 altered the abundance of these p53 target genes that control cell cycle and apoptosis in response to genotoxic stress. Indeed, CSAG2 expression in H460 cells significantly downregulated p53 ac‐K382 levels in response to doxorubicin and importantly decreased levels of p53 targets, Bax, Puma, and p21 (Fig 3D and E). To determine whether regulation of p53 is important for CSAG2 oncogenic activity, we determined whether CSAG2 promotes anchorage‐independent growth in HCT116 p53‐null cells. Unlike in isogenic control HCT116 p53‐wild‐type cells, CSAG2 failed to significantly enhance anchorage‐independent growth of HCT116 p53‐null cells (Fig 3N). Together, these results suggest that CSAG2 interacts with SIRT1, promotes p53 K382 deacetylation, and downregulates p53 target genes that contribute to CSAG2‐induced anchorage‐independent growth.
CSAG2 promotes p53 deacetylation and cell growth through SIRT1 regulation
The reduced acetylation of p53K382 upon CSAG2 expression following DNA damage together with the interaction between CSAG2 and SIRT1 led us to ask whether CSAG2 mediates p53 deacetylation by regulating SIRT1. Therefore, we genetically depleted SIRT1 by siRNA knockdown and monitored the ability of CSAG2 to regulate p53 ac‐K382 levels in HCT116 cells. As expected, SIRT1 knockdown upregulated p53 ac‐K382, consistent with SIRT1 being a major negative regulator of p53 ac‐K382 (Fig 4A). More importantly, CSAG2 failed to alter p53 ac‐K382 levels in SIRT1‐depleted cells (Fig 4A). These results were validated by orthogonal chemical inhibition of SIRT1 with the previously described SIRT1 inhibitor EX‐527 (Gertz et al, 2013). Again, CSAG2 failed to regulate p53 ac‐K382 levels in SIRT1 inhibited (EX‐527 treated) cells (Fig 4B). Additionally, knockdown of CSAG2 failed to upregulate p53 ac‐K382 levels in SIRT1 inhibited (EX‐527 treated) cells (Fig 4C and D). Furthermore, overexpression of catalytically dead SIRT1 H363Y that still binds CSAG2 (Fig 4E) blocked CSAG2 regulation of p53 ac‐K382 levels (Fig 4F). In combination, these results suggest that regulation of p53K382 acetylation by CSAG2 requires SIRT1 activity.
Figure 4. CSAG2 Promotes p53 Deacetylation through SIRT1.
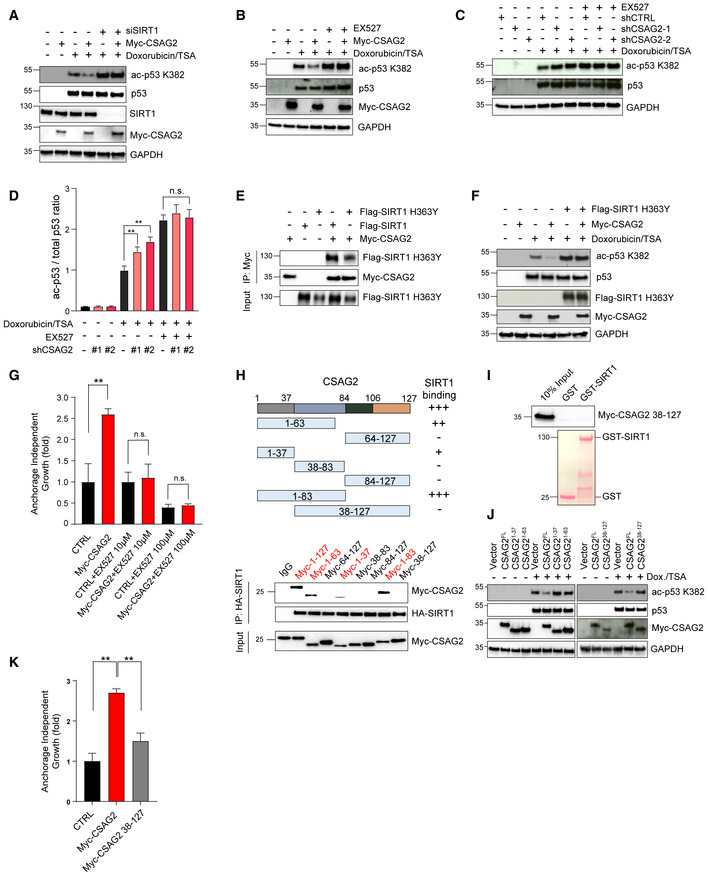
-
AKnockdown of SIRT1 abolished CSAG2‐mediated decrease in ac‐p53K382. CSAG2 overexpressing HCT116 were transfected with SIRT1 siRNA for 72 h before doxorubicin/TSA treatment for 6 h. Cell lysates were blotted for the indicated proteins.
-
BInhibition of SIRT1 enzymatic activity blocked CSAG2‐induced decrease in p53 ac‐K382. CSAG2 overexpressing HCT116 cells were treated with 1 μM doxorubicin/0.4 μM TSA with or without 1 μM EX‐527 for 6 h. Cell lysates were blotted for the indicated proteins.
-
C, DInhibition of SIRT1 enzymatic activity prevents CSAG2‐knockdown increase in p53 ac‐K382. CSAG2‐knockdown stable cells were treated by 1 μM doxorubicin/0.4 μM TSA with or without 1 μM EX‐527 for 6 h. Cells were then harvested and blotted for the indicated proteins (C) and quantitated (D; n = 3 biological replicates). Data shown as mean ± SD.
-
ECSAG2 binds catalytically inactive SIRT1 H363Y. Cells were transfected with the indicated constructs before anti‐Myc IP and blotting for the indicated proteins.
-
FSIRT1 H363Y blocks CSAG2 regulation of p53 ac‐K382 levels. HCT116 cells were transfected with the indicated constructs and blotted for the indicated proteins.
-
GInhibition of SIRT1 activity abolished CSAG2‐induced anchorage‐independent growth. Soft agar colony formation assays were performed in control or CSAG2 overexpressing HCT116 cells with or without EX‐527 treatment. Data are represented as the mean ± SD, n = 3 biological replicates.
-
HSummary of region on CSAG2 required for SIRT1 interaction. HEK293FT cells stably expressing HA‐SIRT1 were transfected with indicated CSAG2 constructs for 48 h before IP with anti‐HA followed by SDS–PAGE and immunoblotting for anti‐Myc.
-
ICSAG2 Δ37 mutant fails to interact with SIRT1 in vitro. Myc‐tagged CSAG2 38–127 fragment was in vitro translated and followed by in vitro binding assay with recombinant GST‐SIRT1, SDS–PAGE, and immunoblotting for anti‐Myc.
-
JCSAG2 Δ37 mutant does not regulate p53 ac‐K382 levels. Cells were transfected with the indicated Myc‐CSAG2 constructs for 48 h before being treated with 1 μM doxorubicin/0.4 μM TSA. Cell lysates were blotted for the indicated proteins.
-
KCSAG2 Δ37 fails to promote anchorage‐independent growth of H460 cells. Soft agar colony formation assays were performed in CSAG2‐negative H460 cells stably expressing CSAG2 wild‐type or CSAG2 Δ37 mutant (n = 3 biological replicates). Data are represented as the mean ± SD.
Next, we investigated whether CSAG2 oncogenic activity is dependent on SIRT1. Therefore, we treated control or CSAG2 overexpressing HCT116 cells with DMSO or EX‐527 and monitored anchorage‐independent growth in soft agar. Consistent with previous results, CSAG2 expression significantly increased anchorage‐independent growth (Fig 4G). However, this effect was abolished in cells treated with EX‐527 SIRT1 inhibitor (Fig 4G). Collectively, these findings indicate that CSAG2 inhibition of p53 acetylation and promotion of anchorage‐independent growth is SIRT1 dependent.
To further determine whether CSAG2 regulation of p53 ac‐K382 and cell growth depends on its ability to interact with SIRT1, we identified the region of CSAG2 required for binding SIRT1. Cell‐based IP assays showed that the N‐terminal 37 amino acids of CSAG2 are required for CSAG2 binding to SIRT1 (Fig 4H). In vitro GST‐pull down assays further confirmed that deletion of CSAG2 N‐terminal 37 amino acid blocked SIRT1 interaction (Fig 4I). These findings are consistent with the inability of CSAG1, that differs from CSAG2 in the absence of the N‐terminal 37 amino acid region (Fig 1B), to bind SIRT1 (Fig 3A and B). Next, we investigated whether the ability of CSAG2 to bind SIRT1 is critical for its ability to regulate p53 ac‐K382 and cell growth. We found that the N‐terminal 1–37 of CSAG2 is necessary, but not sufficient, for downregulation of p53 ac‐K382 levels (Fig 4J). Importantly, CSAG2 Δ37 also failed to induce anchorage‐independent growth of H460 cells in comparison with wild‐type CSAG2 (Fig 4K). Thus, CSAG2 suppression of p53 ac‐K382 and enhancement of cell growth depends on its ability to bind SIRT1.
CSAG2 promotes cellular resistance to genotoxic stress through regulation of SIRT1
Given that CSAG2 inhibits p53 in response to genotoxic stress, we next determined whether CSAG2 may promote chemoresistance to doxorubicin and H2O2. Expression of CSAG2 in CSAG2‐negative H460 cells promoted significant resistance to both doxorubicin and H2O2 as monitored by cell viability (Fig 5A and B). On the contrary, knockdown of endogenous CSAG2 in HCT116 cells increased cellular sensitivity to both of these DNA damaging agents (Fig 5C and D). Consistent with these findings, knockdown of CSAG2 increased the levels of the apoptotic marker cleaved PARP upon doxorubicin treatment of HCT116 (Fig 5E and F) and A375 (Fig EV2I and J) cells. To determine whether CSAG2 promotes cellular resistance to genotoxic stress through regulating SIRT1, we compared the viability of H460 cells expressing wild type or Δ37 CSAG2 after exposure to H2O2. Unlike wild‐type CSAG2, CSAG2 Δ37 that does not bind SIRT1 failed to alter the sensitivity of cells to H2O2 (Fig 5G). Moreover, treatment of SIRT1 inhibitor EX‐527 abolished CSAG2‐induced cell survival upon H2O2 treatment (Fig 5H). Finally, the protective effects of CSAG2 were dependent on p53, as overexpression or knockdown of CSAG2 in p53 null HCT116 cells or p53 mutant H1299 cells did not alter sensitivity to H2O2 or doxorubicin (Fig 5I–N). Collectively, these data suggest that CSAG2, in cooperation with SIRT1, mediates cell survival in response to DNA damage through suppression of p53.
Figure 5. CSAG2 Promotes Cellular Resistance to Genotoxic Stress through Regulation of SIRT1.
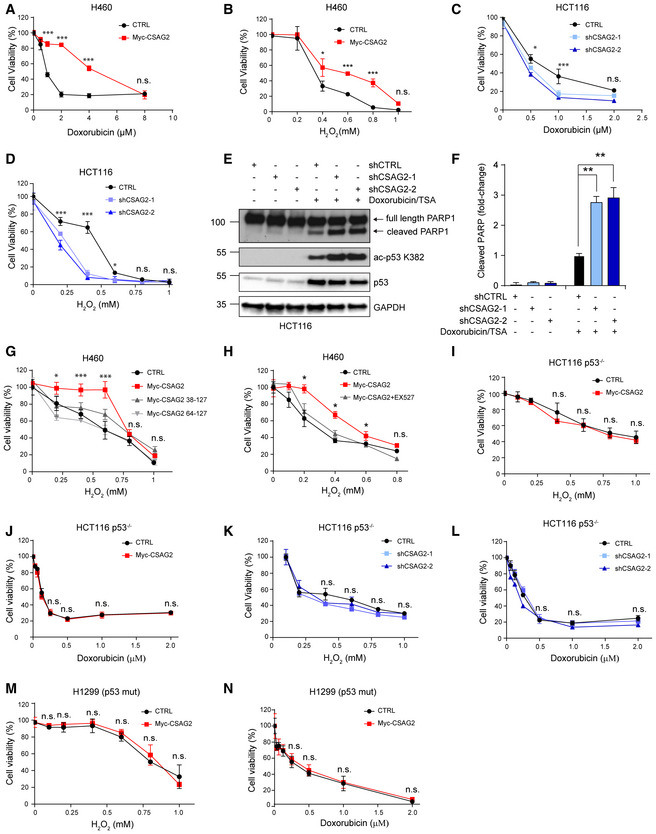
-
A, BControl or CSAG2 expressing H460 cells were treated with the indicated concentrations of doxorubicin (A) or H2O2 (B) for 24 h before cell viability was determined by alamarBlue (n = 6 biological replicates).
-
C, DshCTRL, shCSAG2‐1, or shCSAG2‐2 stable HCT116 cells were treated with the indicated concentrations of doxorubicin (C) or H2O2 (D) for 24 h before cell viability was determined by alamarBlue (n = 6 biological replicates).
-
E, FHCT116 cells were transiently transfected with the indicated shRNAs for 48 h before treatment with 1 μM doxorubicin/0.4 μM TSA for 6 h. Cell lysates were collected and blotted for the indicated proteins (E). Quantitation (n = 3 biological replicates) of normalized cleaved PARP levels is shown (F).
-
GH460 cells were transfected with wild type, Δ37, or Δ63 CSAG2 for 48 h. Cells were then treated with the indicated concentrations of H2O2 for 24 h before cell viability was determined by alamarBlue (n = 6 biological replicates).
-
HInhibition of SIRT1 activity abolished CSAG2 induced cell survival under genotoxic stress. CSAG2 overexpressing H460 cells were treated with the indicated concentrations of H2O2 with or without 1 μM EX‐527 for 24 h before cell viability was determined by alamarBlue (n = 6 biological replicates).
-
I, JControl or Myc‐CSAG2 expressing HCT116 p53−/− cells were treated with the indicated concentrations of H2O2 (I) or doxorubicin (J) for 24 h before cell viability was determined by alamarBlue (n = 6 biological replicates).
-
K, LshCTRL, shCSAG2‐1, or shCSAG2‐2 stable HCT116 p53−/− cells were treated with the indicated concentrations of H2O2 (K) or doxorubicin (L) for 24 h before cell viability was determined by alamarBlue (n = 6 biological replicates).
-
M, NControl or Myc‐CSAG2 expressing H1299 (p53 mutant) cells were treated with the indicated concentrations of H2O2 (M) or doxorubicin (N) for 24 h before cell viability was determined by alamarBlue (n = 6 biological replicates).
CSAG2 enhances SIRT1 enzymatic activity in vitro and alters the acetylation levels of multiple SIRT1 targets
Our previous data showed that CSAG2 enhanced SIRT1 activity by decreasing p53 acetylation. Next, we investigated the molecular mechanism of SIRT1 activation by CSAG2. We explored several possible mechanisms, including altering SIRT1 protein levels, altering SIRT1 subcellular localization, or modifying SIRT1 enzymatic activity. CSAG2 expression had no significant effect on SIRT1 protein levels (Fig 6A) or SIRT1 subcellular localization (Fig 6B). Thus, we focused on elucidating the effect of CSAG2 on SIRT1 enzymatic activity.
Figure 6. CSAG2 directly enhances SIRT1 enzymatic activity.
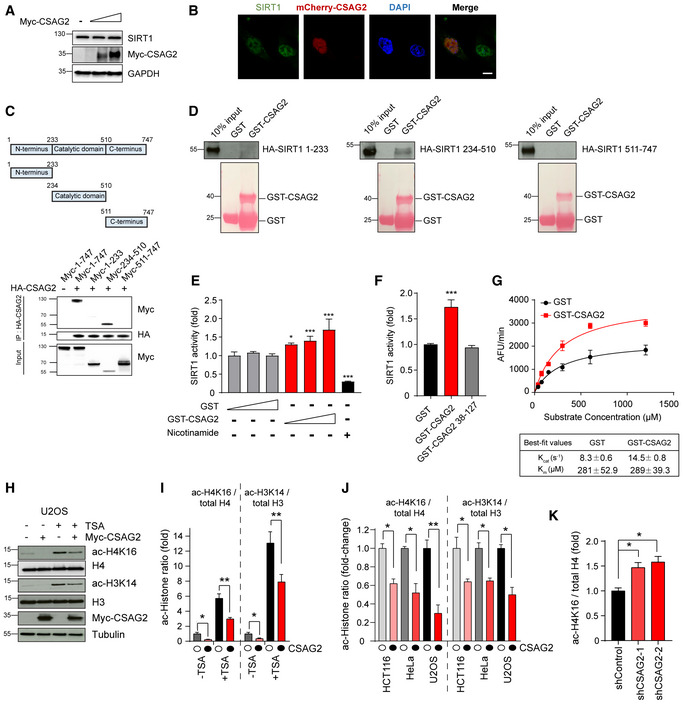
-
ACSAG2 does not alter SIRT1 protein levels. HEK293FT cells were transfected with different amounts of Myc‐CSAG2 for 48 h before cells lysates were harvested and immunoblotted for indicated proteins.
-
BCSAG2 does not alter SIRT1 subcellular location. U2OS cells were transfected with or without mcherry‐CSAG2 for 48 h before fixation, SIRT1 immunostaining, and imaging. Scale bar: 10 μm.
-
CSummary of results mapping interaction region of SIRT1 recognized by CSAG2. HEK293FT cells stably expressing HA‐CSAG2 were transfected with indicated Myc‐SIRT1 constructs for 48 h before IP with anti‐HA followed by SDS–PAGE and immunoblotting for anti‐Myc.
-
DCSAG2 directly binds SIRT1 catalytic domain in vitro. HA‐tagged SIRT1 fragments were in vitro translated. In vitro binding assays with recombinant GST or GST‐CSAG2 were performed followed by SDS–PAGE and immunoblotting for anti‐HA.
-
ECSAG2 enhances SIRT1 activity in vitro. In vitro SIRT1 activity toward p53 ac‐K382 peptide was determined in the presence of GST or GST‐CSAG2, n = 3 biological replicates. Nicotinamide (1 mM) was added to the reaction were indicated to block SIRT1 activity.
-
FCSAG2 Δ37 mutant that does not bind SIRT1 does not enhance SIRT1 activity in vitro. In vitro SIRT1 activity toward p53 ac‐K382 peptide was determined in the presence of GST, GST‐CSAG2 or GST‐CSAG2 38–127, n = 3 biological replicates.
-
GCSAG2 enhances SIRT1 deacetylation of p53 ac‐K382 peptide through increasing kcat, but not Km. not affecting the binding ability with its substrate p53. SIRT1 activity assays were performed as described above, except varying amounts of p53 ac‐K382 fluorometric peptide were used in the presence of saturating NAD+ (3 mM), n = 3 biological replicates.
-
H, IU2OS cells were transfected with Myc‐Vector or Myc‐CSAG2 for 48 h before being treated with or without 0.4 μM TSA for 6 h. Cell lysates were blotted for the indicated proteins (H). Quantitation of expression levels of acetylated histone relative to total histone is shown as the mean ± SD, n = 3 biological replicates. White circles indicate Myc‐Vector and black circles indicate Myc‐CSAG2.
-
JHeLa, U2OS, or HCT116 cells were transfected with Myc‐Vector or Myc‐CSAG2 for 48 h. Cell lysates were blotted for the indicated proteins. Quantitation of expression levels of acetylated Histone relative to total Histone is shown as the mean ± SD, n = 3 biological replicates. White circles indicate Myc‐Vector and black circles indicate Myc‐CSAG2.
-
KKnockdown of endogenous CSAG2 in A375 cells increased ac‐H4K16 levels. Cell lysates were blotted for the indicated proteins. Quantitation of expression levels of ac‐H4K16 relative to total H4 is shown as the mean ± SD, n = 3 biological replicates.
First, we determined which part of SIRT1 is required for CSAG2 binding. Using cell‐based IP (Fig 6C) and in vitro GST‐pull down assays (Fig 6D), we found that CSAG2 directly binds to the catalytic domain, but not the NTD or CTR, of SIRT1. To determine whether CSAG2 directly regulates SIRT1 enzymatic activity, we measured the effect of CSAG2 on SIRT1 deacetylase activity in vitro using recombinant, bacterially expressed CSAG2 and SIRT1 and p53 ac‐K382 reporter peptide as previously described (Milne et al, 2007). Remarkably, GST‐CSAG2, but not GST alone, increased SIRT1‐dependent p53 ac‐K382 deacetylation in a dose‐dependent manner (Fig 6E). To investigate whether CSAG2‐mediated p53 ac‐K382 deacetylation requires interaction with SIRT1, in vitro SIRT1 activity assays were performed comparing CSAG2 wild‐type and CSAG2 Δ37 mutant. In comparison with wild‐type CSAG2, CSAG2 Δ37 failed to enhance SIRT1 enzymatic activity toward p53 ac‐K382 (Fig 6F).
Known regulators of SIRT1 (including STACs; sirtuin‐activating compounds) primarily function through enhancing substrate binding (Km) (Milne et al, 2007). Surprisingly, SIRT1 Km for p53 ac‐K382 peptide was not significantly different with or without CSAG2 (289 μM and 281 μM, respectively) (Fig 6G). However, CSAG2 significantly increased SIRT1 kcat from 8.3/s to 14.5/s (Fig 6G). Thus, CSAG2 has a unique mechanism of action compared to other SIRT1 regulators and promotes SIRT1 enzymatic activity through binding to SIRT1 catalytic domain and increasing intrinsic enzyme rate (kcat) without altering substrate binding (Km).
Finally, these findings suggest that CSAG2 may promote deacetylation of multiple SIRT1 targets. Thus, we examined whether CSAG2 altered the acetylation levels of H3K14 and H4K16 that have previously been established as SIRT1 regulated (Imai et al, 2000; Vaquero et al, 2004; Fraga et al, 2005; Dang et al, 2009). Expression of CSAG2 in U2OS cells significantly decreased both H3K14 and H4K16 acetylation levels (Fig 6H and I). These findings were confirmed in two additional cell lines (Figs 6J and EV2K). Furthermore, knockdown of endogenous CSAG2 increased ac‐H4K16 levels in A375 cells (Figs 6K and EV2L). Collectively, these findings suggest that CSAG2 functions as a SIRT1 activator whose mechanism of action is target agnostic.
Discussion
SIRT1 is a NAD+‐dependent deacetylase that plays important roles in cellular metabolism and stress responses through deacetylating a variety of substrates (Chang & Guarente, 2014). Moreover, SIRT1 has been shown to have essential roles in several biological processes, including aging, and diseases, such as cancer and diabetes (Satoh et al, 2011). As such, much interest in modulating SIRT1 activity has emerged, with several small molecular activators of SIRT1 having been discovered (Sinclair & Guarente, 2014). However, identification of cellular regulators of SIRT1 has been limited. Furthermore, understanding how SIRT1 is altered in disease, including cancer, is unclear. In this study, we identify CSAG2 as a new cancer‐specific SIRT1 activator that stimulates SIRT1 enzymatic activity through direct binding.
CSAG2 is mainly expressed in the testis but is frequently turned on in many tumor types, including colon, lung, and breast tumors. Our findings suggest that activation of CSAG2 in cancer cells is not simply a passenger event during cellular transformation and tumorigenesis, but rather CSAG2 is a driver gene that supports multiple phenotypes associated with tumorigenesis. We propose that one critical oncogenic function of CSAG2 is upregulation of SIRT1 activity and suppression of the p53 tumor suppressor (Fig 7). We show that CSAG2 expression suppresses genotoxic‐induced cell toxicity in a SIRT1‐ and p53‐dependent manner. Consistently, patients expressing high levels of SIRT1 are more resistant to chemotherapy than patients with low SIRT1 expression (Zhang et al, 2013; Chen et al, 2014). In line with this, we show that CSAG2 expression correlates with poor prognosis in several tumor types. Thus, CSAG2 may serve as an important biomarker for patient response to DNA damaging chemotherapeutic. Although we show that CSAG2 suppression of p53 is important for its ability to promote anchorage‐independent growth and chemoresistance, two processes tightly controlled by p53, CSAG2 regulation of SIRT1 likely has a number of impacts on cells that in concert drive tumorigenesis.
Figure 7. Model of CSAG2 regulation of SIRT1 and tumorigenesis.
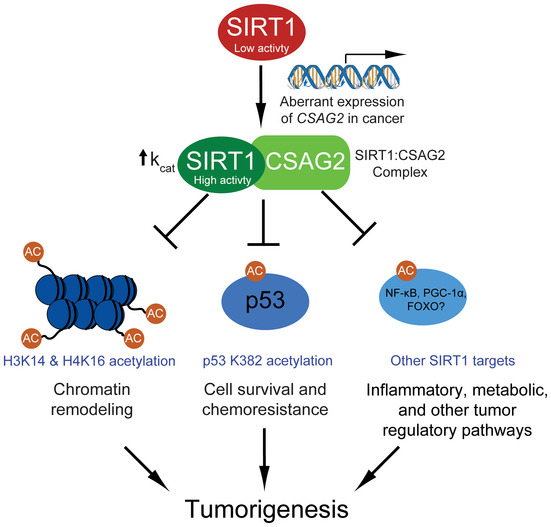
A schematic model for how aberrant transcriptional activation of CSAG2 in cancer leads to enhanced SIRT1 activity and regulation of key tumor regulatory pathways. Note that the oncogenic potential of CSAG2 is likely attribute to multiple SIRT1 targets and pathways.
In recent years, the role of SIRT1 in cancer biology has become increasingly apparent and growing evidence demonstrates an oncogenic function for SIRT1 (Bosch‐Presegue & Vaquero, 2011; Wilking & Ahmad, 2015). The initial connection of SIRT1 to cancer was made when SIRT1 was found to deacetylate and repress the activity of the tumor suppressor p53 (Yi & Luo, 2010). However, SIRT1 has pleiotropic effects on cells, including altering cellular metabolic processes, by deacetylating a number of substrates, such as FOXO, Ku70, NF‐κB, and PGC‐1α (Olmos et al, 2011). Future studies into whether CSAG2 alters SIRT1 deacetylation of these targets will be interesting. Regardless, our study indicates CSAG2 drives tumorigenesis through modulating SIRT1. Interestingly, SIRT1 inhibitors (tenovins, EX‐527, and sirtinol), either alone or in combination, have been shown to reduce malignant growth (Wilking et al, 2014; Carafa et al, 2016). Thus, CSAG2 expression status may be a useful enrollment biomarker to select patients with the greatest potential response to SIRT1 inhibitors.
How does CSAG2 promote the deacetylase activity of SIRT1? Previous structural and biochemical studies have suggested that the central conserved catalytic domain of SIRT1 is subjected to multiple modes of regulation, including NAD+, substrate binding, the NTD, and the CTR ESA (Davenport et al, 2014). We found that CSAG2 bound the SIRT1 catalytic domain in cells and in vitro. Intriguingly, unlike small molecule SIRT1 activators, CSAG2 did not affect SIRT1 Km for the ac‐K382 p53 substrate peptide. Unexpectedly, enzyme kinetics experiment showed that CSAG2 enhanced SIRT1 kcat toward ac‐K382 p53 substrate peptide. Thus, we speculate that CSAG2 binds SIRT1 catalytic domain and triggers a conformational change that stimulates SIRT1 activity, possibly through altering interaction between the catalytic domain and ESA or NTD or increasing affinity for NAD+.
We show that CSAG2 expression correlated with other Xq28 locus MAGE‐A genes, including the related MAGE‐A2, ‐A3, ‐A6, and ‐A12. MAGE‐A3 and ‐A6 have been shown to also drive tumorigenesis through regulation of core cellular metabolic and energy homeostasis pathways, including ubiquitination and degradation of AMP‐activated protein kinase AMPK (Pineda et al, 2015). Intriguingly, AMPK positively regulates SIRT1 through production of NAD+ (Canto et al, 2009). Thus, we hypothesize that the MAGE‐CSAG Xq28 gene cluster evolved to allow simultaneous downregulation of AMPK by MAGE‐As, while maintaining SIRT1 activity by CSAG2 in the face of reduced NAD+. Thus, genes in the MAGE‐CSAG Xq28 gene cluster are functionally linked, similar to bacterial operons. Consistent with this idea, CSAG1 has been suggested to promote mitotic progression specifically in p53 defective tumor cells (Sapkota et al, 2020). Thus, CSAG1 may function to ensure proper cell division upon CSAG2 downregulation of p53.
In summary, our findings illuminate a previously unrecognized regulation of SIRT1 during tumorigenesis by the enigmatic cancer‐specific CSAG2 protein. These results have important implications on understanding how key cellular metabolic pathways are altered in cancer and highlight cancer–testis antigens as prominent players. Furthermore, our findings provide previously unanticipated mechanisms to enhance SIRT1 enzymatic activity that could be of benefit in a number of pathologies.
Materials and Methods
Cell culture and transfections
HEK293FT, HEK293, HCT116, H1299, U2OS, HeLa, and A375 cells (ATCC) were grown in DMEM supplemented with 10% (v/v) fetal bovine serum, 2 mM l‐glutamine, 100 units/ml penicillin, 100 units/ml streptomycin, and 0.25 mg/ml amphotericin B. H460 cells were grown in RPMI supplemented with 5% (v/v) heat‐inactivated fetal bovine serum. HCT116 p53‐null cells were a kind gift from Dr. Bert Vogelstein (Johns Hopkins University). Plasmid transfections were performed using Effectene (QIAGEN) and siRNAs using Lipofectamine RNAiMax (Thermo) according to the manufacturer's protocols.
Generation of stable overexpression and shRNA cell lines
HCT116 and H460 cells were transduced with Myc‐vector or Myc‐CSAG2 lentivirus using polybrene in 6‐well plates. Two days after lentiviral transduction, cells were selected over 2 weeks using 2.5 mg/ml of blasticidin (GIBCO). HCT116 and A375 cells were transduced with pLKO.1 or pLKO.1‐Tet control or CSAG2 shRNA lentivirus using polybrene in 6‐well plates. Two days after lentiviral transduction, cells were selected over 2 weeks using 2.5 mg/ml of blasticidin. The sequences of shRNAs that target CSAG2: shCSAG2‐1, 5′‐GCCCAGAAGCCCTATCAAAGT; shCSAG2‐2, 5′‐CGAACGAGGAACTCAATCAAA. HEK293 cells were transfected with either tandem affinity purification (TAP)‐vector or TAP‐CSAGs using Effectene in 6 cm2 plates. After 48 h, cells were selected with 1 μg/ml of puromycin over 2 weeks.
Clonogenic growth and anchorage‐independent growth soft agar assays
For clonogenic growth assays on plates, cells were plated in 6‐well plates in triplicate. After 1 week, cells were fixed and stained with 0.05% (w/v) crystal violet and counted. For anchorage‐independent growth soft agar assays, cells were suspended in 0.375% Noble agar (Difco) supplemented with regular growth medium and overlaid on 0.5% Noble agar. Cells were incubated for 2 weeks before colonies > 100 μm in size were counted.
Xenograft tumor growth assays
1 × 106 HCT116 or A375 cells with doxycycline‐inducible shRNAs were mixed with Matrigel (Corning) before injection into the flank of NOD scid gamma male mice (Jackson Lab) (n = 6 for each group). Doxycycline (2 mg/ml) was administered via drinking water. Tumor size was measured 2–3 times a week during the duration of the experiment. At the end of the study, tumors were dissected and weighed. All animal studies were approved by the St. Jude Children's Research Hospital Institutional Animal Care and Use Committee (IACUC).
Tandem affinity purification
Ten 15 cm2 plates of HEK293 cells stably expressing TAP‐vector, TAP‐CSAG1, or TAP‐CSAG2 were lysed with TAP lysis buffer (10% (v/v) glycerol, 50 mM HEPES‐KOH pH 7.5, 100 mM KCl, 2 mM EDTA, 0.1% (v/v) NP‐40, 10 mM NaF, 0.25 mM Na3VO4, 50 mM β‐glycerophosphate, 2 mM DTT, and 1X protease inhibitor cocktail (Sigma)), and cleared supernatants were bound to IgG‐Sepharose beads (GE Amersham) and then washed in lysis buffer and TEV buffer (10 mM HEPES‐KOH pH 8.0, 150 mM NaCl, 0.1% (v/v) NP‐40, 0.5 mM EDTA, 1 mM DTT, and 1X protease inhibitor cocktail). Protein complexes were cleaved off the beads by TEV protease (Sigma) and incubated with calmodulin‐Sepharose beads (GE Amersham) in calmodulin binding buffer (10 mM HEPES‐KOH pH 8.0, 150 mM NaCl, 1 mM Mg acetate, 1 mM imidazole, 0.1% (v/v) NP‐40, 6 mM CaCl2, 10 mM 2‐mercaptoethanol) and then washed in calmodulin rinse buffer (50 mM ammonium bicarbonate pH 8.0, 75 mM NaCl, 1 mM Mg acetate, 1 mM imidazole, 2 mM CaCl2) before eluted with SDS sample buffer, subjected to SDS–PAGE, and stained with GelCode Blue stain (Thermo Fisher Scientific) before protein identification by LC‐MS/MS.
Mass spectrometry
The gel bands were reduced with dithiothreitol (DTT) (Sigma) and alkylated by iodoacetamide (IAA) (Sigma), washed, dried down, and rehydrated with a buffer containing trypsin (Promega). Proteolysis was performed overnight at 37°C. Peptides were extracted in acetonitrile, dried down in a speed vacuum, and reconstituted in 5% (v/v) formic acid.
The peptide mixture was separated on a nanoscale capillary reverse phase C18 column (75 id, 10 cm) by a HPLC system (Thermo EASY‐nLC 1000). Buffer A was 0.2% formic acid, and Buffer B was 70% acetonitrile; 0.2% (v/v) formic acid. The peptides were eluted over a 90‐min liquid chromatography gradient by increasing organic from 12–70%. The peptides were ionized by electrospray ionization and detected by an inline mass spectrometer (Thermo LTQ Orbitrap Elite). The mass spectrometer was operated in data‐dependent mode with a high‐resolution survey scan in Orbitrap and 20 low‐resolution MS/MS scans in the ion) for each cycle.
UniProt human database was used for searching raw data with Sequest v.28 (rev. 12) search engine. The human database was concatenated with a reversed decoy database for evaluating false discovery rate. Mass tolerance of 25 ppm for precursor and 0.5 Da for product ions were used for database search. Two maximal missed cleavages and three maximal modification sites were allowed. The assignment of b and y ions was used for identification. Carbamidomethylation of Cysteine (+57.02146 Da) was used for static modifications, and Met oxidation (+15.99492 Da) was considered as a dynamic modification. Mass accuracy and matching scores filters were used for MS/MS spectra to reduce protein false discovery rate to < 1%. Proteins identified in one gel lane were combined together, and the total number of spectra, namely spectral counts (SC), matching to individual proteins may reflect their relative abundance in one sample after normalizing for protein molecular weight. The spectral counts between samples for a given protein were used to calculate the P‐value which is derived by G‐test.
RNA preparation and qRT–PCR
RNA was extracted from cultured cells using RNAStat60 (TelTest) according to manufacturer's instructions. Total RNA was treated with DNase I (Roche) and converted to cDNA using High Capacity Reverse Transcription kit (Life Technologies). cDNA and appropriate primers were plated in triplicate in a 384‐well plate, and gene expression levels were measured using SYBR green master mix (Applied Biosystems). Oligonucleotides used for qRT–PCR: CSAG2 forward, 5′‐AGATGTCCAGGAAACCACGAGC; reverse, 5′‐ TTTCCCTTCCGGGTTGTCTTGG.
Immunoprecipitation and immunoblotting
HEK293FT cells were plated in 6 cm2 plates and transfected 24 h later with Effectene (QIAGEN) according to the manufacturer's protocol. After 48 h, cells were washed and scraped in cold PBS, spun down, and resuspended in lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM DTT, 0.1% (v/v) Triton X‐100, 10 mM N‐Ethylmaleimide (NEM), and 1× protease inhibitor cocktail). Cell lysates were incubated with appropriate antibodies overnight at 4°C and then with protein A beads (Santa Cruz Biotechnology, sc‐2003) for 2 h at 4°C. Beads were then washed with lysis buffer three times and eluted with 2× SDS sample buffer. For immunoblotting, samples in SDS sample buffer were resolved on SDS–PAGE gels (Bio‐rad) and then transferred to nitrocellulose membranes prior to blocking in TBS0T with 5% (w/v) milk powder or 3% (w/v) bovine serum albumin and probing with primary and secondary antibodies (GE Healthcare). Protein signal was visualized after addition of ECL detection reagent (GE Healthcare) according to manufacturer's instructions.
Antibodies and siRNAs
Antibodies used in this study: anti‐GAPDH (Cell Signaling Technology, 2118S), anti‐Myc (Roche, 11666606001), anti‐ Acetyl‐p53 Lys382 (Cell Signaling Technology, 2525S), anti‐p53 (Santa Cruz Biotechnology, sc‐126), anti‐p21 (Cell Signaling Technology, 2947S), anti‐PUMA (Abcam, ab33906), Bax (Cell Signaling Technology, 2772S), anti‐H3 (Abcam, ab1791), anti‐H4 (Abcam, ab10158), anti‐acetylated‐H3K14 (Abcam, ab52946), anti‐acetylated‐H4K16 (Millipore Sigma, 07329), anti‐PARP (Cell Signaling Technology, 9542), donkey anti‐Rabbit IgG (GE, NA934V), and sheep anti‐Mouse IgG (GE, NA931V). siRNA used were as follows: MISSION siRNA Universal Negative Control #1 (Sigma) and SIRT1: SASI_Hs01_00153666 (Sigma).
Cell viability assay
To assess cell viability after genotoxic stress, 5 × 103 cells were seeded in 96‐well plates and incubated with DNA damage agents for 24 h prior to changing the media and adding alamarBlue (Thermo Fisher Scientific). Plates were read 4 h later by measuring the fluorescence with excitation wavelength at 540 nm and emission wavelength at 590 nm on an Enspire plate reader.
Recombinant protein purification and in vitro binding assay
GST‐CSAG2, GST‐CSAG2 (38–127), or GST alone was produced in BL21 (DE3) cells by overnight induction at 16°C with 0.5 mM Isopropyl β‐D‐1‐thiogalactopyranoside (IPTG). GST‐tagged proteins were purified from bacterial lysates with glutathione Sepharose (GE Amersham) and eluted with 10 mM glutathione. In vitro binding assays were performed as described previously. Briefly, 15 μg of purified GST‐tagged proteins was bound to glutathione Sepharose beads (Amersham) in binding buffer (25 mM Tris pH 8.0, 2.7 mM KCl, 137 mM NaCl, 0.05% (v/v) Tween‐20, and 10 mM 2‐mercaptoethanol) for 1 h and then blocked for 1 h in binding buffer containing 5% (w/v) milk powder. In vitro translated proteins (Promega SP6‐TNT Quick rabbit reticulocyte lysate system) were then incubated with the bound beads for 1 h, extensively washed in binding buffer, eluted with 2× SDS sample buffer, boiled, subjected to SDS–PAGE, and immunoblotting.
Immunofluorescence and microscopy
U2OS cells were transfected with or without mcherry‐CSAG2 for 48 h before fixation. Cells then were washed in 1× PBS, fixed in 3% (w/v) paraformaldehyde for 15 min at room temperature, washed twice in 1X PBS, and permeabilized for 20 min at 4°C in 1X PBS containing 100 μg/ml digitonin. After permeabilization, cells were incubated for 60 min with anti‐SIRT1 antibody diluted in 1X PBS containing 100 μg/ml digitonin and 5% (w/v) BSA. Cells were then washed in PBS containing 100 μg/ml digitonin three times and incubated for 30 min in 4 μg/ml Alexa‐488 secondary antibody (Invitrogen Molecular Probes) diluted in PBS containing 100 μg/ml digitonin and 5% (w/v) BSA. Cells were then washed three times in PBS containing 100 μg/ml digitonin, DNA stained with 1 μg/ml DAPI for 2 min, washed in PBS, and mounted.
In vitro SIRT1 activity assays
SIRT1 deacetylase activity was measured with a SIRT1 fluorimetric activity assay/drug discovery kit (catalog #AK555, Biomol International LP). For determination of the relative SIRT1 deacetylase activity in vitro, purified SIRT1 protein was incubated with 25 μM fluorogenic acetylated p53 peptide substrate and 500 μM NAD+ for 20 min at 37°C. For determination of enzyme kinetic parameters, the reaction contained a saturating concentration of NAD+ (3 mM) while varying fluorogenic acetylated p53 peptide concentrations (0–1.25 mM). The reactions were incubated for 10 min at 37°C. Reactions were halted by the addition of 1 mM nicotinamide, and the deacetylation‐dependent fluorescent signal was determined using a 360‐nm excitation laser and a 460‐nm emission filter on a fluorescence plate reader. Background control reactions were performed in the absence of enzyme. All of the reactions were performed in triplicate. Km and kcat values were obtained by fitting the data to the Michaelis–Menten equation using GraphPad Prism software.
Statistics and data analysis
Data are expressed as mean ± SD. Statistical analyses were performed with unpaired, two‐tailed Student's t‐test or ANOVA analysis. Statistical significance is represented in figures by *P < 0.05, **P < 0.01, and ***P < 0.001. Survival data were analyzed by kmplotter (http://kmplot.com/analysis). TCGA gene expression data were visualized by firebrowse (http://firebrowse.org). Gene expression data for normal tissues were analyzed from GTEx (https://www.gtexportal.org).
Author contributions
XY and PRP contributed to experimental design. XY performed experiments, data analysis, and figure composition. XY and PRP wrote the manuscript.
Conflict of interest
P.R.P. is a paid consultant of Levo Therapeutics, Inc. and Amgen, Inc.
Supporting information
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank members of the Potts laboratory for helpful discussions and critical reading of the manuscript. We especially thank Dr. Bert Vogelstein (Johns Hopkins University) for generously providing HCT116 p53‐null cells. This work was partially supported by American Cancer Society Research Scholar Award 181691010 (P.R.P.). P.R.P is a paid consultant for Levo Therapeutics, Inc. and Amgen, Inc.
EMBO Reports (2020) 21: e50912
Data availability
The AP‐MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD020231. https://www.ebi.ac.uk/pride/archive/projects/PXD020231/.
References
- Beckerman R, Prives C (2010) Transcriptional regulation by p53. Csh Perspect Biol 2: a000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch‐Presegue L, Vaquero A (2011) The dual role of sirtuins in cancer. Genes Cancer 2: 648–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM (2005) Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 19: 1751–1759 [DOI] [PubMed] [Google Scholar]
- Bredenbeck A, Hollstein VM, Trefzer U, Sterry W, Walden P, Losch FO (2008) Coordinated expression of clustered cancer/testis genes encoded in a large inverted repeat DNA structure. Gene 415: 68–73 [DOI] [PubMed] [Google Scholar]
- Canto C, Gerhart‐Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, Jarho E, Lahtela‐Kakkonen M, Mai A, Altucci L (2016) Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenet 8: 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L (2015) The multifaceted functions of sirtuins in cancer. Nat Rev Cancer 15: 608–624 [DOI] [PubMed] [Google Scholar]
- Chang HC, Guarente L (2014) SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 25: 138–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Sun K, Jiao SF, Cai N, Zhao X, Zou HB, Xie YX, Wang ZS, Zhong M, Wei LX (2014) High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Sci Rep 4: 7481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF (2003) Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)‐deficient mice. Proc Natl Acad Sci USA 100: 10794–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Case AW, Riera TV, Considine T, Lee JE, Hamuro Y, Zhao H, Jiang Y, Sweitzer SM, Pietrak B et al (2015) Crystallographic structure of a small molecule SIRT1 activator‐enzyme complex. Nat Commun 6: 7645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL (2009) Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459: 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AM, Huber FM, Hoelz A (2014) Structural and functional analysis of human SIRT1. J Mol Biol 426: 526–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan ZF, Feller AJ, Toh HC, Makastorsis T, Seiden MV (1999) TRAG‐3, a novel gene, isolated from a taxol‐resistant ovarian carcinoma cell line. Gene 229: 75–81 [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar‐Garea A, Boix‐Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K et al (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37: 391–400 [DOI] [PubMed] [Google Scholar]
- Gerhart‐Hines Z, Dominy JE Jr, Blattler SM, Jedrychowski MP, Banks AS, Lim JH, Chim H, Gygi SP, Puigserver P (2011) The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol Cell 44: 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M, Fischer F, Nguyen GTT, Lakshminarasimhan M, Schutkowski M, Weyand M, Steegborn C (2013) Ex‐527 inhibits Sirtuins by exploiting their unique NAD(+)‐dependent deacetylation mechanism. Proc Natl Acad Sci USA 110: E2772–E2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisays F, Brace CS, Yackly SM, Kwon HJ, Mills KF, Kashentseva E, Dmitriev IP, Curiel DT, Imai SI, Ellenberger T (2015) The N‐terminal domain of SIRT1 is a positive regulator of endogenous SIRT1‐dependent deacetylation and transcriptional outputs. Cell Rep 10: 1665–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hida Y, Kubo Y, Murao K, Arase S (2007) Strong expression of a longevity‐related protein, SIRT1, in Bowen's disease. Arch Dermatol Res 299: 103–106 [DOI] [PubMed] [Google Scholar]
- Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, Nagy TR (2007) SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res 67: 6612–6618 [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD‐dependent histone deacetylase. Nature 403: 795–800 [DOI] [PubMed] [Google Scholar]
- Karam JA, Huang S, Fan JH, Stanfield J, Schultz RA, Pong RC, Sun XK, Mason RP, Xie XJ, Niu G et al (2011) Upregulation of TRAG3 gene in urothelial carcinoma of the bladder. Int J Cancer 128: 2823–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Kho JH, Kang MR, Um SJ (2007) Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity (vol 28, pg 277, 2007). Mol Cell 28: 513 [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen JJ, Lou ZK (2008) DBC1 is a negative regulator of SIRT1. Nature 451: 583–U510 [DOI] [PubMed] [Google Scholar]
- Knight JR, Allison SJ, Milner J (2013) Active regulator of SIRT1 is required for cancer cell survival but not for SIRT1 activity. Open Biol 3: 130130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkola T, Suuronen T, Molnar F, Maatta J, Salminen A, Jarho EM, Lahtela‐Kakkonen M (2014) AROS has a context‐dependent effect on SIRT1. FEBS Lett 588: 1523–1528 [DOI] [PubMed] [Google Scholar]
- Kwon HS, Ott M (2008) The ups and downs of SIRT1. Trends Biochem Sci 33: 517–525 [DOI] [PubMed] [Google Scholar]
- Lakshminarasimhan M, Curth U, Moniot S, Mosalaganti S, Raunser S, Steegborn C (2013) Molecular architecture of the human protein deacetylase Sirt1 and its regulation by AROS and resveratrol. Biosci Rep 33: e00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Gu W (2013) SIRT1: regulator of p53 Deacetylation. Genes Cancer 4: 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Fang D (2013) The roles of SIRT1 in cancer. Genes Cancer 4: 97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materna V, Surowiak P, Kaplenko I, Spaczynski M, Duan ZF, Zabel M, Dietel M, Lage H (2007) Taxol‐resistance‐associated gene‐3 (TRAG‐3/CSAG2) expression is predictive for clinical outcome in ovarian carcinoma patients. Virchows Arch 450: 187–194 [DOI] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB et al (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450: 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L (2004) Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563 [DOI] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC‐1{alpha}. J Biol Chem 280: 16456–16460 [DOI] [PubMed] [Google Scholar]
- Olmos Y, Brosens JJ, Lam EWF (2011) Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Update 14: 35–44 [DOI] [PubMed] [Google Scholar]
- Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, Kozaki K, Akishita M, Ouchi Y, Kaneki M (2006) Sirt1 inhibitor, Sirtinol, induces senescence‐like growth arrest with attenuated Ras‐MAPK signaling in human cancer cells. Oncogene 25: 176–185 [DOI] [PubMed] [Google Scholar]
- Pan M, Yuan H, Brent M, Ding EC, Marmorstein R (2012) SIRT1 contains N‐ and C‐terminal regions that potentiate deacetylase activity. J Biol Chem 287: 2468–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda CT, Ramanathan S, Tacer KF, Weon JL, Potts MB, Ou YH, White MA, Potts PR (2015) Degradation of AMPK by a cancer‐specific ubiquitin ligase. Cell 160: 715–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SM, Quelle DE (2014) p53 acetylation: regulation and consequences. Cancers 7: 30–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaninejad A, Zamani MR, Pourvahedi M, Golchehre Z, Bereshneh AH, Rezaei N (2016) Cancer/testis antigens: expression, regulation, tumor invasion, and use in immunotherapy of cancers. Immunol Invest 45: 619–640 [DOI] [PubMed] [Google Scholar]
- Sapkota H, Wren JD, Gorbsky GJ (2020) CSAG1 maintains the integrity of the mitotic centrosome in cells with defective P53. J Cell Science 133: jcs239723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Stein L, Imai S (2011) The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handb Exp Pharmacol 206: 125–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, Verdin E (2007) Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 26: 5489–5504 [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L (2014) Small‐molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol 54: 363–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stunkel W, Peh BK, Tan YC, Nayagam VM, Wang X, Salto‐Tellez M, Ni B, Entzeroth M, Wood J (2007) Function of the SIRT1 protein deacetylase in cancer. Biotechnol J 2: 1360–1368 [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument‐Bromage H, Tempst P, Reinberg D (2004) Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 16: 93–105 [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Eagon EN, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA (2001) hSIR2(SIRT1) functions as an NAD‐dependent p53 deacetylase. Cell 107: 149–159 [DOI] [PubMed] [Google Scholar]
- Wilking MJ, Singh CK, Nihal M, Ndiaye MA, Ahmad N (2014) Sirtuin deacetylases: a new target for melanoma management. Cell Cycle 13: 2821–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilking MJ, Ahmad N (2015) The role of SIRT1 in cancer the saga continues. Am J Pathol 185: 26–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Luo J (2010) SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta 1804: 1684–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Rong NN, Chen J, Zou CW, Jing HY, Zhu XL, Zhang WL (2013) SIRT1 expression is associated with the chemotherapy response and prognosis of patients with advanced NSCLC. PLoS ONE 8: e79162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W (2008) Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451: 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The AP‐MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD020231. https://www.ebi.ac.uk/pride/archive/projects/PXD020231/.