

Abstract
Background
Genetic testing is an emerging diagnostic approach in early‐onset epilepsy. Identification of the heterogeneous genetic causes of epilepsy may mitigate unnecessary evaluations and allow more accurate diagnosis and therapy. We aimed to uncover genetic causes of early‐onset epilepsy using next‐generation sequencing (NGS) to elucidate the diagnostic candidates and evaluate the diagnostic yield of targeted gene panel testing.
Methods
We evaluated 116 patients with early‐onset epilepsy developed before 2 years old and normal brain imaging using a NGS‐based targeted gene panel. Variants were classified according to their pathogenicity, and the diagnostic yield of the targeted genes and associated clinical factors were determined.
Results
We detected 40 disease‐causing variants with diagnostic yield of 34.5% (19 pathogenic, 21 likely pathogenic). Twelve variants were novel. The most commonly detected genes were SCN1A, associated with Dravet syndrome, and PRRT2, associated with benign familial infantile epilepsy. Other variants were identified in ARX, SCN2A, KCNQ2, PCDH19, STXBP1, DEPDC5, and SCN8A. The age of seizure onset and family history were associated with disease‐causing variants.
Conclusion
Next‐generation sequencing‐based targeted testing is an effective diagnostic test, with 30%–40% comparable diagnostic yield. Patients with earlier seizure onset and family history of epilepsy were the best candidates for testing. For pediatric patients with early‐onset epilepsy, genetic diagnosis is important for accurate prognosis and treatment.
Keywords: diagnostic yield, early‐onset epilepsy, genetic epilepsy, next‐generation sequencing, targeted epilepsy gene panel
Next‐generation sequencing‐based targeted gene panel is an effective diagnostic test for early‐onset epilepsy with heterogeneous genotypes and phenotypes. The best candidates for genetic testing are patients with earlier seizure onset and family history of epilepsy.

1. INTRODUCTION
Epilepsy is a relatively common neurologic disorder reported in approximately 4–6 cases per 1,000 children, with epileptic seizure occurring frequently during childhood (Waaler, Blom, Skeidsvoll, & Mykletun, 2000; Wiebe, Bellhouse, Fallahay, & Eliasziw, 1999). Epilepsy develops as a result of myriad heterogeneous causes, and the etiology of epilepsy determines the prognosis and the response to treatment (Datta & Wirrell, 2000; Rantala & Ingalsuo, 1999). Traditionally, clinicians perform various evaluations, including laboratory tests that screen for metabolic insults, electroencephalogram (EEG), and brain magnetic resonance imaging (MRI; Berg, Baca, Loddenkemper, Vickrey, & Dlugos, 2013).Based on the results of such diagnostic tests, many cases involving young children with epilepsy are found to be caused by congenital brain lesions or cerebral insults due to hypoxic or metabolic events (Kramer, 1999; Vasudevan & Levene, 2013). However, the etiology and disease course of early‐onset epilepsy for those with negative brain imaging results or no associated medical history remain ambiguous. In recent years, such cases have been considered to arise from genetic causes as diagnostic molecular genetic testing has been introduced (Deprez, Jansen, & De Jonghe, 2009).
Numerous studies have been performed to identify the causes and clinical phenotypes of early‐onset epilepsy with normal brain imaging (Balciuniene et al., 2019; Demos et al., 2019; Trump et al., 2016). These efforts revealed several genetic factors contributing to early‐onset epilepsy and established various genetic epilepsy syndromes in childhood. In addition, the causative gene is the main determinant of the clinical course and prognosis in genetic epilepsy (Covanis, 2012). However, there are many obstacles making it difficult to reach an exact genetic diagnosis for a particular epilepsy. Early‐onset epilepsy is a group of disorders that is heterogeneous in its cause, clinical course, and neurodevelopmental outcome. The relationship between genotype and phenotype in early‐onset epilepsy is not consistent and is relatively weak (Hani, Mikati, & Mikati, 2015).Thus, it is challenging to identify the genetic cause of early‐onset epilepsy using Sanger sequencing of a specific gene.
Genetic epilepsy is now considered a comprehensive concept, including cases related to congenital structural abnormalities and metabolic disorders caused by genetic mutation. It is easy to diagnose epilepsies, where brain structural abnormalities are an obvious cause of seizures, using brain MRI. However, patients with normal brain imaging require an approach that can detect the cytogenetic cause at the molecular level. Recently, genetic defects have been presumed to cause more than one‐third of all epilepsy cases, and the probability is much higher in early‐onset epilepsies occurring at <2 years of age (Hani et al., 2015). Many studies have reported various clinical conditions likely to be diagnosed as a genetic cause in pediatric epilepsy and have revealed new candidate genes that may be a cause of epilepsy (Ream & Patel, 2015; Sands & Choi, 2017). These studies have reported different results for the diagnostic rates and related factors, depending on the study population. Therefore, we selected patients with early‐onset epilepsy having normal brain imaging findings as a target for investigating genetic causes of epilepsy in Korea.
Facilitated by the remarkable advancements in molecular genetic technologies in recent decades, various genetic testing methods have been developed to elucidate the genetic cause of epilepsy (Orsini, Zara, & Striano, 2018). Among them, next‐generation sequencing (NGS) is one of the most useful diagnostic tools because of its cost‐effectiveness and substantial diagnostic yield in early‐onset epilepsy. Moreover, considering the high heterogeneity in genotype and phenotype of the patients with early‐onset epilepsy, NGS may offer significant time‐savings (Lemke et al., 2012). The differentiating point of NGS compared to other genetic tests is its ability to evaluate multiple genes at the same time, conferring a substantial advantage for time to diagnosis (Della Mina et al., 2015; Moller, Dahl, & Helbig, 2015).
The primary objective of this study was to identify genetic variants in patients with early‐onset epilepsy, using NGS‐based targeted gene panel testing. We evaluated the diagnostic yield of targeted gene testing in populations with presumed genetic epilepsy, and determined the most likely causal candidates. Additionally, we demonstrated the genotype‐phenotype correlation of various genes detected in this study.
2. METHODS
2.1. Ethical compliance
This study was approved by the Institutional Review Board of Samsung Seoul Hospital (IRB No. 2014‐07‐001‐004 and 2019‐01‐030), and written informed consent was obtained from the parents of all pediatric patients included in this study.
2.2. Patients
This study included Korean pediatric patients with early‐onset epilepsy between June 2014 and May 2018 at Samsung Medical Center (Seoul, Korea). Cases of early‐onset epilepsy were defined as those where the first seizure developed before 2 years of age. We excluded symptomatic or well‐defined syndromes having epilepsy as one of their symptoms with the following criteria: (a) presence of structural abnormalities on brain MRI or computed tomography (CT), (b) confirmed chromosomal abnormality‐related syndromes accompanying epileptic seizure such as Down syndrome, Angelman syndrome, or Wolf–Hirschhorn syndrome, and (c) insults associated with metabolic disorders or perinatal hypoxic damage.
We reviewed the medical records of all patients retrospectively and collected data on variable clinical characteristics including gender, age of seizure onset, type of seizures, frequency of seizures, antiseizure medications (ASMs), family history of epilepsy or febrile seizure, developmental milestones, and results of EEG and brain MRI. Neurodevelopmental states were assessed clinically and classified by pediatric neurologists at the last follow‐up. Cognitive impairment was assessed according to the full‐scale intelligence quotient from the Korean Wechsler intelligence scale for children (K‐WISC‐IV). In patients whose intelligence assessment was impossible, developmental milestones were divided into four categories based on the parents’ answers in the Korean Developmental Screening Test (K‐DST) administered by pediatric neurologists. Categories included normal, mild delay (milestone delayed by <6 months), moderate delay (milestone delayed by 6 months to 1 year), and severe delay (milestone delayed by >1 year; Suh, Sohn, Kim, Jung, & Eun, 2016).
2.3. Clinical diagnosis of epileptic syndrome
Based on electrical and clinical characteristics, patients with various epilepsies and epilepsy syndromes ranging from self‐limited to intractable epilepsy were included in this study. Clinical diagnosis of epilepsy syndrome was based on the guidelines of the Commission on Classification and Terminology of the International League Against Epilepsy (ILAE; http://www.epilepsydiagnosis.org/; Scheffer et al., 2017).
2.4. DNA extraction, library preparation, and exome sequencing
2.4.1. Variant calling and filtering
Detailed procedures for DNA processing and variant calling and filtering are reported in the Supporting Information.
2.4.2. Targeted epilepsy genes
We performed genetic testing using NGS‐based targeted epilepsy gene panels that varied slightly according to the time of test performance. Given the prolonged period of this study, the gene list was modified to reflect the tendency and newly listed genes. The genes known to be associated with early‐onset epileptic encephalopathy (EOEE), febrile seizure, familial epilepsy, genetic generalized epilepsy, or seizure accompanied by intellectual disabilities were selected based on genes implicated in epilepsy in the literature and online databases (http://www.ncbi.nlm.nih.gov/omim). Among those genes, we selected and analyzed 76 genes that were common to the panels. The list of targeted genes in the panels is provided in Table S1.
2.4.3. Pathogenicity and interpretation of detected variants
Detected variants were classified as “pathogenic (PV),” “likely pathogenic (LPV),” or “uncertain significance (VUS),” according to the international guidelines of the American College of Medical Genetics (ACMG) and Association for Molecular Pathology (AMP; Richards et al., 2015).
2.5. Statistical analysis
Statistical analysis was performed using SPSS for Windows (version 23, SPSS). Fisher's exact test and Mann–Whitney test were used to compare the gender preference, incidence of genetic variant, intractability of epilepsy, presence of severe developmental delays, family history of seizures, prolonged seizures, clustered seizures, types of seizure, number of ASMs, and average age of seizure onset. Logistic regression analysis was used to predict clinical factors affecting the detection of disease‐causing variants. The probability cutoff of statistical significance was set at p < .05.
3. RESULTS
3.1. Clinical characteristics of patients with early‐onset epilepsy
One‐hundred‐sixteen pediatric patients with early‐onset epilepsy were included in this study, consisting of 54 females and 62 males. The average age of seizure onset was 0.58 ± 0.43 (M ± SD) years (range: 1 day after birth–2 years of age). Seventy‐one patients (61.2%) had their first seizure before 6 months of age, 29 patients (25%) between 6 months and 1 year old, and the remainder (n = 16, 13.8%) after 1 year of age. Delayed development was present in 66 patients (56.9%), and 54 of them showed severe global development delay (81.8%). Family history of epilepsy was reported in 31 patients (26.7%). Detailed clinical characteristics of the patients are summarized in Table 1.
Table 1.
Clinical characteristics of the participants for targeted gene panel testing (n = 116)
| Female: Male | 54 (46.6): 62 (53.4) |
|---|---|
| Age of seizure onset (year) |
|
| <6 months | 71 (61.2) |
| 6 months to 1 year | 29 (25.0) |
| 1–2 years | 16 (13.8) |
| Developmental delay | |
| Normal development | 49 (42.2) |
| Mild | 9 (7.8) |
| Moderate | 3 (2.6) |
| Severe | 54 (46.6) |
| Unknown a | 1 (0.9) |
| Family history of epilepsy | 31 (26.7) |
| Electroclinical syndrome | |
| Dravet syndrome | 21 (18.1) |
| EOEE | 29 (25.0) |
| GEFSP | 9 (7.8) |
| BFNS | 4 (3.4) |
| BFIE | 13 (11.2) |
| Unclassified | 33 (28.4) |
The data are described as numbers (%).
Abbreviations: BFIE, benign familial infantile epilepsy; BFNS, benign familial neonatal seizure; EOEE, early‐onset epileptic encephalopathy; GEFSP, generalized epilepsy with febrile seizure plus.
One patient was not exactly evaluated for development due to follow‐up loss.
A wide spectrum of epilepsy syndromes was included in this study. Among self‐limiting epilepsy cases, four patients with benign familial neonatal seizure (BFNS) and 13 patients with benign familial infantile epilepsy (BFIE) were reported. Among intractable epilepsy syndromes, 21 patients with Dravet syndrome and 29 patients with EOEE were included. In patients with EOEE, 10 had infantile spasms in infancy, and seven of them had evolved to Lennox–Gastaut syndrome (LGS). In addition, nine other patients were diagnosed with generalized epilepsy with febrile seizure plus (GEFSP), and the rest were not classified as a specific genetic epilepsy syndrome (Table 1).
3.2. Detection of variants using a targeted gene panel
The mean coverage of candidate genes included in three targeted gene panels used in this study was 99.20%, 99.22%, and 95.12% per base pair with ≥10×, respectively. Presumed disease‐causing variants were identified in 40 cases. Therefore, the overall diagnostic yield of the targeted gene panel testing was 34.5% (Table 2). Nineteen variants were classified as PV, and 21 were interpreted as LPV according to the ACMG guidelines (Figure 1a). There were 12 different novel variants in 13 patients. Figure 1b shows the disease‐causing variants detected in 40 patients and their electroclinical diagnosis. The diagnostic yield varied with each epilepsy syndrome (Figure 1c). We found 11 PVs in 13 patients with BFIE (84.6%), which was the highest among all epilepsy syndromes. Dravet syndrome followed BFIE with 81% (n = 17/21), and BFNS had a 50% positive rate of detection (n = 2/4). With respect to EOEE, approximately 27% of the patients had a disease‐causing variant (n = 8/29).
Table 2.
Clinical characteristics and detected variants of the patients with presumed disease‐causing variants (n = 40)
| Patient | Sex | Onset age | Diagnosis age | Electroclinical syndrome | Type of seizures | Variant | FHx of seizure | DD/ID | Febrile seizure | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Inherit | Refseq a | c.DNA | Amino acid | ACMG b | |||||||||
| E‐001 | F | 4 m | 2 y 10 m | Dravet syndrome |
FT GT |
PCDH19 | XL | NM_001184880.1 | c.370G>T c | p.(Asp124Tyr) | LP | Mother | Mild | Yes |
| E‐002 | F | 4 m | 2 y 9 m | Dravet syndrome |
FIA FHC GAA |
SCN1A | AD | NM_001165963.2 | c.5596del c | p.(Asp1866fIlesTer11) | P | None | Severe | Yes |
| E‐010 | F | 6 m | 7 y | Dravet syndrome |
FHC GAA |
SCN1A | AD | NM_001165963.2 | c.602+1G>C c | NA | LP | None | Severe | Yes |
| E‐037 | M | 5 m | 9 y 9 m | Dravet syndrome |
GM GMA FHC |
SCN1A | AD | NM_001165963.2 | c.1088C>G | p.(Thr363Arg) | P | None | Severe | Yes |
| E‐039 | F | 11 m | 9 y 8 m | Dravet syndrome |
GTC FTC |
PCDH19 | XL | NM_001184880.1 | c.1998del | p.(Asp667MetfsTer9) | LP | None | Mild | Yes |
| E‐044 | M | 5 m | 12 y 7 m | Dravet syndrome |
GTC FHC FIA |
SCN1A | AD | NM_001165963.2 | c.580G>A | p.(Asp194Asn) | LP | None | Severe | Yes |
| E‐051‐P | M | 4 m | 1 y 2 m | BFIE |
FIA GTC |
PRRT2 | AD | NM_145239.2 | c.971del | p.(Gly324GlufsTer13) | P | Sibling | Normal | None |
| E‐051‐S | F | 4 m | 7 y 3 m | BFIE |
FIA GTC |
PRRT2 | AD | NM_145239.2 | c.971del | p.(Gly324GlufsTer13) | P | Sibling | Normal | None |
| E‐076 | M | 6 m | 16 y 4 m | Dravet syndrome |
GTC FHK FIA |
SCN1A | AD | NM_001165963.2 | c.1178G>A | p.(Arg393His) | P | None | Severe | Yes |
| E‐079 | M | 3 d | 2 y 9 m | BFNS | GT | KCNQ2 | AD | NM_172107.3 | c.1771C>T c | p.(Gln591Ter) | P | Sibling | Normal | None |
| E‐092 | F | 6 m | 11 y 4 m | Dravet syndrome |
GT FIA FM w V |
SCN1A | AD | NM_001165963.2 | c.2589+3A>T | NA | P | None | Severe | Yes |
| E‐100 | M | 2 d | 9 y 3 m | Dravet syndrome |
FHC FHK GT |
SCN1A | AD | NM_001165963.2 | c.4723C>T | p.(Arg1575Cys) | LP | None | Severe | Yes |
| E‐103 | F | 6 m | 6 y 2 m | Dravet syndrome |
FHC GM GTC |
SCN1A | AD | NM_001165963.2 | c.264+5G>C | NA | LP | Aunt | Severe | Yes |
| E‐104 | M | 5 m | 8 y 11 m | Dravet syndrome |
FHC FM w V GTC |
SCN1A | AD | NM_001165963.2 | c.4242C>G c | p.(Asn414Lys) | LP | None | Severe | Yes |
| E‐121 | M | 3 m | 20 y | Dravet syndrome |
GM GT |
SCN1A | AD | NM_001165963.2 | c.846del c | p.(Thr283ProfsTer10) | LP | None | Severe | Yes |
| E‐122 | M | 4 m | 10 y 3 m | EOEE |
IS FM GT |
ARX | XLR | NM_139058.2 | c.1146G>C c | p.(Lys382Asn) | LP | Cousin | Severe | None |
| E‐129 | M | 3 m | 17 y 6 m | Dravet syndrome |
GT GM FHC |
SCN1A | AD | NM_001165963.2 | c.2589+3A>T | NA | LP | None | Severe | Yes |
| E‐142 | F | 6 m | 11 m | Dravet syndrome |
GTC FIA |
SCN1A | AD | NM_001165963.2 | c.235G>A | p.(Asp79Asn) | LP | Cousin | Moderate | Yes |
| E‐147 | F | 1 m | 10 y | EOEE |
IS GM FC |
STXBP1 | AD | NM_003165.3 | c.994_1003del c | p.(Lys332LeufsTer21) | P | None | Severe | None |
| E‐153 | F | 5 m | 8 y 4 m | Dravet syndrome |
FHC FIA GTC |
SCN1A | AD | NM_001165963.2 | c.2415+5G>A | NA | LP | None | Severe | Yes |
| E‐155 | M | 5 m | 7 m | BFIE |
FIA FHC |
PRRT2 | AD | NM_145239.2 | c.649dupC | p.(Arg217ProfsTer8) | P | None | Normal | None |
| E‐170 | F | 4 m | 5 m | EOEE | GT | KCNQ2 | AD | NM_172109.3 | c.773A>G | p.(Asn258Ser) | LP | None | Moderate | None |
| E‐180 | F | 6 m | 8 y 5 m | Dravet syndrome |
GT FHC |
SCN1A | AD | NM_001165963.2 | c.2792G>A | p.(Arg931His) | LP | Aunt | Severe | Yes |
| E‐199 | M | 6 m | 10 y | BFIE/PKD |
FIA GT |
PRRT2 | AD | NM_145239.2 | c.649dupC | p.(Arg217ProfsTer8) | P | None | Normal | None |
| E‐200 | F | 6 m | 10 m | BFIE | GT | PRRT2 | AD | NM_145239.2 | c.650del | p.(Arg217GlnfsTer12) | P | None | Normal | None |
| E‐204 | M | 9 m | 1 y 10 m | EOEE |
GA GTC |
SCN8A | AD | NM_014191.3 | c.778T>G c | p.(Phe260Val) | LP | None | Severe | None |
| E‐214 | M | 10 m | 12 y 4 m | EOEE |
GM GT FIA |
ARX | XLR | NM_139058.2 | c.1135C>T c | p.(Arg379Cys) | LP | Father | Severe | None |
| E‐218 | M | 4 m | 5 m | BFIE | GTC | PRRT2 | AD | NM_145239.2 | c.649dupC | p.(Arg217ProfsTer8) | P | Sibling | Normal | None |
| E‐220‐P | F | 3 m | 5 m | BFIE |
GTC FC |
PRRT2 | AD | NM_145239.2 | c.649dupC | p.(Arg217ProfsTer8) | P | Sibling | Normal | None |
| E‐220‐S1 | F | 3 m | 6 y 4 m | BFIE | GT | PRRT2 | AD | NM_145239.2 | c.649dupC | p.(Arg217ProfsTer8) | P | Sibling | Normal | None |
| E‐221 | M | 6 m | 18 y 7 m | Dravet syndrome |
FHC GTC FIA |
SCN1A | AD | NM_001165963.2 | c.5341T>G | p.(Tyr1781Asp) | LP | Cousin | Severe | Yes |
| E‐230 | M | 12 m | 3 y 7 m | GEFSP | GT | SCN1A | AD | NM_001165963.2 | c.5671G>T c | p.(Glu1891Ter) | P | None | Normal | Yes |
| E‐234 | F | 4 m | 3 y | Unclassified |
GT FHC |
DEPDC5 | AD | NM_001242896.1 | c.3802C>T | p.(Arg1268Ter) | LP | None | Normal | Yes |
| E‐244 | M | 5 d | 3 m | BFNS |
FHC GT |
SCN2A | AD | NM_021007.2 | c.4712T>C | p.(Ile1571Thr) | LP | None | Normal | None |
| E‐251 | F | 4 m | 1 y | BFIE |
FIA GT |
PRRT2 | AD | NM_145239.2 | c.649dupC | p.(Arg217ProfsTer8) | P | Aunt | Normal | None |
| E‐258‐P | M | 2 m | 6 m | EOEE |
FIA IS |
ARX | XLR | NM_139058.2 | c.1146G>C c | p.(Lys382Asn) | LP | Cousin | Severe | None |
| E‐283 | M | 4 m | 5 m | BFIE | GT | PRRT2 | AD | NM_145239.2 | c.649dupC | p.(Arg217ProfsTer8) | P |
Mother Cousin |
Normal | None |
| E‐294 | M | 4 m | 4 m | BFIE |
FHC GTC |
PRRT2 | AD | NM_145239.2 | c.649dupC | p.(Arg217ProfsTer8) | P | Sibling | Normal | None |
| E‐391 | M | 2 d | 2 m | EOEE |
FIA GT |
SCN2A | AD | NM_021007.2 | c.5645G>A | p.(Arg1882Gln) | P | None | Severe | None |
| E‐392 | M | 1 d | 1 m | EOEE |
GT FT |
SCN2A | AD | NM_021007.2 | c.4609A>T | p.(Ile537Phe) | LP | None | Severe | None |
Abbreviations: AD, autosomal dominant; BFIE, benign familial infantile epilepsy; BFNS, benign familial neonatal seizure; d, days; DD, developmental delay; EOEE, early‐onset epileptic encephalopathy; FHC, focal hemiclonic; FHK, focal hyperkinetic; FHx, family history; FIA, focal impaired awareness; FM w V, focal motor with version; FM, focal motor; FT, focal tonic; GAA, generalized atypical absence; GEFSP, generalized epilepsy with febrile seizure plus; GM, generalized myoclonic; GMA, generalized myoclonic absence; GT, generalized tonic; GTC, generalized tonic clonic; ID, intellectual disability; IS, infantile spasms; LP, likely pathogenic; m, months; NA, not available; P, pathogenic; PKD, paroxysmal kinesigenic dyskinesia; XL, X‐linked; XLR, X‐linked recessive; y, year.
The sequencing reads were aligned to the human genome reference (GRCh37: Genome Reference Consortium human build 37).
Identified variants were classified according to the standards and guidelines by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Richards et al., 2015).
Novel variant not reported previously.
Figure 1.
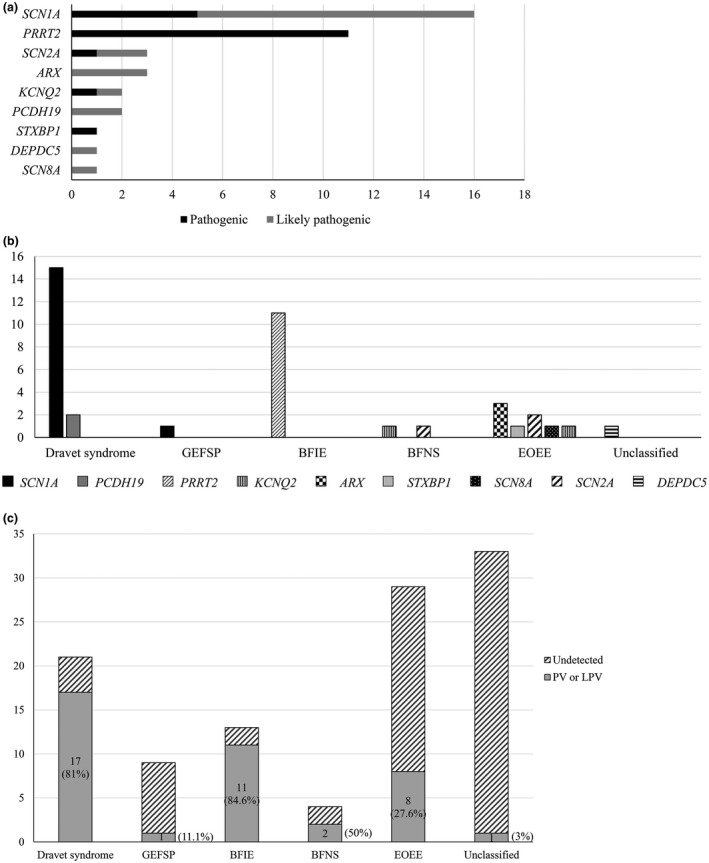
Detection of disease‐causing variants using a targeted gene panel (a) Forty disease‐causing variants were found in 116 patients with early‐onset epilepsy developed before 2 years of age. There were 19 pathogenic variants and 21 likely pathogenic variants. SCN1A variants, which are associated with Dravet syndrome and generalized epilepsy with febrile seizure plus, were most common (n = 16), and PRRT2 variants, which are associated with BFIE, were the second most common (n = 11). (b) Detection of disease‐causing variants and electroclinical syndrome. Among the patients that were genetically confirmed as having Dravet syndrome, 15 had a SCN1A mutation and two had a PCHD19 mutation. All patients having a PRRT2 mutation were diagnosed with BFIE. KCNQ2 mutations were found in patients with BFNS and EOEE. Others included ARX, SCN2A, STXBP1, and SCN8A in patients with EOEE, and a DEPDC5 mutation in a patient with intractable focal epilepsy. (c) Diagnostic yield in each epilepsy syndrome. BFIE showed the highest diagnostic rate with 84.6% (n = 11/13) and Dravet syndrome followed that with 81% (n = 17/21). BFNS had a 50% positive rate of detection (n = 2/4) and about a quarter of patients with EOEE had a disease‐causing variant (n = 8/29, 27.6%). BFIE, benign familial infantile epilepsy; BFNS, benign familial neonatal seizure; GEFSP, generalized epilepsy with febrile seizure plus; EOEE, early‐onset epileptic encephalopathy
In addition, 27 other variants were found and classified as VUS. These included genes related to channelopathy, encoding sodium and potassium channels. Table S2 shows the VUS and electroclinical diagnosis in the patients.
The diagnostic yield differed depending on the age of seizure onset and the developmental delay. In 71 patients whose seizures developed before 6 months of age, 36 disease‐causing variants were found, with a positive hit‐rate of 50.7%. The remaining four disease‐causing variants were found in patients with first seizure onset between 6 months and 1 year of age (4/29, 13.8%). In patients whose seizure onset was before 6 months of age, statistically more frequent disease‐causing variants were found (Figure 2a; p < .001).The presence or absence of developmental delay did not change the detection rate of variants in presumed disease‐causing genes statistically between the two groups (Figure 2b; p = .779). We identified 24 disease‐causing variants in 66 patients with developmental delay (36.4%), and most of them (n = 19) were found in those with severe developmental delays. In 49 patients reaching normal developmental milestones, 16 disease‐causing variants were found (32.7%).
Figure 2.
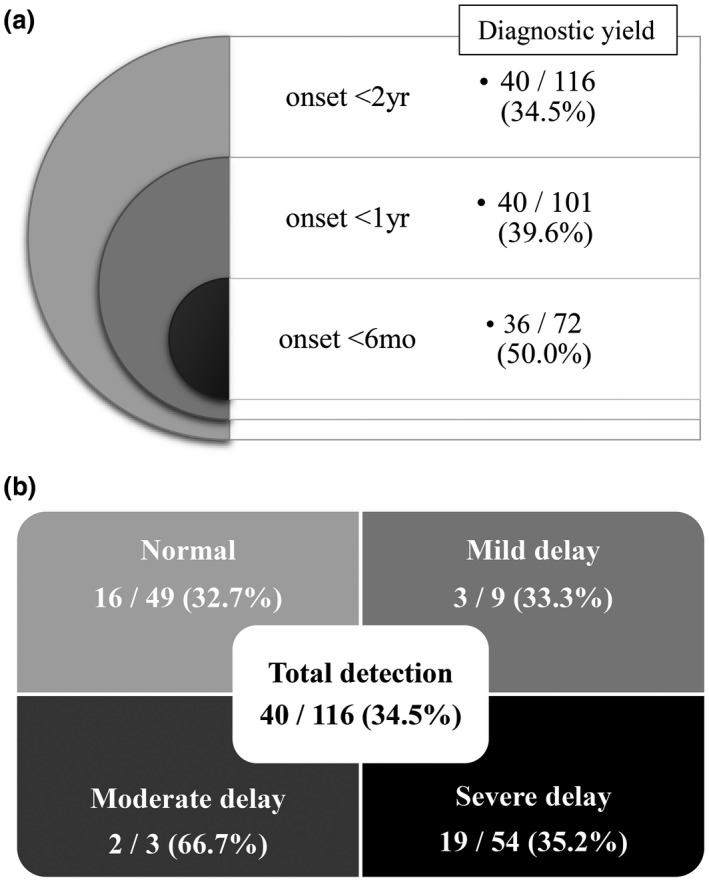
Diagnostic yield depending on the age of seizure onset and the developmental delay (a) Diagnostic yield of the targeted epilepsy gene panel was different among age groups of seizure onset (p < .001). The earlier the age of seizure onset, the higher the diagnostic rate of gene panel testing. Approximately half of patients with seizure onset before the age of 6 months were revealed to have causative genetic variants. (b) The diagnostic yield trended differently according to the degree of developmental delay. Twenty‐four had disease‐causing variants among 66 patients with delayed development (24/66, 36.4%), however, without statistical significance (p = .779)
In addition, we classified the patients according to the intractability of their seizures, defined as having uncontrolled symptoms despite more than two ASMs with reasonable management. We analyzed the difference in the presence of the disease‐causing variants between these classifications. There was no significant relationship between intractable epilepsy and the detection of a disease‐causing variant (p = .663).
Based on the results of multivariate logistic regression analysis, the age of seizure onset (p < .001) and family history of epilepsy (p = .012) showed statistically significant relationships with the presence of disease‐causing variants (Table 3). Among the patients (n = 71) with early infantile onset seizure (onset age ≤6 months old), the diagnostic rate was highest in those accompanied by severe delays in developmental milestones (20/35, 57.1%). This included 14 SCN1A variants, two SCN2A variants, two ARX variants, one KCNQ2 variant, and one STXBP1 variant.
Table 3.
The logistic regression analysis for clinical factors associated with the detection of disease‐causing variants
| Clinical factors | Coefficient | Standard error | p‐value | Odds ratio | OR 95% CI | |
|---|---|---|---|---|---|---|
| Lower | Upper | |||||
| Sex | 0.105 | 0.457 | .818 | 1.111 | 0.454 | 2.718 |
| Onset age | ‐3.207 | 0.900 | .000 | 0.040 | 0.007 | 0.236 |
| Developmental delay | 0.144 | 0.518 | .782 | 1.155 | 0.418 | 3.189 |
| Family history of epilepsy | 1.356 | 0.537 | .012 | 3.880 | 01.355 | 11.115 |
| Intractability | 0.552 | 0.520 | .288 | 1.737 | 0.627 | 4.814 |
3.3. Heterogeneity of genotype and phenotype
We analyzed the correlation of detected genes with the clinical characteristics of patients in this study. In patients with developmental delay, six genes includingSCN1A, SCN2A, KCNQ2, ARX, PCDH19, and STXBP1 were detected. Patients whose first seizure was febrile harbored variants in SCN1A, PCDH19, and DEPDC5 genes, and those with a family history of epilepsy had variants in SCN1A, KCNQ2, ARX, and PRRT2. Patients with early‐onset of seizure before 6 months of age had variants in eight genes including SCN1A, SCN2A, KCNQ2, PCDH19, ARX, PRRT2, DEPDC5, and STXBP1 (Figure 3). Some genes are associated with multiple clinical characteristics and others were exclusive.
Figure 3.
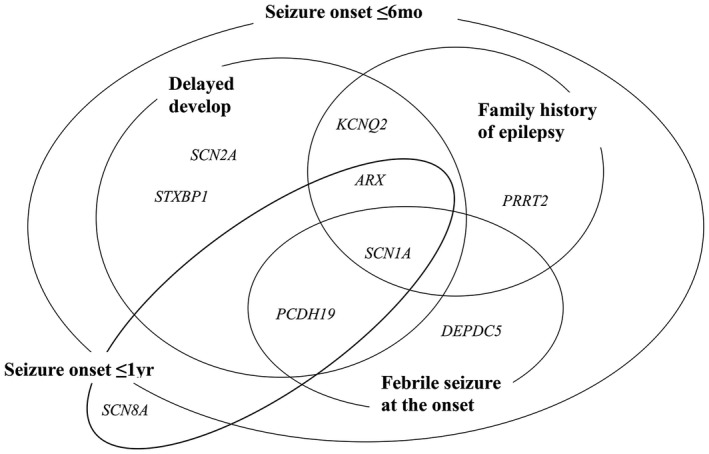
Detected causative genes overlap or show the difference according to clinical characteristics
3.3.1. Dravet syndrome
Dravet syndrome was the most common epilepsy syndrome as a single disorder in this study, affecting 21 patients. Their average age of seizure onset was 5.9 ± 3.0 months old (range: 2 days after birth–16 months). Table 4A summarizes the demographic and clinical data of the patients with Dravet syndrome.
Table 4.
Clinical characteristics of patients with Dravet syndrome and benign familial infantile epilepsy
| Total patients with Dravet syndrome | 21 |
| (A) | |
| Female: Male | 14: 7 |
| Age of seizure onset (months) | 5.9 ± 3.0 (range 2 days–16) |
| SMEI: SMEB | 14: 7 |
| Developmental delay | |
| Normal to mild | 5 (23.8) |
| Severe | 16 (76.2) |
| Numbers of seizure type | |
| 1 | None |
| 2 | 7 (33.3) |
| ≥3 | 14 (66.7) |
| Antiseizure medications | |
| ≤2 | 9 (42.9) |
| ≥3 | 12 (57.1) |
| Initial interictal electroencephalogram | |
| Normal | 12 (57.1) |
| Generalized epileptiform discharges | 1 (4.5) |
| Focal epileptiform discharges | 8 (38.1) |
| Detection of disease‐causing variants | 17 (81.0) |
| Pathogenic | 5 (23.8) |
| Likely pathogenic | 12 (57.4) |
| Total patients with BFIE | 13 |
| (B) | |
| Female: Male | 7: 6 |
| Age of seizure onset (months) | 4.8 ± 3.6 (range 3–8) |
| Family history of seizure | 8 (61.5) |
| Family history of PKD | 3 (23.1) |
| Type of seizure | |
| Focal impaired awareness | 6 (46.2) |
| Focal hemiclonic | 2 (15.4) |
| Generalized tonic or tonic‐clonic | 5 (38.5) |
| Antiseizure medications | 13 (100.0) |
| Monotherapy | 6 (46.2) |
| Levetiracetam | 4 |
| Carbamazepine | 1 |
| Valproic acid | 1 |
| Polytherapy | 7 (54.8) |
| Age of last seizure (months) | 8.1 ± 5.3 (range 4–24) |
| PRRT2 variant | 11 (84.6) |
| c.649dupC; p.(Arg217ProfsTer8) | 8 (61.5) |
| c.971del; p.(Gly324GlufsTer13) | 2 (15.4) |
| c.650del; p.(Arg217GlnfsTer12) | 1 (7.7) |
Data are M ± SD or n (%) values.
Abbreviations: BFIE, benign familial infantile epilepsy; MRI, magnetic resonance imaging; PKD, paroxysmal kinesigenic dyskinesia; SMEB, severe myoclonic epilepsy of infancy‐borderland; SMEI, severe myoclonic epilepsy of infancy.
Four variants in patients E‐002, E‐043, E‐121, E‐221 were novel. Among variant effect classifications, missense variants were the most commonly detected (n = 9) followed by splicing variants (n = 5). More variants were located in a functionally important region of the SCN1A protein, in severe myoclonic epilepsy of infancy (SMEI) patients. In 12 SMEI patients with disease‐causing SCN1Avariant, eight variants (8/12, 66.7%) were located in the S4–S6 region. There was only a single patient (1/3, 33.3%) in the severe myoclonic epilepsy of infancy––borderland (SMEB) group with a truncating variant located in the voltage‐sensor S4 DI region.
3.3.2. Benign familial infantile epilepsy
This study included 13 patients who were diagnosed with BFIE, including two familial cases (four individuals). They had their first seizure at an average age of 4.8 ± 3.6 months (Table 4B). There were eight patients who had family members with infantile epilepsy, and three patients had family members with paroxysmal kinesigenic dyskinesia (PKD). A PV of the PRRT2 gene was found in 11 patients, and c.649dupC; p(Arg217ProfsTer8) was the most common nonsense variant detected in eight patients. Two other frame shift deletions of PRRT2 were detected in three patients, and there was no difference in their clinical manifestations. All patients had normal brain MRIs and developmental milestones. A focal impaired awareness seizure was observed in six patients, and a generalized seizure occurred in five patients. Two other patients showed a focal hemiclonic seizure. All patients were treated with ASMs for the seizures, and seven patients were receiving two or more ASMs. All patients, except E‐199 having coexisting PKD, had their last seizure before 1 year of age. Patient E‐199 had his last seizure at the age of two and was taking oxcarbazepine for PKD that developed in the teen years.
3.3.3. Early‐onset epileptic encephalopathy
Eight PVs or LPVs were detected in 29 patients with EOEE excluding Dravet syndrome. Among them, three patients had ARX variants and the other five had SCN2A (n = 2), KCNQ2, STXBP1, and SCN8A variants (Figure 1b). Causative genes were not found using NGS‐based targeted sequencing in the remaining patients (n = 21).
ARX gene
Three patients with ARX variants were born at term and had an uncomplicated birth. Two children (E‐122 and E‐258‐P) were maternal cousins and shared the same variant of the ARX gene. Both had infantile spasms at the age of 4 months and showed severe developmental delay. We detected a novel LPV (c.1146G>C; p.(Lys382Asn)) in the ARX gene in the patients and their unaffected mothers (Figure S1).
KCNQ2 and SCN2A: Two channelopathy‐related genes
We found two variants in the KCNQ2 gene, which associated with different clinical manifestations in two unrelated patients. The c.1771C>T variant of the KCNQ2 gene was detected in a patient with BFNS (E‐079). The other patient (E‐170) having a LPV (c.773A>G; p.(Asn258Ser)) in the same gene presented with EOEE.
Two variants (c.4712T>C and c.2969A>T) in the SCN2A gene were found in two unrelated individuals with BFNS (E‐244 in Table 2 and E‐203 in Table S2). They had their first seizure on the 5th and 7th day of their life, respectively, which were generalized tonic or focal hemiclonic. Patient E‐203 with the c.2969A>T variant had no more seizure after the neonatal period, while the other patient (E‐244) with the c.4712T>C variant had the last seizure at the age of 4 months. They developed normally. However, the c.2969A>T variant was classified as VUS because the parents’ genotype was not available. The c.4712T>C variant was confirmed as de novo via Sanger sequencing of both parents and was previously reported a PV for Ohtahara syndrome (Liu et al., 2018). In contrast, two patients with EOEE and a LPV or PV (c.5645G>A in E‐391 and c.4609A>T in E‐392) in the SCN2A gene had very frequent seizures beginning within 2 days after birth that were refractory to multiple ASMs. There were intermittent background suppression and multifocal epileptiform discharges on their EEGs in the neonatal period, and the background was disorganized as they aged.
4. DISCUSSION
We performed NGS‐based targeted epilepsy gene panel testing on 116 individuals having early‐onset epilepsy with their first seizure occurring before 2 years of age. Disease‐causing variants were detected in one‐third of subjects. This study showed diagnostic efficacy comparable to other NGS‐based genetic studies performed in Chinese and Danish population and an even higher detection rate in medication‐refractory or early‐onset epilepsy in children(Butler, da Silva, Alexander, Hegde, & Escayg, 2017; de Kovel et al., 2016; Lemke et al., 2012; Segal et al., 2016; Zhang et al., 2017). In particular, the detection rate was higher with a younger age of seizure onset and family history of epilepsy, not influenced by developmental delay or intractability of seizures. This finding identifies an eligible population for genetic testing for epilepsy.
Several previous reports revealed the genetic cause of pediatric epilepsy accompanying developmental disorders, however, few studies were conducted on a population similar to ours, which included patients with epilepsy occurring at an early age and normal brain imaging findings (Heyne et al., 2019; Ortega‐Moreno et al., 2017). Our study provided data from a large patient cohort of a single ethnicity with early‐onset epilepsy before 2 years of age, we believe that the data are worth comparing to those from other studies. Several Korean studies analyzed the genetic causes of pediatric epilepsy without structural brain lesions. Two studies investigated 74 and 278 patients with epileptic encephalopathy that developed before 3 years of age and found, a genetic cause in 37.8% (n = 28/74) and 37.1% (n = 103/278) of the patients, respectively, using a NGS panel including 172 candidate genes (Ko et al., 2018; Rim et al., 2018). Although our diagnostic yield was somewhat lower than that reported in those studies, our patient cohort was comprehensive, including not only patients with intractable epileptic encephalopathy but also those with benign epilepsy. The genes identified as disease‐causing genes in our patients were also different from those identified in other studies. In our study, the PRRT2 gene, which causes BFIE, was noted second most commonly unlike other studies.
In this study, we could deduce that the age of seizure onset was associated with notable differences in the detection rate of causative genes. Patients with an onset of epilepsy before the age of 6 months and having a family member affected by epilepsy had a high probability of finding a genetic cause. In patients with early infantile onset of a seizure before 6 months of age, half (36/72, 50%) possessed disease‐causing variants. Conversely, no disease‐causing variants were detected in epilepsy patients whose first seizures occurred between 1 and 2 years of age. Previous studies showed a similar tendency through subgroup analyses (Parrini et al., 2017). Møller et al. reported that PVs were detected in 57% of neonatal onset epilepsy (Moller et al., 2016), while Trump et al. reported the highest diagnostic rate of causative variants in patients with seizure onset before the age of 2 months (30/77, 39%; Trump et al., 2016). A recent large cohort study analyzing clinical data from the Epi4K Consortium also reported that the age of seizure onset is a heritable trait (Ellis et al., 2019). A prospective study in the United States also emphasized the diagnostic value in the genetic diagnosis of early‐onset epilepsy (Butler et al., 2017). Based on these findings, the author assumed that the age of seizure onset was an important factor influencing the probability of positive results in genetic studies.
Clinical manifestations should be considered to be another factor influencing the detection rate of NGS‐based targeted gene panel testing. Previous studies about genetic evaluation of epilepsy reported the diagnostic yield of NGS was higher (17%–33%) in case with severe symptoms (Sands & Choi, 2017). Ream et al. demonstrated that generalized epilepsy and epileptic encephalopathy were associated with positive results of genetic testing (Ream & Mikati, 2014). However, our results did not show the correlation of higher genetic diagnostic yield according to disease severity or developmental delay. In this study, patients with comparatively obvious symptoms representing their particular electroclinical diagnosis, even if it's not necessarily intractable, had a high probability of possessing the disease‐causing variant. Forty‐seven patients were classified as having a clinically established epilepsy syndrome, such as Dravet syndrome, GEFSP, or benign familial epilepsy. Thirty‐one (66%) of the patients with clinically established epilepsy syndromes were confirmed their genetic diagnosis using the NGS‐based targeted gene panel testing. In other 69 patients, including those with unclassified epilepsy and EOEE not categorized into specific established epilepsy syndromes, nine patients (13%) were revealed to have a disease‐causing variant. This finding was concordant with that in a previous study on Korean patients with seizure onset within 1 year of age conducted by Jang et al. (Jang et al., 2019). They showed a variable diagnostic yield among the subgroups that was not proportional to disease severity. In addition, the patients who were not classified into the established phenotypes were likely to display neurocognitive dysfunction or intractable seizures. These differences in diagnostic yield between studies may be influenced by the inclusion criteria for the participants, depending on whether the studies targeted patients with epileptic encephalopathy or all patients with early‐onset epilepsy. This allowed us to recognize that benign epilepsy also accounts for a considerable proportion of the cases of genetic epilepsy and to keep that in mind in the context of pediatric epilepsy.
Genetic diagnosis of epilepsy is useful not only in the detection of causative gene but also to guide therapy. Demos et al. reported that proper genetic diagnosis guided the treatments in 39% (n = 23/59) of patients with early‐onset epilepsy (onset age ≤ 5 years old; Demos et al., 2019). Our results also changed the therapeutic strategy in eight out of 40 patients with identified disease‐causative genes (20%). These findings may also enable diagnosis of familial epilepsies, including early diagnosis of family members with newly developed symptoms and predicting the presence of genetic mutation in other family members. Two patients with a PV of PRRT2 had a younger sibling with the same clinical manifestations, and a quick diagnosis was possible by Sanger sequencing of the variant found in their older siblings. The other two patients with BFIE caused by the PRRT2 gene were able to taper the ASMs after a certain period in anticipation of a favorable prognosis, although they experienced seizures frequently in the early stage of the disease. In our study, four patients with epileptic encephalopathy benefited from our genetic diagnosis. In two boys diagnosed with SCN2A encephalopathy (E‐391 and E‐392) by NGS‐based gene panel testing, we attempted to control seizures using Na+ channel blockers based on other previous studies(Howell et al., 2015; Wolff et al., 2017). The patients responded to the medication with decreasing frequency of seizures. In the other two patients with Dravet syndrome (E‐044 and E‐129) confirmed genetically, the therapeutic plan was changed after the genetic diagnosis was established. The frequency of seizures significantly decreased in both patients when a new ASM (stiripentol) was introduced in one patient (E‐044) and the use of the Na+ channel blocker (oxcarbazepine) was terminated in the other patient (E‐129).
This study highlighted a heterogeneity of genotype and phenotype in epilepsy that has already been suggested in the literature (Carvill et al., 2013; EPGP Collaborative et al., 2013; Parrini et al., 2017). Although patients with a wide spectrum of early‐onset epilepsies were included, various genes were revealed to be the cause of a single electroclinical syndrome. Five different genes were identified as their genetic cause in eight patients with EOEE and two genes were identified in the patients with Dravet syndrome. Moreover, some patients showed conflicting manifestations of epilepsy, even though they harbored a LPV of the same genes, SCN2A and KCNQ2. Although patient E‐244 had the same variant of the SCN2A gene described as the cause of epileptic encephalopathy in previous studies, he showed a benign course with controlled seizures and normal developmental milestones (Liu et al., 2018). Another patient E‐170 having the same variant of the KCNQ2 gene found in a Turkish BFNS family had clinical features consistent with EOEE (Maljevic et al., 2011; Yalcin et al., 2007). This discrepancy was also found in an infant with KCNQ2 encephalopathy due to the c.881C>T variant, which was previously reported in BFNS (Allen et al., 2014).
Aside from 40patients with confirmed genetic diagnosis, genetic causes of epilepsy were not found in two‐thirds of subjects in this study. Negative results in this evaluation did not mean that the patient did not possess a genetic cause of epilepsy or that the detected variants were not the genetic cause. We found 27VUSs in various genes and the pathogenicity of most of these could not be determined because of insufficient information in the literature (Butler et al., 2017; de Kovel et al., 2016; Lemke et al., 2012; Parrini et al., 2017; Ream & Mikati, 2014). Occasionally, clinical features of the patient are not obvious and it is difficult to interpret the variant or assess its clinical significance (Sands & Choi, 2017). Because there are also limitations in published resources and knowledge regarding these interpretations, some genes are not definitely known to be associated with disease (SoRelle, Thodeson, Arnold, Gotway, & Park, 2019; Traynelis et al., 2017). Although their clinical implication is not clear, these have a potential to be proven as genetic causes of epilepsy in the future. For such cases, additional complementary data, including a segregation test of the patient's family members, an adaptation of cumulative data from different sources, and functional validation are needed.
5. CONCLUSION
In conclusion, early‐onset pediatric epilepsy shows a broad spectrum of genetic causes and phenotypes, from benign and self‐limiting to intractable with a devastating course. Genetic investigation in pediatric epilepsy serves to identify causative genes, predict the prognosis, and offer genetic counseling to family members. While numerous technical methods are currently available for genetic diagnosis of epilepsy, selection of a suitable testing method with the highest diagnostic yield is recommended. To improve diagnostic yield, meticulous and precise analysis of clinical features is indispensable. Considering the heterogeneity of genotype and phenotype in early‐onset pediatric epilepsy, NGS‐based targeted gene panel testing is a useful method with hit‐rate 30%–40%due to its time‐saving and cost‐effective aspects. In addition, an effort to identify the cause of undiagnosed patients through further evaluations, including segregation analysis of family members, whole exome or whole genome sequencing, increased coverage, and periodic reanalysis should be done. In the future, we anticipate that a therapeutic approach will be developed based on the genetic diagnosis of epilepsy.
CONFLICTS OF INTEREST
My colleagues and I declare that there is no conflict of interest regarding this work.
AUTHOR CONTRIBUTION
JWL, C‐SK, and JHL conceived and designed the study. JHL enrolled the participants. JWL and JHL analyzed their clinical data. CL and JWL analyzed the data of genetic evaluation. C‐SK reviewed the analysis of genetic variants. JWL drafted the manuscript. JHL approved the submitted version of the manuscript.
Supporting information
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
The authors are very grateful to the patients and their families for their participation in this study and a research coordinator for collecting samples and processing genetic evaluations. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Ministry of Education (No. NRF‐2017R1D1A1B03034685).
Lee J, Lee C, Ki C‐S, Lee J. Determining the best candidates for next‐generation sequencing‐based gene panel for evaluation of early‐onset epilepsy. Mol Genet Genomic Med. 2020;8:e1376 10.1002/mgg3.1376
DATA AVAILABILITY STATEMENT
All accepted manuscripts are required to publish a data availability statement to confirm the presence or absence of shared data.
REFERENCES
- Allen, N. M. , Mannion, M. , Conroy, J. , Lynch, S. A. , Shahwan, A. , Lynch, B. , & King, M. D. (2014). The variable phenotypes of KCNQ‐related epilepsy. Epilepsia, 55(9), e99–e105. 10.1111/epi.12715 [DOI] [PubMed] [Google Scholar]
- Balciuniene, J. , DeChene, E. T. , Akgumus, G. , Romasko, E. J. , Cao, K. , Dubbs, H. A. , … Abou Tayoun, A. (2019). Use of a dynamic genetic testing approach for childhood‐onset epilepsy. JAMA Network Open, 2(4), e192129 10.1001/jamanetworkopen.2019.2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, A. T. , Baca, C. B. , Loddenkemper, T. , Vickrey, B. G. , & Dlugos, D. (2013). Priorities in pediatric epilepsy research: Improving children's futures today. Neurology, 81(13), 1166–1175. 10.1212/WNL.0b013e3182a55fb9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, K. M. , da Silva, C. , Alexander, J. J. , Hegde, M. , & Escayg, A. (2017). Diagnostic yield from 339 epilepsy patients screened on a clinical gene panel. Pediatric Neurology, 77, 61–66. 10.1016/j.pediatrneurol.2017.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill, G. L. , Heavin, S. B. , Yendle, S. C. , McMahon, J. M. , O'Roak, B. J. , Cook, J. , … Mefford, H. C. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nature Genetics, 45(7), 825–830. 10.1038/ng.2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covanis, A. (2012). Epileptic encephalopathies (including severe epilepsy syndromes). Epilepsia, 53(Suppl 4), 114–126. 10.1111/j.1528-1167.2012.03621.x [DOI] [PubMed] [Google Scholar]
- Datta, A. N. , & Wirrell, E. C. (2000). Prognosis of seizures occurring in the first year. Pediatric Neurology, 22(5), 386–391. 10.1016/S0887-8994(00)00130-2 [DOI] [PubMed] [Google Scholar]
- de Kovel, C. G. F. , Brilstra, E. H. , van Kempen, M. J. A. , van‘t Slot, R. , Nijman, I. J. , Afawi, Z. , … Koeleman, B. P. C. (2016). Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Molecular Genetics & Genomic Medicine, 4(5), 568–580. 10.1002/mgg3.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demos, M. , Guella, I. , DeGuzman, C. , McKenzie, M. B. , Buerki, S. E. , Evans, D. M. , … Farrer, M. J. (2019). Diagnostic Yield and treatment impact of targeted exome sequencing in early‐onset epilepsy. Frontiers in Neurology, 10, 434 10.3389/fneur.2019.00434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deprez, L. , Jansen, A. , & De Jonghe, P. (2009). Genetics of epilepsy syndromes starting in the first year of life. Neurology, 72(3), 273–281. 10.1212/01.wnl.0000339494.76377.d6 [DOI] [PubMed] [Google Scholar]
- Ellis, C. A. , Churilov, L. , Epstein, M. P. , Xie, S. X. , Bellows, S. T. , Ottman, R. , … Epi, K. C. (2019). Epilepsy in families: Age at onset is a familial trait, independent of syndrome. Annals of Neurology, 86(1), 91–98. 10.1002/ana.25499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hani, A. J. , Mikati, H. M. , & Mikati, M. A. (2015). Genetics of pediatric epilepsy. Pediatric Clinics of North America, 62(3), 703–722. 10.1016/j.pcl.2015.03.013 [DOI] [PubMed] [Google Scholar]
- Heyne, H. O. , Artomov, M. , Battke, F. , Bianchini, C. , Smith, D. R. , Liebmann, N. , … Lemke, J. R. (2019). Targeted gene sequencing in 6994 individuals with neurodevelopmental disorder with epilepsy. Genetics in Medicine, 21(11), 2496–2503. 10.1038/s41436-019-0531-0 [DOI] [PubMed] [Google Scholar]
- Howell, K. B. , McMahon, J. M. , Carvill, G. L. , Tambunan, D. , Mackay, M. T. , Rodriguez‐Casero, V. , … Scheffer, I. E. (2015). SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology, 85(11), 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, S. S. , Kim, S. Y. , Kim, H. , Hwang, H. , Chae, J. H. , Kim, K. J. , … Lim, B. C. (2019). Diagnostic yield of epilepsy panel testing in patients with seizure onset within the first year of life. Frontiers in Neurology, 10, 988 10.3389/fneur.2019.00988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, A. , Youn, S. E. , Kim, S. H. , Lee, J. S. , Kim, S. , Choi, J. R. , … Kang, H.‐C. (2018). Targeted gene panel and genotype‐phenotype correlation in children with developmental and epileptic encephalopathy. Epilepsy Research, 141, 48–55. 10.1016/j.eplepsyres.2018.02.003 [DOI] [PubMed] [Google Scholar]
- Kramer, U. (1999). Epilepsy in the first year of life: A review. Journal of Child Neurology, 14(8), 485–489. [DOI] [PubMed] [Google Scholar]
- Lemke, J. R. , Riesch, E. , Scheurenbrand, T. , Schubach, M. , Wilhelm, C. , Steiner, I. , … Biskup, S. (2012). Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia, 53(8), 1387–1398. 10.1111/j.1528-1167.2012.03516.x [DOI] [PubMed] [Google Scholar]
- Liu, J. , Tong, L. , Song, S. , Niu, Y. , Li, J. , Wu, X. , … Li, B. (2018). Novel and de novo mutations in pediatric refractory epilepsy. Molecular Brain, 11(1), 48 10.1186/s13041-018-0392-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic, S. , Naros, G. , Yalcin, O. , Blazevic, D. , Loeffler, H. , Caglayan, H. , … Lerche, H. (2011). Temperature and pharmacological rescue of a folding‐defective, dominant‐negative KV 7.2 mutation associated with neonatal seizures. Human Mutation, 32(10), E2283–E2293. [DOI] [PubMed] [Google Scholar]
- McGovern, K. , Stillman, N. , McKenna, K. , Mays, V. , Williams, M. , Carpenter, A. , … Yourich, A. (2013). The epilepsy phenome/genome project. Clinical Trials, 10(4), 568–586. 10.1177/1740774513484392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina, E. D. , Ciccone, R. , Brustia, F. , Bayindir, B. , Limongelli, I. , Vetro, A. , … Zuffardi, O. (2015). Improving molecular diagnosis in epilepsy by a dedicated high‐throughput sequencing platform. European Journal of Human Genetics, 23(3), 354–362. 10.1038/ejhg.2014.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller, R. S. , Dahl, H. A. , & Helbig, I. (2015). The contribution of next generation sequencing to epilepsy genetics. Expert Review of Molecular Diagnostics, 15(12), 1531–1538. 10.1586/14737159.2015.1113132 [DOI] [PubMed] [Google Scholar]
- Møller, R. S. , Larsen, L. H. G. , Johannesen, K. M. , Talvik, I. , Talvik, T. , Vaher, U. , … Dahl, H. A. (2016). Gene panel testing in epileptic encephalopathies and familial epilepsies. Molecular Syndromology, 7(4), 210–219. 10.1159/000448369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini, A. , Zara, F. , & Striano, P. (2018). Recent advances in epilepsy genetics. Neuroscience Letters, 667, 4–9. 10.1016/j.neulet.2017.05.014 [DOI] [PubMed] [Google Scholar]
- Ortega‐Moreno, L. , Giráldez, B. G. , Soto‐Insuga, V. , Losada‐Del Pozo, R. , Rodrigo‐Moreno, M. , Alarcón‐Morcillo, C. , … Serratosa, J. M. (2017). Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLoS One, 12(11), e0188978 10.1371/journal.pone.0188978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrini, E. , Marini, C. , Mei, D. , Galuppi, A. , Cellini, E. , Pucatti, D. , … Guerrini, R. (2017). Diagnostic targeted resequencing in 349 patients with drug‐resistant pediatric epilepsies identifies causative mutations in 30 different genes. Human Mutation, 38(2), 216–225. 10.1002/humu.23149 [DOI] [PubMed] [Google Scholar]
- Rantala, H. , & Ingalsuo, H. (1999). Occurrence and outcome of epilepsy in children younger than 2 years. Journal of Pediatrics, 135(6), 761–764. 10.1016/S0022-3476(99)70098-3 [DOI] [PubMed] [Google Scholar]
- Ream, M. A. , & Mikati, M. A. (2014). Clinical utility of genetic testing in pediatric drug‐resistant epilepsy: A pilot study. Epilepsy & Behavior, 37, 241–248. 10.1016/j.yebeh.2014.06.018 [DOI] [PubMed] [Google Scholar]
- Ream, M. A. , & Patel, A. D. (2015). Obtaining genetic testing in pediatric epilepsy. Epilepsia, 56(10), 1505–1514. 10.1111/epi.13122 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rim, J. H. , Kim, S. H. , Hwang, I. S. , Kwon, S. S. , Kim, J. , Kim, H. W. , … Kang, H.‐C. (2018). Efficient strategy for the molecular diagnosis of intractable early‐onset epilepsy using targeted gene sequencing. BMC Medical Genomics, 11(1), 6 10.1186/s12920-018-0320-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands, T. T. , & Choi, H. (2017). Genetic testing in pediatric epilepsy. Current Neurology and Neuroscience Reports, 17(5), 45 10.1007/s11910-017-0753-y [DOI] [PubMed] [Google Scholar]
- Scheffer, I. E. , Berkovic, S. , Capovilla, G. , Connolly, M. B. , French, J. , Guilhoto, L. , … Zuberi, S. M. (2017). ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia, 58(4), 512–521. 10.1111/epi.13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal, E. , Pedro, H. , Valdez‐Gonzalez, K. , Parisotto, S. , Gliksman, F. , Thompson, S. , … Fertig, E. (2016). Diagnostic yield of epilepsy panels in children with medication‐refractory epilepsy. Pediatric Neurology, 64, 66–71. 10.1016/j.pediatrneurol.2016.06.019 [DOI] [PubMed] [Google Scholar]
- SoRelle, J. A. , Thodeson, D. M. , Arnold, S. , Gotway, G. , & Park, J. Y. (2019). Clinical utility of reinterpreting previously reported genomic epilepsy test results for pediatric patients. JAMA Pediatrics, 173(1), e182302 10.1001/jamapediatrics.2018.2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh, C. R. , Sohn, S. Y. , Kim, G. H. , Jung, S. K. , & Eun, B. L. (2016). Single‐center experience of the Korean‐Developmental Screening Test for infants and children. Korean Journal of Pediatrics, 59(12), 483–489. 10.3345/kjp.2016.59.12.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis, J. , Silk, M. , Wang, Q. , Berkovic, S. F. , Liu, L. , Ascher, D. B. , … Petrovski, S. (2017). Optimizing genomic medicine in epilepsy through a gene‐customized approach to missense variant interpretation. Genome Research, 27(10), 1715–1729. 10.1101/gr.226589.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trump, N. , McTague, A. , Brittain, H. , Papandreou, A. , Meyer, E. , Ngoh, A. , … Scott, R. H. (2016). Improving diagnosis and broadening the phenotypes in early‐onset seizure and severe developmental delay disorders through gene panel analysis. Journal of Medical Genetics, 53(5), 310–317. 10.1136/jmedgenet-2015-103263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan, C. , & Levene, M. (2013). Epidemiology and aetiology of neonatal seizures. Seminars in Fetal and Neonatal Medicine, 18(4), 185–191. 10.1016/j.siny.2013.05.008 [DOI] [PubMed] [Google Scholar]
- Waaler, P. E. , Blom, B. H. , Skeidsvoll, H. , & Mykletun, A. (2000). Prevalence, classification, and severity of epilepsy in children in western Norway. Epilepsia, 41(7), 802–810. 10.1111/j.1528-1157.2000.tb00246.x [DOI] [PubMed] [Google Scholar]
- Wiebe, S. , Bellhouse, D. R. , Fallahay, C. , & Eliasziw, M. (1999). Burden of epilepsy: The Ontario Health Survey. Canadian Journal of Neurological Sciences, 26(4), 263–270. 10.1017/S0317167100000354 [DOI] [PubMed] [Google Scholar]
- Wolff, M. , Johannesen, K. M. , Hedrich, U. B. S. , Masnada, S. , Rubboli, G. , Gardella, E. , … Møller, R. S. (2017). Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A‐related disorders. Brain, 140(5), 1316–1336. 10.1093/brain/awx054 [DOI] [PubMed] [Google Scholar]
- Yalcin, O. , Caglayan, S. H. , Saltik, S. , Cokar, O. , Agan, K. , Dervent, A. , & Steinlein, O. K. (2007). A novel missense mutation (N258S) in the KCNQ2 gene in a Turkish family afflicted with benign familial neonatal convulsions (BFNC). The Turkish Journal of Pediatrics, 49(4), 385–389. [PubMed] [Google Scholar]
- Zhang, Q. , Li, J. , Zhao, Y. , Bao, X. , Wei, L. , & Wang, J. (2017). Gene mutation analysis of 175 Chinese patients with early‐onset epileptic encephalopathy. Clinical Genetics, 91(5), 717–724. 10.1111/cge.12901 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Data Availability Statement
All accepted manuscripts are required to publish a data availability statement to confirm the presence or absence of shared data.