

Abstract
Background
Hemophilia A (HA) is an X‐linked recessive bleeding disorder caused by pathogenic variants of the coagulation factor VIII gene (F8). Half of the patients with severe HA have a recurrent inversion in the X chromosome, that is, F8 intron 22 or intron 1 inversion. Here, we characterized an abnormal F8 due to atypical complex X chromosome rearrangements in a Japanese patient with severe HA.
Methods
Recurrent F8 inversions were tested with inverse shifting‐PCR. The genomic structure was investigated using PCR‐based direct sequencing or quantitative PCR.
Results
The proband's X chromosome had a 119.5 kb insertion, a reverse duplex of an extragenic sequence on the F8 telomere region into the F8 intron 1 with two breakpoints. The telomeric breakpoint was a joining from the F8 intron 1 to the inverted FUNDC2 via a two‐base microhomology, and the centromeric breakpoint was a recombination between F8 intron 1 homologous sequences. The rearrangement mechanism was suggested as a multi‐step rearrangement with template switching such as fork stalling and template switching (FoSTeS)/microhomology‐mediated break‐induced replication (MMBIR) and/or homologous sequence‐associated recombination during a sister chromatid formation.
Conclusion
We identified the aberrant X chromosome with a split F8 due to a multi‐step rearrangement in a patient with severe HA.
Keywords: chromosomal rearrangement, F8, FoSTeS/MMBIR, hemophilia A, homologous recombination, inversion
Hemophilia A (HA) is an X‐linked recessive bleeding disorder caused by mutations in the coagulation factor VIII gene (F8). We identified the aberrant chromosome X with a split F8 due to a multi‐step rearrangement in a severe HA patient. The rearrangement mechanism was suggested as a multi‐step rearrangement with template switching such as FoSTeS/MMBIR and/or homologous sequence associating recombination during a sister chromatid formation.
1. INTRODUCTION
Hemophilia A (HA) is an X‐linked recessive bleeding disorder caused by pathogenic variants of the coagulation factor VIII gene (F8, OMIM: 300841). Based on the coagulation factor VIII (FVIII) activity (FVIII:C), HA is classified into three phenotypes: severe (FVIII:C <1 IU/dL), moderate (1 ≤ FVIII:C <5 IU/dL), and mild (5 ≤ FVIII:C <40 IU/dL). F8 consists of 26 exons and 25 introns spanning 186 kb at a long arm of the X chromosome (Xq28; Lannoy & Hermans, 2016). Thus far, 3,052 of the various F8 pathogenic variants, including unique structural variations such as a large deletion or duplication, have been reported in the F8 variant database (http://www.factorviii‐db.org/, accessed on 5 April 2020).
Half of the patients with severe HA have a recurrent intra‐chromosomal inversion known as intron 22 inversion (Inv22) caused by nonallelic homologous recombination (NAHR) between homologous sequences: int22h‐1 within F8 intron 22, and either int22h‐2 or int22h‐3 located at 400 kb telomeric distal from F8 (Lakich, Kazazian, Antonarakis, & Gitschier, 1993; Naylor, Brinke, Hassock, Green, & Giannelli, 1993). Moreover, the F8 intron 1 inversion (Inv1) is responsible for the genetic defect in 1%–5% of all severe HA cases. Inv1 is due to the intra‐chromosomal NAHR crossing over two almost identical sequences: int1h‐1 in the F8 intron 1 and int1h‐2 located at approximately 126 kb telomeric distal from F8 (Bagnall, Waseem, Green, & Giannelli, 2002). In addition to the typical F8 Inv22 or Inv1, several studies have pointed out that some cases of severe HA carry an unusual F8 structural abnormality that may associate with inversions. The int22hs‐associated recombination between F8 and neighbor genes causes large deletions or duplications (Chen et al., 2017; Fujita et al., 2012). Moreover, several reports suggested that int1hs is also involved in large duplications and/or deletions within a telomeric region of the X chromosome. (Pio, Oliveira, Soares, & Rezende, 2011; Sanna et al., 2013; Sukarova Stefanovska, Dejanova, Tchakarova, Petkov, & Efremov, 2008; You et al., 2014). However, to our knowledge, the mechanism by which those complex F8 rearrangements occur has not been thoroughly investigated.
In this study, we analyzed the F8 in a Japanese patient with severe HA and characterized the abnormal X chromosomal structure, NC_000023.11:g.155007148_155013478delins155028224_155147780inv, that was an insertion of a large duplicon derived from the F8 extragenic alignment into the F8 intron 1. We thus proposed a hypothetical mechanism by which the aberrant X chromosome was constructed through multi‐step rearrangement with replicative DNA repair models such as the fork stalling and template switching (FoSTeS)/microhomology‐mediated break‐induced replication (MMBIR) and homologous recombination (HR) during a sister chromatid formation.
2. MATERIALS AND METHODS
2.1. Patient and DNA sample
The proband was a 6‐month‐old Japanese boy affected by severe HA (FVIII:C <1 IU/dL, Figure 1a III‐3). The proband's mother showed reduced FVIII activity (FVIII:C = 56.3 IU/dL, Figure 1a II‐2) and had bleeding symptoms at the time of surgery. The proband's father and maternal grandparents did not have any bleeding tendency. The FVIII antigen level (FVIII:Ag) was measured using the VisuLize Factor VIII ELISA Kit (Affinity Biologicals Inc., Ancaster, Canada) and the FVIII:Ag in the proband's plasma was lower than the limit of detection (<0.8%). After informed consent was obtained from the parents, genomic DNA (gDNA) was extracted from peripheral blood cells as described previously (Kojima et al., 1987). This study was approved by the Ethics Committee of Nagoya University School of Medicine (Identification number: 2015‐0391).
Figure 1.
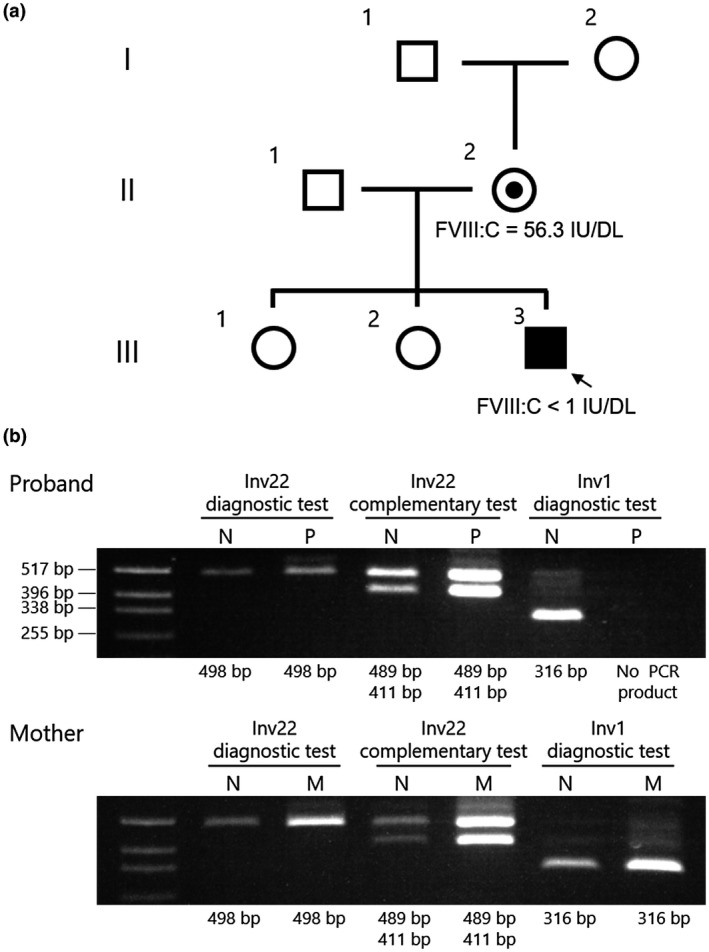
Inverse shifting‐PCR (IS‐PCR) suggested a structural abnormality in the proband's F8 intron 1. (a) Family pedigree. Proband (III‐3) is indicated by an arrow. The proband's mother (II‐2) showed reduced FVIII activity (FVIII:C = 56.3 IU/dL) and had bleeding symptoms at the time of surgery. The proband's father (II‐1) and maternal grandparents (I‐1 and I‐2) did not have any bleeding tendency. (b) Recurrent inversion analysis with IS‐PCR. N, normal male control; P, proband; M, mother. In Inv1 diagnostic test, the proband's PCR product was not observed (expected band sizes are normal: 316 bp and Inv1: 281 bp, respectively). Other tests showed a pattern similar to that in a normal control (Inv22 diagnostic test: 498 bp, Inv22 complementary test: 489 and 411 bp)
2.2. F8 intron 22 and intron 1 inversions analysis by inverse shifting‐PCR (IS‐PCR)
IS‐PCR was performed as previously described (Rossetti, Radic, Larripa, & De Brasi, 2008) with minor modifications. About 2 μg of gDNA was digested with 15 U of BclI (New England Biolabs) for 3 hr in a total reaction volume of 50 μL as per the manufacturer's instruction. The restriction enzyme was inactivated through a phenol/chloroform/isoamyl alcohol extraction followed by ethanol precipitation, and gDNA was dissolved in 4 μL of TE buffer (10‐mM Tris with 1‐mM EDTA, pH 8.0). The digested gDNA samples were self‐ligated using Ligation high ver. 2 (Toyobo Co., Ltd.) for 6 hr at 16°C in a reaction volume of 8 μL (4 μL of the digested gDNA was mixed with an equal amount of Ligation high ver. 2). After ethanol precipitation, ligated samples were resolved with 30 μL of TE buffer. PCR was performed in a reaction volume of 20 μL containing 1 μL of the ligated gDNA, 0.5 μM of each primer, 0.4 U of KOD FX DNA polymerase (Toyobo), and additional PCR reaction reagents for the KOD FX. Primer sets for IS‐PCR are listed in Table S1. Thermal cycling was performed using the step‐down procedure preceded by a hot‐starting step as follows: 94°C for 2 min; five cycles of denaturation at 98°C for 10 s, annealing at 68°C for 30 s, and extension at 68°C for 35 s; then five cycles of denaturation at 98°C for 10 s, annealing at 66°C for 30 s, and extension at 68°C for 35 s; further 25 cycles of denaturation at 98°C for 10 s, annealing at 65°C for 30 s, and extension at 68°C for 35 s; followed by a final extension at 68°C for 2 min. PCR products were analyzed using electrophoresis on a 1.5% agarose gel and stained with 1 μg/mL of ethidium bromide.
2.3. F8 DNA sequencing
All 26 exons, including exon‐intron junctions, promoter region, and untranslated regions, were amplified via PCR using originally designed gene‐specific primers (Table S2). The PCR was performed in reaction volumes of 20 μL containing 80 ng of gDNA, 0.5 μM of each primer, and 10 μL of AmpliTaq Gold 360 Master Mix (Thermo Fisher Scientific Inc.). The cycling conditions were: 30 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s, preceded by 95°C for 10 min, followed by 72°C for 2 min. PCR products were quantified using electrophoresis on 1.5% agarose gels. The DNA sequencing reaction was conducted with the BigDye Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific Inc.) according to the manufacturer's instructions. Samples were analyzed with the ABI PRISM 310 Genetic Analyzer or ABI PRISM 3130 Genetic Analyzer (Thermo Fisher Scientific Inc.). In this study, the nucleotide position at X chromosome was referred in GenBank: NC_000023.11.
2.4. Multiplex ligation‐dependent probe amplification (MLPA) analysis for F8
MLPA was performed using the SALSA MLPA P178 F8 probemix (MRC‐Holland) according to the manufacturer's instructions. DNA fragments were analyzed with the ABI PRISM 310 Genetic Analyzer. The relative quantity of the patient F8 was evaluated in comparison with that of the pooled Japanese healthy male gDNA control.
2.5. Amplification of the F8 intron 1
To analyze the F8 intron 1, a PCR amplicon was designed and divided into four segments (Figure 2a). The F8 intron 1 specific primer sets are listed in Table S3. PCR was performed in reaction volumes of 20 μL containing 80 ng of circularized DNA, 0.3 μM of each primer, 0.4 U of KOD FX Neo DNA polymerase (Toyobo co., Ltd.), and additional PCR reaction reagents for the KOD FX Neo. The cycling conditions were: 25 cycles of denaturation at 98°C for 30 s, annealing at 63°C for 30 s, and extension at 68°C for 5 min, preceded by 98°C for 2 min, followed by 68°C for 7 min. The PCR products were analyzed using electrophoresis on a 1% agarose gel. To detect the breakpoint of large deletions within intron 1, the F8 intron 1 was entirely amplified with the primers seg1‐Fw and seg4‐Rv (Table S3) and the KOD FX Neo DNA polymerase. The PCR cycling conditions were as described above with a modification of the extension time to 14 min.
Figure 2.
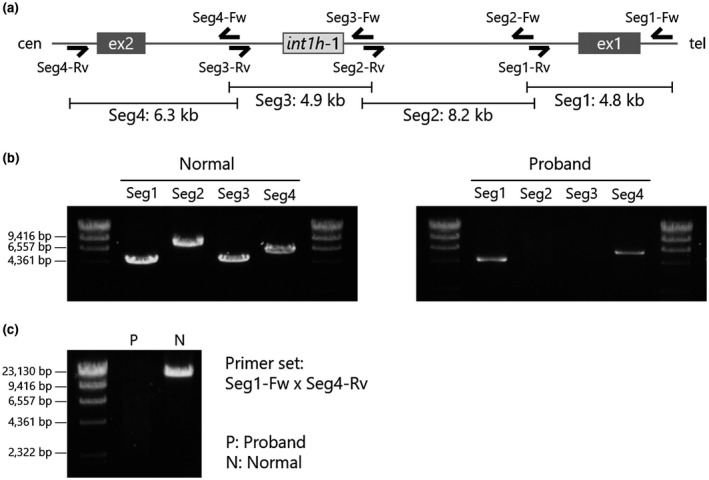
PCR mapping in F8 intron 1. (a) Primer design of mapping PCR in F8 intron 1. F8 intron 1 is divided into four segments, Seg 1 (4.8 kb), Seg 2 (8.2 kb), Seg 3 (4.9 kb), and Seg 4 (6.3 kb). (b) Results of F8 intron 1 mapping in normal male individual (left) and proband (right). Segments 2 and 3 were not detected in the proband's gDNA. (c) Full‐length PCR amplification of F8 intron 1. Long‐range PCR successfully amplified the product in the gDNA of normal male subject (N), while no amplicon was detected in the proband's gDNA (P)
2.6. Inverse PCR
Inverse PCR was used to investigate the genomic structure of the mutated F8. Primers for inverse PCR were designed in opposite orientation in the known regions (Table S3, Figures 3a and 4a). gDNA was digested with PstI (Takara Bio Inc.) or HindIII (New England Biolabs Japan Inc.) at the appropriate temperature, and fragments were circularized with the Ligation high ver. 2. After ethanol precipitation, the circularized gDNA was resolved with 20 μL of TE buffer. PCR was performed in a total volume of 20 μL containing 1 μL of circularized gDNA, 0.4 U of the KOD FX DNA polymerase, 0.3 μM of each primer, and additional PCR reagents for the KOD FX DNA polymerase. PCR was performed in a step‐down procedure as follows: five cycles of denaturation at 98°C for 10 s, annealing at 70°C for 30 s, and extension at 70°C for 15 min. Subsequently, the annealing temperature was modified to 68°C for five cycles, 66°C for five cycles, and 64°C for 25 cycles. Cycling was preceded by 94°C for 2 min, followed by a final extension at 68°C for 2 min. The PCR product was analyzed using electrophoresis on 1% agarose gel and purified with the FastGene Gel/PCR Extraction Kit (Nippon Genetics Co., Ltd.). The direct sequencing of variant‐specific PCR products was performed using the BigDye Terminator v1.1 Cycle Sequencing Kit and ABI PRISM 3130 Genetic Analyzer.
Figure 3.
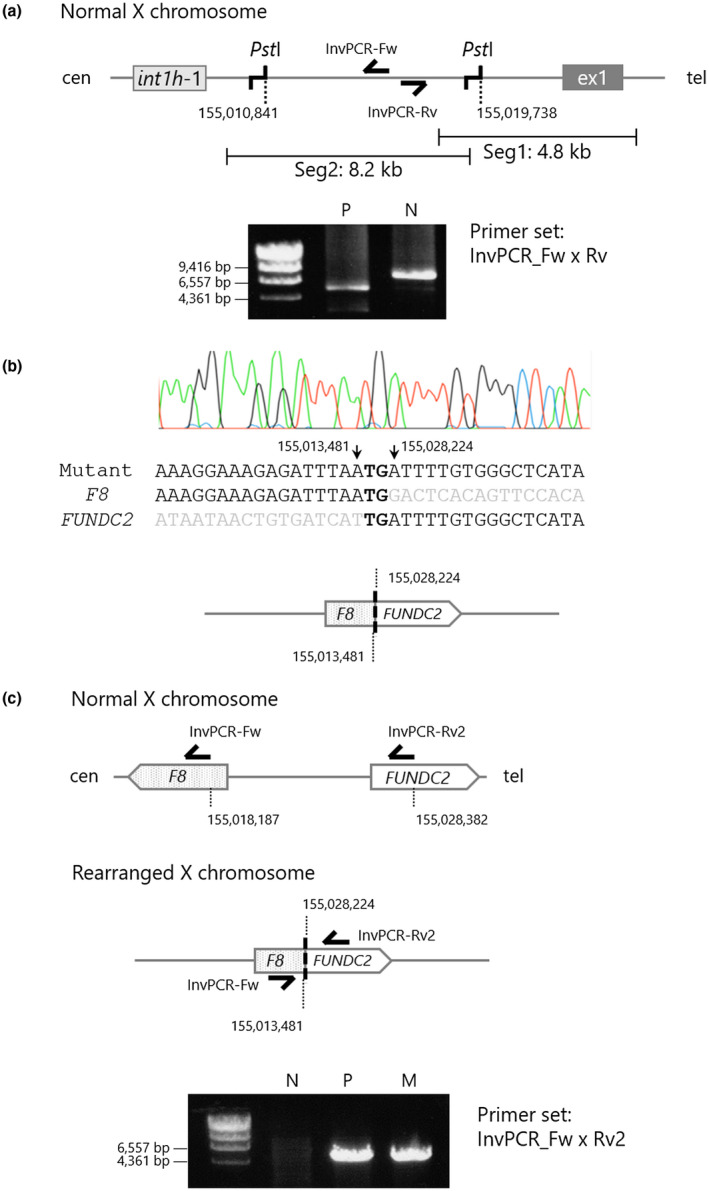
Identification of a telomeric breakpoint in the proband's F8 intron 1. (a) Design of inverse PCR with PstI site within F8 intron 1 segments 1–2 (upper) and PCR results in a normal male subject (N) and the proband (P; lower). PstI‐inverse PCR suggested that the proband's F8 intron 1 had a structure abnormality. (b) Direct sequencing of the abnormal inverse PCR product derived from the proband's gDNA. The electropherogram of a breakpoint connecting F8 intron 1 (NC_000023.11 nt155,013,481) to inverted FUNDC2 (NC_000023.11 nt155,028,224) via two bases of microhomology (upper) and its speculated structural diagram (lower). (c) Mutant‐specific PCR with primer set, InvPCR‐Fw and InvPCR‐Rv2. Design of the mutant‐specific PCR (upper) and the PCR results in a normal male subject, the proband, and the proband's mother. N: normal male subject, P: proband, M: proband's mother
Figure 4.
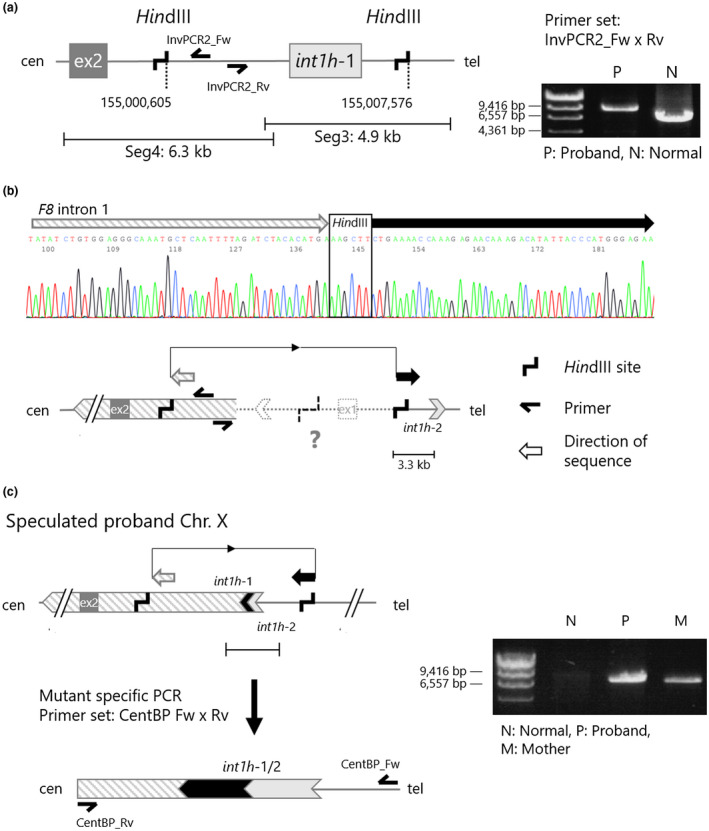
Identification of a centromeric breakpoint in the proband's F8 intron 1. (a) Design of inverse PCR with HindIII site within F8 intron 1 segments 3–4 (left), and the PCR results in a normal male subject (N) and the proband (P; right). HindIII‐inverse PCR suggested that the proband's F8 intron 1 had another structure abnormality at the centromeric side. (b) Direct sequencing result of the abnormal inverse PCR product derived from the proband's gDNA. The structural diagram explains a sequence direction detected in the direct sequencing. (c) Mutant‐specific PCR with primer set, CentBP_Fw and CentBP_Rv. Design of the mutant‐specific PCR (left) and the PCR results in a normal male subject, the proband, and the proband's mother (right). N: normal male subject, P: proband, M: proband's mother
2.7. Long‐range PCR analysis for the F8 variant associated with the intron 1 homologous region (int1h; Inv1 diagnostic long‐PCR)
In order to investigate the F8 structure variation involved with int1h, including Inv1, we originally designed a specific primer set for int1h‐1 and int1h‐2 (Table S1 and Figure S1a). PCR was performed in a reaction volume of 20 μL containing 80 ng of gDNA, 0.3 μM of each primer, 0.4 U of KOD FX Neo DNA polymerase (Toyobo co., Ltd.), and additional PCR reaction reagents for the KOD FX Neo. The thermal cycling conditions were 25 cycles of denaturation at 98°C for 30 s, annealing at 68°C for 30 s, and extension at 68°C for 7 min, preceded by 98°C for 2 min, followed by 68°C for 7 min. The products were analyzed using electrophoresis on 0.6% agarose gels.
2.8. Quantitative gene mapping in the X chromosome
Quantitative gene mapping was performed using quantitative PCR (qPCR). Specific primer sets were originally designed at neighboring genes of the F8 (Table S4 and Figure 5b). qPCR was performed in a total volume of 15 μL containing 15 ng of sonicated gDNA, 0.4 μM of each specific primer, and 7.5 μL SYBR Premix Ex Taq II Tli RNaseH Plus (Takara Bio Inc.) The PCR reaction was performed and analyzed with the Thermal Cycler Dice Real Time System II (Takara Bio Inc.).
Figure 5.
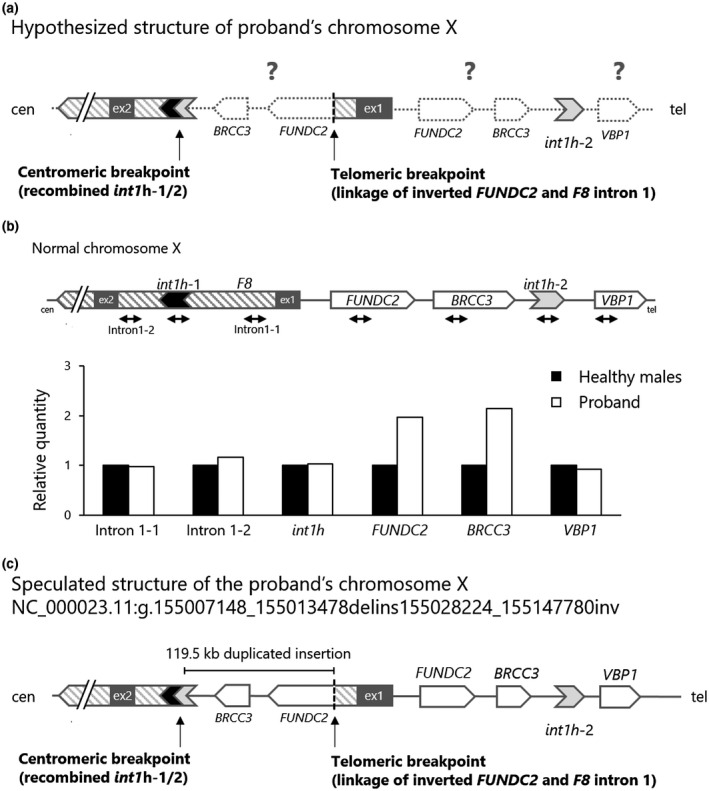
Speculation of the proband's chromosome X structure. (a) Hypothesized structure of proband's chromosome X, based on the observations of two breakpoints and a partial inversion structure. Dotted objects show uninvestigated regions. (b) Quantitative gene mapping analysis. Design of qPCR on the F8 and its neighboring loci (upper) and the results of copy number variations (lower). Gene dosage was normalized to a gDNA pooled from three normal male individuals. FUNDC2 and BRCC3 were assessed as a doubled gene dosage. (c) Speculated structure of the proband's chromosome X (NC_000023.11:g.155007148_155013478delins155028224_155147780inv). The patient F8 was split with an over 119.5 kb of intra‐chromosomal inverted/duplicated alignment
2.9. Reverse transcription (RT)‐PCR analysis of F8 mRNA in PBMCs
Peripheral blood mononuclear cells (PBMCs) were separated from 5 ml of EDTA anticoagulated whole blood using the Ficoll‐Paque Premium (GE Healthcare UK Ltd.). Total mRNA derived from PBMCs was extracted with the RNeasy Mini Kit (Qiagen K.K.). cDNA was synthesized using the PrimeScript II Reverse Transcriptase with oligo dT primer or random 6 mer primer (Takara Bio Inc.), and PCR amplification was performed in reaction volumes of 20 μL containing 1 μL of cDNA, 0.3 μM of each primer, 0.4 U of KOD FX DNA polymerase (Toyobo co., Ltd.), and additional PCR reaction reagents for the KOD FX. The thermal cycling conditions were 30 cycles of denaturation at 98°C for 30 s, annealing at 60°C for 30 s, and extension at 68°C for 90 s, preceded by 98°C for 2 min, followed by 68°C for 7 min. The products were analyzed using electrophoresis on a 1% agarose gel. We used four sets of primers to amplify the F8 cDNA according to a previous report with minor modifications (Table S4; El‐Maarri et al., 2005). GAPDH was used as the RT‐PCR validating internal control. The PCR product was analyzed using electrophoresis on a 1.2% agarose gel.
3. RESULTS
3.1. Conventional F8 variant analysis showed an atypical F8 inversion in proband's X chromosome
We first performed IS‐PCR to investigate F8 recurrent inversions, Inv22 and Inv1 (Figure 1b). A pattern of the proband's PCR product was identical to that of a healthy male subject in Inv22 diagnostic and complementary tests. In contrast, the Inv1 diagnostic test did not detect any PCR product in the proband's gDNA. To investigate an exonic deletion or duplication, we quantitatively analyzed a copy number of F8 exons with MLPA (Figure S2). However, the proband's gDNA did not present any copy number alteration in the F8 exons. Moreover, no single base substitution, small insertion, or deletion was detected in the F8 exons and their flanking splice sites. These results suggested that the proband carried a causative structural abnormality in the F8 intron 1.
3.2. PCR mapping analysis showed a complex structure abnormality in the proband's F8 intron 1
In order to investigate the abnormality of F8 intron 1, we designed the primer sets by dividing the F8 intron 1 into four segments (Figure 2a). In a healthy male subject, we detected the products' size as expected: 4.8 kbp, 8.2 kbp, 4.9 kbp, and 6.3 kbp in segments 1, 2, 3, and 4, respectively (Figure 2b left). In the proband, although the products of segments 1 and 4 were clearly detected, those of segments 2 and 3 were not observed (Figure 2b right) suggesting that an unexpected structural abnormality was present in the region of segments 2 to 3. To investigate the structural abnormality in the F8 intron 1, we amplified the full length of intron 1 using primers Seg1‐Fw and Seg4‐Rv (Figure 2a). However, no PCR product was observed in the patient gDNA (Figure 2c). These results suggested that the proband possessed a gross structural abnormality in the F8 intron 1. In addition, regarding the other F8 introns, no other remarkable structural abnormality was observed (Figure S3).
3.3. Identification of a telomeric breakpoint in the patient F8 intron 1
In order to investigate the structural abnormality observed in segments 2 and 3 of the F8 intron 1, we performed inverse PCR (Figure 3). In a normal F8 intron 1, PstI sites are present at the positions 3.0 and 11.9 kb from the splice donor site of the F8 exon 1. The primer set was designed in an opposite orientation within the PstI‐digested fragment (Figure 3a top). In the proband, PstI‐digested gDNA was amplified as an abnormal product with an approximate size of 6 kbp (Figure 3a bottom). Sequencing of the inverse PCR product revealed that the F8 intron 1 was connected to the inverted FUNDC2 that was at 4 kb telomeric distal from F8 on the reference sequence (NC_000023.11). The direct sequencing demonstrated the breakpoint connecting F8 intron 1 (NC_000023.11 nt155,013,481) to the inverted FUNDC2 (NC_000023.11 nt155,028,224) via two bases of microhomology (Figure 3b). In order to confirm the unusual structure detected in the proband's X chromosome, we designed a variant‐specific PCR that detected only the rearranged chromosome (Figure 3c). A reverse primer, named InvPCR‐Rv2, was designed in the FUNDC2 intron 1, which had the same orientation to a counterpart forward primer, InvPCR‐Fw (Figure 3c top and middle). This primer set successfully amplified a product only in the proband and his mother gDNA (Figure 3c bottom).
3.4. Identification of a centromeric breakpoint in the patient F8 intron 1
To further investigate the structure abnormality of the proband's F8, we examined segment 4 in the F8 intron 1 with inverse PCR (Figure 4). In a normal X chromosome, HindIII sites are present in segments 3 and 4 (positioned at 15.1 kb and 22.1 kb away from the splice donor site of the F8 exon 1). The primer set was designed at the centromeric side within the HindIII‐digested fragment (Figure 4a, left). The inverse PCR with the HindIII‐digested patient gDNA showed an abnormal PCR product (8.5 kbp) as compared to that of a healthy male subject (6.5 kbp; Figure 4a, right). Direct sequencing of the abnormal product showed that segment 4 of the proband F8 intron 1 was connected to an inverted DNA alignment that was approximately 3.3 kb centromeric away from int1h‐2 (Figure 4b). This result suggested an inversion structure of the F8 intron 1 via int1h‐1 and 2.
In order to identify a centromeric breakpoint in the patient F8 intron 1, we designed a mutant‐specific PCR with a primer set, CentBP_Fw and CentBP_Rv (Figure 4c left). The mutant‐specific PCR successfully amplified a product from the proband's and mother's gDNA, but not from a healthy subject (Figure 4c right). The direct sequencing denoted that the mutant‐specific amplicon contained a DNA alignment of int1h‐1 and int1h‐2 (Figure S4). These observations indicated that the proband's X chromosome had an inversion structure with a recombination between F8 int1h‐1 and 2.
3.5. Putative proband's X chromosome structure
To further investigate the structure of the proband's X chromosome, we performed F8 Inv1 diagnostic long‐PCR (Figure S1). In a normal control, 12 kbp and 8 kbp PCR products were amplified with primer sets 1‐A/1‐B and 1‐P/1‐Q, respectively. In contrast, in a patient with HA and F8 Inv1, 11 kbp and 9 kbp PCR products were amplified with a primer set 1‐A/1‐Q and 1‐B/1‐P, respectively. In the proband, the PCR using primer sets 1‐A/1‐B and 1‐A/1‐Q amplified products identical in size to 1‐A/1‐B in the normal control and to 1‐A/1‐Q in Inv1 as in the positive HA. However, the primer sets, 1‐P/1‐Q and 1‐B/1‐P, did not detect any amplicons. These observations indicated that the proband's X chromosome had an intact int1h‐2 (positive with primer set 1‐A/1‐B) and an inverted type of int1h‐1/2 (positive with primer set 1‐A/1‐Q).
Given the observations of the two breakpoints and a partial inversion structure in the proband's X chromosome, we hypothesized that the X chromosome possessed a rearranged structure with an intra‐chromosomal inversion and/or duplication (Figure 5a). In order to determine the abnormal structure of the patient's X chromosome, we performed a quantitative gene mapping analysis (Figure 5b). Copy number variations of F8 and its neighboring loci were quantitatively examined. The patient's F8 intron 1, int1h‐1, int1h‐2, and VBP1 were in equal amounts compared to the ones in a pooled healthy male gDNA. In contrast, FUNDC2 and BRCC3 were assessed to have doubled gene dosage, indicating that a genomic sequence that ranged from a part of the FUNDC2 to BRCC3 was duplicated in the patient X chromosome.
Consequently, we suggested that the structure of the X chromosome in the proband was as follows: the patient F8 was split with an over 119.5 kb of intra‐chromosomal inverted/duplicated alignment, described as NC_000023.11:g.155007148_155013478delins155028224_155147780inv (Figure 5c). The large inset broke the F8 intron 1 via two unrelated breakpoints, the telomeric breakpoint with two base homology and the centromeric breakpoint associated with recombination of int1h‐1/2. This recombination resulted in a deletion of 6.3 kb alignment (NC_000023.11 nt155,007,148 to 155,013,480) in the F8 intron 1.
4. DISCUSSION
In this study, we investigated the X chromosome structure in a patient with severe HA and demonstrated the complex genomic rearrangement with a split F8 due to a 119.5 kb duplicated insertion in the F8 intron 1. The telomeric breakpoint was a linkage of the inverted FUNDC2 (nt155,028,224) to the F8 intron 1 (nt155,013,481) via two bases of microhomology (Figure 3). The centromeric breakpoint was a recombination of int1h‐1 and ‐2 (Figure 4).
Several studies have pointed out unusual patterns of the F8 Inv1 in patients with severe HA. Pio et al. (Pio et al., 2011) and Stefanovska et al. reported the case of a patient with severe HA who had intact and inverted types of int1hs (Sukarova Stefanovska et al., 2008). Sanna et al. reported the case of a patient with severe HA who carried a structural rearrangement with both normal and inverted int1h‐1 with 19.32 kb of duplication spanning the F8 exon 2 to exon 6 (Sanna et al., 2013). Moreover, You et al. reported a mutated X chromosome with an extragenic segmental duplicon of 227.3 kb and a deletion in the F8 intron 1 of 2.56 kb (You et al., 2014).
In order to explain the structural abnormality of the proband's X chromosome, we propose a multi‐step rearrangement mechanism (Figure 6). In this model, a telomeric end of the X chromosome is flipped prior to DNA replication (Figure 6a). During DNA replication and sister chromatid formation, a replication error occurs in the DNA replication fork at NC_000023.11 nt155,013,481 (F8 intron 1) followed by an intra‐chromosomal template switching into NC_000023.11 nt155,028,224 (FUNDC2; Figure 6b,c) with two bases of microhomology that forms the telomeric breakpoint. This strand switching may be explained using FoSTeS or MMBIR (Hastings, Ira, & Lupski, 2009; Lee, Carvalho, & Lupski, 2007). After synthesizing a duplicon strand, the abnormal strand switches back to the original strand via an int1h‐1 on the original strand and an int1h‐2 on the abnormal strand (Figure 6d,e). The recombination machinery between the int1h‐1 and ‐2 might have occurred because of a HR or a template switching. In agreement with the present model, a few reports also mentioned that similar chromosomal rearrangements associated with F8 possibly resulted from a combination with the FoSTeS/MMBIR and HR (Chen et al., 2017; Sanna et al., 2013; You et al., 2014).
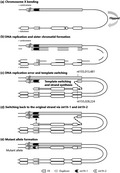
Figure 6.
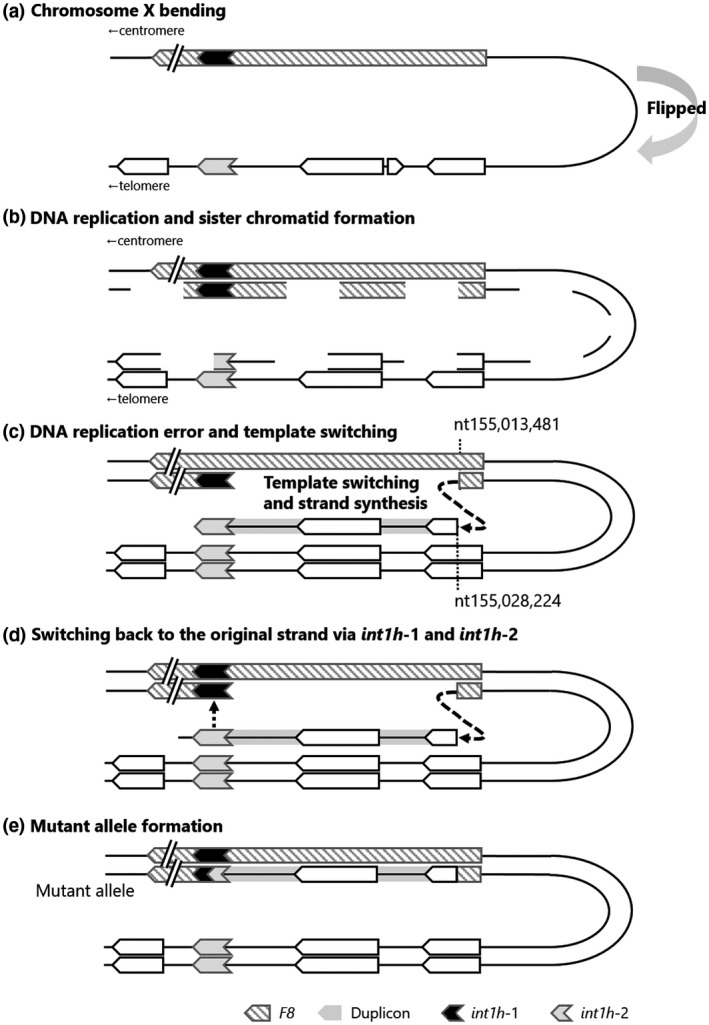
Hypothetical multi‐step mechanism forming the abnormal chromosome X structure. (a) Chromosome X bending. A telomeric end of chromosome X is flipped prior to DNA replication of chromosome X. (b and c) Template switching during DNA replication. A replication error occurred in a DNA replication fork at NC_000023.11 nt155,013,481 (F8 intron 1) followed by an intra‐chromosomal template switching into NC_000023.11 nt155,028,224 (FUNDC2) with two bases of microhomology. This strand switching could be explained by FoSTeS or MMBIR. (d and e) Template switching back and mutant allele formation. After synthesizing a duplicon strand, the abnormal strand switched back to the original strand via an int1h‐1 on the original strand and an int1h‐2 on the abnormal strand. The recombination machinery between the int1h‐1 and ‐2 could have occurred due to homologous recombination or FoSTeS/MMBIR
The proband's mother somatically carries the same abnormal X chromosome, which suggests two conceivable periods of rearrangement occurrence: an early stage of the meiotic phase in either maternal grandparents or an early embryogenesis of the proband's mother. Considering that a recurrent X chromosome inversion, such as F8 Inv22, frequently occurs more in male meiosis than in female meiosis (Rossiter et al., 1994), the present rearrangement might have occurred during spermatogenesis in the maternal grandfather. In contrast, Chen et al described that the F8 pathogenic variant due to a complex recombination mediated by sequential MMBIR and BIR may occur during an early period of embryogenesis in the proband's mother, which results in a somatic mosaicism (Chen et al., 2017). However, in the present study, since we only obtained gDNA extracted from peripheral blood cells from the patient, we were not able to investigate whether the proband's mother possessed a mosaicism with aberrant X chromosome.
In addition to the genomic investigations, we also analyzed F8 mRNA in the proband (Figure S5). FVIII is synthesized in the liver sinusoidal endothelial cells (Do, Healey, Waller, & Lollar, 1999; Pan et al., 2016). In contrast, it is known that F8 mRNA is expressed in PBMCs (Naylor, Green, Montandon, Rizza, & Giannelli, 1991), and the F8 mRNA in PBMCs has been used to analyze a transcript abnormality in patients with HA (Chelly, Concordet, Kaplan, & Kahn, 1989; El‐Maarri et al., 2005; Naylor et al., 1991). We therefore analyzed the F8 transcript in PBMCs from the proband. However, no F8 transcript and not even a truncated fragment that might have been responsible for the proband's FVIII:Ag null phenotype were observed. It is possible that the inverted duplication insertion created novel splicing sites leading to aberrant transcripts that were rapidly degraded by mRNA decay mechanisms such as nonsense‐mediated mRNA decay (Byers, 2002).
In conclusion, we identified the complex abnormal X chromosome in the patient with severe HA, described as NC_000023.11:g.155007148_155013478delins155028224_155147780inv. The 119.5 kb duplicon derived from the F8 extragenic alignment was inserted into the F8 intron 1 with two breakpoints, resulting in thorough splitting of F8. We considered that the aberrant X chromosome was constructed through multi‐step rearrangement with FoSTeS/MMBIR and the int1h‐1/2‐associated recombination during a sister chromatid formation in a spermatogenesis of the maternal grandfather or an early embryogenesis of the proband's mother. This study provides new insights into the mechanisms responsible for several genomic repair machineries involved in deleterious F8 rearrangement and contributes to precise genetic diagnostics in patients with HA.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this manuscript.
AUTHOR CONTRIBUTION
M. T. and S. T. designed and performed the research, analyzed data, and drafted the manuscript. N. S. designed the project, collected, and analyzed the clinical data. M. K., Y. H., K. O., and S. S. conducted the research and analyzed data. A.T. developed and supervised the project. A. K., F. H., S. O., A. S., T. Kanematsu., and T. M. developed the project and collected and analyzed the clinical data. T. Kojima designed the project, analyzed data, and drafted the manuscript.
Supporting information
Figure S1‐S5
Table S1‐S4
ACKNOWLEDGMENTS
We thank Mrs. Wakamatsu for her excellent technical assistance. This study was supported in part by grants‐in‐aid provided by the Japanese Ministry of Education, Culture, Sports, Science, and Technology (Grant No. 16K09825: T. Kojima) and the Japanese Ministry of Health, Labor, and Welfare (Research on Measures for Intractable Diseases, Grant No. 2017‐012: T. Kojima). We thank Editage (www.editage.com) for English language editing.
Tokoro M, Tamura S, Suzuki N, et al. Aberrant X chromosomal rearrangement through multi‐step template switching during sister chromatid formation in a patient with severe hemophilia A. Mol Genet Genomic Med. 2020;8:e1390 10.1002/mgg3.1390
Mahiru Tokoro and Shogo Tamura contributed equally to this work, and both have the right to be listed first in bibliographic documents.
Contributor Information
Shogo Tamura, Email: stamura@met.nagoya-u.ac.jp.
Tetsuhito Kojima, Email: kojima@met.nagoya-u.ac.jp.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Bagnall, R. D. , Waseem, N. , Green, P. M. , & Giannelli, F. (2002). Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood, 99(1), 168–174. 10.1182/blood.v99.1.168 [DOI] [PubMed] [Google Scholar]
- Byers, P. H. (2002). Killing the messenger: New insights into nonsense‐mediated mRNA decay. Journal of Clinical Investigation, 109(1), 3–6. 10.1172/JCI14841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelly, J. , Concordet, J. P. , Kaplan, J. C. , & Kahn, A. (1989). Illegitimate transcription: Transcription of any gene in any cell type. Proceedings of the National Academy of Sciences of the United States of America, 86(8), 2617–2621. 10.1073/pnas.86.8.2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Xie, X. , Wu, X. I. , Lu, Y. , Wang, X. , Wu, W. , … Ding, Q. (2017). Complex recombination with deletion in the F8 and duplication in the TMLHE mediated by int22h copies during early embryogenesis. Thrombosis and Haemostasis, 117(8), 1478–1485. 10.1160/TH17-01-0046 [DOI] [PubMed] [Google Scholar]
- Do, H. , Healey, J. F. , Waller, E. K. , & Lollar, P. (1999). Expression of factor VIII by murine liver sinusoidal endothelial cells. Journal of Biological Chemistry, 274(28), 19587–19592. 10.1074/jbc.274.28.19587 [DOI] [PubMed] [Google Scholar]
- El‐maarri, O. , Herbiniaux, U. , Graw, J. , Schroder, J. , Terzic, A. , Watzka, M. , … Oldenburg, J. (2005). Analysis of mRNA in hemophilia A patients with undetectable mutations reveals normal splicing in the factor VIII gene. Journal of Thrombosis and Haemostasis, 3(2), 332–339. 10.1111/j.1538-7836.2005.01140.x [DOI] [PubMed] [Google Scholar]
- Fujita, J. , Miyawaki, Y. , Suzuki, A. , Maki, A. , Okuyama, E. , Murata, M. , … Kojima, T. (2012). A possible mechanism for Inv22‐related F8 large deletions in severe hemophilia A patients with high responding factor VIII inhibitors. Journal of Thrombosis and Haemostasis, 10(10), 2099–2107. 10.1111/j.1538-7836.2012.04897.x [DOI] [PubMed] [Google Scholar]
- Hastings, P. J. , Ira, G. , & Lupski, J. R. (2009). A microhomology‐mediated break‐induced replication model for the origin of human copy number variation. PLoS Genetics, 5(1), e1000327 10.1371/journal.pgen.1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima, T. , Tanimoto, M. , Kamiya, T. , Obata, Y. , Takahashi, T. , Ohno, R. , … Saito, H. (1987). Possible absence of common polymorphisms in coagulation factor IX gene in Japanese subjects. Blood, 69(1), 349–352. 10.1182/blood.V69.1.349.349 [DOI] [PubMed] [Google Scholar]
- Lakich, D. , Kazazian, H. H. , Antonarakis, S. E. , & Gitschier, J. (1993). Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nature Genetics, 5(3), 236–241. 10.1038/ng1193-236 [DOI] [PubMed] [Google Scholar]
- Lannoy, N. , & Hermans, C. (2016). Principles of genetic variations and molecular diseases: Applications in hemophilia A. Critical Reviews in Oncology Hematology, 104, 1–8. 10.1016/j.critrevonc.2016.04.005 [DOI] [PubMed] [Google Scholar]
- Lee, J. A. , Carvalho, C. M. , & Lupski, J. R. (2007). A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell, 131(7), 1235–1247. 10.1016/j.cell.2007.11.037 [DOI] [PubMed] [Google Scholar]
- Naylor, J. , Brinke, A. , Hassock, S. , Green, P. M. , & Giannelli, F. (1993). Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Human Molecular Genetics, 2(11), 1773–1778. 10.1093/hmg/2.11.1773 [DOI] [PubMed] [Google Scholar]
- Naylor, J. A. , Green, P. M. , Montandon, A. J. , Rizza, C. R. , & Giannelli, F. (1991). Detection of three novel mutations in two haemophilia A patients by rapid screening of whole essential region of factor VIII gene. Lancet, 337(8742), 635–639. 10.1016/0140-6736(91)92450-g [DOI] [PubMed] [Google Scholar]
- Pan, J. , Dinh, T. T. , Rajaraman, A. , Lee, M. , Scholz, A. , Czupalla, C. J. , … Butcher, E. C. (2016). Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo. Blood, 128(1), 104–109. 10.1182/blood-2015-12-684688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pio, S. F. , Oliveira, G. C. , Soares, S. , & Rezende, S. M. (2011). An aberrant pattern for intron 1 inversion of factor VIII gene. Haemophilia, 17(4), 708–709. 10.1111/j.1365-2516.2010.02482.x [DOI] [PubMed] [Google Scholar]
- Rossetti, L. C. , Radic, C. P. , Larripa, I. B. , & De Brasi, C. D. (2008). Developing a new generation of tests for genotyping hemophilia‐causative rearrangements involving int22h and int1h hotspots in the factor VIII gene. Journal of Thrombosis and Haemostasis, 6(5), 830–836. 10.1111/j.1538-7836.2008.02926.x [DOI] [PubMed] [Google Scholar]
- Rosslter, J. P. , Young, M. , Kimberland, M. L. , Hutter, P. , Ketterling, R. P. , Gitschier, J. , Horst, J. , Morris, M. A. , Schaid, D. J. , de Moerloose, P. , Sommer, S. S. , Kazazian, H. H. , … Antonarakis, S. E. (1994). Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells. Human Molecular Genetics, 3(7), 1035–1039. 10.1093/hmg/3.7.1035 [DOI] [PubMed] [Google Scholar]
- Sanna, V. , Ceglia, C. , Tarsitano, M. , Lombardo, B. , Coppola, A. , Zarrilli, F. , … Di Minno, G. (2013). Aberrant F8 gene intron 1 inversion with concomitant duplication and deletion in a severe hemophilia A patient from Southern Italy. Journal of Thrombosis and Haemostasis, 11(1), 195–197. 10.1111/jth.12061 [DOI] [PubMed] [Google Scholar]
- Sukarova Stefanovska, E. , Dejanova, V. , Tchakarova, P. , Petkov, G. , & Efremov, G. D. (2008). Genetic inversions among hemophilia A patients from Macedonia and Bulgaria. Acta Haematologica, 120(3), 192–194. 10.1159/000187648 [DOI] [PubMed] [Google Scholar]
- You, G. , Chi, K. , Lu, Y. , Ding, Q. , Dai, J. , Xi, X. , … Wang, X. (2014). Identification and characterisation of a novel aberrant pattern of intron 1 inversion with concomitant large insertion and deletion within the F8 gene. Thrombosis and Haemostasis, 112(2), 264–270. 10.1160/TH13-10-0892 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1‐S5
Table S1‐S4
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.