

Abstract
Introduction
We sought lipid‐metabolic biomarkers involved in the processes underlying cognitive decline and detected them in association with Alzheimer's disease (AD) phenotypes.
Methods
A least absolute shrinkage and selection operator logistic regression model was used to select lipids that best classified cognitive decline defined by a fast‐annual rate of cognition. Lipid summary scores were constructed as predictors of cognitive decline by using this model. Multivariable‐adjusted models tested the associations of risk score with AD phenotypes.
Results
A model incorporating 17 selected lipids showed good discrimination and calibration. The lipid risk score was positively associated with the baseline Alzheimer Disease Assessment Scale—13‐item cognitive subscale (ADAS‐Cog13) score and cerebrospinal tau protein level, and predicted cognitive diagnoses. Additional results showing that individuals with increased lipid risk scores had rapid change rates of ADAS‐Cog13 and brain atrophy further corroborated the predictive role of lipids.
Discussion
A panel of blood lipids instead of individual lipid molecules could better diagnose and predict cognitive decline.
Keywords: biomarkers, cognitive decline, lipidomics, prediction
1. INTRODUCTION
Cognitive decline is a gradually accumulating pathological process involving silent progressions into mild cognitive impairment (MCI) and dementia. 1 Data‐driven investigations have revealed that the prevalence of cognitive decline involving mild deficits and dementia among individuals aged over 65 years was approximately 20% to 30% in America and 8% in China. 2 In addition, the rapid increase in the aging population worldwide has also accelerated the prevalence with cognitive impairment. 3 , 4 However, there is still a lack of convenient pathways or biomarkers to identify cognitive decline, especially during the early stages of the Alzheimer's disease (AD) spectrum. 5 Notably, blood metabolites have been suggested as promising biomarkers of pre‐symptomatic pathological processes to facilitate identification of individuals with worse cognitive trajectories in early or later life, 6 , 7 , 8 , 9 thereby enhancing the efficiency of recruitment into clinical trials and early intervention.
Polymorphisms at apolipoprotein‐encoding genes involved in lipid transport and metabolism, such as apolipoprotein E (APOE) and clusterin (CLU), are deemed to be associated with an increased risk of AD, indicating the underlying predictive roles of lipid metabolism in early diagnosis. 10 , 11 , 12 With advances in mass spectrometry technology and analytical software, some novel platforms have used untargeted or targeted lipidomics approaches for analyses of disease processes and relevant biomarkers. 13 Multiplatform screening investigations have determined the roles of phosphatidylcholine (PC) molecules in diagnosing MCI and AD, 14 supporting specific associations of abnormal PC metabolism with poor memory performance and cognitive function in normal aging. 15 Similarly, a lipidomic study suggested that the plasma metabolite molecules distinguishing AD patients from controls were mostly derived from lipids, such as cholesteryl esters (CE). 16 Individuals with AD show a comparative decrease in plasma sphingolipid (SP16) levels and an increase in ceramide (CM16) levels. 17 Collectively, these studies support the hypothesis that lipid‐metabolic dysregulations are present in the early and late phases of AD.
Invasive procedures for sampling cerebrospinal fluid (CSF) biomarkers and expensive imaging examinations, such as positron emission tomography (PET) for amyloid imaging, have generally not been applied for screening or determining at‐risk populations with cognitive decline to date. Therefore, we aimed to identify a comprehensive panel of lipid signatures, instead of individual molecules, as a predictor of cognitive decline via tracing the trajectory of cognitive aging. We analyzed the data for 579 plasma lipids of 374 non‐demented participants from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database. To additionally verify the effectiveness of these reliable plasma lipids in predicting poor cognition, we examined the associations of the selected lipids with AD phenotypes.
2. METHODS
2.1. Participants
All data were obtained from the ADNI database (http://adni.loni.usc.edu). The ADNI was launched in 2003 as a public‐private partnership led by principal investigator Michael W. Weiner. Its primary goal is to test whether serial magnetic resonance imaging (MRI), PET, biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. For up‐to‐date information, see www.adni-info.org. ADNI was approved by the institutional review boards of all participating institutions. Written informed consent was obtained from all participants and collateral informants at each site.
Highlights
Seventeen lipids were identified with favorable prediction and calibration efficacy.
A panel of selected lipids independently predicted accelerated cognitive decline.
Lipids associated with change rates of cognitive performance and brain atrophy.
Selected lipids predicted diverse diagnoses and a trend for progressive outcomes.
RESEARCH IN CONTEXT
Systematic review: We systematically searched PubMed and primary literature about the roles of blood‐based lipid signatures in Alzheimer's disease (AD) neuropathology and phenotypes. Relatively heterogeneous studies focusing on AD metabolism relevant to lipid signatures suggest a pressing need to figure out whether lipids are promising diagnostic and predictive biomarkers of cognitive decline.
Interpretation: The study uncovered a panel of lipids with favorable predictive and calibrated efficacy that identified cognitively declined individuals, and reported some novel associations of lipid signature with worse cognitive performance, brain atrophy, progressive cognitive decline, and cognitive diagnoses for non‐dementia elders. These findings deepen our knowledge about the roles of lipids in the early stage of AD.
Future directions: Further research is warranted to focus on blood lipids in the associations with longitudinal changes in AD neuropathology. Integrating genetic variations and other biological information in the future may enhance the adaptability of the existing models.
We studied 374 ADNI‐1 non‐demented elders with available data for plasma lipid molecules, CSF core biomarkers, and cognitive performance and neuroimaging assessments. These included 128 cognitively normal (CN) individuals and 246 persons with MCI. We further classified MCI patients as stable MCI (sMCI) and progressive MCI (pMCI) patients on the basis of diagnosis of conversion to AD during at least one follow‐up year (see detailed flowchart in Figure S1 in supporting information). The mean and maximum follow‐up period was 5.06 years (standard deviation = 2.91) and 10 years, respectively. Table S1 in supporting information shows the detailed follow‐up durations of different cognitive statuses. ADNI evaluated the general cognition of these participants annually using the Alzheimer Disease Assessment Scale–13‐itemcognitive subscale (ADAS‐Cog13) and the Mini‐Mental State Examination (MMSE). All available data were obtained from the ADNI database.
2.2. Plasma lipid signature measurements
The Alzheimer Disease Metabolomics Consortium (ADMC) deposited the generated baseline targeted lipidomic data for ADNI plasma samples to the Laboratory of Neuroimaging (LONI). ADMC uses metabolomics and lipidomics platforms to cover a broad area of biochemistry and to enable interrogation of the AD metabolome with the goal of building a metabolomics database for ADNI. Analysis of plasma extracts was performed on an Agilent 6490 QQQ mass spectrometer with an Agilent 1290 series high‐performance liquid chromatography (HPLC) system and a ZORBAX eclipse plus C18 column (2.1 × 100 mm 1.8 μm, Agilent). Mass spectrometry analysis was performed in the positive ion mode with dynamic scheduled multiple reaction monitoring (MRM). Mass spectrometry results were integrated using Agilent software (MassHunter B9.00). The ADMC lab eventually examined a total of 579 plasma lipid molecules.
2.3. CSF measurements and classification
In the ADNI database, CSF amyloid beta (Aβ42), phosphorylated tau (p‐tau), and total tau (t‐tau) concentrations were measured by the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with research‐use‐only INNOBIA AlzBio3 (Ghent, Belgium) immunoassay kit‐based reagents. ADNI participants with available CSF biomarkers data at baseline were grouped according to the CSF levels of Aβ42 and p‐tau. Amyloid abnormal (A+) or normal (A–) statuses were defined by a cutoff value of 1.11 for the florbetapir standardized uptake value ratio (SUVR) and 192 pg/mL for CSF Aβ42. Tau pathological abnormal (T+) or normal (T–) statuses were defined by a cutoff value of 23 pg/mL for CSF p‐tau level. 18 In addition, borderline cases, which were ± 5% from the original cutoffs, were excluded to avoid reaching conclusions derived from those cases. 19 , 20
2.4. Neuroimaging assessments
Structural MRI was conducted using a Siemens Trio 3.0 tesla (T) scanner or 1.5 T scanner (Siemens, Erlangen, Germany). For the 3.0 and 1.5 T MRI images, Free‐surfer software version 5.1 and 4.3 image processing frameworks were used to estimate regional volume, respectively. For amyloid PET imaging, the averaged 18F‐florbetapir retention ratio of four cortical gray matter regions (frontal, precuneus, parietal cortex, and anterior‐cingulate) was applied for the global 18F‐florbetapir SUVR, and the cerebellum was considered the reference region.
2.5. Analytic methods
2.5.1. Training and validation sets
All participants were randomly divided into two independent sets to facilitate internal validation: the training set consisted of 299 individuals, and the never‐before‐seen validation set contained the remaining 75 persons. Between‐group differences in demographic, neuropsychologic, and neuroimaging data were tested by chi‐squared tests for categorical variables and Mann‐Whitney U tests for continuous non‐normally distributed variables.
2.5.2. Cases and controls
We used the linear mixed effect model to estimate the individual's change rate of cognitive decline by calculating the rate of change in the MMSE score (see Method section in supporting information). The MMSE score, as the dependent variable, was log‐transformed to ensure normality. Each individual's rate of change in MMSE was extracted from the model for subsequent analyses. Of note, we constructed a case‐control design to classify participants into two groups according to cognitive changes in MMSE: individuals with rates of change worse than the mean were assigned into case group with rapid cognitive progression, and others were classified as controls with slow cognitive decline.
2.5.3. The selection and assessment of predictive lipids
The least absolute shrinkage and selection operator (LASSO) logistic regression model was applied for selection of a subset of baseline plasma lipids related to cognitive decline (see Method section in supporting information). 21 This machine‐learning algorithm identifies specific variables that predict a given dependent variable, and allows optimal variable weights for this prediction. In the training set, 579 lipids were incorporated into the LASSO model we developed for predicting cognitive decline (ie, well‐defined case events). A 10‐fold cross‐validation procedure served to optimize the penalization and weight parameters of this LASSO model. 6 Finally, we estimated the discriminatory efficacy of this model in the training set by quantifying the area under the receiver‐operator characteristic (ROC) curve, and similarly tested the generalizability of this model in the validation set. In addition, the Hosmer‐Lemeshow tests and calibration curves were used to estimate the calibration efficacy of this model in both sets.
2.5.4. Construction of the lipidomic risk score
To explore the impact of all selected lipid profiles on cognitive decline, we constructed a lipidomic summary risk score for each participant by multiplying the z‐scored baseline lipid level times the corresponding standardized weights from LASSO regression. Discrimination and calibration performance for the model incorporating clinical variables (age, sex, years of education, and the status of APOE‐ε4 and lipid‐lowering medication), and the risk score was further assessed in the training set and confirmed in the validation set. The Delong test was conducted to estimate the statistical differences in discriminatory efficacy among different models by plotting ROC curves. 22 Multivariable logistic regression models combining clinical variables and CSF core biomarkers detected the independently predictive effect of the lipidomic risk score.
2.5.5. Analyses of cross‐sectional and longitudinal associations
We applied multivariable linear regression to test cross‐sectional associations of the lipidomic risk score with baseline ADAS‐Cog13 score and CSF core biomarkers. We used a linear mixed effect model to assess the longitudinal associations of baseline risk score with the change rates of cognitive performance (ADAS‐Cog13) and brain structural measures. A significant value for the interaction term between time and risk score in the linear mixed model indicated that lipids affected the rate of cognitive change (see Method section in supporting information). The Cox proportional hazards model was used to assess the impact of the lipidomic risk score on the time of conversion from MCI to AD. Logistic regression analysis was used to assess the predictive effects of the lipidomic risk score on diagnostic categories. All regression analyses were adjusted for clinical variables. Analyses of brain structural measures were additionally adjusted for total intracranial volume to account for variations of head size.
Statistical significance was set at a two‐sided P < .05. The following variables were z‐scored to ensure normality prior to subsequent analyses: lipidomic risk score, ADAS‐Cog13 score, CSF Aβ42, CSF p‐tau, CSF t‐tau, as well as hippocampal and lateral ventricular volumes. All statistical analyses were performed using R version 3.5.1 software program and IBM SPSS Statistics 25.
3. RESULTS
3.1. Demographic and clinical characteristics of the study population
In the training set, individuals in the case group included a higher proportion of APOE‐ε4 carriers, and were more likely to show worse cognition (ADAS‐Cog13 and MMSE scores) and brain atrophy (lateral ventricular and hippocampal volumes) than controls (All P < .05; Table S2 in supporting information). Similarly, in the validation set, differences were still significant in terms of the proportion of APOE‐ε4 carriers, cognitive performance, and hippocampal volume between cases and controls.
3.2. LASSO‐weighted lipid signature and cognitive decline
The LASSO approach identified 17 lipids in a sparse, high‐dimensional logistic model in the training set (Figure 1A and B). These lipids displayed favorable prediction efficacy, with an area under the curve (AUC) of 0.768 (95% confidence interval [CI], 0.715–0.821) in the training set and 0.747 (95% CI, 0.627–0.867) in the validation set (Figure 1C and D). Figure 2 provides an overview of the weighted coefficients of the 17 lipids. The lipidomic risk scores were calculated by a linear combination of 17 lipids that reflected the risk of cognitive decline. In the training set, the cases showing fast cognitive decline presented higher lipidomic risk scores than controls (0.509 ± 0.196 vs −0.051 ± 0.156, P < .001), which was further confirmed in the validation set (0.097 ± 0.208 vs −0.060 ± 0.132, P < .001). Differences in the 17 plasma lipids between participants with or without cognitive decline are shown in detail in Table S3 in supporting information.
FIGURE 1.
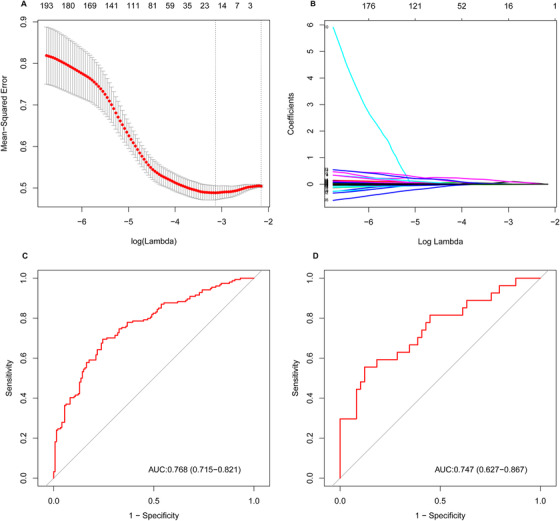
Potential lipid predictor selection using least absolute shrinkage and selection operator (LASSO) logistic regression and the predictive accuracy of lipid signatures. A, Selection of the parameters in the LASSO model by 10‐fold cross‐validation based on minimum criteria. The vertical dotted line points to the optimal lambda value and the number of optimal predictors. B, The pathway of coefficients among all lipid signatures. The 17 lipid signatures were selected via LASSO regression. C and D, Receiver operating characteristic curves of the lipid signatures in the training and validation sets, respectively
Abbreviation: AUC, area under the curve.
FIGURE 2.
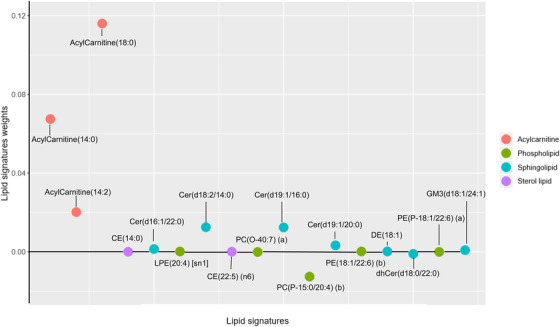
Seventeen lipid predictors of cognitive decline weighted by least absolute shrinkage and selection operator (LASSO) logistic regression. Color‐coded with the same classification annotated in the figure
Abbreviations: CE, cholesteryl ester; LPE, lysophosphatidylethanolamine; PC, phosphatidylcholine; Cer, ceramide; dhCer, dihydroceramide; GM3, ganglioside.
Subsequently, the Hosmer‐Lemeshow test and ROC analysis were used to assess the calibration and discriminatory efficacies of a model with clinical variables and risk score as independent variables. The model presented favorable calibration (P = .809; Figure S2A in supporting information) and prediction efficacy (AUC = 0.789, 95% CI = 0.737–0.840; Figure S2B) both in the training and the validation sets (Figure S2C and D).
The discriminatory efficacy of the lipidomic risk score was further detected among all participants (Figure S3 in supporting information). Addition of the risk score to the model significantly improved the capacity to predict cognitive declined outcomes (AUC = 0.779, 95% CI = 0.712–0.845) in comparison to the reference model with clinical variables (P = .014). The polygenic hazard score (PHS) has been generally applied for prediction of cognitive decline via evaluation of APOE and 31 other genetic variants among the risk population. The reference model assessing PHS yielded an AUC of 0.766 (95% CI = 0.715–0.818). As expected, the model including PHS, lipid signature, and clinical variables yielded the best improvement in prediction (Figure 3).
FIGURE 3.
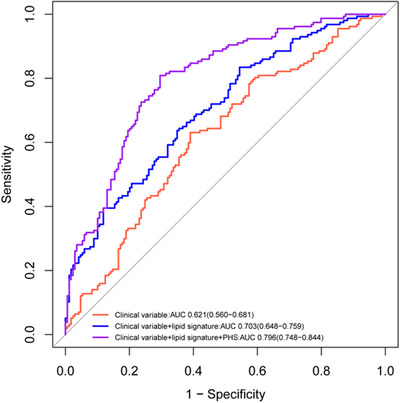
Receiver operating characteristic curve analysis for prediction of cognitive decline
Abbreviation: AUC, area under the curve; PHS, polygenic hazard score.
The logistic regression model showed that the lipidomic risk score was associated with cognitive decline independent of clinical covariates, especially the levels of CSF Aβ42, p‐tau, and t‐tau (odds ratio [OR] = 3.15, 95% CI = 1.73–5.77; Table S4 in supporting information).
3.3. Lipid risk score was associated with neuropsychiatric assessments, CSF biomarkers, and brain structural measures
Cross‐sectional analyses showed the positive associations of the risk score with ADAS‐Cog13 (β = 0.134, P = .007; Table 1), CSF p‐tau level (β = .161, P = .025), and CSF t‐tau level (β = 0.185, P = .010). Longitudinal investigations indicated interactions between lipidomic risk score and time with a higher risk score as predictors of faster annual worsening in ADAS‐Cog13 and neuroimaging MRI findings (ADAS‐Cog13: β = 0.045, P = .001; hippocampus: β = −0.014, P = .007; lateral ventricles: β = 0.020, P < .001).
TABLE 1.
Modeling the association of lipid risk score on clinical outcomes
| Model | β Coefficient | P |
|---|---|---|
| Cross‐sectional | ||
| ADAS‐Cog13 | 0.134 | 0.007 |
| CSF Aβ42 | −0.124 | 0.055 |
| CSF t‐tau | 0.185 | 0.010 |
| CSF p‐tau | 0.161 | 0.025 |
| Longitudinal | ||
| ADAS‐Cog13 | ||
| Main effect | 0.091 | 0.002 |
| Interaction time | 0.045 | 0.001 |
| Hippocampus | ||
| Main effect | −0.121 | 0.003 |
| Interaction time | −0.014 | 0.007 |
| Lateral ventricles | ||
| Main effect | 0.073 | 0.078 |
| Interaction time | 0.020 | <0.001 |
| Cox (Hazard ratio) | Statistic | p‐value |
| MCI conversion to AD dementia | 1.124(0.944‐1.340) | 0.190 |
Abbreviations: Aβ42, 42‐aminoacid isoform of amyloid beta; AD, Alzheimer's disease; ADAS‐Cog, the Alzheimer's Disease Assessment Scale‐13‐item cognitive subscale; Cox, Cox proportional hazard model; CSF, cerebrospinal fluid; MCI, mild cognitive impairment; p‐tau, hyperphosphorylated tau protein; t‐tau, total tau protein.
3.4. Lipidomic risk score associated with diagnostic categories
Statistical comparisons indicated the possible predictive roles of the lipidomic risk score in the following diagnostic groups: case versus control, sMCI versus CN, pMCI versus CN, A+ versus A–, and A+T+ versus A–T– (Figure 4). The increased lipidomic risk score was correlated with a 1.86‐fold increase in the odds of cognitive decline (95% CI = 1.428–2.432; Figure 5). Similarly, a higher lipidomic risk score produced a 1.75‐fold increase in the odds of pMCI versus CN (95% CI = 1.259–2.436) and a 1.34‐fold increase in the odds of sMCI versus CN (95% CI = 1.015–1.811). There was a modest trend toward a 1.39‐fold increase in the probability of A+T+ with the increase in lipid risk score compared to A–T– (95% CI = 0.920–2.095). No correlations were found between the lipid risk score and other diagnostic categories.
FIGURE 4.
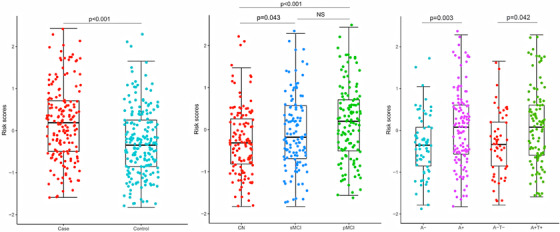
The boxplot of the lipid risk score in different diagnostic groups at baseline
Abbreviations: sMCI, stable mild cognitive impairment; pMCI, progressive mild cognitive impairment; CN, cognitively normal; A+T+, amyloid abnormal using CSF Aβ and tau abnormal using CSF p‐tau; A–T–, amyloid normal using CSF Aβ and tau normal using CSF p‐tau.
FIGURE 5.
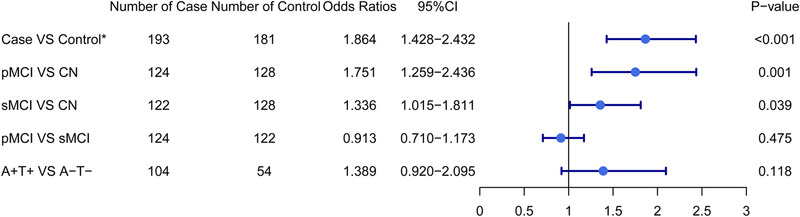
The risk effect estimates among four diagnostic groups by multivariate logistic regression. Models are adjusted for age at baseline, sex, years of education, status of APOE‐ε4, and lipid medications. * The case is defined as participant with worse cognitive decline according to the rate of change in Mini‐Mental State Examination
Abbreviations: sMCI, stable mild cognitive impairment; pMCI, progressive mild cognitive impairment; CN, cognitively normal; A+T+, amyloid abnormal using CSF Aβ and tau abnormal using CSF p‐tau; A–T–, amyloid normal using CSF Aβ and tau normal using CSF p‐tau.
The Cox proportional hazard model was then used in individuals with a baseline diagnosis of MCI to determine the predictive role of the lipidomic risk score in the conversion to AD. Individuals with an increased lipid risk score had a modest trend toward disease progression (hazard ratio [HR] = 1.124, 95% CI = 0.944–1.340).
4. DISCUSSION
The ideal peripheral biomarkers would provide useful indications of cognitive decline at an early stage, because irreversible shifts would have developed silently in the brain by the time perceivable clinical symptoms appear. In this longitudinal study of non‐demented elderly participants, a panel of 17 plasma lipid molecules was identified as a potential predictor of cognitive decline with a good trade‐off between predictive ability (high AUC) and sparsity (low number of lipids). Furthermore, this 17‐lipid signature was positively associated with the baseline ADAS‐Cog13 score and levels of CSF tau protein and the risk of diverse diagnostic categories. Individuals with higher lipid risk scores showed faster change rates of ADAS‐Cog13 and brain atrophy (hippocampus and lateral ventricles) during follow‐up. These results indicated that the baseline plasma lipid combination, but not individual lipids, should be considered a valuable noninvasive tool to assess and predict cognitive decline. By summarizing the findings for multiple lipids, our study expanded the possible role of aberrant lipids metabolism in the cognitive decline, presenting a major advantage over previous explorations.
The selected lipid signatures covered sterol lipids, acylcarnitine, phospholipids, and sphingolipids. Several previous studies examined the underlying mechanisms of lipid signatures in their associations with cognitive decline. Acyl‐coenzyme A: cholesterol acyltransferases (ACAT) are critical coenzymes in the cholesterol esterification pathway. Animal studies have shown improvements in the cognitive functions of AD mice with increased levels of CEs via inhibition of ACAT. 16 Plasma acylcarnitines play a role in damaged fatty acid beta‐oxidation in mitochondria and are positively correlated with total tau proteins, 23 , 24 which support the adverse effects of mitochondrial dysfunctions in tau mediated neurodegeneration. 25 Ceramides can exacerbate the Aβ production by stabilizing the Aβ precursor protein‐cleaving enzyme. 25 In addition, ceramides can induce mitochondrial impairment and cell apoptosis, which might contribute to neurodegenerative changes. 8 , 26 Similar studies revealed that lower levels of PC were associated with increased AD risk and accelerated cognitive decline. 13 , 14 , 25 This association has been deemed to relate to dysfunction of cellular lipid production. The essential elements in the synthesis of certain phospholipids are the contacts between endoplasmic reticulum and mitochondria. 27 , 28 Previous studies found that these contacts were impaired in AD, and various APOE alleles interfered with these contacts. 29 , 30 , 31
We investigated a panel of plasma lipids as a potential diagnostic and predictive biomarker of cognitive decline in the early stage of dementia by incorporating non‐demented individuals. These findings were thus less driven by the complex status of dementia. A second strength is the usage of change rates of cognitive performance for classifications of individuals with or without rapid cognitive decline. This approach facilitated selection of lipids that were closely associated with longitudinal trajectories of cognition based on a robust LASSO‐logistic model. Of note, the discriminatory and calibration efficacies of these lipids were well validated in an unseen data set. Subsequent analyses of the associations between the panel of lipids and AD phenotypes enhanced the possible roles of lipids in prediction of cognitive decline. Consistent with our subsequent findings, previous cross‐sectional studies focused on associations of sets of coregulated plasma lipids with AD phenotypes in a mixed population including individuals with AD. 25 However, our primary findings were based on longitudinal changes in cognition and our objective was to identify some lipids that could facilitate the diagnosis or prediction of cognitive decline, which was different from previous efforts. The contributions of individual lipids may be small, but the summation of several lipids could improve prediction and reflect the lipid metabolism of each person, highlighting the immense significance of this study. In addition, the risk estimates of the established lipid risk score remained significant after adjustment for clinical variables, which underscored the independent effect of the lipid signature on cognitive decline.
The current study had several limitations. Although common vascular factors and body mass index may disturb lipid metabolism, the relatively small sample sizes of the analysis groups precluded full adjustment for potential variables, except for lipid‐lowering medication. Moreover, estimation of the rate of cognitive decline using the rate of change in the MMSE score is a crude approach. Finally, we failed to clarify the roles of lipids in distinguishing pMCI from sMCI, which may be caused by the existing aberrant lipid metabolism both in sMCI and pMCI individuals.
5. CONCLUSION
Our findings suggest that a combination of lipids, including acylcarnitine, sterol lipid, phospholipid, and sphingolipid, may have a valuable role in predicting aggravated cognitive performance, brain atrophy, and different diagnostic categories in the early phase. A comprehensive panel of blood lipids instead of evaluations of individual lipid molecules can more effectively identify individuals effectively who are undergoing or have the potential to suffer from subsequent cognitive decline. Further integration of genetic variations and other biological information may enhance the adaptability of the existing models and lead to a better understanding of the biological context in which relevant molecules act.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (91849126). Data collection and sharing for this project was funded by Alzheimer's Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Ma Y‐H, Shen X‐N, Xu W, et al. A panel of blood lipids associated with cognitive performance, brain atrophy, and Alzheimer's diagnosis: A longitudinal study of elders without dementia. Alzheimer's Dement. 2020;12:e12041 10.1002/dad2.12041
REFERENCES
- 1. Bettio LEB, Rajendran L, Gil‐Mohapel J. The effects of aging in the hippocampus and cognitive decline. Neurosci Biobehav Rev. 2017;79:66‐86. [DOI] [PubMed] [Google Scholar]
- 2. Ma C, Yin Z, Zhu P, Luo J, Shi X, Gao X. Blood cholesterol in late‐life and cognitive decline: a longitudinal study of the Chinese elderly. Mol Neurodegener. 2017;12:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kivipelto M, Mangialasche F, Ngandu T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol. 2018;14:653‐666. [DOI] [PubMed] [Google Scholar]
- 4. Cooper C, Sommerlad A, Lyketsos CG, Livingston G. Modifiable predictors of dementia in mild cognitive impairment: a systematic review and meta‐analysis. Am J Psychiatry. 2015;172:323‐334. [DOI] [PubMed] [Google Scholar]
- 5. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta‐analysis. Lancet Neurol. 2016;15:673‐684. [DOI] [PubMed] [Google Scholar]
- 6. Varma VR, Oommen AM, Varma S, et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: a targeted metabolomics study. PLoS Med. 2018;15:e1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Low DY, Lefèvre‐Arbogast S, González‐Domínguez R, et al. Diet‐related metabolites associated with cognitive decline revealed by untargeted metabolomics in a prospective cohort. Mol Nutr Food Res. 2019;63:e1900177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wong MW, Braidy N, Poljak A, Pickford R, Thambisetty M, Sachdev PS. Dysregulation of lipids in Alzheimer's disease and their role as potential biomarkers. Alzheimers Dement. 2017;13:810‐827. [DOI] [PubMed] [Google Scholar]
- 9. Kim M, Snowden S, Suvitaival T, et al. Primary fatty amides in plasma associated with brain amyloid burden, hippocampal volume, and memory in the European Medical Information Framework for Alzheimer's Disease biomarker discovery cohort. Alzheimers Dement. 2019;15:817‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buckley RF, Mormino EC, Amariglio RE, et al. Sex, amyloid, and APOE epsilon4 and risk of cognitive decline in preclinical Alzheimer's disease: Findings from three well‐characterized cohorts. Alzheimers Dement. 2018;14:1193‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenberg RN, Lambracht‐Washington D, Yu G, Xia W. Genomics of Alzheimer disease: a review. JAMA Neurol. 2016;73:867‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer's disease: an update. Annu Rev Neurosci. 2014;37:79‐100. [DOI] [PubMed] [Google Scholar]
- 13. Proitsi P, Kim M, Whiley L, et al. Association of blood lipids with Alzheimer's disease: a comprehensive lipidomics analysis. Alzheimers Dement. 2017;13:140‐151. [DOI] [PubMed] [Google Scholar]
- 14. Whiley L, Sen A, Heaton J, et al. Evidence of altered phosphatidylcholine metabolism in Alzheimer's disease. Neurobiol Aging. 2014;35:271‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perez‐Galvez A, Jaren‐Galan M, Garrido‐Fernandez J, Calvo MV, Visioli F, Fontecha J. Activities, bioavailability, and metabolism of lipids from structural membranes and oils: Promising research on mild cognitive impairment. Pharmacol Res. 2018;134:299‐304. [DOI] [PubMed] [Google Scholar]
- 16. Proitsi P, Kim M, Whiley L, et al. Plasma lipidomics analysis finds long chain cholesteryl esters to be associated with Alzheimer's disease. Transl Psychiatry. 2015;5:e494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han X, Rozen S, Boyle SH, et al. Metabolomics in early Alzheimer's disease: identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS One. 2011;6:e21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palmqvist S, Mattsson N, Hansson O, Alzheimer's Disease Neuroimaging I . Cerebrospinal fluid analysis detects cerebral amyloid‐beta accumulation earlier than positron emission tomography. Brain. 2016;139:1226‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu JT, Li JQ, Suckling J, et al. Frequency and longitudinal clinical outcomes of Alzheimer's AT(N) biomarker profiles: a longitudinal study. Alzheimers Dement. 2019;15:1208‐1217. [DOI] [PubMed] [Google Scholar]
- 21. Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16:385‐395. [DOI] [PubMed] [Google Scholar]
- 22. DeLong ER, DeLong DM, Clarke‐Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837‐845. [PubMed] [Google Scholar]
- 23. Ciavardelli D, Piras F, Consalvo A, et al. Medium‐chain plasma acylcarnitines, ketone levels, cognition, and gray matter volumes in healthy elderly, mildly cognitively impaired, or Alzheimer's disease subjects. Neurobiol Aging. 2016;43:1‐12. [DOI] [PubMed] [Google Scholar]
- 24. Adams SH, Hoppel CL, Lok KH, et al. Plasma acylcarnitine profiles suggest incomplete long‐chain fatty acid beta‐oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African‐American women. J Nutr. 2009;139:1073‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barupal DK, Baillie R, Fan S, et al. Sets of coregulated serum lipids are associated with Alzheimer's disease pathophysiology. Alzheimers Dement (Amst). 2019;11:619‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magtanong L, Ko PJ, Dixon SJ. Emerging roles for lipids in non‐apoptotic cell death. Cell Death Differ. 2016;23:1099‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rowland AA, Voeltz GK. Endoplasmic reticulum‐mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843:2253‐2262. [DOI] [PubMed] [Google Scholar]
- 29. Tambini MD, Pera M, Kanter E, et al. ApoE4 upregulates the activity of mitochondria‐associated ER membranes. EMBO Rep. 2016;17:27‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hedskog L, Pinho CM, Filadi R, et al. Modulation of the endoplasmic reticulum‐mitochondria interface in Alzheimer's disease and related models. Proc Natl Acad Sci U S A. 2013;110:7916‐7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Area‐Gomez E, Del Carmen Lara Castillo M, Tambini MD, et al. Upregulated function of mitochondria‐associated ER membranes in Alzheimer disease. EMBO J. 2012;31:4106‐4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.