

Abstract
Background
Primary microcephaly (MCPH) is a congenital neurodevelopmental disorder manifesting as small brain and intellectual disability. It underlies isolated reduction of the cerebral cortex that is reminiscent of early hominids which makes it suitable model disease to study the hominin‐specific volumetric expansion of brain. Mutations in 25 genes have been reported to cause this disorder. Although majority of these genes were discovered in the Pakistani population, still a significant proportion of these families remains uninvestigated.
Methods
We studied a cohort of 32 MCPH families from different regions of Pakistan. For disease gene identification, genome‐wide linkage analysis, Sanger sequencing, gene panel, and whole‐exome sequencing were performed.
Results
By employing these techniques individually or in combination, we were able to discern relevant disease‐causing DNA variants. Collectively, 15 novel mutations were observed in five different MCPH genes; ASPM (10), WDR62 (1), CDK5RAP2 (1), STIL (2), and CEP135 (1). In addition, 16 known mutations were also verified. We reviewed the literature and documented the published mutations in six MCPH genes. Intriguingly, our cohort also revealed a recurrent mutation, c.7782_7783delGA;p.(Lys2595Serfs*6), of ASPM reported worldwide. Drawing from this collective data, we propose two founder mutations, ASPM:c.9557C>G;p.(Ser3186*) and CENPJ:c.18delC;p.(Ser7Profs*2), in the Pakistani population.
Conclusions
We discovered novel DNA variants, impairing the function of genes indispensable to build a proper functioning brain. Our study expands the mutational spectra of known MCPH genes and also provides supporting evidence to the pathogenicity of previously reported mutations. These novel DNA variants will be helpful for the clinicians and geneticists for establishing reliable diagnostic strategies for MCPH families.
Keywords: ASPM, CDK5RAP2, CENPJ, CEP135, MCPH, STIL
Genomic analyses of 32 Pakistani families manifesting primary microcephaly.
1. INTRODUCTION
Autosomal recessive primary microcephaly (MCPH, MIM #251200) is a rare neurodevelopmental disorder that is diagnosed by simultaneous observation of reduced head circumference (HC) of −3 SD (standard deviation)—−2 SD at birth—below the expected mean and impaired cognition. Neuroimaging of MCPH patients shows isolated cortical hypoplasia with preserved architecture and simplified gyration (Shaheen et al., 2019; Zaqout, Morris‐Rosendahl, & Kaindl, 2017). MCPH is recognized as a rare disorder, though it is highly prevalent in countries with a high rate of consanguinity, like Pakistan (1/10,000), while it is only sporadically observed (1/1,000,000) in European populations (Cox, Jackson, Bond, & Woods, 2006). The genetic etiology of MCPH is heterogeneous with mutations reported in as many as 25 different genes playing roles in diverse cellular pathways (Table S1). Among these, ASPM (MIM#605481) alone accounts for 68% of cases followed by 14% by WDR62 (MIM#613583) and 8% by MCPH1 (MIM#607117) (Zaqout et al., 2017), whereas other genes were reported only in a few families. The most frequently observed pathomechanism is abnormal spindle orientation in neural progenitor cells (NPCs), resulting in premature switching from both symmetric proliferative and asymmetric self‐renewing to symmetric consumptive division (Fei, Haffner, & Huttner, 2014; Fish, Kosodo, Enard, Paabo, & Huttner, 2006). It has been proposed that the dysfunction of the MCPH proteins either dysregulates cell cycle dynamics or increases apoptosis, both of which might disrupt mitotic neurogenesis leading to the MCPH phenotype (Cox et al., 2006; Zaqout et al., 2017).
2. MATERIALS AND METHODS
2.1. Patient consent and ethics approval
With informed consent from parent(s)/guardians, blood samples and clinical information were collected following the rules described in the Declaration of Helsinki. The study was approved by the ethics committee of the National Institute for Biotechnology and Genetic Engineering (NIBGE) in Faisalabad, Pakistan and University of Punjab, Lahore, Pakistan.
2.2. Genomic analyses
For disease gene identification, a stepwise approach was followed; first, a few families were directed for Sanger sequencing of ASPM due to the fact that it is the most common cause of MCPH in Pakistan as well as worldwide (Ahmad et al., 2017). Second, a few families were sequenced for gene panel customized for screening the reported genes of MCPH (Table S2). Third, families excluded from mutations in ASPM were subjected to whole‐exome sequencing (WES) using the Agilent (Santa Clara, CA) version 6 enrichment kit and the Illumina HiSeq 4000 sequencing system (paired‐end reads, 2 x75 bp). WES data were analyzed using our in‐house VARBANK database (http://varbank.ccg.uni‐koeln.de). In addition to the above‐mentioned approaches, a few families were also investigated by genome‐wide linkage analysis using Illumina (San Diego, CA) HumanCoreExome 12 v1.1 array and Affymetrix (Santa Clara, CA) Axiom Precision Medicine Research Array (PMRA), as described earlier (Moawia et al., 2017). WES data analysis provided us with the information of potentially pathogenic variants which were further validated for co‐segregation by Sanger sequencing (Figure S3). Data obtained by linkage analysis by genotyping of only a few selected families corroborated the involvement of the identified gene (Figure S4). The identified variants were further consulted to calculate the allele frequency in multiple databases such as in‐house database of Cologne Center for Genomics with >1600 exomes, dbSNP151, 1000 Genomes (build 20110521), the public Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle (build ESP6500), gnomAD, Iranome, and the Greater Middle Eastern Variome.
3. RESULTS
In this study, we report mutational findings of 32 consanguineous Pakistani families with 98 affected individuals (57 males and 41 females)—70 of them were analyzed in this study. The patients were recruited according to the diagnostic criteria of reduced HC (≥3 SD), intellectual disability, and absence of brain atrophy/indicative symptoms. Clinical investigation of all affected individuals revealed reduced skull size (head circumference below −5 SD) and mild to profound intellectual disability. In addition, speech impairment, aggressive behavior, and self‐care deficits were notable features for most of the affected individuals (Figures S1 and S2, and Table S3).
Genetic investigations conducted on 32 families divulged that 22 families had mutations in ASPM, 5 in WDR62, 1 in CDK5RAP2 (MIM#608201), 1 in CENPJ (MIM#609279), 2 in STIL (MIM#181590), and 1 in CEP135 (MIM#611423) (Figure 1 and Table 1). This information supported the previous observation of ASPM and WDR62 being the most commonly involved genes in MCPH (Ahmad et al., 2017). Notably, our genetic investigations revealed 15 novel mutations in five different MCPH genes; ASPM (10), WDR62 (1), CDK5RAP2 (1), STIL (2), and CEP135 (1) (Figure 1 and Table 1).
Figure 1.
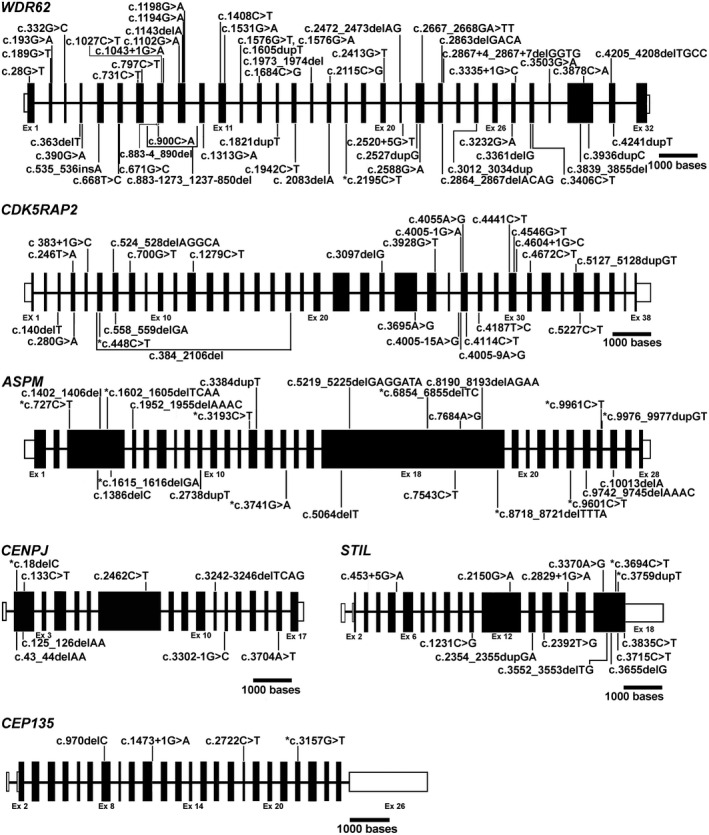
Schematic representation of the genomic structure of human WDR62, CDK5RAP2, ASPM, CENPJ, and STIL along with the causal variants. Figure shows all the known and novel mutations of WDR62, CDK5RAP2, ASPM, CENPJ, STIL, and CEP135 causing primary and syndromic microcephaly due to the mutations in the respective genes. Exons are drawn according to the given scale whereas introns are shown as arbitrary lines. White boxes denote untranslated regions. Notice that asterisk (*) represents the novel mutations found in this study. ASPM variant;c.3384dupT, previously known as c.3384_3385insT, is renamed according to the HGVS (Human Genome Variation Society) guidelines
Table 1.
List of MCPH mutations identified in this study
| Gene | Exon | Ref. Seq. ID | cDNA mutation | Protein mutation | gnomAD Frequency c | Family ID | Reference |
|---|---|---|---|---|---|---|---|
| ASPM | 3 | NM_018136.4 | c.727C>T | p.(Arg243*) | 3.98e−6/1,245848,0 | Family 20 | Novel |
| 3 | NM_018136.4 | c.1260_1266delTCAAGTC | p.(Gln421Hisfs*32) | 4e−6/1,245792,0 | Family 7,13 | Gul et al. (2006) | |
| 3 | NM_018136.4 | c.1602_1605delTCAA | p.(Asn534Lysfs*14) | 0 | Family 14 | Novel | |
| 3 | NM_018136.4 | c.1615_1616delGA | p.(Glu539Argfs*15) | 0 | Family 25 | Novel | |
| 13 | NM_018136.4 | c.3193C>T | p.(Gln1065*) | 0 | Family 27 | Novel | |
| 18 | NM_018136.4 | c.4212G>A | p.(Trp1404*) | 0 | Family 18 | Ahmad et al. (2017) | |
| 18 | NM_018136.4 | c.5149delA | p.(Ile1717*) | 0 | Family 12,19 | Gul et al. (2007) | |
| 18 | NM_018136.4 | c.5959C>T | p.(Gln1987*) | 0 | Family 30 | Ahmad et al. (2017) | |
| 18 | NM_018136.4 | c.7782_7783delGA | p.(Lys2595Serfs*6) | 2.23e−4/62,278516,0 | Family 31 | Passemard et al. (2016) | |
| 18 | NM_018136.4 | c.8190_8193delAGAA | p.(Arg2732Lysfs*4) | 8.09e−6/2,242788,0 | Family 6 | Passemard et al. (2009) | |
| 18 | NM_018136.4 | c.8508_8509delGA | p.(Lys2837Metfs*34) | 8.02e−6/2,244284,0 | Family 1,5 | Bond et al. (2003) | |
| 18 | NM_018136.4 | c.8718_8721delTTTA | p.(Leu2907Argfs*30) | 0 | Family 26 | Novel | |
| 21 | NM_018136.4 | c.9190C>T | p.(Arg3064*) | 2.8e−5/7,244828,0 | Family 15 | Nicholas et al. (2009) | |
| 21 | NM_018136.4 | c.9286C>T | p.(Arg3096*) | 4.089e−6/1,249754,0 | Family 28 | Darvish et al. (2010) | |
| 23 | NM_018136.4 | c.9557C>G | p.(Ser3186*) | 3.99e−6/1,245312,0 | Family 17 | Gul et al. (2006) | |
| 23 | NM_018136.4 | c.9601C>T | p.(Gln3201*) | 0 | Family 22 | Novel | |
| 24 | NM_018136.4 | c.9789 T > A | p.(Tyr3263*) | 0 | Family 2 | Nicholas et al. (2009) | |
| 25 | NM_018136.4 | c.9961C>T | p.(Gln3321*) | 3.99e−6/1,245316,0 | Family 16 | Novel | |
| 18 | NM_018136.4 | c.6854_6855delTC | p.(Leu2285Glnfs*32) | 2.13e−5/6,281042,0 | Family 32 | Novel | |
| 25 | c.9976_9977dupGT | p.(Ser3327 Tyrfs*14) | NA | Novel | |||
| 15 | c.3741G>A | p.(Lys1247=) | NA | Novel | |||
| WDR62 | 9 | NM_001083961.1 | c.1194G>A | p.(Trp398*) | 1.22e−5/3,245910,0 | Family 21 | Sajid Hussain et al. (2013) |
| 18 | NM_001083961.1 | c.2195C>T | p.(Thr732Ile) | 0 | Family 4 | Novel | |
| 28 | NM_001083961.1 | c.3361delG | p.(Ala1121Glnfs*6) | 0 | Family 10 | Sajid Hussain et al. (2013) | |
| 30 | NM_001083961.1 | c.3936dupC a | p.(Val1313Argfs*18) | 0 | Family 8 | Yu et al. (2010) | |
| 31 | NM_001083961.1 | c.4241dupT b | p.(Ser1415Glufs*40) | 0 | Family 9 | Nicholas et al. (2010) | |
| CDK5RAP2 | 6 | NM_018249.5 | c.448C>T | p.(Arg150*) | 1.59e−5/4,246038,0 | Family 3 | Novel |
| CENPJ | 2 | NM_018451.4 | c.18delC | p.(Ser7Profs*2) | 0 | Family 11 | Bond et al. (2005) |
| STIL | 17 | NM_001048166.1 | c.3694C>T | p.(Arg1232*) | 1.99e−5/5,246162,0 | Family 24 | Novel |
| 17 | NM_001048166.1 | c.3759dupT | p.(Pro1254Serfs*2) | 0 | Family 29 | Novel | |
| CEP135 | 23 | NM_025009.4 | c.3157G>T | p.(Glu1053*) | 3.99e−6/1,245562,0 | Family 23 | Novel |
This mutation was previously reported as c.3936_3937insC.
c.4241_4242insT, both of them were corrected according to the HGVS (Human Genome Variation Society) guidelines.
Allele frequency observed in gnomAD/allele count, total allele number, number of homozygotes.
Out of 22 ASPM linked families, 8 carried 10 novel mutations—seven homozygous in seven families and three heterozygous in one family. All of the seven homozygous mutations were protein‐truncating—four nonsense and three frameshifts—which may result in the complete loss‐of‐function of ASPM. Interestingly, a family with three heterozygous mutations carried two frameshifts (p.(Leu2285Glnfs*32); p.(Ser3327 Tyrfs*14)) and one apparently synonymous substitution (p.(Lys1247=)). We speculate that this synonymous substitution may also disrupt splicing as the altered nucleotide (c.3741G>A) is present at the 5′ end of exon 15 of ASPM. The effect of this variation on splicing, however, could not be verified due to the unavailability of RNA from the patient. The remaining 14 families segregated already reported 11 mutations (Table 1) (Ahmad et al., 2017; Bond et al., 2003; Darvish et al., 2010; Gul et al., 2006, 2007; Nicholas et al., 2009; Passemard et al., 2009, 2016). Three of these known mutations were found in two families each (Table 1). The homozygous frameshift mutation c.8190_8193AGAA;p.(Arg2732Lysfs*4) has been previously reported in the compound heterozygous state together with c.3945_3946delATCTT;p.Arg1315Serfs*2 (Passemard et al., 2009). All the patients carrying ASPM mutation(s) manifested one or more comorbidities such as seizures, stuttering, hypersalivation, and cup‐shaped ears. One notable additional feature was bilateral hearing impairment (HI), observed in two (out of three) patients of family 14 who carry a protein‐truncating ASPM mutation (Table S3). Sensorineural hearing loss has previously been reported in MCPH patients carrying mutations in ASPM and CDK5RAP2 (Darvish et al., 2010; Pagnamenta et al., 2012). To check for the possibility of an independent mutation causing HI in these patients, we interviewed the extended family for a history of HI segregating independently of MCPH, but it was found unremarkable. Parallel to the sequencing approach, genome‐wide linkage analysis was also performed in five selected families on the Axiom Precision Medicine Research Array (PMRA) from Affymetrix (Santa Clara, CA), which delineated the homozygous segments on chromosome 1 harboring ASPM (Figure S4), thus providing further evidence for the involvement of this gene in disease causation.
The second most frequently mutated gene (15.62%) in our cohort was WDR62. In this gene, five mutations were identified which included one novel missense and four known protein‐truncating variants (Table 1 and Figure 1) (Nicholas et al., 2010; Sajid Hussain et al., 2013; Yu et al., 2010). The missense mutation c.2195C>T;p.(Thr732Ile), identified in family 4, is predicted to be deleterious and disease causing by a number of bioinformatics tools (CADD score = 30, PROVEAN = −4.262, Mutation Taster = 89, PolyPhen‐2 = 0.99 and SIFT = 0.01). Further, this mutation is not documented in any of the public databases of genomic variants. Genome‐wide linkage analysis for selected members of this family revealed a homozygous segment on chromosome 19 at cytoband 19q13.1‐13.3 with a LOD score of 5.4. This homozygous segment (rs73039760‐32,418,599 bp to rs4141695‐44,985,674 bp) contains WDR62, the only gene in which a deleterious mutation was identified (Figure S4). In addition to core symptoms of microcephaly (mean HC = −10 SD), mild to moderate intellectual disability, the patients also presented speech impairment, short stature (in V‐6), and seizures (in IV‐1) (Table S3).
Family 3 from our cohort was identified with a novel mutation in CDK5RAP2. Affected individuals of this family presented severe microcephaly with HC ranging −11 to −12 SD, speech impairment, and short stature ranging from −5 SD to −6 SD (Table S3). Linkage analysis of this family indicated four peaks reaching the theoretical maximum LOD score of 3.01 on chromosome 8, 9, 13, and 20. The candidate variant obtained from exome sequencing was located in the linkage region on chromosome 9 (Figure S4). It was a homozygous truncating mutation in exon 6 of CDK5RAP2 (NM_018249.5;c.448C>T;(p.Arg150*)) (Table 1).
One family of our cohort carried a previously reported homozygous frameshift mutation c.18delC;p.(Ser7Profs*2) of CENPJ (Bond et al., 2005). We also identified two novel mutations in STIL; one nonsense mutation c.3694C>T;p.(Arg1232*) in family 24 and a frameshift mutation c.3759dupT;p.(Pro1254Serfs*2) in family 29 (Figure 1 and Table 1). One family was identified with the nonsense mutation c.3157G>T;p.(Glu1053*) in CEP135 (Table 1).
4. DISCUSSION
ASPM encodes abnormal spindle‐like microcephaly‐associated protein and is the most common cause of MCPH contributing 40% to 68% of MCPH cases (Letard et al., 2018; Zaqout et al., 2017). The findings of this study are in line with previous reports because we have also observed 68.75% (22/32) families carrying a mutation in ASPM. Building on another elegant study by Letard and colleagues (Letard et al., 2018)—summarizing 189 ASPM mutations in 282 previously reported and 39 new families—we reviewed the literature published thereafter and extended the mutational spectrum to 211 in 381 families (Ahmed et al., 2019; Bazgir, Agha Gholizadeh, Sarvar, & Pakzad, 2019; Bhargav, Sreedevi, Swapna, Vivek, & Kovvali, 2017; Boonsawat et al., 2019; Khan, Wang, Han, Ahmad, & Zhang, 2018; Kvarnung et al., 2018; Li et al., 2017; Marakhonov et al., 2018; McSherry et al., 2018; Moriwaki et al., 2019; Okamoto, Kohmoto, Naruto, Masuda, & Imoto, 2018; Shaheen et al., 2019). This number also includes 10 novel mutations of 8 families identified in this study (Figure 1 and Table S4).
Intriguingly, our cohort did not show the previously identified founder mutation, c.3978G; p.(Trp1326*), of Northern Pakistani Pashtun ethnicity (Ahmad et al., 2017). One explanation for this discrepancy is that none of the families recruited for this study belonged to Pashtun ethnicity; rather they originated from a region with population composition of diverse ethnic background. Thus our findings support this mutation to be a founder event, rather than a recurrent mutation. It is, however, noteworthy that our cohort contains a family carrying another recurrent mutation—c.7782_7783delGA;p.(Lys2595Serfs*6)—of ASPM, which has been reported in several different ethnic groups originating from Europe, Africa, and Asia (Letard et al., 2018). Another nonsense mutation, ASPM:c.9557C>G;p.(Ser3186*), reported exclusively in even Pakistani families, is also found in a family from our cohort (Table 1 and Figure 1), which increases the number of families with this particular mutation from 7 to 8. This observation indicated that it could be another founder mutation in the Pakistani population.
Our data also supported the notion that WDR62 (MIM# 613583) is the second most frequently mutated gene (14%) of MCPH (Zaqout et al., 2017), because we have found a nearly similar number of MCPH families (15.62%) with mutations in this gene. After a thorough literature review of WDR62 mutations by Poulton and colleagues (Poulton et al., 2014), summarizing a total of 24 mutations, 29 new mutations were published thereafter (Kvarnung et al., 2018; Yi et al., 2019; Zombor et al., 2019). The novel missense mutation reported in our study increased the total number of identified mutations to 54 (Figure 1 and Table S5).
So far, 24 mutations of 24 families have been reported in CDK5RAP2 and the identification of the novel nonsense mutation in this study increased this number to 25 (Figure 1 and Table S6) (Ahmad et al., 2017; Issa et al., 2013; Shaheen et al., 2019). A few cases of ASPM and CDK5RAP2‐related MCPH have been reported with short stature at birth, but the majority of them attained normal height at a later age (Issa et al., 2013; Passemard et al., 2009). Our patients show short stature at the age of 10 years, which adds to the phenotypic spectrum due to CDK5RAP2 dysfunction. Notably, one of the patients (V‐6) of family 4 mutated with WDR62 also show short stature clinically diagnosed only at the age of 18 years. This is another example of a patient unable to attain normal height at adult age. The literature survey revealed only eight mutations of CENPJ found in 17 families, including one reported in this study, manifesting the MCPH phenotype (Figure 1 and Table S7) (Darvish et al., 2010; Sajid Hussain et al., 2013; Shaheen et al., 2019). This frameshift mutation could reasonably be a founder mutation of the Pakistani population as this is the 5th consecutive Pakistani family reported with this mutation. Analysis of published data about disease‐causing STIL mutations and by including two novel mutations found in this study raise the total number of STIL mutations to 13 in 12 families (Figure 1 and Table S8) (Cristofoli, De Keersmaecker, De Catte, Vermeesch, & Van Esch, 2017; Shaheen et al., 2019). Nonsense mutation of CEP135 could most likely cause nonsense‐mediated decay of the mutant transcript; else it would result in a truncated protein, lacking the C‐terminus. Hitherto, only three mutations have been reported in CEP135 and our report of this additional one raises this number to 4 in four families (Figure 1 and Table S9) (Farooq et al., 2016; Hussain et al., 2012; Shaheen et al., 2019).
Conclusively, in 32 families from Pakistan, we have identified 15 novel variants and 16 previously reported ones in well‐characterized MCPH genes (Table 1). Functional evaluation of the novel variants will elucidate their effect on neurogenesis and the development of the MCPH phenotype. It will also help to develop correlation with the type of mutation and the severity of the disease. Finally, the review of published variants and addition of pathogenic variants of MCPH genes provided by this study will be helpful for devising diagnostic strategies for MCPH.
CONFLICT OF INTEREST
No competing interests.
AUTHOR CONTRIBUTIONS
Sajida Rasool, Jamshaid Mahmood Baig, Abubakar Moawia: Methodology, investigation, and validation. Ilyas Ahmad, Maria Iqbal, Syeda Seema Waseem, Maria Asif, Emrah Kaygusuz: Genomic investigation. Uzma Abdullah: Data acquisition, manuscript review, and editing. Ehtisham Ul Haq Makhdoom, Muhammad Zakaria, Shafaq Ramzan, Saif ul Haque, Asif Mir, Iram Anjum, Mehak Fiaz, Zafar Ali, Muhammad Tariq, Neelam Saba, Wajid Hussain, Saba Irshad: Clinical data acquisition. Birgit Budde: Genomic investigations and validation. Angelika Anna Noegel, Stefan Höning, Shahid Mahmood Baig, Peter Nürnberg: Supervision, funding acquisition, and manuscript review and editing. Muhammad Sajid Hussain: Supervision, conceptualization, project administration, visualization and interpretation of data, and manuscript writing.
Supporting information
Fig S1‐S4
Table S1‐S9
ACKNOWLEDGMENT
We pay our gratitude to the participating patients and their families for their cooperation to conduct this study. We are thankful to the staff members of the Cologne Center for Genomics for their technical assistance.
Rasool S, Baig JM, Moawia A, et al. An update of pathogenic variants in ASPM, WDR62, CDK5RAP2, STIL, CENPJ, and CEP135 underlying autosomal recessive primary microcephaly in 32 consanguineous families from Pakistan. Mol Genet Genomic Med. 2020;8:e1408 10.1002/mgg3.1407
Sajida Rasool, Jamshaid Mahmood Baig, and Abubakar Moawia contributed equally to this study.
FUNDING INFORMATION
This study was funded by the Higher Education of Commission (HEC) of Pakistan (SR, JMB, and AM) and the Center for Molecular Medicine Cologne (PN and MSH)
[Correction added on 18 August 2020, after first online publication: affiliation 13 has been added and linked to Emrah Kaygusuz.]
REFERENCES
- Ahmad, I. , Baig, S. M. , Abdulkareem, A. R. , Hussain, M. S. , Sur, I. , Toliat, M. R. , … Nürnberg, P. (2017). Genetic heterogeneity in Pakistani microcephaly families revisited. Clinical Genetics, 92(1), 62–68. 10.1111/cge.12955 [DOI] [PubMed] [Google Scholar]
- Ahmed, J. , Windpassinger, C. , Salim, M. , Wiener, M. , Petek, E. , Schaflinger, E. , … Khan, M. (2019). Genetic study of Khyber‐Pukhtunkhwa resident Pakistani families presenting primary microcephaly with intellectual disability. The Journal of the Pakistan Medical Association, 69(12), 1812–1816. 10.5455/JPMA.300681 [DOI] [PubMed] [Google Scholar]
- Bazgir, A. , Agha Gholizadeh, M. , Sarvar, F. , & Pakzad, Z. (2019). A novel frameshift mutation in abnormal spindle‐like microcephaly (ASPM) gene in an Iranian patient with primary microcephaly: A case report. Iranian Journal of Public Health, 48(11), 2074–2078. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/31970108 [PMC free article] [PubMed] [Google Scholar]
- Bhargav, D. S. , Sreedevi, N. , Swapna, N. , Vivek, S. , & Kovvali, S. (2017). Whole exome sequencing identifies a novel homozygous frameshift mutation in the ASPM gene, which causes microcephaly 5, primary, autosomal recessive. F1000Research, 6, 2163 10.12688/f1000research.12102.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond, J. , Roberts, E. , Springell, K. , Lizarraga, S. , Scott, S. , Higgins, J. , … Woods, C. G. (2005). A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nature Genetics, 37(4), 353–355. 10.1038/ng1539 [DOI] [PubMed] [Google Scholar]
- Bond, J. , Scott, S. , Hampshire, D. J. , Springell, K. , Corry, P. , Abramowicz, M. J. , … Woods, C. G. (2003). Protein‐truncating mutations in ASPM cause variable reduction in brain size. American Journal of Human Genetics, 73(5), 1170–1177. 10.1086/379085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonsawat, P. , Joset, P. , Steindl, K. , Oneda, B. , Gogoll, L. , Azzarello‐Burri, S. , … Rauch, A. (2019). Elucidation of the phenotypic spectrum and genetic landscape in primary and secondary microcephaly. Genetics in Medicine, 21(9), 2043–2058. 10.1038/s41436-019-0464-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, J. , Jackson, A. P. , Bond, J. , & Woods, C. G. (2006). What primary microcephaly can tell us about brain growth. Trends in Molecular Medicine, 12(8), 358–366. 10.1016/j.molmed.2006.06.006 [DOI] [PubMed] [Google Scholar]
- Cristofoli, F. , De Keersmaecker, B. , De Catte, L. , Vermeesch, J. R. , & Van Esch, H. (2017). Novel STIL compound heterozygous mutations cause severe fetal microcephaly and centriolar lengthening. Molecular Syndromology, 8(6), 282–293. 10.1159/000479666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvish, H. , Esmaeeli‐Nieh, S. , Monajemi, G. B. , Mohseni, M. , Ghasemi‐Firouzabadi, S. , Abedini, S. S. , … Najmabadi, H. (2010). A clinical and molecular genetic study of 112 Iranian families with primary microcephaly. Journal of Medical Genetics, 47(12), 823–828. 10.1136/jmg.2009.076398 [DOI] [PubMed] [Google Scholar]
- Farooq, M. , Fatima, A. , Mang, Y. , Hansen, L. , Kjaer, K. W. , Baig, S. M. , … Tommerup, N. (2016). A novel splice site mutation in CEP135 is associated with primary microcephaly in a Pakistani family. Journal of Human Genetics, 61(3), 271–273. 10.1038/jhg.2015.138 [DOI] [PubMed] [Google Scholar]
- Fei, J. F. , Haffner, C. , & Huttner, W. B. (2014). 3′ UTR‐dependent, miR‐92‐mediated restriction of Tis21 expression maintains asymmetric neural stem cell division to ensure proper neocortex size. Cell Reports, 7(2), 398–411. 10.1016/j.celrep.2014.03.033 [DOI] [PubMed] [Google Scholar]
- Fish, J. L. , Kosodo, Y. , Enard, W. , Paabo, S. , & Huttner, W. B. (2006). Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proceedings of the National Academy of Sciences of the United States of America, 103(27), 10438–10443. 10.1073/pnas.0604066103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gul, A. , Hassan, M. J. , Mahmood, S. , Chen, W. , Rahmani, S. , Naseer, M. I. , … Ahmad, W. (2006). Genetic studies of autosomal recessive primary microcephaly in 33 Pakistani families: Novel sequence variants in ASPM gene. Neurogenetics, 7(2), 105–110. 10.1007/s10048-006-0042-4 [DOI] [PubMed] [Google Scholar]
- Gul, A. , Tariq, M. , Khan, M. N. , Hassan, M. J. , Ali, G. , & Ahmad, W. (2007). Novel protein‐truncating mutations in the ASPM gene in families with autosomal recessive primary microcephaly. Journal of Neurogenetics, 21(3), 153–163. 10.1080/01677060701508594 [DOI] [PubMed] [Google Scholar]
- Hussain, M. S. , Baig, S. M. , Neumann, S. , Nürnberg, G. , Farooq, M. , Ahmad, I. , … Nürnberg, P. (2012). A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. American Journal of Human Genetics, 90(5), 871–878. 10.1016/j.ajhg.2012.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa, L. , Mueller, K. , Seufert, K. , Kraemer, N. , Rosenkotter, H. , Ninnemann, O. , … Morris‐Rosendahl, D. J. (2013). Clinical and cellular features in patients with primary autosomal recessive microcephaly and a novel CDK5RAP2 mutation. Orphanet Journal of Rare Diseases, 8, 59 10.1186/1750-1172-8-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. , Wang, R. , Han, S. , Ahmad, W. , & Zhang, X. (2018). Identification of a novel nonsense ASPM mutation in a large consanguineous pakistani family using targeted next‐generation sequencing. Genetic Testing and Molecular Biomarkers, 22(3), 159–164. 10.1089/gtmb.2017.0229 [DOI] [PubMed] [Google Scholar]
- Kvarnung, M. , Taylan, F. , Nilsson, D. , Anderlid, B. M. , Malmgren, H. , Lagerstedt‐Robinson, K. , … Lundberg, E. S. (2018). Genomic screening in rare disorders: New mutations and phenotypes, highlighting ALG14 as a novel cause of severe intellectual disability. Clinical Genetics, 94(6), 528–537. 10.1111/cge.13448 [DOI] [PubMed] [Google Scholar]
- Letard, P. , Drunat, S. , Vial, Y. , Duerinckx, S. , Ernault, A. , Amram, D. , … Passemard, S. (2018). Autosomal recessive primary microcephaly due to ASPM mutations: An update. Human Mutation, 39(3), 319–332. 10.1002/humu.23381 [DOI] [PubMed] [Google Scholar]
- Li, R. , Sun, L. , Fang, A. , Li, P. , Wu, Q. , & Wang, X. (2017). Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle‐like (ASPM related primary) microcephaly disease. Protein & Cell, 8(11), 823–833. 10.1007/s13238-017-0479-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marakhonov, A. V. , Konovalov, F. A. , Makaov, A. K. , Vasilyeva, T. A. , Kadyshev, V. V. , Galkina, V. A. , … Zinchenko, R. A. (2018). Primary microcephaly case from the Karachay‐Cherkess Republic poses an additional support for microcephaly and Seckel syndrome spectrum disorders. BMC Medical Genomics, 11(Suppl 1), 8 10.1186/s12920-018-0326-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSherry, M. , Masih, K. E. , Elcioglu, N. H. , Celik, P. , Balci, O. , Cengiz, F. B. , … Tekin, M. (2018). Identification of candidate gene FAM183A and novel pathogenic variants in known genes: High genetic heterogeneity for autosomal recessive intellectual disability. PLoS One, 13(11), e0208324 10.1371/journal.pone.0208324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moawia, A. , Shaheen, R. , Rasool, S. , Waseem, S. S. , Ewida, N. , Budde, B. , … Hussain, M. S. (2017). Mutations of KIF14 cause primary microcephaly by impairing cytokinesis. Annals of Neurology, 82(4), 562–577. 10.1002/ana.25044 [DOI] [PubMed] [Google Scholar]
- Moriwaki, T. , Yamazaki, N. , So, T. , Kosuga, M. , Miyazaki, O. , Narumi‐Kishimoto, Y. , … Fukuhara, Y. (2019). Normal early development in siblings with novel compound heterozygous variants in ASPM. Human Genome Variation, 6, 56 10.1038/s41439-019-0088-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas, A. K. , Khurshid, M. , Désir, J. , Carvalho, O. P. , Cox, J. J. , Thornton, G. , … Woods, C. G. (2010). WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nature Genetics, 42(11), 1010–1014. 10.1038/ng.682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas, A. K. , Swanson, E. A. , Cox, J. J. , Karbani, G. , Malik, S. , Springell, K. , … Woods, C. G. (2009). The molecular landscape of ASPM mutations in primary microcephaly. Journal of Medical Genetics, 46(4), 249–253. 10.1136/jmg.2008.062380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, N. , Kohmoto, T. , Naruto, T. , Masuda, K. , & Imoto, I. (2018). Primary microcephaly caused by novel compound heterozygous mutations in ASPM. Human Genome Variation, 5, 18015 10.1038/hgv.2018.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnamenta, A. T. , Murray, J. E. , Yoon, G. , Sadighi Akha, E. , Harrison, V. , Bicknell, L. S. , … Knight, S. J. (2012). A novel nonsense CDK5RAP2 mutation in a Somali child with primary microcephaly and sensorineural hearing loss. American Journal of Medical Genetics. Part A, 158A(10), 2577–2582. 10.1002/ajmg.a.35558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passemard, S. , Titomanlio, L. , Elmaleh, M. , Afenjar, A. , Alessandri, J.‐L. , Andria, G. , … Verloes, A. (2009). Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology, 73(12), 962–969. 10.1212/WNL.0b013e3181b8799a [DOI] [PubMed] [Google Scholar]
- Passemard, S. , Verloes, A. , Billette de Villemeur, T. , Boespflug‐Tanguy, O. , Hernandez, K. , Laurent, M. , … Schaer, M. (2016). Abnormal spindle‐like microcephaly‐associated (ASPM) mutations strongly disrupt neocortical structure but spare the hippocampus and long‐term memory. Cortex, 74, 158–176. 10.1016/j.cortex.2015.10.010 [DOI] [PubMed] [Google Scholar]
- Poulton, C. J. , Schot, R. , Seufert, K. , Lequin, M. H. , Accogli, A. , Annunzio, G. D. , … Mancini, G. M. (2014). Severe presentation of WDR62 mutation: Is there a role for modifying genetic factors? American Journal of Medical Genetics. Part A, 164A(9), 2161–2171. 10.1002/ajmg.a.36611 [DOI] [PubMed] [Google Scholar]
- Sajid Hussain, M. , Marriam Bakhtiar, S. , Farooq, M. , Anjum, I. , Janzen, E. , Reza Toliat, M. , … Hansen, L. (2013). Genetic heterogeneity in Pakistani microcephaly families. Clinical Genetics, 83(5), 446–451. 10.1111/j.1399-0004.2012.01932.x [DOI] [PubMed] [Google Scholar]
- Shaheen, R. , Maddirevula, S. , Ewida, N. , Alsahli, S. , Abdel‐Salam, G. M. H. , Zaki, M. S. , … Alkuraya, F. S. (2019). Genomic and phenotypic delineation of congenital microcephaly. Genetics in Medicine, 21(3), 545–552. 10.1038/s41436-018-0140-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, Y. G. , Lee, D. W. , Kim, J. , Jang, J. H. , Lee, S. M. , & Jang, D. H. (2019). Two novel mutations (c.883‐4_890del and c.1684C>G) of WDR62 gene associated with autosomal recessive primary microcephaly: A case report. Frontiers in Pediatrics, 7, 457 10.3389/fped.2019.00457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, T. W. , Mochida, G. H. , Tischfield, D. J. , Sgaier, S. K. , Flores‐Sarnat, L. , Sergi, C. M. , … Walsh, C. A. (2010). Mutations in WDR62, encoding a centrosome‐associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nature Genetics, 42(11), 1015–1020. 10.1038/ng.683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaqout, S. , Morris‐Rosendahl, D. , & Kaindl, A. M. (2017). Autosomal Recessive Primary Microcephaly (MCPH): An update. Neuropediatrics, 48(3), 135–142. 10.1055/s-0037-1601448 [DOI] [PubMed] [Google Scholar]
- Zombor, M. , Kalmár, T. , Nagy, N. , Berényi, M. , Telcs, B. , Maróti, Z. , … Sztriha, L. (2019). A novel WDR62 missense mutation in microcephaly with abnormal cortical architecture and review of the literature. Journal of Applied Genetics, 60(2), 151–162. 10.1007/s13353-019-00486-y [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4
Table S1‐S9