

Abstract
Background
Necrotrophic effector proteins secreted by fungal pathogens are important virulence factors that mediate the development of disease in wheat. Pyrenophora tritici-repentis (Ptr), the causal agent of wheat tan spot, has a race structure dependent on the combination of effectors. In Ptr, ToxA and ToxB are known proteinaceous effectors responsible for necrosis and chlorosis respectively. While Ptr ToxA is encoded by the single gene ToxA, ToxB has multiple loci in the Ptr genome, which is postulated to be directly related to the level of ToxB production and leaf chlorosis. Although previous analysis has indicated that the majority of the ToxB loci lie on a single chromosome, the exact number and chromosomal locations for all the ToxB loci have not been fully identified.
Results
In this study, we have sequenced the genome of a race 5 ToxB-producing isolate (DW5), using PacBio long read technology, and found that ToxB duplications are nested in the complex subtelomeric chromosomal regions. A total of ten identical ToxB gene copies were identified and based on flanking sequence identity, nine loci appeared associated with chromosome 11 and a single copy with chromosome 5. Chromosome 11 multiple ToxB gene loci were separated by large sequence regions between 31 and 66 kb within larger segmental duplications in an alternating pattern related to loci strand, and flanked by transposable elements.
Conclusion
This work provides for the first time the full accompaniment of ToxB loci and surrounding regions, and identifies the organization and distribution of ten ToxB loci to subtelomeric regions. To our knowledge, this is the first report of an interwoven strand-related duplication pattern event. This study further highlights the importance of resolving the highly complex distal chromosomal regions, that remain difficult to assemble, and can harbour important effectors and virulence factors.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-020-07029-4.
Keywords: Triticum aestivum, Fungal pathogen, Tan spot, Necrotroph, Effector, Host-selective toxin
Background
The inverse gene-for-gene interactions between host plants and necrotrophic fungal pathogen typically involve pathogen effectors, which interact with a compatible locus in the host leading to toxin sensitivity and disease susceptibility.
Pyrenophora tritici-repentis (Ptr) a necrotrophic fungal pathogen and the causal agent of wheat (Triticum aestivum L) tan spot, produces a number of effectors that mediate the development of foliar disease on susceptible wheat genotypes. Tan spot has two distinct leaf symptoms, which are necrosis and chlorosis [1]. These symptoms are the result of secreted effectors ToxA, ToxB and ToxC [2–4] and other as yet uncharacterised effectors [5, 6]. ToxA and ToxB, are characterised as small effector proteins that produce necrosis and chlorosis symptoms, respectively [2, 4]. While ToxC, which also causes chlorosis, has not been characterised and may be the product of a secondary metabolite gene cluster [3].
For the two proteinaceous toxins, ToxA reacts with a corresponding susceptibility gene in wheat (Tsn1), which makes the host sensitive to the effector [7], while the corresponding host gene for ToxB remains as yet unknown but is associated with the Tsc2 locus on chromosome 2B [8].
In the Ptr genome, ToxA is a single locus gene, the result of a horizontal gene transfer from another fungal pathogen species [9]. While in contrast, there are multiple identical gene copies of ToxB [10, 11], in which the copy number variation has been shown to have an association with virulence. Nine copies of ToxB in race 5 isolates (DW2, DW7, DW13 and DW16), were estimated by phosphoimage analysis, and of these six copies were individually cloned and sequenced from DW7 (1-3 kb in length) [10]. Southern analysis indicated that the ToxB loci reside on two unknown chromosomes, approximately 3.5 and 2.7 Mb in length, with the majority located on the smaller chromosome [10].
To date a number of Ptr whole genome sequencing projects involving race 5 isolates (ToxB-producing) have not been able to determine if the ToxB loci are clustered or dispersed [12, 13] in the genome. We therefore undertook genome sequencing via PacBio long read technology to resolve the number, organization and distribution of ToxB loci within the genome of a race 5 isolate (DW5). A comparative analysis of these ToxB regions to a race 1 isolate (ToxB non-producing), which was previously assembled from PacBio long read technology and optical mapping [12], was undertaken to identify any flanking sequence conservation.
Results
Ptr isolate DW5 whole genome assembly analysis
The Ptr race 5 isolate DW5 was sequenced using long read single molecule PacBio technology and the error corrected reads were assembled and annotated (Table 1). Furthermore, a previous PacBio sequenced Ptr race 1 isolate (M4), which was scaffolded into chromosomes based on an optical map, but not annotated at the time [12], was also annotated during this study. The DW5 genome assembly size was 40.87 Mb, close to the genome size of M4 at 40.92 Mb [12], however DW5 was slightly more fragmented with 60 contigs as compared to the 50 contigs for M4 [12]. This fragmentation may be directly related to a slightly higher repeat content in DW5 and the slightly smaller content of protein coding genes compared to M4 (Table 1). Protein coding gene predictions for the DW5 contigs and M4 scaffold assemblies were 14,276 and 15,466, respectively. The DW5 annotated genome has been deposited at DDBJ/ENA/GenBank under the accession MUXC00000000. The version described in this paper is version MUXC02000000. The annotated M4 genome has been deposited in DDBJ/ENA/GenBank under accession NQIK00000000. The version described in this paper is version NQIK02000000.
Table 1.
Ptr isolate sequence, assembly and annotation statistics
| M4a | DW5 | |
|---|---|---|
| Isolate information | ||
| Race | 1 | 5 |
| Effectors | AC | B |
| Collection site | Western Australia | North Dakota |
| Collection year | 2009 | 1998 |
| Sequencing | ||
| Sequencing Platform | PacBio RSII | PacBio Sequel |
| Number of reads | 594,877 | 1,306,274 |
| Total sequence (Mbp) | 3500 | 14,225 |
| Sequencing coverage | 75 | 355.62 |
| Error correction | ||
| Number of reads | 56,338 | 302,147 |
| Error corrected sequence (Mbp) | 758 | 3085 |
| Genome coverage | 19 | 77.13 |
| Assembly statistics | ||
| NCBI accession | NQIK00000000 | MUXC00000000 |
| Number of contigs | 50 | 60 |
| Number of scaffolds* | 41* | NA |
| Total length contigs/scaffolds* (Mb) | 40.92* | 40.87 |
| N50 (Mb) | 3.65 | 3.13 |
| L50 | 4 | 5 |
| Mean size (kb) | 998 | 681 |
| GC % | 50.73 | 50.21 |
| Repeat % | 7.96 | 8.33 |
| Gene predictions | M4b | |
| Gene number | 15,443 | 14,276 |
| Total CDS (Mb) | 20.7 | 17.9 |
| Average size (kb) | 1.3 | 1.2 |
| Predicted effectors | ||
| Number of predicted effectors | 445 | 401 |
| Total length (aa) | 61,737 | 60,150 |
| Mean gene size (aa) | 138 | 150 |
| Median gene size (aa) | 117 | 123 |
a Genome was sourced from previous optically mapped assembly [12]. b M4 genome annotation this study. * scaffolds
Whole genome comparative analysis between Ptr races 1 and 5
The genome sequence of DW5 (race 5) was aligned to M4 (race 1) [12] to determine sequence conservation at a chromosome level. Thirteen DW5 contigs showed colinear alignment to the scaffolded M4 chromosomes at greater than 98% sequence identity (Fig. 1) with no large-scale chromosomal rearrangements. DW5 contigs 3, 5, 7 and 8 were sequenced from 5′ telomere to 3′ telomere informed by the presence of the tandem telomere repeat motifs (CCCTAA)n/(TTAGGG)n.
Fig. 1.

Ptr nucleotide sequence alignment for race 5 isolate DW5 contigs (vertical axis) to race 1 M4 chromosomes (horizontal axis). The sequence dot plot shows the percent sequence similarity between the two genomes. The diagonal red line is the maximal colinear alignment between the two genomes. Individual contigs and chromosomes are delineated by grey lines in both axes of the sequence dot plot. *DW5 contigs are reverse complemented. On the vertical axis, DW5 contigs that represent chromosomes are informed by telomere repeat motifs, single (green circle) and both (red circles connected by a red bar)
Based on M4 chromosomes, thirteen DW5 assembled contigs matched nine chromosomes, which included chromosomes 1–9 (Table 2). A chromosome fusion between chromosome 10 and 11 (referred to as chromosome 10) in Australian isolate M4 resolved by optical mapping [12] was not observed for DW5, where DW5 contig 8 possessed both 5′ and 3′ telomere motifs (Table 2), which would represent chromosome a (telomere to telomere).
Table 2.
DW5 genome assembly relative to M4
| M4 chromosome | M4 chromosome length (Mb) | DW5 contig | DW5 contig length (Mb) | DW5 contig 5′ and 3′ telomere motifs |
|---|---|---|---|---|
| 1 | 9.91 |
1 12a |
8.11 1.82 |
Yes |
| 2 | 5.07 | 2a | 4.42 | No |
| 3 | 3.65 | 3 | 3.65 | Yes |
| 4 | 3.15 | 5 | 3.13 | No |
| 5 | 3.38 | 4a | 3.36 | Yes |
| 6 | 3.05 | 6 | 2.73 | No |
| 7 | 2.76 |
13 11a |
0.83 1.96 |
Yes |
| 8 | 2.40 | 7 | 2.68 | Yes |
| 9 | 2.17 | 10 | 2.12 | No |
| 10 | 4.30 | 9a | 2.18 | No |
| 10 | 4.30 | 8 | 2.18 | Yes |
aReverse complemented sequence
Multiple ToxB loci have alternate strand positions
The DW5 assembly was searched for ToxB homologs and 10 copies were identified across 5 contigs (DW5_contig_0004, DW5_contig_0009, DW5_contig_00015, DW5_contig_00016 and DW5_contig_00018). A single ToxB loci was found for each of the larger two contigs DW5_contig_0004 (3.65 Mb) and DW5_contig_0009 (2.18 Mb), labelled here as ToxB1 and ToxB2, respectively (Table 3). Multiple ToxB loci were located on the smaller contigs DW5_contig_00015 (ToxB3, ToxB4 and ToxB5), DW5_contig_00016 (ToxB6, ToxB7 and ToxB8) and DW5_contig_00018 (ToxB9 and ToxB10), sized 126, 123 and 99 kb, respectively. ToxB genes were not immediate neighbours and loci appeared to locate in alternate strand positions separated by relatively large distances that ranged between 31 and 66 kb in size. This pattern was observed across the three contigs (DW5_contig_00015, DW5_contig_00016 and DW5_contig_00018) harbouring multi-loci ToxB (Fig. 2).
Table 3.
Ptr isolate DW5 ToxB subtelomeric gene locations, chromosomes relative to M4 and sequence identity to DW7 ToxB cloned sequence
| Ptr DW5 | Ptr M4 | Ptr DW7 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ToxBa | Contig | Contig size (Mb) | ToxB Gene start | ToxB Gene end | ToxB Strand | Loci Label | M4b | DW7c | DW7 cloned sequence length (bp) | DW5 ToxB locus % Sequence identityd |
| A1F99_069381 | DW5_contig_00004 | 3.365 | 3,306,588 | 3,306,848 | + | ToxB1 | Chr5 | AY425485 (ToxB6) | 1769 | 100.00 |
| A1F99_114980 | DW5_contig_00009 | 2.180 | 2,152,566 | 2,152,826 | – | ToxB2 | Chr10 | AY425481 (ToxB2) | 3563 | 99.99 |
| A1F99_139310 | DW5_contig_00015 | 0.126 | 10,557 | 10,817 | – | ToxB3 | None | AY425481 (ToxB2) | 3563 | 99.99 |
| A1F99_139400 | DW5_contig_00015 | “” | 41,790 | 42,050 | + | ToxB4 | AY425480 (ToxB1) | 4471 | 100.00 | |
| A1F99_139580 | DW5_contig_00015 | “” | 103,025 | 103,285 | – | ToxB5 | AY425483 (ToxB4) | 1696 | 100.00 | |
| A1F99_139650 | DW5_contig_00016 | 0.123 | 4627 | 4887 | + | ToxB6 | None | AY425484 (ToxB6) | 2494 | 100.00 |
| A1F99_139840 | DW5_contig_00016 | “” | 70,550 | 70,810 | – | ToxB7 | AY425480 (ToxB1) | 4471 | 99.44 | |
| A1F99_139950 | DW5_contig_00016 | “” | 112,439 | 112,699 | + | ToxB8 | AY425480 (ToxB1) | 4471 | 100.00 | |
| A1F99_140280 | DW5_contig_00018 | 0.099 | 11,326 | 11,586 | – | ToxB9 | None | AY425483 (ToxB4) | 1696 | 100.00 |
| A1F99_140440 | DW5_contig_00018 | “” | 62,388 | 62,648 | + | ToxB10 | AY425482 (ToxB4) | 4007 | 100.00 |
Fig. 2.

Alternating strand positions of ToxB loci Ptr isolate DW5 in contigs 15, 16 and 18. ToxB loci are shown as blue arrow on the forward strand and green on the reverse strand. The coding sequence is shown in yellow
Multiple ToxB loci are associated with subtelomeric chromosomal regions
Based on genome alignments to M4, two contigs (DW5_contig_0004 and DW5_contig_0009) with single ToxB loci were syntenic with the subtelomeric regions of M4 chromosomes 5 and 10, respectively (Fig. 1 and Table 3). No significant alignments were identified for the three smaller multiple ToxB loci contigs (DW5_contig_0015, DW5_contig_0016 and DW5_contig_0018) to the genome of M4. However, a search back to the DW5 genome (self-search) identified alignments for all three contigs to chromosome 10 (DW5_contig_0009) (Fig. 3), sequence breaks can be seen where regions of paralogous sequence are interspersed with repeat elements. No other alignments to the DW5 genome were found except for self-contig alignments. The alignment of the fragmented ToxB contigs with the 5′ subtelomeric region of chromosome 10 (reverse complemented DW5_contig_0009) and the presence of a 5′ telomere motif (TTAGGG)n in chromosome 5 (reverse complemented DW5_contig_0004) (Table 4), weighted chromosome 10 as the possible origin of ToxB3–10 loci and chromosome 5 (DW5_contig_0004) as the only source for the ToxB1 locus. The alignment of the 5′ telomere region of chromosome 10 and ToxB loci (ToxB3 to ToxB10) thus implied that contigs 15, 16 and 18 could be the fragmented regions not assembled from the 5′ telomere region of chromosome 10 (Fig. 4).
Fig. 3.

DW5 ToxB contig nucleotide sequence alignments to terminal chromosome regions. DW5 sub-telomeric regions (120 kb) for contig 4: 3,245,792-3,365,792 bp (chromosomes 5) and contig 9: 2,060,091-2,180,091 bp (chromosome 10) (vertical axis). DW5 contigs 15, 16 and 18 are represented on the horizontal axis with gene annotations below (yellow boxes). Sequence alignments are plotted in the sequence plot in grey. ToxB loci positions for chromosome 5 and 10 are indicated with horizontal blue dashed lines for B1 and B2. The remaining locations of ToxB in the contigs are shown with vertical dashed blue lines for contigs 15, 16 and 18. To view any repeat element patterns major elements are highlighted for contigs 15, 16 and 18, as Tnp-hAT transposons, Gypsy and Copia retrotransposons, Bipolaris maydis I-2-CH element and a region containing the (TTAGGG)n telomere motif
Table 4.
ToxB contigs and telomere motifs
| Contig | Contig size (Mb) | 5′ Motif (ccctaa)n |
3′ Motif (ttaggg)n |
M4a |
|---|---|---|---|---|
| DW5_contig_00004 (reverse complement) | 3.365 | + | – | Chr5 |
| DW5_contig_00009 (reverse complement) | 2.180 | – | + | Chr10 |
| DW5_contig_00015 | 0.126 | – | – | None |
| DW5_contig_00016 | 0.123 | – | – | None |
| DW5_contig_00018 | 0.099 | – | + | None |
a M4 chromosome with DW5 ToxB contig alignment
Fig. 4.

DW5 contig sequence homology between 5′ subtelomere regions of chromosome 10 and ToxB loci. Nucleotide sequences are from top to bottom, chromosome 10 (zoomed on reverse complemented contig 9:2.0–2.18 Mb), contigs 15, 16 and 18. ToxB loci (green triangles) and surrounding genes (red arrows) are shown for each contig. Telomere motifs (brown circles) are shown on the 3′ and 5′ end of chromosome 10 and reverse complemented contig 18, respectively. Sequence homology between contigs is shown on the same strand (blue) and complementary (red)
All ToxB loci, except ToxB6, which was truncated in the 5′ region upstream of ToxB, were co-located with dimer Tnp-hAT repeat genes. The dimer Tnp-hAT genes were located 10–15 kb upstream of the ToxB loci.
Larger groups of conserved regions are found between the ToxB loci based on strand positions
The ToxB loci and flanking sequence regions of 5 kb upstream and downstream were extracted (including ToxB mRNA transcript) for a nucleotide multiple sequence alignment to determine sequence conservation between the ten loci. Only ToxB6 was truncated in the 5′ sequence region due to the locus location (contig16:4,627–4,887 bp). The ToxB 10 kb multiple sequence alignment showed a highly conserved region of 3,170 bp with a large proportion (2.5 kb) highly conserved upstream of ToxB for all ten loci (Fig. 5a). On closer examination, the ToxB 10 kb regions could be grouped by their locus strand (Fig. 5b). The full 10 kb regions were highly conserved for ToxB loci B4, B6 and B8 on the forward strands of contigs 15 and 16 (group 1). While further conservation was found for reversed stranded ToxB loci B5, B7 and B9 (group 2) on contigs 15, 16, and 18, and to a lesser extent for reverse strand ToxB loci B2 and B3 (group 3) on contigs 9 and 15 (not shown in Fig. 5).
Fig. 5.

Multiple nucleotide sequence alignment of the ten ToxB 10 kb regions. a Conserved sequence (blue), sequence not conserved (white) and sequence gaps (grey) are shown for the ten ToxB 10 kb regions. A highly conserved region (2508–5478 bp) was mainly upstream of ToxB (red) and the complete coding sequence (CDS) (green). b Sequence alignment overview (not to scale) show the 10 kb ToxB region conserved sequences grouped by the locus strand. Group 1 (B4, B6 and B8) forward strand ToxB loci and Group 2 (B5, B7 and B9) reverse strand ToxB loci
When the homology between the ten ToxB 10 kb regions was summarized for conserved and distinctive regions (Fig. 6), the 10 kb regions surrounding ToxB1 on chromosome 5 were found to be more divergent than the remaining loci proposed to be from chromosome 10. It was also noted that a small hypothetical protein (128 bp) was conserved 288 bp downstream of the ToxB loci in all forward stranded positions except ToxB1 and only in reverse positioned ToxB2 and ToxB3.
Fig. 6.

DW5 ToxB 10 kb locus region (B1-B10) sequence homology. Regions in common are represented by the same colours relative to the ToxB gene position (red). For example, a small 5′ region is shared by B5, B7 and B9 and is shown in purple. Grey regions are common to all ten loci and gaps are shown in white. The hypothetical protein (HP) is found downstream of ToxB in B2–4, 6, 8 and 10. Regions of similarity are shown between forward sequence (forward arrow) and reverse sequence (reverse arrow). Short regions of similarity contained within a larger region are secondary alignments
ToxB and promoter region
All ten copies of the 261 bp ToxB protein coding sequence are identical, as found previously for six of the sequenced copies [10]. Based on DW5 mRNA transcript from a previous study [12], ToxB has a two exon gene structure of 533 bp in length. ToxB exon1 (94 bp) and exon 2 (439 bp) flank an intron 52 bp in size. The exon 1 5′ UTR and exon 2 3′ UTR have lengths of 99 bp and 172 bp, respectively (Additional file 1). Previously, the ToxB promoter was reported to be greater than 300 bp upstream of the coding sequence [10]. The upstream region from ToxB (2 kb) was then searched for transcription binding site motifs. A DNA binding site was predicted upstream of ToxB, 847 bp from the starting codon of ToxB2–9, and 644 bp for ToxB1 and ToxB10 at an expected value of 4.9e-178. The most significant motif profile MA0320.1 (IME1) was identified with a probability value of 2.20e-06 (Additional file 2).
Discussion
Ptr ToxB multiloci analysis
This is the first genome sequence investigation into the distribution of ToxB loci in Ptr using long read sequencing technologies. A previous study for race 5 isolate DW7 found that six of the sequenced copies, all had identical protein coding sequence identity [10]. In this study, all the the ToxB loci (585 bp) identified have identical sequence, including exon and intron sequences. It was previously suggested that DW7 ToxB loci resided on two unknown chromosomes, approximately 3.35 and 2.7 Mb in size, with the majority of the loci on the smaller chromosome [10]. In this study, the ToxB loci were located on chromosome 5 and 11, which had assembly sizes of 3.36 and 2.18 Mb respectively, which are close to the previously estimated chromosome sizes by Martinez et al., (2004). Of the ten ToxB loci, nine appeared to be associated with the smaller chromosome 11 located in the 3′ distal region. A Ptr chromosome noted for a chromosome fusion event for a race 1 isolate M4 [12]. The telomere to telomere support for eleven DW5 chromosomes is similar to the findings for another American race 1 isolate Ptr Pt-1C-BFP [13], unlike the 10 chromosome genome of Australian isolate M4 [12] (Fig. 7). Large scale segmental rearrangements have been frequently identified in the subtelomere regions of fungal chromosomes, where breakage/fusion events and large-scale rearrangements frequently occur [12, 14, 15]. During meiosis the subtelomeric regions have instability often referred to as plasticity [16]. In these regions, chromosome breakage fusion cycles begin with the loss of telomeres which causes instability and potential fusion of sister chromatids. During the breakage fusion cycle, the site of breakage during separation in erroneously fused sister chromatids can lead to sequence duplication, deletion and rearrangement [16]. It is therefore probable that the recent highly conserved duplications of loci in race 5 have occurred through multiple breakage fusion events between the distal chromosome regions and may have at one stage been potentially lost from race 1 isolates.
Fig. 7.
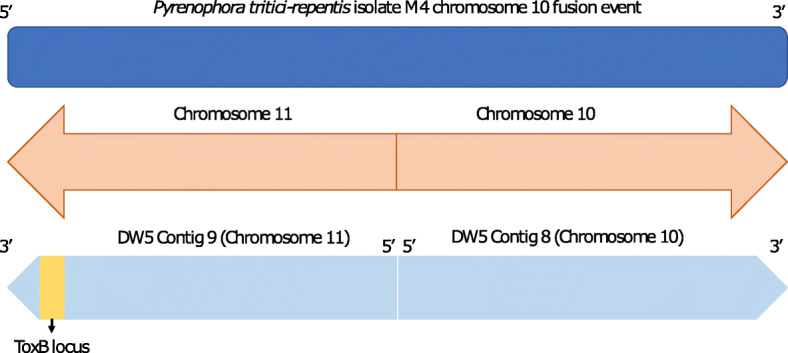
Overview of ToxB locus in DW5 relative to the M4 chromosome 10 fusion event. M4 chromosome 10 (top) is the result of a fusion between chromosomes 10 and 11 (shown in the middle). DW5 Contig 9 (chromosome 11) and Contig 8 (chromosome 10) (bottom) are shown relative to M4 chromosome 10. ToxB locus (yellow) which aligns to the 5’ distal region of M4 chromosome 10 is shown in the 3’ distal region of DW5 Contig 9 (chromosome 11)
Genome plasticity in distal chromosome regions can contribute to rapid fungal diversification, especially for Ptr [11]. In this study the subtelomeric ToxB loci location within Ptr DW5 provided a favourable environment for duplication, which may have provided this isolate a potential advantage for survival.
Ptr ToxB patterns of duplication
In addition to the positioning of the ToxB duplication within the distal region of chromosome 11, ToxB loci were located equidistant downstream from dimer Tnp-haT transposases, a familiar gene found coupled to Ptr ToxA and within the horizontally transferred region, also found in Parastagonospora nodorum and Bipolaris sorokiniana [9, 17]. It is therefore possible that the dimer Tnp-hAT transposases observed in DW5 may have played a self-complementing role in the duplication of ToxB, providing regions of homology between flanking regions, resulting in larger regions of homology as observed between the multiple DW5 ToxB copies. Our data found that multiple ToxB gene duplication events involved much larger segmental duplications, flanked by transposable elements, than previously identified [10]. Here, we also identified that larger homologous regions could be grouped by the strand from which the duplicated ToxB is transcribed. Furthermore, we believe this is first reporting of a potential interwoven strand-related duplication pattern/event of a necrotrophic effector gene.
ToxB transcription factor binding site analysis
The binding of transcription factors to specific DNA binding sites (identified by a DNA motif) is key for the transcriptional regulation of genes, here a transcription factor binding motif IME1 profile was identified upstream of the multiple ToxB loci. The motif of IME1 is a conserved regulatory site for Saccharomyces cerevisiae, previously identified from ChIP-chip data [18]. Although the IME1 transcription factor protein (UniProt accession P21190) is required for sporulation and early sporulation-specific genes expression, further experimental validation would be required in Ptr race 5 isolates to determine if the potential transcription factor is indeed involved in the regulation of ToxB.
Conclusions
Our findings provided insights into the unique nature of the multicopy ToxB organisation in the Ptr genome and revealed a potentially complex effector gene regulatory network. This study directly works towards a better understanding of genome plasticity events in fungal adaptation and effector gene evolution.
Material and methods
Ptr race 5 isolate DW5 collection and sequencing
The Ptr race 5 isolate DW5 was collected in 1998 from North Dakota, USA and was kindly provided by Tim Friesen (North Dakota, USA).
Isolate genomic DNA was extracted from 3-day old mycelia grown in Fries 3 medium using the BioSprint 15 automated workstation according to the manufacturer’s instruction (Qiagen, Germany). DNA was then treated with 50 μg/ml of RNase enzyme (Qiagen, Hilden, Germany) for 1 h followed by phenol/chloroform extraction. DNA was precipitated with sodium acetate and ethanol, and resuspended in TE buffer [14].
The DW5 genome was sequenced using PacBio Sequel technology (https://www.pacb.com) by Novogene (China, https://en.novogene.com/). The PacBio sequence coverage for isolate DW5 was 77x. The DW5 genome was also Illumina sequenced (www.illumina.com) for 150 PE reads at 100x coverage by Novogene (China, (https://en.novogene.com/)). The Illumina data was used for post-genome assembly error correction (polishing).
Ptr isolate DW5 whole genome assembly
The DW5 PacBio sequence data was error corrected and assembled using Canu version 1.9 [19] with pacbio-raw and genome size of 40 Mb parameter settings on a heterogeneous Hewlett Packard Enterprise Linux cluster (Zeus, https://pawsey.org.au). The DW5 assembled PacBio contigs were then indexed using BWA index version 0.7.17-r1188 [20]. The DW5 genomic Illumina read data, sequenced in this study, was then aligned to the indexed DW5 assembled PacBio contigs using BWA mem version 0.7.17-r1188 [20] (−t 16). The alignment file (BAM format) was then filtered for concordant read alignments using SAMTools version 1.7 view (−f 0 × 2) and sorted [21] for further genome error correction (polishing). The DW5 PacBio assembly was then error corrected using Pilon version 1.23 [22] (--changes --tracks --output DW5_pilon --defaultqual 20 --threads 16 --frags ‘DW5 sorted BAM file’).
The DW5 PacBio assembled genome was then masked for low complexity sequence and known fungal repeats using RepeatMasker (RM) [23] version 2.9.0+, Dfam 3.0 [24, 25] and Repbase 20,181,026 [26] with taxon fungi parameter available through a docker image (https://hub.docker.com/r/taavipall/repeatmasker-image).
DW5 and M4 gene prediction and annotation
The PacBio DW5 assembled contigs and a previously assembled Ptr race 1 isolate M4 scaffold assembly [12] were indexed using bowtie2-build version 2.3.4.1 [27]. Previously sequenced stranded RNA-seq Illumina read data [12] for DW5 and M4 were aligned to the respective indexed genomes DW5 (DDBJ/ENA/GenBank accession MUXC02000000) and M4 (DDBJ/ENA/GenBank accession NQIK02000000) using TopHat2 version 2.1.1 [28] (--no-discordant -N 0 -i 10 -I 5000 -p 16 --library-type fr-firststrand). Based on the accepted TopHat2 alignments (BAM file), mRNA transcripts, in GTF format, were then generated using CuffLinks version 2.2.1 [29] (-p -library-type fr-firststrand). The transcript GTF file format was then converted to GFF3 using GenomeTools gtf_to_gff3 version 1.5.10 [30] to provide transcript support (evidence) towards the ab initio gene predictions.
Ab initio gene predictions were made with GeneMark-ES v 4.33 (--ES --fungus --cores 16 --evidence) [31] and Coding Quarry v2.0 [32] (-p 16 -t) in pathogen mode (PM), both ab initio gene predictions were supported by the transcript GFF3 file. Published Ptr protein FASTA sequences were downloaded from NCBI using NCBI txid45151 on the 20th January 2020 and aligned to the genomes using Exonerate v2.2.0 [33] (--showvulgar no --showalignment no --minintron 10 --maxintron 2000 --percent 90) mode protein2genome. The final gene prediction sets were then merged via EvidenceModeller v1.1.1 [34] using a combination of protein alignments and the two ab initio predictions on the genome, with a minimum intron length of 10 bp and evidence weights [31] CodingQuarry:10, GeneMark.hmm:10, Exonerate:5 and CuffLinks:10.
Gene annotations were assigned from BLASTX (v2.2.26) [35] searches (expected value ≤ 1e-05) against the following databases Uniref90 (October, 2019), NCBI Refseq (taxon = Ascomycota) (October, 2019) and sequence domains were assigned by RPS-BLAST (v2.2.26) against Pfam (October, 2019), Smart (October, 2019) and CDD (October, 2019). The blast protein and domain searches were then summarised using AutoFACT version 3.4 [36].
The annotated proteins were searched for signal peptides using SignalP version 5.0b [37] (-format short -gff3 -mature -org euk). Those identified with signal peptides were then searched for predicted effectors using EffectorP version 2.0 [38]. EffectorP 2.0 has a low false positive rate of 11.2% and a high accuracy of 88.8% for effector prediction [38].
DW5 ToxB identification and analyses
All published ToxB sequences, 76 in total, were downloaded from NCBI GenBank nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore) with the text search (ToxB) AND “Pyrenophora tritici-repentis”[porgn:__txid45151] (Additional file 3) and searched against the DW5 genome using BLATX v3.5 [39] (-maxIntron = 5000 -minIdentity = 70) and ≥ 50% query coverage (to detect any truncated genes).
Sequence flanking the identified ToxB loci, a total length of 10 kb, were then extracted using EMBOSS extractseq version 6.6.0.0 [40] and aligned with ToxB mRNA and CDS using Muscle [41] (-clwstrict). The multiple sequence alignment was then visualised in JalView version 2.10.5 [42], figures were created using the alignment overview.
To obtain a better view of sequence regions shared between the ten DW5 ToxB 10 kb regions, each sequence was aligned to each other at greater than 70% sequence identity, using BLAT version 3.5 [39] fastMap option, all coordinates were then used to create a bed file for visualisation using GenomeTools (gt) sketch version 1.5.10 [30].
The 2 kb sequence region upstream of ToxB was submitted to MEME Suite 5.1.1 [43] for motif discovery with classic discovery mode, site distribution zero or one occurrence and motif width between 6 and 50 inclusive. The most significant motif was submitted to TOMTOM [44] to identify similar motifs in the published nonredundant database JASPAR CORE 2018 [45] for eukaryotes.
Whole genome alignment
DW5 PacBio assembled contigs were aligned to the optically mapped M4 chromosome scaffold [12] reference using NUCmer v3.1 (--maxmatch --coords) [46]. The sequence dot plot figure (Fig. 1) was generated using MUMmerplot v3.1 [46] with option for color plot line with percentage similarity gradient. EMBOSS revseq version 6.6.0.0 [40] was used for the reverse complementation of sequence.
The sequence dot plot of smaller regions (Fig. 3) were generated using Dotter version 4.44.1 [47].
The alignment and visualisation (Fig. 5) of the multiple ToxB loci regions for Contigs 9, 15, 16 and 18 was conducted using Easyfig (−blastn) linux version 2.2.2 [48].
Supplementary information
Additional file 1. Nucleotide multiple sequence alignment of the ten ToxB loci regions.
Additional file 2. Predicted DNA binding site motif.
Additional file 3.Pyrenophora tritici-repentis sequence accessions downloaded from NCBI GenBank nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore) on the 27/02/2020.
Acknowledgements
We thank the Australian grain growers for their continued support of research through the Grains Research and Development Corporation (GRDC) and the Australian Government National Collaborative Research Infrastructure Strategy (NCRIS) for providing access to Pawsey Supercomputing under a National Computational Merit Allocation Scheme (NCMAS), Nectar Research/Pawsey Nimbus Cloud resources. We would also like to thank Prof. Tim Friesen, Department of Plant Pathology, North Dakota State University, Fargo, ND for supplying the isolate DW5.
Authors’ contributions
PM conducted the bioinformatics analysis and wrote the draft manuscript. PTS and CM conducted the molecular analysis. All authors contributed to reviewing and editing this manuscript. CM led the project conceptualization. The authors read and approved the final manuscript.
Funding
This work was generously supported through co-investment by Grains Research and Development Corporation (GRDC) and Curtin University (project code CUR00023) as well as Australian Government National Collaborative Research Infrastructure Strategy and Education Investment Fund Super Science Initiative. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Availability of data and materials
All data generated or analyzed during this study are included and can be accessed in this published article (and in Additional file 3). The annotated genome of DW5 has been deposited at DDBJ/ENA/GenBank repository under accession MUXC00000000. The DW5 version described in this paper is MUXC02000000. The annotated genome of M4 has been deposited at DDBJ/ENA/GenBank repository under accession NQIK00000000. The M4 version described in this paper is version NQIK02000000.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors have read the manuscript and declare that they have no competing interests.
Footnotes
The original version of this article was revised: an update on the location of DW5 ToxB cluster relative to the M4 chromosome 10 fusion event was required.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
8/9/2021
A Correction to this paper has been published: 10.1186/s12864-021-07836-3
References
- 1.Lamari L, Bernier CC. Evaluation of wheat lines and cultivars to Tan spot [Pyrenophora-Tritici-Repentis] based on lesion type. Can J Plant Pathol. 1989;11(1):49–56. [Google Scholar]
- 2.Strelkov SE, Lamari L, Ballance GM. Characterization of a host-specific protein toxin (Ptr ToxB) from Pyrenophora tritici-repentis. Mol Plant Microbe In. 1999;12(8):728–732. [Google Scholar]
- 3.Effertz RJ, Meinhardt SW, Anderson JA, Jordahl JG, Francl LJ. Identification of a Chlorosis-inducing toxin from Pyrenophora tritici-repentis and the chromosomal location of an insensitivity locus in wheat. Phytopathology. 2002;92(5):527–533. doi: 10.1094/PHYTO.2002.92.5.527. [DOI] [PubMed] [Google Scholar]
- 4.Ciuffetti LM, Tuori RP, Gaventa JM. A single gene encodes a selective toxin causal to the development of tan spot of wheat. Plant Cell. 1997;9(2):135–144. doi: 10.1105/tpc.9.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali S, Gurung S, Adhikari TB. Identification and characterization of novel isolates of Pyrenophora tritici-repentis from Arkansas. APS. 2010;94(2):229–235. doi: 10.1094/PDIS-94-2-0229. [DOI] [PubMed] [Google Scholar]
- 6.See PT, Marathamuthu KA, Iagallo EM, Oliver RP, Moffat CS. Evaluating the importance of the tan spot ToxA-Tsn1 interaction in Australian wheat varieties. Plant Pathol. 2018;67(5):1066–1075. [Google Scholar]
- 7.Liu Z, Friesen TL, Ling H, Meinhardt SW, Oliver RP, Rasmussen JB, et al. The Tsn1-ToxA interaction in the wheat-Stagonospora nodorum pathosystem parallels that of the wheat-tan spot system. Genome. 2006;49(10):1265–1273. doi: 10.1139/g06-088. [DOI] [PubMed] [Google Scholar]
- 8.Corsi B, Percival-Alwyn L, Downie RC, Venturini L, Iagallo EM, Campos Mantello C, et al. Genetic analysis of wheat sensitivity to the ToxB fungal effector from Pyrenophora tritici-repentis, the causal agent of tan spot. Theor Appl Genet. 2020;133(3):935–950. doi: 10.1007/s00122-019-03517-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, Ling H, Faris JD, et al. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet. 2006;38(8):953–956. doi: 10.1038/ng1839. [DOI] [PubMed] [Google Scholar]
- 10.Martinez JP, Oesch NW, Ciuffetti LM. Characterization of the multiple-copy host-selective toxin gene, ToxB, in pathogenic and nonpathogenic isolates of Pyrenophora tritici-repentis. Mol Plant-Microbe Interact. 2004;17(5):467–474. doi: 10.1094/MPMI.2004.17.5.467. [DOI] [PubMed] [Google Scholar]
- 11.Aboukhaddour R, Cloutier S, Ballance GM, Lamari L. Genome characterization of Pyrenophora tritici-repentis isolates reveals high plasticity and independent chromosomal location of ToxA and ToxB. Mol Plant Pathol. 2009;10(2):201–212. doi: 10.1111/j.1364-3703.2008.00520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moolhuijzen P, See PT, Hane JK, Shi G, Liu Z, Oliver RP, et al. Comparative genomics of the wheat fungal pathogen Pyrenophora tritici-repentis reveals chromosomal variations and genome plasticity. BMC Genomics. 2018;19(1):279. doi: 10.1186/s12864-018-4680-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manning VA, Pandelova I, Dhillon B, Wilhelm LJ, Goodwin SB, Berlin AM, et al. Comparative genomics of a plant-pathogenic fungus, Pyrenophora tritici-repentis, reveals transduplication and the impact of repeat elements on pathogenicity and population divergence. G3 (Bethesda) 2013;3(1):41–63. doi: 10.1534/g3.112.004044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moolhuijzen P, See PT, Moffat CS. A new PacBio genome sequence of an Australian Pyrenophora tritici-repentis race 1 isolate. BMC Res Notes. 2019;12(1):642. doi: 10.1186/s13104-019-4681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertazzoni S, Williams AH, Jones DA, Syme RA, Tan KC, Hane JK. Accessories make the outfit: accessory chromosomes and other dispensable DNA regions in plant-pathogenic Fungi. Mol Plant Microbe In. 2018;31(8):779–788. doi: 10.1094/MPMI-06-17-0135-FI. [DOI] [PubMed] [Google Scholar]
- 16.Chuma I, Hotta Y, Tosa Y. Instability of subtelomeric regions during meiosis in Magnaporthe oryzae. J Gen Plant Pathol. 2011;77:317–25.
- 17.McDonald MC, Taranto AP, Hill E, Schwessinger B, Liu Z, Simpfendorfer S, et al. Transposon-Mediated Horizontal Transfer of the Host-Specific Virulence Protein ToxA between Three Fungal Wheat Pathogens. MBio. 2019;10(5):e01515–19. [DOI] [PMC free article] [PubMed]
- 18.MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Fast and accurate short read alignment with burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2004;Chapter 4:Unit 4 10. doi: 10.1002/0471250953.bi0410s05. [DOI] [PubMed] [Google Scholar]
- 24.Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016;44(D1):D81–D89. doi: 10.1093/nar/gkv1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wheeler TJ, Clements J, Eddy SR, Hubley R, Jones TA, Jurka J, et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 2013;41(Database issue):D70–D82. doi: 10.1093/nar/gks1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langdon WB. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015;8(1):1. doi: 10.1186/s13040-014-0034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gremme G, Steinbiss S, Kurtz S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013;10(3):645–656. doi: 10.1109/TCBB.2013.68. [DOI] [PubMed] [Google Scholar]
- 31.Borodovsky M, Lomsadze A. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr Protoc Bioinformatics. 2011;Chapter 4:Unit 4 6 1–10. doi: 10.1002/0471250953.bi0406s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Testa AC, Hane JK, Ellwood SR, Oliver RP. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics. 2015;16:170. doi: 10.1186/s12864-015-1344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9(1):R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiryev SA, Papadopoulos JS, Schaffer AA, Agarwala R. Improved BLAST searches using longer words for protein seeding. Bioinformatics. 2007;23(21):2949–2951. doi: 10.1093/bioinformatics/btm479. [DOI] [PubMed] [Google Scholar]
- 36.Koski LB, Gray MW, Lang BF, Burger G. AutoFACT: an automatic functional annotation and classification tool. BMC Bioinformatics. 2005;6:151. doi: 10.1186/1471-2105-6-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 38.Sperschneider J, Dodds PN, Gardiner DM, Singh KB, Taylor JM. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol Plant Pathol. 2018;19(9):2094–2110. doi: 10.1111/mpp.12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olson SA. EMBOSS opens up sequence analysis. European molecular biology open software suite. Brief Bioinform. 2002;3(1):87–91. doi: 10.1093/bib/3.1.87. [DOI] [PubMed] [Google Scholar]
- 41.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 44.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8(2):R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018;46(D1):D260–D2D6. doi: 10.1093/nar/gkx1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delcher AL, Salzberg SL, Phillippy AM. Using MUMmer to identify similar regions in large sequence sets. Curr Protoc Bioinformatics. 2003;Chapter 10:Unit 10 3. doi: 10.1002/0471250953.bi1003s00. [DOI] [PubMed] [Google Scholar]
- 47.Sonnhammer EL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167(1–2):GC1–G10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- 48.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Nucleotide multiple sequence alignment of the ten ToxB loci regions.
Additional file 2. Predicted DNA binding site motif.
Additional file 3.Pyrenophora tritici-repentis sequence accessions downloaded from NCBI GenBank nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore) on the 27/02/2020.
Data Availability Statement
All data generated or analyzed during this study are included and can be accessed in this published article (and in Additional file 3). The annotated genome of DW5 has been deposited at DDBJ/ENA/GenBank repository under accession MUXC00000000. The DW5 version described in this paper is MUXC02000000. The annotated genome of M4 has been deposited at DDBJ/ENA/GenBank repository under accession NQIK00000000. The M4 version described in this paper is version NQIK02000000.