

Abstract
The inhibition of the DNA damage response (DDR) pathway in the treatment of cancer has recently gained interest, and different DDR inhibitors have been developed. Among them, the most promising ones target the WEE1 kinase family, which has a crucial role in cell cycle regulation and DNA damage identification and repair in both nonmalignant and cancer cells. This review recapitulates and discusses the most recent findings on the biological function of WEE1/PKMYT1 during the cell cycle and in the DNA damage repair, with a focus on their dual role as tumor suppressors in nonmalignant cells and pseudo-oncogenes in cancer cells. We here report the available data on the molecular and functional alterations of WEE1/PKMYT1 kinases in both hematological and solid tumors. Moreover, we summarize the preclinical information on 36 chemo/radiotherapy agents, and in particular their effect on cell cycle checkpoints and on the cellular WEE1/PKMYT1-dependent response. Finally, this review outlines the most important pre-clinical and clinical data available on the efficacy of WEE1/PKMYT1 inhibitors in monotherapy and in combination with chemo/radiotherapy agents or with other selective inhibitors currently used or under evaluation for the treatment of cancer patients.
Keywords: WEE1 family kinases, WEE1, PKMYT1, Cell cycle, DNA repair, Pseudo-oncogene, Tumor suppressor
Background
The WEE1 kinase family consists of three serine/threonine kinases sharing conserved molecular structures and encoded by the following genes: WEE1 (WEE1 G2 Checkpoint Kinase), PKMYT1 (membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase), and WEE1B (WEE2 oocyte meiosis inhibiting kinase). In eukaryotic somatic cells, WEE1 and PKMYT1 play a key role in cell cycle regulation and, in particular, they are involved in the entry into mitosis [1]. Their role as regulators is crucial during normal cell cycle progression and in response to DNA damages, as part of the DNA damage response (DDR) pathways. Similarly, WEE2 regulates cell cycle progression and, in particular, meiosis [2]. Briefly, WEE2 plays a dual regulatory role in oocyte meiosis by preventing premature restart prior to ovulation and permitting metaphase II exit at fertilization [3]. Despite the identification of WEE2 somatic mutations (1.9% of cases) and copy number (CN) alterations (22.5% of patients with CN loss and 22.5% with CN gain) across several cancer types (https://portal.gdc.cancer.gov), they have not been functionally linked to tumor development so far. Therefore, the following sections will be focused on WEE1 and PKMYT1 kinases that have a well-recognized role in oncology and hemato-oncology.
WEE1 and PKMYT1 in cell cycle regulation
WEE1 and PKMYT1 act as tumor suppressors in non-malignant eukaryotic somatic cells. Similarly to other DDR-related kinases, their main biological function is to prevent replication of cells with altered DNA. The main downstream target of WEE1 family kinases is the cyclin-dependent kinase 1 (CDK1)-cyclin B1 complex, also known as mitotic-promoting factor (MPF). WEE1 phosphorylates CDK1 on Tyr15 while PKMYT1 has a dual activity on Tyr15 and Thr14 [4] (Fig. 1a). The phosphorylation of those residues keeps the MPF complex inhibited until the cell approaches mitosis. WEE1 is located in the nucleus, while PKMYT1 is associated with the endoplasmic reticulum and Golgi apparatus [5, 6], and regulates Golgi membrane reassembly following mitosis [7]. Together, WEE1 and PKMYT1 ensure that CDK1 remains inactive as it shuttles into and out of the nucleus [8]. Through its extra-nuclear localization, PKMYT1 can also promote CDK1 cytosolic segregation. At the G2/M border, if no DNA damage has been detected, CDK1 phosphorylation on Tyr15 and Thr14 is rapidly removed by CDC25C phosphatase. In the nucleus, the CDK-activating kinase (CAK) complex composed by cyclin-dependent kinase 7 (CDK7), cyclin H1, and MAT1 promotes MPF complex activation through the phosphorylation of CDK1(Thr161) [9, 10]. The active MPF complex is then imported into the nucleus through phosphorylation of cyclin B1 (Ser126, Ser128, Ser133, and Ser147) [11]. This event is required to enter mitosis. The relevance of WEE1 and PKMYT1 regulation of CDK1 has been recently confirmed by in vivo studies. Indeed, the replacement of the CDK1 inhibitory phosphorylation sites with non-phosphorylatable amino acids (CDK1T14A/Y15F) was embryonic lethal in mice [12]. Once activated, the MPF complex can phosphorylate WEE1 and PKMYT1 to promote their inactivation via different cascades [5, 13, 14]. WEE1 is phosphorylated (Ser123) by CDK1 at the onset of mitosis, thereby generating a binding motif for polo like kinase 1 (PLK1) and casein kinase 2 (CK2), that in turn phosphorylate WEE1 (Ser53 and Ser121, respectively) [14, 15]. Together, the phosphorylation of the three Ser residues serves as a tag for the degradation of WEE1 by the ubiquitin ligase SCFβ-TrCP [13]. PKMYT1 is also phosphorylated by CDK1 and PLK1 and this event promotes its degradation [16]. In addition to the checkpoint function at the G2/M border, recent findings highlighted a role of WEE1 in the regulation of replication dynamics during S phase (intra S phase checkpoint). When cells reach the S phase, replication is initiated from a large number of replication origins triggered through the activation of the pre-replication complex [17] and following the activation of S phase specific CDK, primarily CDK2 [18, 19]. Similarly to CDK1, CDK2 regulation is controlled through Tyr15 phosphorylation status, that is balanced by WEE1 (Fig. 1a) and cell division cycle 25A (CDC25A) activity [20]. Both WEE1 and CDC25A/C have been shown to modulate unperturbed replication through regulating CDK1/CDK2 activity. Monoallelic expression of CDK1T14A/Y15F induced replication stress and S phase arrest in mouse embryonic fibroblasts (MEFs), with substantial increase of γH2AX levels, chromosomal fragmentation, and DDR activation, as a consequence of intra-S phase DNA damage [12]. Moreover, unscheduled origin firing due to loss of WEE1 leads to exhaustion of the replication protein A1 (RPA1) pool and, as a consequence, to death during DNA replication (replication catastrophe). The intra S phase activity of WEE1 is independent from PKMYT1 that is unable to phosphorylate CDK2 [5]. In addition, WEE1, but not PKMYT1, contributes to the control of mitosis exit. Indeed, Wee1-deficient MEFs showed mitotic defects (e.g., in the number and position of centrosomes) that induce arrest in mitosis or, in the majority of cells, mitotic slippage [21, 22]. At the end of mitosis, WEE1 inhibits CDK1 through phosphorylation of its Tyr15 residue (Fig. 1a). This event is dependent on the activation of the CTD phosphatase subunit 1 (FCP1) that dephosphorylates and activates WEE1 and other crucial component of the spindle assembly checkpoint (SAC) complex [23]. Although the precise mechanisms that regulate FCP1 activity is still unknown, it has been showed that FCP1 promotes the dephosphorylation of crucial SAC components, including cell division cycle 20 (CDC20) and ubiquitin specific peptidase 44 (USP44), thus promoting APC/CCdc20 activation and chromosome segregation [24–26]. Moreover, WEE1 directly interacts with APC/C components, including fizzy and cell division cycle 20 related 1 (CDH1), CDC20, cell division cycle 27 (CDC27), and its deletion enforced APC/C activity, resulting in alterations of the level of APC/C substrates and mitosis progression at the expense of genomic stability [21].
Fig. 1.

WEE1 and PKMYT1 biological functions. a Schematic representation of WEE1 and PKMYT1 involvement in cell cycle checkpoints. WEE1 regulates the activity of both CDK1 and CDK2 kinases (trough phosphorylation of Tyr15) and is involved in the regulation of intra-S, G2/M, and M phase cell cycle checkpoints. PKMYT1 selectively regulates CDK1 (through phosphorylation of Tyr15 and Thr14) and is plays a role in the G2/M phase checkpoint. b Schematic representation of the regulation of MUS81-EME1/2 endonuclease complexes by WEE1 during S and G2/M cell cycle phases. By inhibiting CDK2 or CDK1, WEE1 prevents MUS81 activation and the generation of DNA damages during S phase, and chromosomes pulverization during G2/M phase
WEE1 regulates replication forks and genome stability
The activity of WEE1 through the cell cycle can explain its tumor suppressor function, at least in nonmalignant cells. This observation was confirmed and disentangled in preclinical studies. Indeed, conditional Wee1 heterozygous deletion in the murine mammary epithelium caused enhanced proliferation, with cells progressing into mitosis while still undergoing DNA replication, and consequent accumulation of DNA damage, resulting in genomic instability and, ultimately, in tumor development [21]. Biological processes such as DNA replication and homologous recombination involve the formation of branched DNA structures that physically link chromosomes. Such DNA structures needs to be disengaged prior to entry into mitosis, in order to ensure proper chromosome segregation. Eukaryotic cells evolved different mechanisms to identify and process branched DNA structures (Y-shape DNA) and the most important one involves the structure-selective endonuclease MUS81. MUS81 forms heterodimeric complexes with the non-catalytic subunits EME1 or EME2 and recognizes Y-shape DNA structures during DNA replication or during mitosis (homologous recombination). The activity of MUS81-EME1/2 complex is crucial to recover stalled replication forks, during prolonged S phase arrest, and to reset DNA junction between twin chromatids during homologous recombination [27]. In unperturbed cells, WEE1 protects replication forks and prevents the generation DNA damages and chromosome pulverization through an indirect inhibition of MUS81 functionality [28]. Indeed, WEE1 phosphorylates CDK1 and CDK2, thus preventing the CDK-mediated phosphorylation and activation of MUS81-EME1/2 complexes [29]. Lack of WEE1-dependent regulation of MUS81-EME1/2 endonucleases may lead to cleavage of unwanted DNA structure (excessive replication forks), which would slow down replication progression and increase genomic instability [27, 28] (Fig. 1b).
WEE1 and PKMYT1 deregulation in cancer cells
WEE1 and PKMYT1 act like oncogenes
The biological role of WEE1 and PKMYT1 in cancer cells is not fully understood. Reduced WEE1 expression has been detected in breast cancer compared with normal tissues, independently of the tumor grade [21]. However, most findings suggest that both kinases act like oncogenes rather than tumor suppressors. Indeed, they are frequently overexpressed in both solid and hematological tumors and a genome-wide CRISPR screen of 563 cancer cell lines, showed that they are essential for the cell viability of almost all cell lines [30]. The dependency of cancer cells on WEE1 family proteins may be linked to the following mechanisms (Fig. 2): (i) the high proliferation rate of cancer cells that follows the activation of driver oncogenes (e.g. RAS, MYC) needs to be sustained by a strong cell cycle regulation machinery; (ii) cancer cells frequently inactivate p53, which is a key gatekeeper of G0/G1 and S phases and, as a consequence, the regulation of cell cycle is sustained entirely by the G2/M checkpoint; (iii) the over-expression of DDR-related kinases is fundamental to maintain a tolerable level of genetic instability, an intrinsic feature of cancer cells [31, 32]. Therefore, we can speculate that, once the malignant transformation process has been induced, WEE1 upregulation exerts a pro-tumorigenic functions by securing a tolerable level of genomic instability to cancer cells. The following sections summarize the current knowledge on the molecular and functional alterations of WEE1 and PKMYT1 in hematological and solid tumors.
Fig. 2.
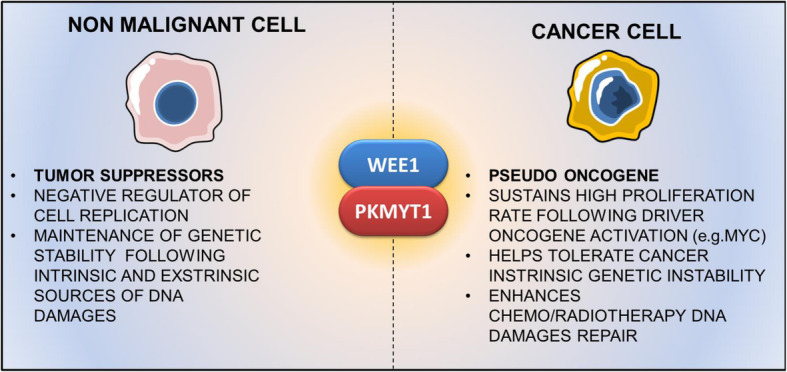
WEE1 family proteins role as tumor suppressors or pseudo-oncogenes in non-malignant and cancer cells
WEE1 and PKMYT1 genetic lesions in cancer
WEE1 and PKMYT1 are rarely mutated in cancer patients, with an overall mutation frequency of 1.2% and 0.2%, respectively (https://portal.gdc.cancer.gov). The distribution of somatic mutations is highly heterogeneous across cancer types (WEE1: 0.2–7.6%; PKMYT1:0.1–3.6%), with a higher frequency in uterine corpus endometrial carcinoma (UCEC) and tumors of the gastrointestinal tract (stomach and colon adenocarcinoma, Fig. 3a, b). In particular, WEE1 mutations have been reported in 7.6% of UCEC cases. Moreover, PKMYT1 lesions have been also detected in 2.7% of diffuse large B cell lymphoma (DLBC). Conversely, both kinase genes are rarely mutated in brain lower grade glioma (LGG), ovarian serous cystadenocarcinoma (OV), prostate adenocarcinoma (PRAD), and sarcoma (SARC), with a frequency lower than 0.5%. Both genes are mainly targeted by missense mutations that preferentially cluster in the region encoding the WEE1 kinase domain and its surroundings (Fig. 3c), suggesting a potential gain of function effect of the kinase activity. Conversely, the mutations are scattered throughout the PKMYT1 sequence (Fig. 3d). Little is known about the functional consequences of WEE1 and PKMYT1 mutations. In the majority of cancer types, the transcript expression in the mutated cases is higher than the median value of the entire cohort (https://www.cbioportal.org), supporting once more an oncogenic function. In pancreatic adenocarcinoma (PA) patients and cell lines, an insertion was identified in the WEE1 poly-T track, which contains the binding site of the HuR RNA binding protein [33]. The insertion resulted in decreased WEE1 expression upon mitomycin-induced DNA damage, which would argue against a protective effect of the mutation. Copy number alterations (CNAs) represent a more frequent event compared with mutations, with the WEE1 gene being predominantly involved in CN loss (23.7% of cases versus 7.8% of patients with CN gains), while PKMYT1 showing a higher percentage of CN gain (15.9% versus 12.0% of CN loss, Fig. 3e, f). The predominance of WEE1 deletion events (6.3% versus 3.25% of cases with amplification) was also observed in breast cancer, in line with its reduced expression, as mentioned above [21]. Overall, cancer types showing the highest recurrence (> 10%) of CNAs were OV (27.7%), lung squamous cell carcinoma (LUSC, 14.8%), uterine carcinosarcoma (UCS, 12.5%), and SARC (11.2%) for WEE1 and OV (18.8%), bladder urothelial carcinoma (BLCA, 13.7%), and esophageal carcinoma (ESCA, 10.3%) for PKMYT1. Of note, OV and LUSC have been classified as tumors with multiple recurrent chromosomal gains and losses [34], which may suggest a bystander effect related to chromosomal instability in these tumor types, especially in the case of WEE1 deletion, that is unexpected, based on the general oncogenic function exerted by the kinase.
Fig. 3.

WEE1 and PKMYT1 mutations and copy number alterations (CNAs) in cancer. a Frequency of patients with WEE1 or b PKMYT1 gene mutations across cancers from TCGA cohorts. c Distribution of mutations according to the WEE1 and d PKMYT1 amino acid (aa) sequence and protein domains (WEE1 transcript ENST00000450114, 646 aa; PKMYT1 transcript ENST00000262300, 499 aa). e Frequency of patients with copy number gain or loss in WEE1 or f PKMYT1 across cancers (https://portal.gdc.cancer.gov; ACC adrenocortical carcinoma, BLCA bladder urothelial carcinoma, BRCA breast invasive carcinoma, CESC cervical squamous cell carcinoma and endocervical adenocarcinoma, COAD colon adenocarcinoma, CHOL cholangiocarcinoma, DLBC diffuse large B cell lymphoma, ESCA esophageal carcinoma, GBM glioblastoma multiforme, HNSC head and neck squamous cell carcinoma, KICH kidney chromophobe, KIRK kidney renal clear cell carcinoma, KIRP kidney renal papillary cell carcinoma, LGG brain lower grade glioma, LIHC liver hepatocellular carcinoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, MESO mesothelioma, OV ovarian serous cystadenocarcinoma, PAAD pancreatic adenocarcinoma, PCPG pheochromocytoma and paraganglioma, PRAD prostate adenocarcinoma, READ rectum adenocarcinoma, SARC sarcoma, SKCM skin cutaneous melanoma, STAD stomach adenocarcinoma, TGCT testicular germ cell tumors, THYM thymoma, UCS uterine carcinosarcoma, UCEC uterine corpus endometrial carcinoma)
WEE1 and PKMYT1 functional role in hematological and solid tumors
Few studies have analyzed WEE1 and PKMYT1 expression in hematological malignancies. Our group showed that WEE1 kinase is highly expressed in acute lymphoblastic leukemia (ALL) cell lines and primary cells in comparison with normal hematopoietic cells, and that PKMYT1 is upregulated in relapsed ALL samples compared with nonmalignant hematopoietic cells [35]. Moreover, we demonstrated that ALL cells are dependent on WEE1 functionality for their survival and proliferation and that PKMYT1 levels may influence the sensitivity to the WEE1 inhibitor AZD-1775 [35]. Similar results on the role of WEE1 were obtained in multiple myeloma (MM), acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and chronic lymphocyte leukemia (CLL) [36–39]. In AML cells, WEE1 and PKMYT1 are key gene discriminating between FLT3-ITD, FLT3-TKD, and NRAS-mutated samples. They were expressed at lower levels selectively in FLT3-ITD specimens in comparison with wild-type cells, suggesting either a tumor suppressor role in the leukemogenic process or a potential vulnerability n this AML subtype [40]. Pharmacological WEE1 inhibition alone or in combination with histone deacetylase inhibitors showed therapeutic potential in FLT3-ITD AML, confirming their dependency on WEE1 activity [41]. Since FLT3-ITD AML have intrinsic homologous recombination repair defects [42]. WEE1 inhibition may exacerbate the cell genotoxic stress by disrupting multiple cell cycle checkpoints. WEE1 has been showed to be a valuable target also for lymphoma patients [43]. In parallel, PKMYT1 proved to be essential for MM cell line viability, since its downregulation strongly decreased cell growth, while inducing apoptosis [44].
WEE1 and PKMYT1 are also over-expressed in solid tumors, including hepatocellular carcinoma, colon cancer, glioblastoma, non-small-cell lung cancer (NSCLS), neuroblastoma, and gastric cancers [31, 32, 45–47]. High WEE1 expression has been associated with negative prognostic factors including lymph node involvement, induction of metastasis, increased biomarkers of proliferation (CCND1, Ki67, or CCNA1) and resistance to treatments (radiotherapy or chemotherapy) [48–51]. Elevated PKMYT1 levels have been associated with tumor progression, a more aggressive disease, the induction of metastasis at least in NSCLS patients [45] and, generally, with poor prognosis. Depending on the cancer subtype, the expression of WEE1 and PKMYT1 has been linked with the activation of cellular pathways crucial for the specific disease. In melanoma cells, WEE1 silencing caused an increase of phospho p38 protein levels, indicating a role in the regulation of p38/MAPK pathway activation during p53-independent DNA damage response [49]. In hepatocellular carcinoma and colorectal cancers, PKMYT1 regulates epithelial-mesenchymal transition (EMT), a process relevant to tumor progression, invasion, metastasis, and drug resistance, through the activation of the beta-catenin/TCF signaling [32, 46], while PKMYT1 has been reported to control Notch pathway in NSCLC [45]. In particular, crucial component of the pathway, including NOTCH1, p21, and HES1 are downregulated by chemical inhibition of PKMYT1 [45]. In neuroblastic tumors, PKMYT1 is required to stabilize MYCN protein, which is a crucial proto-oncogene for this cancer types [52]. Moreover, in esophageal squamous cell carcinoma (ESCC) cell lines and primary cells, the expression of PKMYT1 is associated with and regulates the activation of the AKY/mTOR pathway [53] (Table 1). Taken together, this evidence suggests a broad role of WEE1/PKMYT1 besides the DNA damage response pathway that may increase the interest towards its therapeutic targeting.
Table 1.
WEE1 and PKMYT1 molecular alterations in hematological and solid tumors according to literature
| Gene | Genetic alteration | Disease | Effect/prognostic value | Reference |
|---|---|---|---|---|
| Hematological tumors | ||||
| WEE1 | Over-expression | ALL; AML; MM; CML; CLL; DLBCL | Crucial for cell viability of cancer cells (experimentally proven). | [35–40, 43, 54] |
| Copy number Gain | AML | Biological effect or prognostic value unknown | [55] | |
| PKMYT1 | Over-expression | ALL; MM | Crucial for cell viability of cancer cells (experimentally proven). | [35, 44] |
| Solid tumors | ||||
| WEE1 | Over-expression | GC; MaM; GL; OC; CC | Associated with lymph node involvement, induction of metastasis, increased biomarkers of proliferation (CCND1, Ki67 or CCNA1), resistance to treatment and poor overall survival. | [48–51, 56–60] |
| Mutation | PA | Insertion causing decrease WEE1 expression upon DNA damage | [33] | |
| PKMYT1 | Over-expression | HC; CC; GLB; NSCLC; N; GS | Associated with tumor progression, aggressive disease and poor overall survival. | [31, 32, 45–47] |
| Mutation | N | Biological effect or prognostic value unknown | [61] | |
ALL acute lymphoblastic leukemia, AML acute myeloid leukemia, MM multiple myeloma, CML chronic myeloid leukemia, CLL chronic lymphocyte leukemia, DLBCL diffuse large B cell lymphoma, GC gastric cancer, MaM malignant melanoma, GL gliomas, OC ovarian cancer, CC colorectal cancer, PA pancreatic adenocarcinoma, HC hepatocellular carcinoma, GLB glioblastoma, NSCLC non-small-cell lung cancer, N neuroblastoma
Development of WEE1 and PKMYT1 inhibitors
WEE1 and PKMYT1 inhibitors have single agent and chemo-sensitizer effects
Due to their potential oncogenic role, WEE1 and PKMYT1 have been investigated as therapeutic targets for hematological and solid tumors. Several pharmacological inhibitors have been designed and subsequently validated in different cancer models. The available literature highlights a common mechanism of action of WEE1/PKMYT1 inhibitors in cancer cells either in single agent or in combination with DNA damaging agents (chemotherapy/radiotherapy). WEE1/PKMYT1 kinase inhibition causes G2/M cell cycle checkpoint override, premature mitotic entry, and cell death during mitosis, through a mechanism generally known as mitotic catastrophe (Fig. 4a). From a biological point of view, the inhibition of WEE1 kinase causes a significant reduction of phospho-CDK1 (Tyr15), thus promoting the accumulation of active CDK1-cyclin B1 complex and, consequently, mitotic entry. The beginning of mitosis is also associated with a progressive accumulation of DNA damages and the degeneration in mitotic catastrophe. The sensitivity to WEE1 kinase inhibitors in relation to TP53 mutational status remains controversial. Indeed, some studies reported increased sensitivity of TP53 mutant cell lines to WEE1 inhibitors in comparison to TP53 wild-type ones [62, 63], while others showed no association between p53 functionality and the effectiveness of WEE1 inhibition [35, 64]. These discrepancies may be linked to the intrinsic chromosomal instability of the analyzed tumors and to additional alterations deregulating the G1 checkpoint in TP53 wild-type cases that may enhance the sensitivity to WEE1 targeting.
Fig. 4.

Mechanism of action of WEE1/PKMYT1 inhibitors for the treatment of cancer cells. a Schematic representation of WEE1/PKMYT1 inhibition as monotherapy. In cancer cells, oncogenes promote high rate of proliferation, replication stress and the over-expression of WEE1/PKMYT1 kinases. In this scenario, cancer cells need WEE1 and PKMYT1 to sustain replication stress and proliferation. The inhibition of WEE1/PKMYT1 results in the accumulation of DNA damages, the increase of genetic instability and induction of apoptosis. b Schematic representation of WEE1/PKMYT1 inhibition in combination with DNA damaging agents. Cancer cells respond to DNA damages by activating WEE1/PKMYT1 kinases. The inhibition of WEE1/PKMYT1 enhances the cytotoxicity of DNA damaging agents by inhibiting DNA repair and promoting cell cycle progression even in the presence of DNA damages. Therefore, cancer cells accumulate massive DNA damages until a point of no return
Regarding the role of WEE1 inhibitors as chemo-sensitizer agents, a large number of studies demonstrated a synergistic activity between DNA damaging agents (chemotherapy including doxorubicin, cytarabine, methotrexate, cisplatin, clofarabine, etoposide, 5-fluorouracil, and radiotherapy) and different WEE1/PKMYT1 inhibitors in preclinical models [48, 56, 65–69]. The mechanism of action of the combination is based on the inhibition of the DDR pathway following induction of DNA damage induced by the chemotherapy or radiotherapy agents. In this scenario, cancer cells with damaged DNA fail to arrest cell cycle, continue to proliferate, and accumulate massive DNA damage until a point of no return (Fig. 4b). Indeed, several DNA damaging agent promote the indirect activation of WEE1 and PKMYT1 kinases, as showed mostly by the activation of cell cycle checkpoints (S and G2/M checkpoints) in cancer cells. We summarized in Table 2 the results of preclinical studies in which the effect of different chemotherapy agents or radiotherapy has been evaluated in terms of cell cycle perturbation and altered expression of WEE1 or PKMYT1 following in vitro or in vivo treatment. Taken together, the abovementioned data prove that WEE1 and PKYMYT1 are ideal targets to override cell cycle checkpoint regulation and to improve the efficacy of DNA-damaging agents. In particular, tumors with a high level of chromosomal instability may respond to WEE1/PKMYT1 inhibition per se, while cases with a more stable genomic asset may benefit of the combination between DNA-damaging agents and WEE1 family kinase inhibitors. The following sections reports the main preclinical and clinical findings obtained using small molecules inhibitors of WEE1 and PKMYT1 kinases.
Table 2.
Effects of standard of care chemo/radiotherapy agents on cell cycle checkpoints activation
| Chemotherapy agents/radiotherapy | Intra S checkpoint | G2/M checkpoint | WEE1 and/or PKMYT1 experimentally proven involvement in cancer model |
|---|---|---|---|
| Actinomycin | No | Yes [70, 71] | WEE1 upregulation [71] |
| Azacitidine | No | Yes [72] | NA |
| Bleomycin | No | Yes [73] | NA |
| Carboplatin | No | Yes [74] | NA |
| Cisplatin | No | Yes [75, 76] | WEE1 inhibition enhanced cytotoxicity [76, 77] |
| Cyclophosphamide | NA | NA | WEE1 upregulation [78] |
| Cytarabine | Yes [79, 80] | Yes [79, 80] | WEE1 upregulation [38, 81] |
| Clofarabine | Yes [35] | No | WEE1 inhibition enhanced cytotoxicity [35] |
| Daunorubicin | Yes [82] | Yes [82] | NA |
| Decitabine | No | Yes [83] | NA |
| Docetaxel | No | Yes [84, 85] | NA |
| Doxorubicin | No | Yes [86] | WEE1 upregulation [86]; WEE1 inhibition enhanced cytotoxicity [35] |
| Epirubicin | No | Yes [87, 88] | WEE1 inhibition enhanced cytotoxicity [89] |
| Epothilone | No | Yes [90] | NA |
| Etoposide | No | Yes [91] | WEE1 inhibition enhanced cytotoxicity [67] |
| Fluorouracil | Yes [92] | No | WEE1 inhibition enhanced cytotoxicity [92] |
| Fludarabine | Yes [80] | No | NA |
| Gemcitabine | Yes [80] | No | WEE1 upregulation [93]; WEE1 inhibition enhanced cytotoxicity [94] |
| Hydroxyurea | Yes [95] | No | WEE1 inhibition enhanced cytotoxicity [96] |
| Idarubicin | No | Yes [97] | NA |
| Irinotecan | No | Yes [98] | WEE1 inhibition enhanced cytotoxicity [99] |
| Mechlorethamine | Yes [100] | Yes [100] | NA |
| Mercaptopurine | Yes [101] | No | NA |
| Methotrexate | Yes [102] | No | WEE1 inhibition enhanced cytotoxicity [103] |
| Mitoxantrone | No | Yes [104] | WEE1 inhibition enhanced cytotoxicity [78] |
| Oxaliplatin | No | Yes [105] | WEE1 inhibition enhanced cytotoxicity [106] |
| Paclitaxel | No | Yes [107] | WEE1 inhibition enhanced cytotoxicity [108] |
| Pemetrexed | Yes [109] | No | WEE1 inhibition enhanced cytotoxicity [110] |
| Radiotherapy (ionizing radiation) | No | Yes [111] | WEE1 inhibition enhanced cytotoxicity [68] |
| Teniposide | Yes [112] | Yes [112] | NA |
| Thioguanine | Yes [113] | Yes [113] | WEE1 inhibition enhanced cytotoxicity [65] |
| Topotecan | No | Yes [114] | WEE1 inhibition enhanced cytotoxicity [115] |
| Vinblastine | No | Yes [116] | NA |
| Vincristine | No | Yes [117] | WEE1 inhibition enhanced cytotoxicity [118] |
Preclinical studies of WEE1 and PKMYT1 inhibitors
Several targeted compounds showed an inhibitory activity on WEE1 and PKMYT1 kinases and their efficacy was proven in a number of tumor types. Table 3 shows the main preclinical studies that used WEE1/PKMYT1 inhibitors in single agent or in combination with chemo/radiotherapy agents in different tumor types.
Table 3.
Preclinical studies evaluating the effect of WEE1 inhibitors in monotherapy or in combination with chemotherapy/radiotherapy in cancer
| Inhibitor | Treatment | Cancer model | Main biological effect | References |
|---|---|---|---|---|
| PD0166285 | M | GBM-astrocytoma |
-G2/M checkpoint override -Forced mitotic entry |
[51] |
| Adavosertib | M | MM, ALL, AML TNBC, DLBCL, MCL |
-G2/M checkpoint override -Forced mitotic entry -Mitotic catastrophe -Replicative catastrophe |
[35, 99, 119–122] |
| PD0166285 | +R | GBM-astrocytoma |
-Mitotic catastrophe -Inhibition of DNA repair |
[51] |
| Adavosertib | +R | CC, LC, BC, PC, OC, DLBCL, ES |
-Increased DNA damage -Induction of apoptosis -Mitotic catastrophe |
[78, 99, 123–126] |
| Adavosertib | +C | AML, ALL, MM, BC, CC, GC, DLBCL |
-S or G2/M checkpoint override -Increased DNA damaged -Induction of apoptosis |
[35, 37, 38, 76, 92, 99, 121, 127–129] |
| Adavosertib | +HDAC i | AML, HNSCC |
-Replication stress -Replicative catastrophe -Increased DNA damage -Inhibition of DNA repair |
[41, 130, 131] |
| Adavosertib | +ATR i | AML, DLBCL, MCL, BC |
-Replication stress -Replicative catastrophe -Increased DNA damaged -Inhibition of DNA repair |
[132–135] |
| Adavosertib | +mTOR i | AML, ALL, OC, NSCLC | -Inhibition of DNA repair | [136–139] |
| Adavosertib | +CHK1 i | MCL, DLBCL, ALL, AML |
-Replication stress -Increased DNA damage -Replicative catastrophe |
[103, 140–142] |
| Adavosertib | +BCL2i/MCL-1 i | DLBCL |
-Force mitotic entry -Increase DNA damage -INDUCTION of apoptosis |
[143] |
| Adavosertib | +PARP1 i | NSCLC, AML, ALL |
-G2/M checkpoint override -Replication stress -Increased DNA damage -Inhibition of DNA repair |
[126, 144–146] |
| Adavosertib | +AURORA A i | HNSCC |
-Forced mitotic entry -Mitotic catastrophe |
[147] |
| Adavosertib | +CDK2 i | BC |
-Replication stress -Replicative catastrophe |
[89] |
| Adavosertib | +SIRT1 i | LC | -Inhibition of DNA repair | [148] |
| Adavosertib | +CDK4-6 i | S | -Replication stress | [149] |
| Adavosertib | +BCR-ABL1 i | ALL | -Inhibition of DNA repair -G2/M checkpoint override | [35] |
| Adavosertib | +Proteasome i | MM |
-G2/M checkpoint override -Forced mitotic entry -Inhibition of DNA repair |
[36] |
| Adavosertib | +BET i | NSCLC |
-Inhibition of DNA repair -Forced mitotic entry -Mitotic catastrophe |
[150] |
M monotherapy, R radiotherapy, C chemotherapy, ALL acute lymphoblastic leukemia, AML acute myeloid leukemia, MM multiple myeloma, DLBCL diffuse large B cell lymphoma, MCL mantle cell lymphoma, GC gastric cancer, GL gliomas, OC ovarian cancer, CC colorectal cancer, PC pancreatic cancer, ES esophageal cancer, HC hepatocellular carcinoma, GLB glioblastoma, NSCLC non-small-cell lung cancer, N neuroblastoma, S sarcomas, LC lung cancer, BC breast cancer, HNSCC head and neck squamous cell carcinoma, TNBC triple negative breast cancer
PD0166285 is the first reported drug, with an inhibitory activity against WEE1, PKMYT1, and a range of other kinases including c-Src, EGFR, FGFR1, CHK1, and PDGFRb [151].
Adavosertib (AZD-1775) is the first highly potent and selective WEE1 inhibitor. A large number of preclinical studies evaluated its efficacy in single agent and in combinatory approaches. Regarding the mechanism of action, adavosertib induces S and/or G2/M cell cycle checkpoints override, depending on cancer types, when used in monotherapy. Cell cycle perturbation is associated with a progressive accumulation of DNA damages and by the induction of apoptosis [35, 99, 119–122]. This last event is cell cycle phase-dependent and can occur (i) as a consequence of S phase checkpoint override, when cancer cells start DNA replication even in the presence of DNA damages (replicative catastrophe); (ii) following G2/M phase checkpoint override, that results in forced entry into mitosis, even in the presence of DNA damages (mitotic catastrophe).
In combination strategies, adavosertib was able to enhance the cytotoxicity of chemo/radiotherapy agents, by inducing cell cycle checkpoint override, inhibition of DNA damage repair, and induction of apoptosis [35, 37, 38, 92, 121, 127–129]. The chemo-sensitizer efficacy of DDR inhibitors has been linked to drug scheduling [94, 152, 153]. Recently in pancreatic adenocarcinoma cells, it has been reported that the efficacy of a triple regimen combining gemcitabine, CHK1, and WEE1 inhibitors is strictly dependent on the timing of drug administration. Indeed, the maximum effect of the combination is obtained when gemcitabine and CHK1 inhibitors are administered simultaneously (thus inducing replicative stress) and adavosertib is added at a later time [94].
Moreover, strong synergism has been observed by combining adavosertib with small molecules, including DDR-related inhibitors (CDK2 [89], CDK4-6 [149], CHK1 [103, 140–142], ATM [132–135], AURORA A [147], PARP1 [144], SIRT1 [148] inhibitors), histone deacetylase (HDAC) inhibitors [41, 130, 131], tyrosine kinase inhibitors (BCR-ABL1 inhibitors [35]), anti-apoptotic protein inhibitors (BCL2 and MCL1 inhibitors [143]), mTOR inhibitor [136–139], and proteasome inhibitors [36].
We have recently reported synergistic effects of adavosertib in combination with different tyrosine kinase inhibitors in both BCR-ABL1-positive and -negative ALL cell lines and primary cells. Interestingly, strong synergism was found in BCR-ABL1-negative ALL cell lines treated with adavosertib in combination with bosutinib isomer. In the study, we speculated that the strong cytotoxic effect of the combination was due to the concomitant inhibition of WEE1 and PKMYT1 kinases [35]. Indeed, no selective inhibitor has been currently developed to target its functionality. However, several known tyrosine kinase inhibitors have an inhibitory off-target effect on PKMYT1. Among them, compounds commonly used for the treatment of BCR-ABL1-positive CML and ALL, as dasatinib and bosutinib (and a structural isomer of bosutinib [154, 155]) were shown to inhibit PKMYT1 activity.
Overall, the data suggest that WEE1/PKMYT1 inhibition is a suitable pharmacological target for combination strategies in cancer. The broad spectrum of activities exerted by the two kinases, and especially by WEE1, across the cell cycle, makes them good candidates for a number of diverse therapeutic combinations.
WEE1 inhibitors from bench to bedside
Several clinical studies are currently evaluating the efficacy of adavosertib on different aggressive and advanced tumors (Table 4).
Table 4.
Clinical trials evaluating WEE1/PKMYT1 inhibitor in monotherapy or in combination for cancer therapy
| Study ID | Study title | Tumor | Interventions | Status | Phase |
|---|---|---|---|---|---|
| NCT02610075 | Phase Ib Study to Determine MTD of AZD1775 Monotherapy in Patients With Locally Advanced or Metastatic Solid Tumours. | S | AZD1775 | C | 1 |
| NCT03668340 | AZD1775 in Women With Recurrent or Persistent Uterine Serous Carcinoma | S | AZD1775 | R | 2 |
| NCT02482311 | Safety, Tolerance, PK, and Anti-tumour Activity of AZD1775 Monotherapy in Patients With Advanced Solid Tumours | S | AZD 1775 | C | 1 |
| NCT02207010 | A Phase 0 Study of AZD1775 in Recurrent GBM Patients | S | AZD1775 | NA | 1 |
| NCT03315091 | Phase I Study to Assess the Effect of Food on AZD1775 Pharmacokinetics in Patients With Advanced Solid Tumours | S | AZD1775 | C | 1 |
| NCT01748825 | AZD1775 for Advanced Solid Tumors | S/H | AZD1775 | ANR | 1 |
| NCT02511795 | AZD1775 Combined With Olaparib in Patients With Refractory Solid Tumors | S | AZD1775 + Olaparib | C | 1 |
| NCT03313557 | AZD1775 Continued Access Study to Assess Safety and Tolerability for Patients Enrolled in AZD1775 Clinical Pharmacology Studies | S | AZD1775 | C | 1 |
| NCT02593019 | Phase II, Single-arm Study of AZD1775 Monotherapy in Relapsed Small Cell Lung Cancer Patients | S | AZD1775 | NA | 2 |
| NCT02688907 | Phase II, Single-arm Study of AZD1775 Monotherapy in Relapsed Small Cell Lung Cancer Patients With MYC Family Amplification or CDKN2A Mutation Combined With TP53 Mutation | S | AZD1775 | T | 2 |
| NCT02087176 | A Placebo Controlled Study Comparing AZD1775 + Docetaxel Versus Placebo + Docetaxel to Treat Lung Cancer | S | AZD1775 + Docetaxel | T | 2 |
| NCT03012477 | CISPLATIN + AZD-1775 In Breast Cancer | S | AZD1775 + Cisplatin | ANR | 2 |
| NCT02341456 | Phase Ib Study AZD1775 in Combination With Carboplatin and Paclitaxel in Adult Asian Patients With Solid Tumours | S | AZD1775 + Carboplatin or Paclitaxel | C | 1 |
| NCT02791919 | Wee1 Kinase Inhibitor AZD1775 and Combination Chemotherapy in Treating Children, Adolescents and Young Adults With Relapsed or Refractory Acute Myeloid Leukemia | H | AZD1775 + Cytarabine or Filgrastim or Fludarabine Phosphate | W | 1 |
| NCT02513563 | AZD1775 Plus Carboplatin-Paclitaxel in Squamous Cell Lung Cancer | S | AZD1775 + Carboplatin or Paclitaxel | R | 2 |
| NCT03718143 | AZD1775 in Advanced Acute Myeloid Leukemia, Myelodysplastic Syndrome and Myelofibrosis | H | AZD1775 + Cytarabine | T | 2 |
| NCT02585973 | Dose-escalating AZD1775 + Concurrent Radiation + Cisplatin for Intermediate/High Risk HNSCC | S | AZD1775 + Cisplatin + Radiation | R | 1 |
| NCT02087241 | Ph II Trial of Carboplatin and Pemetrexed With or Without AZD1775 for Untreated Lung Cancer | S | AZD1775 + pemetrexed or carboplatin | T | 2 |
| NCT02381548 | Phase I Trial of AZD1775 and Belinostat in Treating Patients With Relapsed or Refractory Myeloid Malignancies or Untreated Acute Myeloid Leukemia | H | AZD1775 + Belinostat | T | 1 |
| NCT03333824 | Effects of AZD1775 on the PK Substrates for CYP3A, CYP2C19, CYP1A2 and on QT Interval in Patients With Advanced Cancer | S | AZD1775 | C | 1 |
| NCT02906059 | Study of Irinotecan and AZD1775, a Selective Wee 1 Inhibitor, in RAS or BRAF Mutated, Second-line Metastatic Colorectal Cancer | S | AZD1775 + Irinotecan | R | 1 |
| NCT02037230 | Dose Escalation Trial of AZD1775 and Gemcitabine (+Radiation) for Unresectable Adenocarcinoma of the Pancreas | S | AZD1775 + Gemcitabine+ Radiation Therapy | C | 1,2 |
| NCT02617277 | Safety, Tolerability and Pharmacokinetics of AZD1775 (Adavosertib) Plus MEDI4736 (Durvalumab) in Patients With Advanced Solid Tumours | S | AZD1775 + Durvalumab | ANR | 1 |
| NCT02666950 | WEE1 Inhibitor AZD1775 With or Without Cytarabine in Treating Patients With Advanced Acute Myeloid Leukemia or Myelodysplastic Syndrome | H | AZD1775 + Cytarabine | C | 2 |
| NCT01047007 | A Dose Escalation Study of MK1775 in Combination With 5-FU or 5-FU/CDDP in Patients With Advanced Solid Tumor (1775-005) | S | AZD1775 + 5-FU or 5-FU/CDDP | T | 1 |
| NCT01164995 | Study With Wee-1 Inhibitor MK-1775 and Carboplatin to Treat p53 Mutated Refractory and Resistant Ovarian Cancer | S | AZD1775 + carboplatin | NA | 2 |
| NCT02448329 | Study of AZD1775 in Combination With Paclitaxel, in Advanced Gastric Adenocarcinoma Patients Harboring TP53 Mutation as a Second-line Chemotherapy | S | AZD1775 + paclitaxel | R | 2 |
| NCT02508246 | WEE1 Inhibitor MK-1775, Docetaxel, and Cisplatin Before Surgery in Treating Patients With Borderline Resectable Stage III-IVB Squamous Cell Carcinoma of the Head and Neck | S | AZD1775 + Cisplatin + Docetaxel | C | 1 |
| NCT03253679 | AZD1775 in Treating Patients With Advanced Refractory Solid Tumors With CCNE1 Amplification | S | AZD1775 | R | 2 |
| NCT01076400 | A Study of MK-1775 in Combination With Topotecan/Cisplatin in Participants With Cervical Cancer (MK-1775-008) | S | AZD1775 + Topotecan or Cisplatin | T | 1,2 |
| NCT02196168 | Cisplatin With or Without WEE1 Inhibitor MK-1775 in Treating Patients With Recurrent or Metastatic Head and Neck Cancer | S | AZD1775 +Cisplatin | T | 2 |
| NCT02101775 | Gemcitabine Hydrochloride With or Without WEE1 Inhibitor MK-1775 in Treating Patients With Recurrent Ovarian, Primary Peritoneal, or Fallopian Tube Cancer | S | AZD1775 + Gemcitabine | ANR | 2 |
| NCT03028766 | WEE1 Inhibitor With Cisplatin and Radiotherapy: A Trial in Head and Neck Cancer | S | AZD1775 + Cisplatin + Radio therapy | ANR | 1 |
| NCT01357161 | A Study of MK-1775 in Combination With Paclitaxel and Carboplatin Versus Paclitaxel and Carboplatin Alone for Participants With Platinum-Sensitive Ovarian Tumors With the P53 Gene Mutation (MK-1775-004) | S | AZD1775 + paclitaxel + carboplation | C | 2 |
| NCT03284385 | Testing AZD1775 in Advanced Solid Tumors That Have a Mutation Called SETD2 | S | AZD1775 | R | 2 |
| NCT00648648 | A Dose Escalation Study of MK-1775 in Combination With Either Gemcitabine, Cisplatin, or Carboplatin in Adults With Advanced Solid Tumors (MK-1775-001) | S | AZD1775 + Gemcitabine or Cisplatin or Carboplatin | C | 1 |
| NCT02194829 | Paclitaxel Albumin-Stabilized Nanoparticle Formulation and Gemcitabine Hydrochloride With or Without WEE1 Inhibitor MK-1775 in Treating Patients With Previously Untreated Pancreatic Cancer That Is Metastatic or Cannot Be Removed by Surgery | S | AZD-1775 + Gemcitabine + paclitaxel | ANR | 1,2 |
| NCT02576444 | Olaparib Combinations | S | AZD1775 + olaparib | ANR | 2 |
| NCT04197713 | Testing the Sequential Combination of the Anti-cancer Drugs Olaparib Followed by Adavosertib (AZD1775) in Patients With Advanced Solid Tumors With Selected Mutations and PARP Resistance, STAR Study | S | AZD1775 + olaparib | ANR | 1 |
| NCT01922076 | Adavosertib and Local Radiation Therapy in Treating Children With Newly Diagnosed Diffuse Intrinsic Pontine Gliomas | S | AZD1775 + Radiation Therapy | ANR | 1 |
| NCT03579316 | Adavosertib With or Without Olaparib in Treating Patients With Recurrent Ovarian, Primary Peritoneal, or Fallopian Tube Cancer | S | AZD1775 + olaparib | R | 2 |
| NCT02095132 | Adavosertib and Irinotecan Hydrochloride in Treating Younger Patients With Relapsed or Refractory Solid Tumors | S | AZD1775 + Irinotecan or Irinotecan Hydrochloride | R | 1,2 |
| NCT03345784 | Adavosertib, External Beam Radiation Therapy, and Cisplatin in Treating Patients With Cervical, Vaginal, or Uterine Cancer | S | AZD1775 +Cisplatin + Radiation (External Beam Radiation Therapy) | R | 1 |
| NCT01849146 | Adavosertib, Radiation Therapy, and Temozolomide in Treating Patients With Newly Diagnosed or Recurrent Glioblastoma | S | AZD1775 + Radiation Therapy + Temozolomide | R | 1 |
| NCT02937818 | A Phase II, Study to Determine the Preliminary Efficacy of Novel Combinations of Treatment in Patients With Platinum Refractory Extensive-Stage Small-Cell Lung Cancer | S | AZD1775 + carboplatin | ANR | 2 |
| NCT02546661 | Open-Label, Randomised, Multi-Drug, Biomarker-Directed, Phase 1b Study in Pts w/ Muscle Invasive Bladder Cancer | S | AZD1775 + Durvalumab | ANR | 1 |
| NCT02659241 | Adavosertib Before Surgery in Treating Patients With Advanced High Grade Ovarian, Fallopian Tube, or Primary Peritoneal Cancer | S | AZD1775 | R | 1 |
| NCT02272790 | Adavosertib Plus Chemotherapy in Platinum-Resistant Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer | S | AZD1775 + Paclitaxel or Carboplatin or Gemcitabine or pegylated liposomal doxorubicin | ANR | 2 |
| NCT02813135 | European Proof-of-Concept Therapeutic Stratification Trial of Molecular Anomalies in Relapsed or Refractory Tumors | S/H | AZD1775 + carboplatin | R | 1,2 |
| NCT03330847 | To Assess Safety and Efficacy of Agents Targeting DNA Damage Repair With Olaparib Versus Olaparib Monotherapy. | S | AZD1775 + olaparib | R | 2 |
| NCT01827384 | MPACT Study to Compare Effects of Targeted Drugs on Tumor Gene Variations | S | AZD1775 + carboplatin | R | 2 |
| NCT02465060 | Targeted Therapy Directed by Genetic Testing in Treating Patients With Advanced Refractory Solid Tumors, Lymphomas, or Multiple Myeloma (The MATCH Screening Trial) | S/H | AZD1775 | R | 2 |
S solid tumor, H hematological tumor, C completed, R recruiting, W withdraw, ANR active not recruiting, T terminated, NA status unknown (last update 04/22/2020)
The results of phase I trials showed that adavosertib is well tolerated both in single agent and in combination. Depending on the study, the maximum tolerated dose (MTD) was established between 150 and 225 mg orally twice per day for 2.5 days per 2 weeks [156–158]. The most common adverse events reported in the abovementioned studies were fatigue, nausea, vomiting, diarrhea, and hematologic toxicity. Moreover, correlative studies performed on tumor biopsies confirmed in vivo the mechanism of action of adavosertib. Indeed, immunohistochemistry analyses showed a reduction of phospho-CDK1 (Tyr15) and an increase of DNA damages (phospho-γH2AX) in cancer cells [156, 157].
The phase II studies confirmed that adavosertib sensitizes cancer patients to different chemotherapy agents. Interestingly, adavosertib showed efficacy when combined with carboplatin in TP53-mutated ovarian cancer patients, refractory or resistant to first-line platinum-based chemotherapy [159]. Similar results were reported in platinum-resistant primary ovarian cancer patients after treatment with the combination of adavosertib and a single chemotherapeutic agent (carboplatin, paclitaxel, gemcitabine, or pegylated liposomal doxorubicin) [160].
Primary resistance and predictive markers of response to WEE1/PKMYT1 based therapies
Several DDR inhibitors have proved their efficacy against different cancer types in the preclinical and clinical settings [161–165]. Among them, WEE1 inhibitor seems to be the most effective ones, also favored by a relative low off-target toxicity. However, despite the number of studies and the promising results, few predictive markers of response have been identified. Recently, cyclin E level has been linked to the efficacy of adavosertib in breast cancer models [89], with cyclin E-high cells, that generally show elevated chromosome instability, being more sensitive compared with cyclin E-low ones. Despite the reported low levels of WEE1 expression in breast cancer, chromosome instability, that has also prognostic potential mainly in grade 2 tumors [89], may explain the effectiveness of WEE1 inhibitors, as supported by the predictive role of cyclin E. Our group and others showed that high PKMYT1 expression associates with reduced sensitivity to adavosertib, indicating a potential compensatory effect [35, 166]. Moreover, high-throughput proteomic profiling demonstrated that small cell lung cancer and ovarian cancer models with primary resistance to adavosertib express high levels of AKT/mTOR pathway molecules and phosphorylated S6 ribosomal protein [137, 138]. In acute leukemia models, the sensitivity to adavosertib has been recently linked to HDAC and MYC regulation. Indeed, by generating adavosertib-resistant models, the researchers found that resistant acute leukemia cell lines are dependent on increased HDAC activity for their survival, partly due to increased KDM5A function. In addition, gene expression analyses demonstrated a HDAC-dependent expression of MYC in the adavosertib-resistant cell lines [167]. These observations support the success of preclinical studies combining WEE1 and HDAC [41, 130, 131] or bromodomain inhibitors [150].
Conclusion
Thanks to a constantly growing amount of preclinical and clinical data, our knowledge on cancer biology is increasing and, consequently, the list of cancer hallmarks has been progressively expanding. Recent findings demonstrated that cancer cells are characterized by functional and molecular alterations in crucial genes involved in the DDR pathway, which is fundamental for cell cycle regulation, DNA damages recognition, and repair. Functional alterations of DDR-gene have a deep impact on tumor progression and on the clinical outcome of cancer patients. Indeed, the efficacy of standard of care chemo/radiotherapy regimens depends on the generation of DNA damages in proliferating malignant cells. In this scenario, the overexpression or uncontrolled activation of DDR pathways has been showed to protect cancer cells from the therapeutic effect of DNA damaging agents. Moreover, a large number of preclinical studies highlighted that cancer cells depend on the functionality of DDR pathways in order to survive, to tolerate the replicative stress induced by the high proliferative rate and to sustain the intrinsic genetic instability. For these reasons, selective inhibitors have been developed in order to exploit cancer cells’ dependency on DDR-gene functionality. Pre-clinical data has proven the efficacy of DDR inhibition in different kinds of hematological and solid tumors, both as monotherapy and in combination with a wide number of DNA damaging agents. Among DDR inhibitors, the most effective once are those targeting PARP1 and WEE1 family kinases. The effectiveness of PARP1 inhibitors is however dependent on homologous recombination (HR) repair deficiency while WEE1 family kinases inhibitors seems to have a widespread efficacy independently from a specific the genetic background. Indeed, cancer cells seem to be strictly dependent on the functionality of WEE1/PKMYT1 kinases to survive, especially those with alterations targeting the G1 checkpoint. WEE1/PKMYT1 kinases are involved in different biological processes and they seem to play diverse roles in nonmalignant and in cancer cells. Indeed, they control cell cycle regulation and genetic stability in nonmalignant cells and for these reasons act as tumor suppressor genes. Conversely, their ability of promote DNA damages repair and cell cycle control makes them act as pseudo-oncogenes in cancer cells. Several molecular studies showed that malignant cells have high expression level of WEE1 and PKMYT1, which has become a good prognostic biomarker for chemo/radiotherapy regimens. However, we currently lack information regarding predictive markers of response to WEE1/PKMYT1 inhibitors. Large preclinical and clinical studies should be conducted in order to identify specific molecular backgrounds in which the use of WEE1/PKYMT1 inhibitors may be recommended. The identification of molecular vulnerabilities in cancer patients will be fundamental to design novel therapeutic regimens using WEE1/PKMYT1 inhibitors in a chemo/radiotherapy-free, synthetic lethality-based approach.
Acknowledgements
Not applicable.
Abbreviations
- ALL
Acute lymphoblastic leukemia
- AML
Acute myeloid leukemia
- BC
Breast cancer
- C
Chemotherapy
- CC
Colorectal cancer
- CN
Copy number
- CNA
Copy number alteration
- CLL
Chronic lymphocyte leukemia
- CML
Chronic myeloid leukemia
- DDR
DNA damage response
- DLBCL
Diffuse large B cell lymphoma
- ES
Esophageal cancer
- GC
Gastric cancer
- GL
Gliomas
- GLB
Glioblastoma
- HC
Hepatocellular carcinoma
- HR
Homologous recombination
- HNSCC
Head and neck squamous cell carcinoma
- LC
Lung cancer
- M
Monotherapy
- MaM
Malignant melanoma
- MCL
Mantle cell lymphoma
- MM
Multiple myeloma
- MPF
Mitotic promoting factor
- N
Neuroblastoma
- NSCLC
Non-small-cell lung cancer
- OC
Ovarian cancer
- PC
Pancreatic cancer
- R
Radiotherapy
- S
Sarcomas
- SAC
Spindle assembly checkpoint
- TNBC
Triple-negative breast cancer
Authors’ contributions
AGLDR, CC, and GS drafted the first version of the manuscript and created the figures. GM critically revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Funding
This work was supported by ERA-Per-Med (reference number: ERAPERMED2018-275).
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors read and approved the final manuscript.
Competing interests
GM has competing interests with Novartis, BMS, Roche, Pfizer, ARIAD, and MSD. The other authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Schmidt M, Rohe A, Platzer C, et al. Regulation of G2/M transition by inhibition of WEE1 and PKMYT1 Kinases. Molecules. 2017;22:2045. doi: 10.3390/molecules22122045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solc P, Schultz RM, Motlik J. Prophase I arrest and progression to metaphase I in mouse oocytes: Comparison of resumption of meiosis and recovery from G2-arrest in somatic cells. Mol Hum Reprod. 2010;16:654–664. doi: 10.1093/molehr/gaq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakanishi M, Ando H, Watanabe N, et al. Identification and characterization of human Wee1B, a new member of the Wee1 family of Cdk-inhibitory kinases. Genes Cells. 2000;5(10):839–847. doi: 10.1046/j.1365-2443.2000.00367.x. [DOI] [PubMed] [Google Scholar]
- 4.Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: A membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science. 1995;270(5233):86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- 5.Booher RN, Holman PS, Fattaey A. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J Biol Chem. 1997;272(35):22300–22306. doi: 10.1074/jbc.272.35.22300. [DOI] [PubMed] [Google Scholar]
- 6.Liu F, Stanton JJ, Wu Z, Piwnica-Worms H. The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol Cell Biol. 1997;17(2):571–583. doi: 10.1128/MCB.17.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakajima H, Yonemura S, Murata M, et al. Myt1 protein kinase is essential for Golgi and ER assembly during mitotic exit. J Cell Biol. 2008;181(1):89–103. doi: 10.1083/jcb.200708176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow JPH, Poon RYC, Ma HT. Inhibitory phosphorylation of cyclin-dependent kinase 1 as a compensatory mechanism for mitosis exit. Mol Cell Biol. 2011;31(7):1478–1491. doi: 10.1128/MCB.00891-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solomon MJ, Harper JW, Shuttleworth J. CAK, the p34cdc2 activating kinase, contains a protein identical or closely related to p40MO15. EMBO J. 1993;12(8):3133–3142. doi: 10.1002/j.1460-2075.1993.tb05982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lolli G, Johnson LN. CAK-Cyclin-dependent activating kinase: a key kinase in cell cycle control and a target for Drugs? Cell Cycle. 2005;4(4):572–577. doi: 10.4161/cc.4.4.1607. [DOI] [PubMed] [Google Scholar]
- 11.Walsh S, Margolis SS, Kornbluth S. Phosphorylation of the cyclin B1 cytoplasmic retention sequence by mitogen-activated protein kinase and Plx. Mol Cancer Res. 2003;1(4):280–289. [PubMed] [Google Scholar]
- 12.Szmyd R, Niska-Blakie J, Diril MK, et al. Premature activation of Cdk1 leads to mitotic events in S phase and embryonic lethality. Oncogene. 2019;38(7):998–1018. doi: 10.1038/s41388-018-0464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe N, Arai H, Nishihara Y, et al. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A. 2004;101(13):4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toyoshima-Morimoto F, Taniguchi E, Shinya N, et al. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature. 2001;410(6825):215–220. doi: 10.1038/35065617. [DOI] [PubMed] [Google Scholar]
- 15.Van Vugt MATM, Brás A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. 2004;15(5):799–811. doi: 10.1016/j.molcel.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 16.Nakojima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E. Identification of a consensus motif for PlK (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem. 2003;278(28):25277–25280. doi: 10.1074/jbc.C300126200. [DOI] [PubMed] [Google Scholar]
- 17.Takisawa H, Mimura S, Kubota Y. Eukaryotic DNA replication: from pre-replication complex to initiation complex. Curr Opin Cell Biol. 2000;12(6):690–696. doi: 10.1016/S0955-0674(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 18.Heller RC, Kang S, Lam WM, et al. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell. 2011;146(1):80–91. doi: 10.1016/j.cell.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labib K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 2010;24(12):1208–1219. doi: 10.1101/gad.1933010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu Y, Rosenblatt J, Morgan DO. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992;11(11):3995–4005. doi: 10.1002/j.1460-2075.1992.tb05493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vassilopoulos A, Tominaga Y, Kim HS, et al. WEE1 murine deficiency induces hyper-activation of APC/C and results in genomic instability and carcinogenesis. Oncogene. 2015;34(23):3023–3035. doi: 10.1038/onc.2014.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghelli Luserna Di Rorà A, Martinelli G, Simonetti G. The balance between mitotic death and mitotic slippage in acute leukemia: a new therapeutic window? J Hematol Oncol. 2019;12(1):123. doi: 10.1186/s13045-019-0808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Visconti R, Grieco D. Fighting tubulin-targeting anticancer drug toxicity and resistance. Endocr Relat Cancer. 2017;24(9):T107–T117. doi: 10.1530/ERC-17-0120. [DOI] [PubMed] [Google Scholar]
- 24.Visconti R, Palazzo L, Della Monica R, Grieco D. Fcp1-dependent dephosphorylation is required for M-phase-promoting factor inactivation at mitosis exit. Nat Commun. 2012;3:894. doi: 10.1038/ncomms1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8(5):379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 26.Visconti R, Palazzo L, Pepe A, et al. The end of mitosis from a phosphatase perspective. Cell Cycle. 2013;12(1):17–19. doi: 10.4161/cc.22875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martín Y, Domínguez-Kelly R, Freire R. Novel insights into maintaining genomic integrity: Wee1 regulating Mus81/Eme1. Cell Div. 2011;6:21. doi: 10.1186/1747-1028-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domínguez-Kelly R, Martín Y, Koundrioukoff S, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194(4):567–579. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duda H, Arter M, Gloggnitzer J, et al. A mechanism for controlled breakage of under-replicated chromosomes during mitosis. Dev Cell. 2016;39(6):740–755. doi: 10.1016/j.devcel.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 30.Asquith CRM, Laitinen T, East MP. PKMYT1: a forgotten member of the WEE1 family. Nat Rev Drug Discov. 2020;19(3):157. doi: 10.1038/d41573-019-00202-9. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Qi J, Dou Z, et al. Systematic expression analysis of WEE family kinases reveals the importance of PKMYT1 in breast carcinogenesis. Cell Prolif. 2020;53(2):e12741. doi: 10.1111/cpr.12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong D, Kim H, Kim D, et al. Protein kinase, membrane-associated tyrosine/threonine 1 is associated with the progression of colorectal cancer. Oncol Rep. 2018;39(6):2829–2836. doi: 10.3892/or.2018.6371. [DOI] [PubMed] [Google Scholar]
- 33.Lal S, Cozzitorto JA, Blanco F, et al. 988 Sequence alterations in the WEE1 non-coding region is a facilitator and marker for pancreatic tumorigenesis. Gastroenterology. 2014;S-1034.
- 34.Ciriello G, Miller ML, Aksoy BA, et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghelli Luserna Di Rorà A, Beeharry N, Imbrogno E, et al. Targeting WEE1 to enhance conventional therapies for acute lymphoblastic leukemia. J Hematol Oncol. 2018;11(1):99. doi: 10.1186/s13045-018-0641-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barbosa RSS, Dantonio PM, Guimarães T, et al. Sequential combination of bortezomib and WEE1 inhibitor, MK-1775, induced apoptosis in multiple myeloma cell lines. Biochem Biophys Res Commun. 2019;519(3):597–604. doi: 10.1016/j.bbrc.2019.08.163. [DOI] [PubMed] [Google Scholar]
- 37.Van Linden AA, Baturin D, Ford JB, et al. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther. 2013;12(12):2675–2684. doi: 10.1158/1535-7163.MCT-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Porter CC, Kim J, Fosmire S, et al. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia. 2012;26(6):1266–1276. doi: 10.1038/leu.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnston HE, Carter MJ, Larrayoz M, et al. Proteomics profiling of CLL versus healthy B-cells identifies putative therapeutic targets and a subtype-independent signature of spliceosome dysregulation. Mol Cell Proteomics. 2018;17(4):776–791. doi: 10.1074/mcp.RA117.000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neben K, Schnittger S, Brors B, et al. Distinct gene expression patterns associated with FLT3- and NRAS-activating mutations in acute myeloid leukemia with normal karyotype. Oncogene. 2005;24(9):1580–1588. doi: 10.1038/sj.onc.1208344. [DOI] [PubMed] [Google Scholar]
- 41.Zhou L, Zhang Y, Chen S, et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia. 2015;29(4):807–818. doi: 10.1038/leu.2014.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan J, Li L, Small D, Rassool F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: implications for genomic instability and therapy. Blood. 2010;116(24):5298–5305. doi: 10.1182/blood-2010-03-272591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Jong MRW, Visser L, Huls G, et al. Identification of relevant drugable targets in diffuse large B-cell lymphoma using a genome-wide unbiased CD20 guilt-by association approach. PLoS One. 2018;13(2):e0193098. doi: 10.1371/journal.pone.0193098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolomsky A, Gruber F, Stangelberger K, et al. Preclinical validation studies support causal machine learning based identification of novel drug targets for high-risk multiple myeloma. Blood. 2018;132(Supplement 1):3210. doi: 10.1182/blood-2018-99-117886. [DOI] [Google Scholar]
- 45.Sun QS, Luo M, Zhao HM, Sun H. Overexpression of PKMYT1 indicates the poor prognosis and enhances proliferation and tumorigenesis in non-small cell lung cancer via activation of Notch signal pathway. Eur Rev Med Pharmacol Sci. 2019;23(10):4210–4219. doi: 10.26355/eurrev_201905_17925. [DOI] [PubMed] [Google Scholar]
- 46.Liu L, Wu J, Wang S, et al. PKMYT1 promoted the growth and motility of hepatocellular carcinoma cells by activating beta-catenin/TCF signaling. Exp Cell Res. 2017;358(2):209–216. doi: 10.1016/j.yexcr.2017.06.014. [DOI] [PubMed] [Google Scholar]
- 47.Wang XM, Li QY, Ren LL, et al. Effects of MCRS1 on proliferation, migration, invasion, and epithelial mesenchymal transition of gastric cancer cells by interacting with Pkmyt1 protein kinase. Cell Signal. 2019;59:171–181. doi: 10.1016/j.cellsig.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Kim HY, Cho Y, Kang HG, et al. Targeting the WEE1 kinase as a molecular targeted therapy for gastric cancer. Oncotarget. 2016;7(31):49902–49916. doi: 10.18632/oncotarget.10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magnussen GI, Holm R, Emilsen E, et al. High expression of Wee1 is associated with poor disease-free survival in Malignant Melanoma: Potential for targeted therapy. PLoS One. 2012;7(6):e38254. doi: 10.1371/journal.pone.0038254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mueller S, Hashizume R, Yang X, et al. Targeting wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014;16(3):352–360. doi: 10.1093/neuonc/not220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mir SE, De Witt Hamer PC, Krawczyk PM, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18(3):244–257. doi: 10.1016/j.ccr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chayka O, D’Acunto CW, Middleton O, et al. Identification and pharmacological inactivation of the MYCN gene network as a therapeutic strategy for neuroblastic tumor cells. J Biol Chem. 2015;290(4):2198–2212. doi: 10.1074/jbc.M114.624056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q, Zhao X, Zhang C, et al. Overexpressed PKMYT1 promotes tumor progression and associates with poor survival in esophageal squamous cell carcinoma. Cancer Manag Res. 2019;11:7813–7824. doi: 10.2147/CMAR.S214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tibes R, Bogenberger JM, Chaudhuri L, et al. RNAi screening of the kinome with cytarabine in leukemias. Blood. 2012;119(12):2863–2872. doi: 10.1182/blood-2011-07-367557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simonetti G, Padella A, do Valle IF, et al. Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery. Cancer. 2018;125:1–14. [DOI] [PMC free article] [PubMed]
- 56.Caretti V, Hiddingh L, Lagerweij T, et al. WEE1 kinase inhibition enhances the radiation response of diffuse intrinsic pontine gliomas. Mol Cancer Ther. 2013;12(2):141–150. doi: 10.1158/1535-7163.MCT-12-0735. [DOI] [PubMed] [Google Scholar]
- 57.Music D, Dahlrot RH, Hermansen SK, et al. Expression and prognostic value of the WEE1 kinase in gliomas. J Neurooncol. 2016;127(2):381–389. doi: 10.1007/s11060-015-2050-4. [DOI] [PubMed] [Google Scholar]
- 58.Egeland EV, Flatmark K, Nesland JM, et al. Expression and clinical significance of Wee1 in colorectal cancer. Tumor Biol. 2016;37(9):12133–12140. doi: 10.1007/s13277-016-5081-3. [DOI] [PubMed] [Google Scholar]
- 59.Slipicevic A, Holth A, Hellesylt E, et al. Wee1 is a novel independent prognostic marker of poor survival in post-chemotherapy ovarian carcinoma effusions. Gynecol Oncol. 2014;135(1):118–124. doi: 10.1016/j.ygyno.2014.07.102. [DOI] [PubMed] [Google Scholar]
- 60.Shu C, Wang Q, Yan X, Wang J. Whole-genome expression microarray combined with machine learning to identify prognostic biomarkers for high-grade glioma. J Mol Neurosci. 2018;64(4):491–500. doi: 10.1007/s12031-018-1049-7. [DOI] [PubMed] [Google Scholar]
- 61.Novak EM, Halley NS, Gimenez TM, et al. BLM germline and somatic PKMYT1 and AHCY mutations: genetic variations beyond MYCN and prognosis in neuroblastoma. Med Hypotheses. 2016;97:22–25. doi: 10.1016/j.mehy.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 62.Ku BM, Bae Y-H, Koh J, et al. Mutational status of TP53 defines the efficacy of Wee1 inhibitor AZD1775 in KRAS -mutant non-small cell lung cancer. Oncotarget. 2017;8(40):67526–67537. doi: 10.18632/oncotarget.18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bauman JE, Chung CH. CHK it out! Blocking WEE kinase routs TP53 mutant cancer. Clin Cancer Res. 2014;20(16):4173–4175. doi: 10.1158/1078-0432.CCR-14-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kreahling JM, Foroutan P, Reed D, et al. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS One. 2013;8(3):e57523. doi: 10.1371/journal.pone.0057523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ford JB, Baturin D, Burleson TM, et al. AZD1775 sensitizes T cell acute lymphoblastic leukemia cells to cytarabine by promoting apoptosis over DNA repair. Oncotarget. 2015;6(29):28001–28010. doi: 10.18632/oncotarget.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Webster PJ, Littlejohns AT, Gaunt HJ, et al. AZD1775 induces toxicity through double-stranded DNA breaks independently of chemotherapeutic agents in p53-mutated colorectal cancer cells. Cell Cycle. 2017;16(22):2176–2182. doi: 10.1080/15384101.2017.1301329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kahen E, Yu D, Harrison DJ, et al. Identification of clinically achievable combination therapies in childhood rhabdomyosarcoma. Cancer Chemother Pharmacol. 2016;78(2):313–323. doi: 10.1007/s00280-016-3077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee YY, Cho YJ, Shin SW, et al. Anti-tumor effects of Wee1 kinase inhibitor with radiotherapy in human cervical cancer. Sci Rep. 2019;9(1):15394. doi: 10.1038/s41598-019-51959-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.PosthumaDeBoer J, Würdinger T, Graat HCA, et al. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer. 2011;11(1):156. doi: 10.1186/1471-2407-11-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu H, Krystal GW. Actinomycin D decreases Mcl-1 expression and acts synergistically with ABT-737 against small cell lung cancer cell lines. Clin Cancer Res. 2010;16(17):4392–4400. doi: 10.1158/1078-0432.CCR-10-0640. [DOI] [PubMed] [Google Scholar]
- 71.Hayashi Y, Fujimura A, Kato K, et al. Nucleolar integrity during interphase supports faithful Cdk1 activation and mitotic entry. Sci Adv. 2018;4(6):eaap7777. doi: 10.1126/sciadv.aap7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alexander VM, Roy M, Steffens KA, et al. Azacytidine induces cell cycle arrest and suppression of neuroendocrine markers in carcinoids. Int J Clin Exp Med. 2010;3(2):95–102. [PMC free article] [PubMed] [Google Scholar]
- 73.Uchida R, Yokota S, Matsuda D, et al. Habiterpenol, a novel abrogator of bleomycin-induced G2 arrest in Jurkat cells, produced by Phytohabitans suffuscus 3787-5. J Antibiot (Tokyo) 2014;67(11):777–781. doi: 10.1038/ja.2014.62. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Z, Zhang H, Hu Z, et al. Synergy of 1,25-dihydroxyvitamin D3 and carboplatin in growth suppression of SKOV-3 cells. Oncol Lett. 2014;8(3):1348–1354. doi: 10.3892/ol.2014.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sarin N, Engel F, Kalayda GV, et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS One. 2017;12(7):e0181081. doi: 10.1371/journal.pone.0181081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen D, Lin X, Gao J, et al. Wee1 Inhibitor AZD1775 Combined with cisplatin potentiates anticancer activity against gastric cancer by increasing DNA damage and cell apoptosis. Biomed Res Int. 2018;2018:5813292. doi: 10.1155/2018/5813292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng H, Shao F, Martin S, et al. WEE1 inhibition targets cell cycle checkpoints for triple negative breast cancers to overcome cisplatin resistance. Sci Rep. 2017;7:43517. doi: 10.1038/srep43517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Jong MRW, Langendonk M, Reitsma B, et al. WEE1 inhibition synergizes with CHOP chemotherapy and radiation therapy through induction of premature mitotic entry and DNA damage in diffuse large B-cell lymphoma. Ther Adv Hematol. 2020;11:2040620719898373. doi: 10.1177/2040620719898373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma J, Li X, Su Y, et al. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017;7:41950. doi: 10.1038/srep41950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi Z, Azuma A, Sampath D, et al. S-phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine. Cancer Res. 2001;61(3):1065–1072. [PubMed] [Google Scholar]
- 81.Garcia TB, Fosmire SP, Porter CC. Increased activity of both CDK1 and CDK2 is necessary for the combinatorial activity of WEE1 inhibition and cytarabine. Leuk Res. 2018;64:30–33. doi: 10.1016/j.leukres.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Al-Aamri HM, Ku H, Irving HR, et al. Time dependent response of daunorubicin on cytotoxicity, cell cycle and DNA repair in acute lymphoblastic leukaemia. BMC Cancer. 2019;19(1):179. doi: 10.1186/s12885-019-5377-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shang D, Han T, Xu X, Liu Y. Decitabine induces G2/M cell cycle arrest by suppressing p38/NF-κB signaling in human renal clear cell carcinoma. Int J Clin Exp Pathol. 2015;8(9):11140–11148. [PMC free article] [PubMed] [Google Scholar]
- 84.Singh SK, Banerjee S, Acosta EP, et al. Resveratrol induces cell cycle arrest and apoptosis with docetaxel in prostate cancer cells via a p53/p21WAF1/CIP1 and p27KIP1 pathway. Oncotarget. 2017;8(10):17216–17228. doi: 10.18632/oncotarget.15303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morse DL, Gray H, Payne CM, Gillies RJ. Docetaxel induces cell death through mitotic catastrophe in human breast cancer cells. Mol Cancer Ther. 2005;4(10):1495–1504. doi: 10.1158/1535-7163.MCT-05-0130. [DOI] [PubMed] [Google Scholar]
- 86.Vera J, Raatz Y, Wolkenhauer O, et al. Chk1 and Wee1 control genotoxic-stress induced G2-M arrest in melanoma cells. Cell Signal. 2015;27(5):951–960. doi: 10.1016/j.cellsig.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 87.Wu CL, Ping SY, Yu CP, Yu DS. Tyrosine kinase receptor inhibitor-targeted combined chemotherapy for metastatic bladder cancer. Kaohsiung J Med Sci. 2012;28(4):194–203. doi: 10.1016/j.kjms.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 88.Senthebane DA, Jonker T, Rowe A, et al. The role of tumor microenvironment in chemoresistance: 3D extracellular matrices as accomplices. Int J Mol Sci. 2018;19(10):2861. doi: 10.3390/ijms19102861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen X, Low KH, Alexander A, et al. Cyclin E overexpression sensitizes triple-negative breast cancer to Wee1 kinase inhibition. Clin Cancer Res. 2018;24(24):6594–6610. doi: 10.1158/1078-0432.CCR-18-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Seung HL, Seung MS, Dong JS, et al. Epothilones induce human colon cancer SW620 cell apoptosis via the tubulin polymerization-independent activation of the nuclear factor-κB/IκB kinase signal pathway. Mol Cancer Ther. 2007;6(10):2786–2797. doi: 10.1158/1535-7163.MCT-07-0002. [DOI] [PubMed] [Google Scholar]
- 91.Zhang R, Zhu L, Zhang L, et al. PTEN enhances G2/M arrest in etoposide-treated MCF-7 cells through activation of the ATM pathway. Oncol Rep. 2016;35(5):2707–2714. doi: 10.3892/or.2016.4674. [DOI] [PubMed] [Google Scholar]
- 92.Pitts TM, Simmons DM, Bagby SM, et al. Wee1 inhibition enhances the anti-tumor effects of capecitabine in preclinical models of triple-negative breast cancer. Cancers. 2020;12(3):719. doi: 10.3390/cancers12030719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17(9):2799–2806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koh SB, Wallez Y, Dunlop CR, et al. Mechanistic distinctions between CHK1 and WEE1 inhibition guide the scheduling of triple therapy with gemcitabine. Cancer Res. 2018;78(11):3054–3066. doi: 10.1158/0008-5472.CAN-17-3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xu B, Sun Z, Liu Z, et al. Replication stress induces micronuclei comprising of aggregated DNA double-strand breaks. PLoS One. 2011;6(4):e18618. doi: 10.1371/journal.pone.0018618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aarts M, Sharpe R, Garcia-Murillas I, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2(6):524–539. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 97.Morgan MA, Onono FO, Spielmann HP, et al. Modulation of anthracycline-induced cytotoxicity by targeting the prenylated proteome in myeloid leukemia cells. J Mol Med. 2012;90(2):149–161. doi: 10.1007/s00109-011-0814-7. [DOI] [PubMed] [Google Scholar]
- 98.Subhash VV, Tan SH, Yeo MS, et al. ATM expression predicts veliparib and irinotecan sensitivity in gastric cancer by mediating p53-independent regulation of cell cycle and apoptosis. Mol Cancer Ther. 2016;15(12):3087–3096. doi: 10.1158/1535-7163.MCT-15-1002. [DOI] [PubMed] [Google Scholar]
- 99.Yin Y, Shen Q, Tao R, et al. Wee1 inhibition can suppress tumor proliferation and sensitize p53 mutant colonic cancer cells to the anticancer effect of irinotecan. Mol Med Rep. 2018;17(2):3344–3349. doi: 10.3892/mmr.2017.8230. [DOI] [PubMed] [Google Scholar]
- 100.Jan YH, Heck DE, Laskin DL, Laskin JD. Sulfur mustard analog mechlorethamine (Bis(2-chloroethyl)methylamine) modulates cell cycle progression via the DNA damage response in human lung epithelial A549 cells. Chem Res Toxicol. 2019;32(6):1123–1133. doi: 10.1021/acs.chemrestox.8b00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mahbub A, Le Maitre C, Haywood-Small S, et al. Dietary polyphenols influence antimetabolite agents: Methotrexate, 6-mercaptopurine and 5-fluorouracil in leukemia cell lines. Oncotarget. 2017;8(62):104877–104893. doi: 10.18632/oncotarget.20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Costantini DL, Villani DF, Vallis KA, Reilly RM. Methotrexate, paclitaxel, and doxorubicin radiosensitize HER2-amplified human breast cancer cells to the auger electron-emitting radiotherapeutic agent 111In-NLS-trastuzumab. J Nucl Med. 2010;51(3):477–483. doi: 10.2967/jnumed.109.069716. [DOI] [PubMed] [Google Scholar]
- 103.Di Rorà AGL, Bocconcelli M, Ferrari A, et al. Synergism through WEE1 and CHK1 inhibition in acute lymphoblastic leukemia. Cancers (Basel) 2019;11(11):1654. doi: 10.3390/cancers11111654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guerriero E, Sorice A, Capone F, et al. Vitamin C effect on mitoxantrone-induced cytotoxicity in human breast cancer cell lines. PLoS One. 2014;9(12):e115287. doi: 10.1371/journal.pone.0115287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Voland C, Bord A, Péleraux A, et al. Repression of cell cycle-related proteins by oxaliplatin but not cisplatin in human colon cancer cells. Mol Cancer Ther. 2006;5(9):2149–2157. doi: 10.1158/1535-7163.MCT-05-0212. [DOI] [PubMed] [Google Scholar]
- 106.Lal S, Zarei M, Chand SN, et al. WEE1 inhibition in pancreatic cancer cells is dependent on DNA repair status in a context dependent manner. Sci Rep. 2016;6:33323. doi: 10.1038/srep33323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.George J, Banik NL, Ray SK. Molecular mechanisms of taxol for induction of cell death in glioblastomas. In: Ray S, editor. Glioblastoma. New York: Springer; 2010. 10.1007/978-1-4419-0410-2_14.
- 108.Lewis CW, Jin Z, Macdonald D, et al. Prolonged mitotic arrest induced by Wee1 inhibition sensitizes breast cancer cells to paclitaxel. Oncotarget. 2017;8(43):73705–73722. doi: 10.18632/oncotarget.17848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen KC, Yang TY, Wu CC, et al. Pemetrexed induces S-phase arrest and apoptosis via a deregulated activation of Akt signaling pathway. PLoS One. 2014;9(5):e97888. doi: 10.1371/journal.pone.0097888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hirai H, Arai T, Okada M, et al. MK-1775, a small molecule Wee1 inhibitor, enhances antitumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9(7):514–522. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 111.Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci. 2016;17(1):102. doi: 10.3390/ijms17010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li J, Chen W, Zhang P, Li N. Topoisomerase II trapping agent teniposide induces apoptosis and G2/M or S phase arrest of oral squamous cell carcinoma. World J Surg Oncol. 2006;4:41. doi: 10.1186/1477-7819-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wotring LL, Roti Roti JL. Thioguanine-induced S and G2 blocks and their significance to the mechanism of cytotoxicity. Cancer Res. 1980;40(5):1458–1462. [PubMed] [Google Scholar]
- 114.Nguyen D, Zajac-Kaye M, Rubinstein L, et al. Poly(ADP-ribose) polymerase inhibition enhances p53-dependent and -independent DNA damage responses induced by DNA damaging agent. Cell Cycle. 2011;10(23):4074–4082. doi: 10.4161/cc.10.23.18170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shumway SD, Kubica JL, Guertin AD, et al. Abstract 2969: a Wee1 kinase inhibitor, MK-1775, sensitizes cervical carcinoma cell lines to cisplatin and topotecan. Cancer Res. 2011;71(8 Supplement):2969. [Google Scholar]
- 116.Brandl MB, Pasquier E, Li F, et al. Computational analysis of image-based drug profiling predicts synergistic drug combinations: applications in triple-negative breast cancer. Mol Oncol. 2014;8(8):1548–1560. doi: 10.1016/j.molonc.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tu Y, Cheng S, Zhang S, et al. Vincristine induces cell cycle arrest and apoptosis in SH-SY5Y human neuroblastoma cells. Int J Mol Med. 2013;31(1):113–119. doi: 10.3892/ijmm.2012.1167. [DOI] [PubMed] [Google Scholar]
- 118.Visconti R, Della Monica R, Palazzo L, et al. The Fcp1-Wee1-Cdk1 axis affects spindle assembly checkpoint robustness and sensitivity to antimicrotubule cancer drugs. Cell Death Differ. 2015;22(9):1551–1560. doi: 10.1038/cdd.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhu JY, Cuellar RA, Berndt N, et al. Structural basis of Wee kinases functionality and inactivation by diverse small molecule inhibitors. J Med Chem. 2017;60(18):7863–7875. doi: 10.1021/acs.jmedchem.7b00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Restelli V, Chilà R, Lupi M, et al. Characterization of a mantle cell lymphoma cell line resistant to the Chk1 inhibitor PF-00477736. Oncotarget. 2015;6(35):37229–37240. doi: 10.18632/oncotarget.5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Qi W, Xie C, Li C, et al. CHK1 plays a critical role in the anti-leukemic activity of the wee1 inhibitor MK-1775 in acute myeloid leukemia cells. J Hematol Oncol. 2014;7(1):53. doi: 10.1186/s13045-014-0053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Young LA, O’Connor LO, de Renty C, et al. Differential activity of ATR and Wee1 inhibitors in a highly sensitive subpopulation of DLBCL linked to replication stress. Cancer Res. 2019;79(14):3762–3775. doi: 10.1158/0008-5472.CAN-18-2480. [DOI] [PubMed] [Google Scholar]
- 123.Bridges KA, Hirai H, Buser CA, et al. MK-1775, a novel wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17(17):5638–5648. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ma H, Takahashi A, Sejimo Y, et al. Targeting of carbon ion-induced G2 checkpoint activation in lung cancer cells using Wee-1 inhibitor MK-1775. Radiat Res. 2016;185(2):e52. doi: 10.1667/RR4171. [DOI] [PubMed] [Google Scholar]
- 125.Lindenblatt D, Terraneo N, Pellegrini G, et al. Combination of lutetium-177 labelled anti-L1CAM antibody chCE7 with the clinically relevant protein kinase inhibitor MK1775: a novel combination against human ovarian carcinoma. BMC Cancer. 2018;18(1):922. doi: 10.1186/s12885-018-4836-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Parsels LA, Karnak D, Parsels JD, et al. PARP1 Trapping and DNA replication stress enhance radiosensitization with combined WEE1 and PARP inhibitors. Mol Cancer Res. 2018;16(2):222–232. doi: 10.1158/1541-7786.MCR-17-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Caldwell JT, Edwards H, Buck SA, et al. Targeting the wee1 kinase for treatment of pediatric Down syndrome acute myeloid leukemia. Pediatr Blood Cancer. 2014;61(10):1767–1773. doi: 10.1002/pbc.25081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xing L, Lin L, Yu T, et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia. 2020. 10.1038/s41375-020-0745-9. [DOI] [PMC free article] [PubMed]
- 129.Tibes R, Ferreira Coutinho D, Tuen MT, et al. DNA damage repair interference By WEE1 inhibition with AZD1775 overcomes combined azacitidine and Venetoclax resistance in acute myeloid leukmeia (AML) Blood. 2019;134(Supplement_1):2559. doi: 10.1182/blood-2019-130931. [DOI] [Google Scholar]
- 130.Qi W, Zhang W, Edwards H, et al. Synergistic anti-leukemic interactions between panobinostat and MK-1775 in acute myeloid leukemia ex vivo. Cancer Biol Ther. 2015;16(12):1784–1793. doi: 10.1080/15384047.2015.1095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tanaka N, Patel AA, Tang L, et al. Replication stress leading to apoptosis within the S-phase contributes to synergism between vorinostat and AZD1775 in HNSCC harboring high-risk TP53 mutation. Clin Cancer Res. 2017;23(21):6541–6554. doi: 10.1158/1078-0432.CCR-17-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Qi W, Xu X, Wang M, et al. Inhibition of Wee1 sensitizes AML cells to ATR inhibitor VE-822-induced DNA damage and apoptosis. Biochem Pharmacol. 2019;164:273–282. doi: 10.1016/j.bcp.2019.04.022. [DOI] [PubMed] [Google Scholar]
- 133.Restelli V, Lupi M, Chila R, et al. DNA damage response inhibitor combinations exert synergistic antitumor activity in aggressive B-cell lymphomas. Mol Cancer Ther. 2019;18(7):1255–1264. doi: 10.1158/1535-7163.MCT-18-0919. [DOI] [PubMed] [Google Scholar]
- 134.Bukhari AB, Lewis CW, Pearce JJ, et al. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J Clin Invest. 2019;129(3):1329–1344. doi: 10.1172/JCI122622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jin J, Fang H, Yang F, et al. Combined inhibition of ATR and WEE1 as a novel therapeutic strategy in triple-negative breast cancer. Neoplasia (United States) 2018;20(5):478–488. doi: 10.1016/j.neo.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weisberg E, Nonami A, Chen Z, et al. Identification of Wee1 as a novel therapeutic target for mutant RAS-driven acute leukemia and other malignancies. Leukemia. 2014;29(1):27–37. doi: 10.1038/leu.2014.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li F, Guo E, Huang J, et al. mTOR inhibition overcomes primary and acquired resistance to Wee1 inhibition by augmenting replication stress in epithelial ovarian cancers. Am J Cancer Res. 2020;10(3):908–924. [PMC free article] [PubMed] [Google Scholar]
- 138.Sen T, Tong P, Diao L, et al. Targeting AXL and mTOR pathway overcomes primary and acquired resistance to WEE1 inhibition in small-cell lung cancer. Clin Cancer Res. 2017;23(20):6239–6254. doi: 10.1158/1078-0432.CCR-17-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hai J, Liu S, Bufe L, et al. Synergy of WEE1 and mTOR inhibition in mutant KRAS-driven lung cancers. Clin Cancer Res. 2017;23(22):6993–7005. doi: 10.1158/1078-0432.CCR-17-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chila R, Basana A, Lupi M, et al. Combined inhibition of Chk1 and Wee1 as a new therapeutic strategy for mantle cell lymphoma. Oncotarget. 2015;6(5):3394–3408. doi: 10.18632/oncotarget.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Restelli V, Vagni M, Arribas AJ, et al. Inhibition of CHK1 and WEE1 as a new therapeutic approach in diffuse large B cell lymphomas with MYC deregulation. Br J Haematol. 2018;181(1):129–133. doi: 10.1111/bjh.14506. [DOI] [PubMed] [Google Scholar]
- 142.Chaudhuri L, Vincelette ND, Koh BD, et al. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica. 2014;99(4):688–696. doi: 10.3324/haematol.2013.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.De Jong MRW, Langendonk M, Reitsma B, et al. WEE1 inhibition enhances anti-apoptotic dependency as a result of premature mitotic entry and DNA damage. Cancers (Basel) 2019;11(11):1743. doi: 10.3390/cancers11111743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fang Y, McGrail DJ, Sun C, et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35(6):851–867.e7. doi: 10.1016/j.ccell.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lallo A, Frese KK, Morrow CJ, et al. The combination of the PARP inhibitor olaparib and the WEE1 Inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res. 2018;24(20):5153–5164. doi: 10.1158/1078-0432.CCR-17-2805. [DOI] [PubMed] [Google Scholar]
- 146.Garcia TB, Snedeker JC, Baturin D, et al. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol Cancer Ther. 2017;16(10):2058–2068. doi: 10.1158/1535-7163.MCT-16-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lee JW, Parameswaran J, Sandoval-Schaefer T, et al. Combined aurora kinase A (AURKA) and WEE1 inhibition demonstrates synergistic antitumor effect in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2019;25(11):3430–3442. doi: 10.1158/1078-0432.CCR-18-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen G, Zhang B, Xu H, et al. Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene. 2017;36(50):6863–6872. doi: 10.1038/onc.2017.297. [DOI] [PubMed] [Google Scholar]
- 149.Francis AM, Alexander A, Liu Y, et al. CDK4/6 inhibitors sensitize Rb-positive sarcoma cells to Wee1 kinase inhibition through reversible cell-cycle arrest. Mol Cancer Ther. 2017;16(9):1751–1764. doi: 10.1158/1535-7163.MCT-17-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Takashima Y, Kikuchi E, Kikuchi J, et al. Bromodomain and extraterminal domain inhibition synergizes with WEE1-inhibitor AZD1775 effect by impairing nonhomologous end joining and enhancing DNA damage in nonsmall cell lung cancer. Int J Cancer. 2020;146(4):1114–1124. doi: 10.1002/ijc.32515. [DOI] [PubMed] [Google Scholar]
- 151.Panek RL, Lu GH, Klutchko SR, et al. In vitro pharmacological characterization of PD 166285, a new nanomolar potent and broadly active protein tyrosine kinase inhibitor. J Pharmacol Exp Ther. 1997;283(3):1433–1444. [PubMed] [Google Scholar]
- 152.Duan L, Perez RE, Hansen M, et al. Increasing cisplatin sensitivity by scheduledependent inhibition of AKT and Chk1. Cancer Biol Ther. 2014;15(12):1600–1612. doi: 10.4161/15384047.2014.961876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Blackwood E, Epler J, Yen I, et al. Combination drug scheduling defines a “window of opportunity” for chemopotentiation of gemcitabine by an orally bioavailable, selective ChK1 inhibitor, GNE-900. Mol Cancer Ther. 2013;12(10):1968–1980. doi: 10.1158/1535-7163.MCT-12-1218. [DOI] [PubMed] [Google Scholar]
- 154.Levinson NM, Boxer SG. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS One. 2012;7(4):e29828. doi: 10.1371/journal.pone.0029828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Beeharry N, Banina E, Hittle J, et al. Re-purposing clinical kinase inhibitors to enhance chemosensitivity by overriding checkpoints. Cell Cycle. 2014;13(14):2172–2191. doi: 10.4161/cc.29214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mendez E, Rodriguez CP, Kao MC, et al. A phase I clinical trial of AZD1775 in combination with neoadjuvant weekly docetaxel and cisplatin before definitive therapy in head and neck squamous cell carcinoma. Clin Cancer Res. 2018;24(12):2740–2748. doi: 10.1158/1078-0432.CCR-17-3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Do K, Wilsker D, Ji J, et al. Phase I study of single-agent AZD1775 (MK-1775), a wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol. 2015;33(30):3409–3415. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Leijen S, Van Geel RMJM, Pavlick AC, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34(36):4371–4380. doi: 10.1200/JCO.2016.67.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Leijen S, Van Geel RMJM, Sonke GS, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patientswith tp53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol. 2016;34(36):4354–4361. doi: 10.1200/JCO.2016.67.5942. [DOI] [PubMed] [Google Scholar]
- 160.Moore KN, Chambers SK, Hamilton EP, et al. Adavosertib with chemotherapy (CT) in patients (pts) with platinum-resistant ovarian cancer (PPROC): an open label, four-arm, phase II study. J Clin Oncol. 2019;5(_suppl):5513. doi: 10.1200/JCO.2019.37.15_suppl.5513. [DOI] [Google Scholar]
- 161.Yap TA, Plummer R, Azad NS, Helleday T. The DNA damaging revolution: PARP inhibitors and beyond. Am Soc Clin Oncol Educ B. 2019;39:185–195. doi: 10.1200/EDBK_238473. [DOI] [PubMed] [Google Scholar]
- 162.Forment JV, O’Connor MJ. Targeting the replication stress response in cancer. Pharmacol Ther. 2018;188:155–167. doi: 10.1016/j.pharmthera.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 163.Fu S, Wang Y, Keyomarsi K, Meric-Bernstein F. Strategic development of AZD1775, a Wee1 kinase inhibitor, for cancer therapy. Expert Opin Investig Drugs. 2018;27(9):741–751. doi: 10.1080/13543784.2018.1511700. [DOI] [PubMed] [Google Scholar]
- 164.Qiu Z, Oleinick NL, Zhang J. ATR/CHK1 inhibitors and cancer therapy. Radiother Oncol. 2018;126(3):450–464. doi: 10.1016/j.radonc.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ghelli Luserna Di Rora A, Iacobucci I, Martinelli G. The cell cycle checkpoint inhibitors in the treatment of leukemias. J Hematol Oncol. 2017;10(1):77. doi: 10.1186/s13045-017-0443-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lewis CW, Bukhari AB, Xiao EJ, et al. Upregulation of MyT1 promotes acquired resistance of cancer cells to WEE1 inhibition. Cancer Res. 2019;79(23):5971–5985. doi: 10.1158/0008-5472.CAN-19-1961. [DOI] [PubMed] [Google Scholar]
- 167.Garcia TB, Uluisik RC, van Linden AA, et al. Increased HDAC activity and c-MYC expression mediate acquired resistance to WEE1 inhibition in acute leukemia. Front Oncol. 2020;10:296. doi: 10.3389/fonc.2020.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.