

Abstract
Background
The most frequent and common form of Krabbe disease (KD) is early‐onset KD in infants, and late‐onset KD has been reported to be a rare disease. In the present study, we reported an adult‐onset KD patient in a consanguineous Chinese family.
Methods
Clinical and radiological data were collected for a family pedigree. The patient was diagnosed with late‐onset KD through next‐generation sequencing. The result was confirmed by Sanger sequencing. GALC enzyme activity was also examined by the colorimetry method. Both the grey matter volume (GMV) and white matter volume values were examined and compared with the average values from ten age‐matched normal controls. Moreover, we reviewed all the available KD studies on PubMed to understand the correlation between the phenotype and genotype of the identified mutation.
Results
The main manifestations of the proband were sudden onset seizures and cognitive decline. Mutation analysis of the GALC revealed a homozygous c.1901T>C mutation in exon 16, which resulted in an amino acid change in p.L634S. Sanger sequencing results showed that the homozygous mutation was inherited from the patient's parents, both of whom were revealed to be heterozygous carriers. Moreover, a decrease in GALC enzyme activity was also detected. However, no abnormal signals were found in the brain MRI. Further structural MRI analysis revealed a significantly decreased GMV in the proband compared to the normal controls. Moreover, it is of interest that all patients with the c.1901T>C mutation had late‐onset KD and were selected from Asian countries, especially Japan and China.
Conclusions
This patient with a homozygous GALC mutation expands the clinical presentation and characteristics of adult‐onset KD, as indicated by grey matter atrophy without abnormal white matter signals.
Keywords: GALC, grey matter atrophy, Krabbe disease
GALC gene analysis revealed a rare homozygous c.1901T>C mutation in exon 16 of the patient.
No abnormal signals were found in the brain MRI,but a significantly decreased GMV in the proband compared to the normal controls, which is different from previous cases.
It is of interest that all patients with c.1901T>C mutation were late‐onset type and selectively from Asian countries, especially Japan and China.
![]()
1. INTRODUCTION
Krabbe disease (KD), or globoid cell leukodystrophy (OMIM #245200), is a rare inherited autosomal recessive lysosomal storage disorder caused by galactocerebrosidase (GALC) gene mutations (Suzuki & Suzuki, 1970). GALC mutations lead to defective functions of the GALC enzyme, resulting in demyelination of both the central and peripheral nervous systems.
Based on the age of onset, KD is usually categorized as early‐onset KD (infantile form: <6 months) and late‐onset KD. The latter type includes a spectrum of late infantile‐onset (7 months to 2 years), juvenile‐onset (3‐8 years) and adult‐onset (>9 years) onset KD (Xu, Sakai, Taniike, Inui, & Ozono, 2006). The late‐onset forms of KD are rare and are clinically more heterogeneous, with signs that vary from hemiparesis, cerebellar ataxia, and seizures to progressive motor impairment, mental retardation, and behaviour changes; however, this form progresses more slowly than the early‐onset form (Sakai & Otomo, 2016; Yang et al., 2013). The diagnosis of KD is made by measuring GALC enzyme activity and identifying the presence of theGALC mutations (Adachi et al., 2016; Madsen et al., 2019). The typical imaging feature of KD is involvement of the corticospinal tracts or periventricular white matter (Cousyn et al., 2019). In the present report, we describe an adult‐onset KD patient without abnormal signals on an MRI in a consanguineous Chinese family with a homozygous mutation discovered by next‐generation sequencing (NGS), and we summarized all studies of the same gene mutations (Furuya et al., 1997; Hossain et al., 2014; Lim et al., 2016; Satoh et al., 1997; Xu et al., 2006; Yoshimura, Kibe, Irahara, Sakai, & Yokochi, 2016; Zhang et al., 2018; Zhao et al., 2018).
2. CLINICAL REPORT
A 22‐year‐old Chinese woman was referred to our hospital for further neurologic evaluation of neurological symptoms that included psychotic symptoms, such as forced weeping and laughing, and cognitive impairment. She had been repeatedly in and out of another local hospital one‐month prior because of several episodes of generalized convulsions. Her growth and development had been normal until the age of 13 years, and she had begun to feel excessively fatigued after exercise.
On admission, a general physical examination showed no abnormalities. In the neurologic examination, she was alert, oriented, and cooperative. A neuropsychological battery that included the Mini‐Mental State Examination (MMSE), the Cambridge Cognitive Examination‐Chinese version (Camcog‐C), the Hamilton Anxiety Scale (HAMA) and the Hamilton Depression Scale (HAMD), was performed to evaluate her cognition and emotion status. She presented MMSE 23/30, Camcog‐C 66/108 (praxis 5), HAMA 8, HAMD 12, which indicated a decrease in global cognition, especially in executive function and slight depression. Cranial nerves, muscle strength, finger‐to‐nose tests and sensation were normal. Bilateral pathological reflexes were negative. There were no sensory deficits.
Routine laboratory examinations of blood and urine were normal. Other items that were examined for a differential diagnosis, such as serum levels of folic acid, vitamin B12, total homocysteine, lactic acid, endocrinologic items, oligoclonal bands, and autoimmune encephalitis antibodies in the cerebrospinal fluid were also in the normal range. The motor nerve conduction velocity results were in the reference range. The sensory nerve conduction velocity was 34.9 m/s on the left side and 35.2 m/s on the right side of the peroneal nerve and 36.5 m/s on the left side and 35.7 m/s on the right side of the tibial nerve, suggesting the presence of sensory neuropathy. An electroencephalogram (EEG) test presented no epileptic discharge. Evoked potentials, including auditory evoked responses, visual evoked potentials, and somatosensory evoked potentials, were unremarkable.
3. METHODS AND RESULTS
3.1. Ethical compliance
The Institutional Review Board of the First Affiliated Hospital of Anhui Medical University Subcommittee on Human Studies approved this study, and the approval number is Quick‐PJ 2020‐01‐12. Informed written consent was obtained from the patient's family and the volunteers for this study.
3.2. MRI findings
MRI results showed mild brain atrophy but no significant hyperintensities in the white matter on the T2‐weighted and fluid‐attenuated inversion recovery (FLAIR) images (Figures 1 and 2). Brain atrophy was evaluated by a previously established clinical rating method (Besteher et al., 2017; Victoroff, Mack, Grafton, Schreiber, & Chui, 1994). Both of the patient's parents presented no abnormal signals on brain MRI. To obtain accurate grey matter values, ten normal controls were recruited for this study by advertisements (age: 23.2 ± 1.03 years (20–24)). Voxel‐based morphometry (VBM) analysis results showed that the grey matter value of the patient (502 mm3) was more than two standard deviations (SDs) beyond that of the controls (667 ± 71.27 mm3). We also found a tendency toward decreased white matter volume in the patient, although the differences did not reach 2 SDs (415 mm3 vs. 547.5 ± 75.36 mm3).
Figure 1.
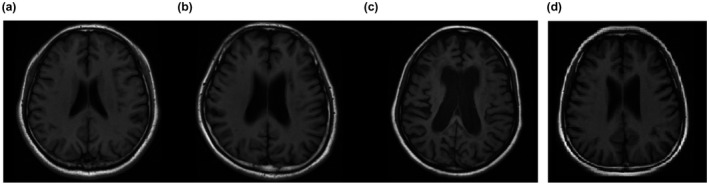
Standard T1 axial images of different severities of frontal lobe atrophy: (a) none; (b) mild; (c) severe; and (d) the mild brain atrophy of the proband according to the standard image
Figure 2.

T2‐weighted magnetic resonance imaging (MRI), and axial fluid‐attenuated inversion recovery (FLAIR) imaging showed no abnormal signals in the proband
3.3. Mutation analysis and GALC enzyme activity test
The molecular analysis of the GALC (reference mRNA sequence: NM_000153.3) was performed by next‐generation sequencing (NGS) technology and revealed that the patient had a homozygous point mutation of c.1901T>C in exon 16 (Figure 3). This mutation results in a single amino acid substitution at position 634 from leucine to serine (Leu634Ser). Sanger sequencing results showed that the homozygous mutations were inherited from the patient's parents, both of whom were revealed as heterozygous carriers (Figure 2).
Figure 3.
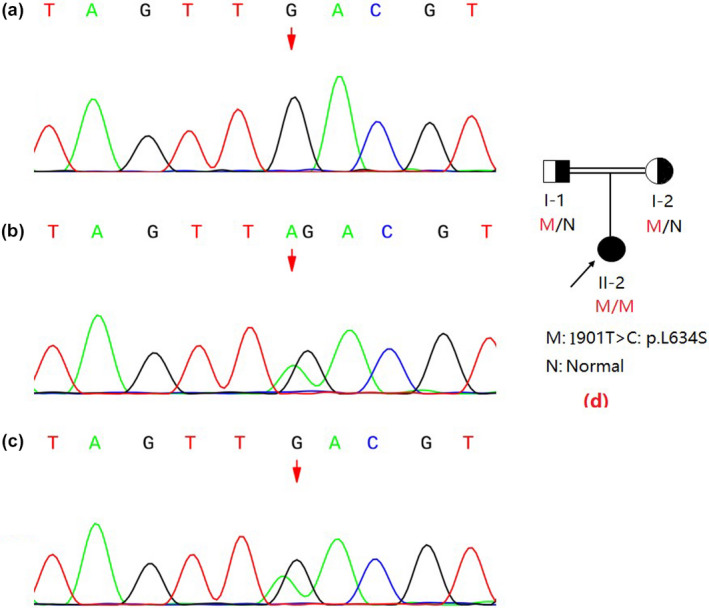
Molecular genetic analysis of the GALC (Reference mRNA sequence: NM_000153.3) showed the c.1901T>C, p.L634S mutation; this patient (a) was homozygous for this mutation, and her father (b) and mother (c) were heterozygous. The proband is indicated by an arrow in the pedigree chart of the family. M: 1901T>C: p.L634S, N: normal
The results for patient with the c.1901T>C mutation are shown in Table 1. The GALC activity of the 18 patients included in the available studies was consistently decreased in the lymphocytes, the skin or both. All patients had late‐onset KD. It is of interest that all 18 patients were from Asian countries, especially Japan and China. The clinical manifestations mainly included spastic paraparesis, gait disturbance, mental deterioration, hearing and vision impairments and language problems. The typical imaging features in the reported studies were predominant corticospinal tract involvement.
Table 1.
Summary of clinical information and mutations identified in KD patients with p.L634S (p.L618S) mutation in previous case reports
| No. | Age at onset | Phenotype | Genotype | Clinical symptomsa | Brain MRIa | Country | Abnormal GALC activitya | Author, years of publication |
|---|---|---|---|---|---|---|---|---|
| 1 | 51 y | Adult | p.[L618S]; [L618S] | Slowly progressive spastic paraparesis and diminished vibration sense | Bilateral frontoparietal subcortical white matter and centrum semiovale | Japan | LYM+SF | Satoh et al. (1997) |
| 2 | 10–20 y | Adult | p.[L618S]; [IVS6 + 5G‐A] | Spastic gait, mental deterioration | Selective pyramidal tract | Japan | LYM | Furuya et al. (1997) |
| 3 | 8 m | Late‐infantile | p.[P302A]; [L618S] | N | N | Japan | LYM+SF | Xu et al. (2006) |
| 4 | 11 m | Late‐infantile | c.[1719dupT]; p.[L618S] | N | N | Japan | LYM | Hossain et al. (2014) |
| 5 | 1 y and 2 m | Late‐infantile | c.[1719dupT]; p.[L618S] | N | N | Japan | LYM | Hossain et al. (2014) |
| 6 | 2 y | Late‐infantile | p.[L618S]; [?] | N | N | Japan | LYM | Hossain et al. (2014) |
| 7 | 3 y | Juvenile | p.[P302A]; [L618S] | N | N | Japan | SF | Hossain et al. (2014) |
| 8 | 14 y | Adult | p.[L618S]; [?] | N | N | Japan | LYM | Hossain et al. (2014) |
| 9 | 35 y | Adult | p.[L618S]; [G646A] | N | N | Japan | SF | Hossain et al. (2014) |
| 10 | 56 y | Adult | p.[L618S]; [?] | N | N | Japan | LYM | Hossain et al. (2014) |
| 11 | 11 m | Late‐infantile | c.635_646 delinsCTC, [p.L618S] | Developmental regression and spastic paraparesis | Predominantly the corticospinal tract | Japan | LYM+SF | Yoshimura et al. (2016) |
| 12 | 12 y | Adult | c.[1687A<T];[1901T>C] | Slow, progressive spastic gait disturbance | Precentral motor cortex, corona radiata, posterior limb of the internal capsule, cerebral peduncle of the bilateral pyramidal tracts and optic radiation | Korea | LYM | Lim et al. (2016) |
| 13 | 10 m | Late‐infantile | p. [F350 V]; [L618S] | Motor regression, language development delay, hearing and vision impairment | N | China | LYM | Zhao et al. (2018) |
| 14 | 2 y | Late‐infantile | p.[R129X]; [L618S] | Motor regression | N | China | LYM | Zhao et al. (2018) |
| 15 | 8 y and 10 m | Juvenile | c.[195+1G>A]; p.[L618S] | Loss of vision, unstable walking | N | China | LYM | Zhao et al. (2018) |
| 16 | <5 y | Juvenile | p. [P318 L]; [L618S] | Left limb movement disorder | N | China | LYM | Zhao et al. (2018) |
| 17 | 20 y | Adult | p.[L618S]; [?] | Psychomotor regression | N | China | LYM | Zhao et al. (2018) |
| 18 | 20 y | Adult | c.1901T>C : c.1901delT | Aphasia | Selective pyramidal tract | China | LYM | Zhang et al. (2018) |
a GALC activity is measured in lymphocytes and skin fibroblasts, respectively. LYM: lymphocyte; SF: skin fibroblast. N: not given in the studies
In the patient in this case,GALC enzyme activity level was decreased at 3.3 nmol/17 h/mg (>12.7 nmol/17 h/mg) according to colorimetry performed with a SPECTRA MAX GEMINI XPS fluorescent enzyme labelling instrument. Both the patient's father and mother showed normal GALC activity levels (123.5 nmol/17 h/mg and 56.93 nmol/17 h/mg, respectively).
4. DISCUSSION
To the best of our knowledge, this is the first report to describe the phenotype of adult‐onset KD associated with the homozygous mutation c.1901T>G (p.Leu618Ser) in China and is also the second reported case of a homozygous mutation (c.1901T>G). There is only one previously published report; it describes a Japanese adult‐onset KD patient, and was initially reported in 1997. Moreover, grey matter atrophy, especially in the frontal lobe, seemed to be the unique change in the MRI of this patient, which is different from previous studies, in which the patients presented with selective pyramidal tract impairment.
The mutation located at c.1901T>G has previously been reported and is considered to be predominantly related to late‐onset KD (Zhang et al., 2018). In the current report, the patient was 13 years old when she began experiencing a slow gait, which is a symptom of adult‐onset KD. When the data for all the patients with the c.1901T>G mutation were summarized, the age of onset was found to range from 8 months to 51 years. These observations, combined with our report, indicate that the c.1901T>G mutation may selectively exist in patients with late‐onset KD. In previous studies, the c.1901T>G mutation was also regarded as a mild mutation type due to its slight symptoms, which include gait disturbance, cognitive impairment, peripheral disease, vision loss, and so on (Satoh et al., 1997; Zhang et al., 2018). Notably, the present patient exhibited most of these clinical symptoms but with insidious onset and slow progress, further supporting the possibility that the c.1901T>G mutation contributes to a more benign clinical outcome than infantile early‐onset KD. Moreover, it was also observed that the c.1901T>G mutation was found only in Asian countries; in contrast, Caucasian patients primarily have a 30‐kb deletion mutation (Hossain et al., 2014). This result raises the possibility that the KD mutation has a regional tendency. Therefore, this summary, along with our case study may aid in elucidating and extending the genotype‐phenotype correlation of mutations in KD.
Selective demyelinating lesions involving the corticospinal tracts or periventricular white matter are the usual imaging characteristics. Myelin impairment in KD largely includes increases in globoid cells and macrophages and proliferating astrocytes, according to the post‐mortem findings. However, in our report, no significant white matter hyperintensities (WMHs) were found on the MRI, and the patient had only slight brain atrophy, especially in the frontal lobe. To the best of our knowledge, there have been no previous reports of imaging findings with grey matter atrophy as a unique characteristic in this disease. Only one report of an adult‐onset KD patient described a normal brain MRI, suggesting that these clinical symptoms may precede imaging changes (Bajaj, Waldman, Orrell, Wood, & Bhatia, 2002). However, in previous studies, some KD patients' symptoms were accompanied by grey matter atrophy in the late phases along with typical abnormal signals on MRIs (Wang, Melberg, Weis, Månsson, & Raininko, 2007). The pathogenetic basis of this finding is unknown and requires further study. It is suspected that brain atrophy is the prophase of KD, while typical WMHs may be found in subsequent years. In the current study, we also found that there was a decreasing trend in white matter atrophy, although the differences were not two SDs greater than those of the controls, which may provide insight for further speculation. It has been suggested that brain volume may be associated with cognition (Marino et al., 2019). Therefore, the cognitive impairment of the proband may be attributed to the decreased grey matter and white matter volume. Specifically, frontal lobe atrophy not only contributed to the symptoms of forced weeping and laughing but also to the praxis deficit. These clinical and brain structural features were different from those of the only reported case of a homozygous mutation (c.1901T>G) in adults. We thus claim that searching for subtle changes in MRIs will improve the ability to diagnose KD earlier, especially when structural MRIs based on the CAT method are used to accurately assess the brain when there is only slight brain atrophy. Clinical variabilities, especially in MRIs, have been referred to in previous studies and these were partly explained by the differences in the activation of enzyme systems that control the level of galactosylsphingosine (Barone et al., 1996). However, the exact pathogenesis mechanism is still beyond our understanding, and this needs to be further explored in future case studies.
This case report and summary demonstrated that the c.1901T>G mutation preferentially existed in patients in Asian countries and selectively involved mild forms of late‐onset KD. The atypical MRI presentation with of brain atrophy in KD in our report expanded the possible phenotypes by providing a clinical presentation. The early diagnosis and management of patients with KD may lead to improved outcomes. Further clinical and neuroradiological studies could be helpful in providing insights into the pathogenesis of the disorder.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
Zhou Xia, Yin Wenwen, Yu Xianfeng, Hu Panpan, Zhu Xiaoqun, and Sun Zhongwu clinically characterized the patient. Zhou Xia and Yin Wenwen performed the experiments. Zhou Xia and Sun Zhongwu planned and supervised the study, wrote the article and provided the funding. All authors approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank all the volunteers, the patient and her family for their agreement to publish this case report. This work was supported by grants from the National Natural Science Foundation of China (81771154) and the Natural Science Foundation of Anhui province (1908085QH322).
Xia Z , Wenwen Y, Xianfeng Y, et al. Adult‐onset Krabbe disease due to a homozygous GALC mutation without abnormal signals on an MRI in a consanguineous family: A case report. Mol Genet Genomic Med. 2020;8:e1407 10.1002/mgg3.1407
FUNDING INFORMATION
The National Natural Science Foundation of China (81771154) and the Natural Science Foundation of Anhui province (1908085QH322)
REFERENCES
- Adachi, H. , Ishihara, K. , Tachibana, H. , Oka, N. , Higuchi, Y. , Takashima, H. , … Kageyama, Y. (2016). Adult‐onset Krabbe disease presenting with an isolated form of peripheral neuropathy. Muscle and Nerve, 54, 152–157. [DOI] [PubMed] [Google Scholar]
- Bajaj, N. P. , Waldman, A. , Orrell, R. , Wood, N. W. , & Bhatia, K. P. (2002). Familial adult onset of Krabbe's disease resembling hereditary spastic paraplegia with normal neuroimaging. Journal of Neurology, Neurosurgery and Psychiatry, 72, 635–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone, R. , Brühl, K. , Stoeter, P. , Fiumara, A. , Pavone, L. , & Beck, M. (1996). Clinical and neuroradiological findings in classic infantile and late‐onset globoid‐cell leukodystrophy (Krabbe disease). American Journal of Medical Genetics, 63, 209–217. [DOI] [PubMed] [Google Scholar]
- Besteher, B. , Squarcina, L. , Spalthoff, R. , Bellani, M. , Gaser, C. , Brambilla, P. , & Nenadić, I. (2017). Brain structural correlates of irritability: Findings in a large healthy cohort. Human Brain Mapping, 38, 6230–6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousyn, L. , Law‐Ye, B. , Pyatigorskaya, N. , Debs, R. , Froissart, R. , Piraud, M. , … Nadjar, Y. (2019). Brain MRI features and scoring of leukodystrophy in adult‐onset Krabbe disease. Neurology, 93, e647–e652. [DOI] [PubMed] [Google Scholar]
- Furuya, H. , Kukita, Y.‐J. , Nagano, S. , Sakai, Y. , Yamashita, Y. , Fukuyama, H. , … Kobayashi, T. (1997). Adult onset globoid cell leukodystrophy (Krabbe disease): Analysis of galactosylceramidase cDNA from four Japanese patients. Human Genetics, 100, 450–456. [DOI] [PubMed] [Google Scholar]
- Hossain, M. A. , Otomo, T. , Saito, S. , Ohno, K. , Sakuraba, H. , Hamada, Y. , … Sakai, N. (2014). Late‐onset Krabbe disease is predominant in Japan and its mutant precursor protein undergoes more effective processing than the infantile‐onset form. Gene, 534, 144–154. [DOI] [PubMed] [Google Scholar]
- Lim, S. M. , Choi, B.‐O. , Oh, S.‐I. , Choi, W. J. , Oh, K.‐W. , Nahm, M. , … Kim, S. H. (2016). Patient fibroblasts‐derived induced neurons demonstrate autonomous neuronal defects in adult‐onset Krabbe disease. Oncotarget, 7, 74496–74509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen, A. , Wibrand, F. , Lund, A. M. , Ek, J. , Dunø, M. , & Østergaard, E. (2019). Genotype and phenotype classification of 29 patients affected by Krabbe disease. JIMD Reports, 46, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino, S. , Bonanno, L. , Lo Buono, V. , Ciurleo, R. , Corallo, F. , Morabito, R. , … Bramanti, P. (2019). Longitudinal analysis of brain atrophy in Alzheimer's disease and frontotemporal dementia. Journal of International Medical Research, 47, 5019–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, N. , & Otomo, T. (2016). Challenge of phenotype estimation for optimal treatment of Krabbe disease. Journal of Neuroscience Research, 94, 1025–1030. [DOI] [PubMed] [Google Scholar]
- Satoh, J.‐I. , Tokumoto, H. , Kurohara, K. , Yukitake, M. , Matsui, M. , Kuroda, Y. , … Hayashi, K. (1997). Adult‐onset Krabbe disease with homozygous T1853C mutation in the galactocerebrosidase gene. Unusual MRI findings of corticospinal tract demyelination. Neurology, 49, 1392–1399. [DOI] [PubMed] [Google Scholar]
- Suzuki, K. , & Suzuki, Y. (1970). Globoid cell leucodystrophy (Krabbe's disease): Deficiency of galactocerebroside beta‐galactosidase. Proceedings of the National Academy of Sciences of the United States of America, 66, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoroff, J. , Mack, W. J. , Grafton, S. T. , Schreiber, S. S. , & Chui, H. C. (1994). A method to improve interrater reliability of visual inspection of brain MRI scans in dementia. Neurology, 44, 2267–2276. [DOI] [PubMed] [Google Scholar]
- Wang, C. , Melberg, A. , Weis, J. , Månsson, J.‐E. , & Raininko, R. (2007). The earliest MR imaging and proton MR spectroscopy abnormalities in adult‐onset Krabbe disease. Acta Neurologica Scandinavica, 116, 268–272. [DOI] [PubMed] [Google Scholar]
- Xu, C. , Sakai, N. , Taniike, M. , Inui, K. , & Ozono, K. (2006). Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype‐phenotype correlation. Journal of Human Genetics, 51, 548–554. [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Ren, X. , Xu, Q. , Wang, C. , Liu, H. , & He, X. (2013). Four novel GALC gene mutations in two Chinese patients with Krabbe disease. Gene, 519, 381–384. [DOI] [PubMed] [Google Scholar]
- Yoshimura, A. , Kibe, T. , Irahara, K. , Sakai, N. , & Yokochi, K. (2016). Predominant corticospinal tract involvement in a late infant with Krabbe disease. Japanese Clinical Medicine, 7, 23–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Yan, C. , Ji, K. , Lin, P. , Chi, L. , Zhao, X. , & Zhao, Y. (2018). Adult‐onset Krabbe disease in two generations of a Chinese family. Annals of Translational Medicine, 6, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, S. , Zhan, X. , Wang, Y. , Ye, J. , Han, L. , Qiu, W. , … Zhang, H. (2018). Large‐scale study of clinical and biochemical characteristics of Chinese patients diagnosed with Krabbe disease. Clinical Genetics, 93, 248–254. [DOI] [PubMed] [Google Scholar]