

Abstract
Ozone (O3) is a criteria air pollutant that exacerbates and increases the incidence of chronic pulmonary diseases. O3 exposure is known to induce pulmonary inflammation, but little is known regarding how exposure alters processes important to the resolution of inflammation. Efferocytosis is a resolution process, whereby macrophages phagocytize apoptotic cells. The purpose of this protocol is to measure alveolar macrophage efferocytosis following O3-induced lung injury and inflammation. Several methods have been described for measuring efferocytosis; however, most require ex vivo manipulations. Described in detail here is a protocol to measure in vivo alveolar macrophage efferocytosis 24 h after O3 exposure, which avoids ex vivo manipulation of macrophages and serves as a simple technique that can be used to accurately represent perturbations in this resolution process. The protocol is a technically non-intensive and relatively inexpensive method that involves whole-body O3 inhalation followed by oropharyngeal aspiration of apoptotic cells (i.e., Jurkat T cells) while under general anesthesia. Alveolar macrophage efferocytosis is then measured by light microscopy evaluation of macrophages collected from bronchoalveolar (BAL) lavage. Efferocytosis is finally measured by calculating an efferocytic index. Collectively, the outlined methods quantify efferocytic activity in the lung in vivo while also serving to analyze the negative health effects of O3 or other inhaled insults.
Keywords: Immunology and Infection, Issue 152, air pollution, ozone, lung, inflammation, alveolar macrophage, efferocytosis
Introduction
The lung is constantly exposed to environmental insults, including air particulates, viruses, bacteria, and oxidant gases that trigger pulmonary inflammation1,2,3. These insults can compromise gas exchange and induce irreversible tissue injury4,5. Alveolar macrophages, which constitute approximately 95% of the immune cells found in murine and human lungs at homeostasis, are critical regulators of pulmonary inflammation after environmental insults1,2,3,4,5. Alveolar macrophages are essential during the host defense by phagocytizing and eliminating pathogens. Recently, alveolar macrophages have been shown to promote tissue homeostasis and the resolution of inflammation through efferocytosis6,7. Efferocytosis is a phagocytic process in which macrophages engulf and eliminate apoptotic cells8,9,10. Efferocytosis also results in the production of mediators (i.e., IL-10, TGF-β, PGE2, and nitric oxide) that further augment the process, resulting in the resolution of inflammation9,10, 11,12,16,18. This process is necessary for preventing secondary necrosis and promoting tissue homeostasis12,13,14. Several studies have linked impaired efferocytosis with various chronic lung diseases, including asthma, chronic obstructive pulmonary disease, and idiopathic pulmonary fibrosis8,9,15,16,17.
O3 is a criteria air pollutant that exacerbates and increases the incidence of chronic pulmonary diseases19,20,21. O3 induces pulmonary inflammation and injury and is known to impair alveolar macrophage phagocytosis of bacterial pathogens22,23. However, it is unknown whether O3 impairs alveolar macrophage efferocytosis. Investigating O3-induced alterations in alveolar macrophage efferocytosis will provide potential insight into how exposure can lead to chronic pulmonary disease incidence and exacerbation. Described below is a simple method to evaluate alveolar macrophage efferocytosis in the lungs of female mice after acute O3 exposure.
The outlined method posseses several advantages over other efferocytosis protocols commonly used in the field by eliminating the use of costly fluorescent dyes, extensive flow cytometry measurements, and ex vivo manipulation of alveolar macrophages24,25. Additionally, this protocol measures alveolar macrophage efferocytosis in the context of the lung microenvironment, which can influence macrophage function.
Protocol
All methods have been approved by the Institutional Animal Care and Use Committee (IACUC) of East Carolina University.
1. Ozone (O3) and filtered air exposures (Day 1)
Place a maximum of 12 female C57BL/6J mice, 8-12 weeks old, in a steel cage (with 12 separate compartments) with wire mesh lids into an O3 exposure chamber.
Place the thermometer in the exposure chamber with the cage to accurately record the temperature and humidity.
-
Turn on the oxygen and ultraviolet (UV) light that is attached to the apparatus.
NOTE: Regulated airflow (>30 air changes/h) with controlled temperature (22-23 °C) and relative humidity (45%-50%) is obtained by the O3 apparatus. O3 is generated by the system in the exposure chamber by directing 100% oxygen through a UV light generator, then mixing with a filtered air supply.
-
Adjust the O3 concentration to 1 ppm and regularly record O3 levels every 10 min for 3 h. Continuously monitor the temperature and humidity of chamber air, as is the O3 concentration with a UV light photometer.
NOTE: Filtered air exposures are performed in a similar apparatus, with only a filtered air supply flowing through the exposure chamber.
Return the animals to their respective cages with bedding, food, and water ad libitum after 3 h of O3/filtered air exposure.
2. Preparation of Jurkat T cell line (Day 2)
NOTE: All procedures should be conducted in a class II biological safety cabinet.
Culture Jurkat T cells in 24 mL of basal cell culture medium + 10% FBS + 5% penicillin/streptomycin at 37 °C + 5% CO2 (Table of Material). Jurkat T cells are a suspension cell line that can be maintained through passaging 1:6-1:8 into pre-warmed culturing media every 3 days. Do not shake.
-
To prepare apoptotic cells, grow them to 90% confluency in each flask (which takes 3-4 days to achieve after passaging). For this study, use cells from five T75 flasks to obtain the sufficient number of cells used in this protocol.
NOTE: A confluent flask contains about 20-24 million cells.
Pipette up cells (which is the entire flask) from each flask (approximately 24 mL) and transfer cells to a sterile 50 mL conical tube using a serological pipette. Use multiple conical tubes for multiple flasks.
Count cells by removing an 11 μL aliquot of cells from the 50 mL conical tube and mix with 11 μL of trypan blue stain and pipette 11 μL onto hemocytometer slides.
Insert slide into an automated cell counter and record the number of live cells to calculate the total cell count in each flask by multiplying the number of live cells by 24, as each flask contains 24 mL of media.
Centrifuge the cell suspension at 271 x g for 5 min at room temperature (RT) to pellet cells.
Discard the supernatant by aspiration and resuspend the cell pellet in media to obtain 3.0 x 106 cells per mL.
Aliquot 5 mL of cells in 100 mm x 20 mm tissue culture dishes (approximately nine dishes will be used; the total amount of cells in each dish should be ~15 x 106).
Use one dish for control/unexposed, and the remaining dishes will be exposed to UV.
-
Set the UV crosslinker to the correct energy level, press the energy button, and enter "600" using the number pad, which the machine will read as 600 μJ/cm2 x 100.
NOTE: UV crosslinker energy units is in μJ/cm2 x 100; therefore, to achieve 60 millijoules/cm2, convert units to match the UV crosslinker.
Irradiate all dishes with cells, not including the control, at 60 millijoules (mJ)/cm2 using the UV crosslinker. Remove the top cover of the tissue culture dishes during UV exposure, as UV light will not penetrate the plastic cover.
Incubate all the dishes in a cell culture incubator, including unexposed control, at 37 °C at 5% CO2for 4 h.
-
Confirm apoptosis by flow cytometry using an apoptosis assay detection kit containing annexin V and propidium iodide (PI) (markers for apoptosis and necrosis, respectively) after 4 h of incubation, per the manufacturer's instructions26,27.
NOTE: Irradiating Jurkat T cells in the UV crosslinker at an energy level of 600 μJ/cm2, following a 4 h incubation will lead to ≥75% of apoptotic (both early and late) cells having more early apoptotic phenotype than the late apoptotic phenotype. This makes it easier for alveolar macrophages to recognize them and engulf as their membranes are uncompromised, unlike late apoptotic cells, leading to a higher efferocytic index and more accurate imaging of alveolar macrophage efferocytosis in this study.- Pool 333 μL (1 x 106 cells) of Jurkat T cells from several dishes (both "no UV" and "UV-exposed") together to use for compensation analysis tubes.
- Aliquot 333 μL of Jurkat T cells in an unstained, annexin V single stain, PI single stain, no UV control, and 600 μJ/cm2 UV-exposed labeled flow cytometry tubes.
- Centrifuge tubes at 188 x g for 5 min at RT and decant the supernatant.
- Wash cells by resuspending in 500 μL of cold, 1x phosphate-buffered saline (PBS).
- Centrifuge and pellet cells at 188 x g for 5 min at RT. Discard the supernatant after centrifugation.
- Prepare 400 μL of 1x binding buffer per flow tube by diluting 10x binding buffer with distilled water while cells are centrifuging.
- Prepare annexin V and PI incubation reagent (100 μL per sample/tube) per the manufacturer's instructions.
- Decant the supernatant after centrifugation and gently resuspend all tubes in 400 μL of 1x binding buffer, then add 100 μL of annexin V incubation reagent to each sample tube. Lastly, add 100 μL of annexin V single stain and PI single stain to their respective tubes, but do not add anything beyond the 1x binding buffer to the unstained tube.
- Incubate tubes in the dark for 15 min at RT.
- Centrifuge all cells at 188 x g for 5 min at RT and decant supernatant.
- Resuspend cells in 400 μL of 1x binding buffer, then analyze samples for apoptosis by flow cytometry. Collect at least 10,000 events per tube to allow accurate representation of staining.
Combine all the irradiated cells from dishes into a 50 mL conical tube and pellet cells by centrifugation at 271 x g for 5 min at RT.
Discard the supernatant from the tube by aspiration and resuspend cells in 24 mL of sterile phosphate buffered saline (PBS) and pellet cells by centrifugation at 271 x g for 5 min at room temperature.
-
Discard the supernatant from the tube by aspiration and resuspend cells in the amount of PBS used for dosing mice approved by IACUC. The dose used is between 5-10 x 106 cells/50 μL per mouse; therefore, for 10 mice, resuspend in 500 μL (number of cells in each dose varies depending on how many cells are cultured for irradiation).
NOTE: Makeup at least two additional doses to account for any liquid that may stick to the sides of the pipette tip resulting in the loss of cells.
3. Murine oropharyngeal instillation of apoptotic cells (Day 2)
Prepare dosing inoculum of apoptotic cells using a P200 pipette prior to anesthetizing mice to expedite the procedure. As per the institutional guidelines, a volume of 50 μL containing approximately 5-10 x 106 cells is utilized for oropharyngeal (o.p.) instillation to ensure best results.
Anesthetize mice in a clear chamber with 2% isoflurane at a flow rate of 1 L/min or as per the institutional guidelines. Anesthetize one to two mice at a time; the number is determined by the comfort level of the experimenter. Observe the breathing pattern and confirm deep breaths are visible with 2–3 s counts between breaths. Check for the depth of anesthesia by the lack of response to the toe pinch.
Position the mouse in a semi-recumbent supine position. Use a surgical string tied between pegs on a slanted acrylic sheet board to suspend by the maxillary incisors.
-
Using a pair of blunt non-ridged forceps, lightly grab and pull the mouse tongue. Instill the apoptotic cells into the oral cavity with a P200 pipette. Dosing is successful when the mice make a crackling noise 1–2 s after giving the dose.
NOTE: Take care to avoid inducing trauma either to the tongue or oropharynx before the apoptotic cell instillation.
-
With a gloved finger, gently block the nose until the mouse inhales while the tongue is retracted. Cover the nose until no liquid is visible in the oral cavity and the mouse has taken two or more inhalations.
NOTE: As mice are obligate nose breathers, covering the nose helps ensure that the mouse will inhale the apoptotic cells into the lungs.
Remove the mouse from the inoculation board and return it to the cage to allow recovery from anesthesia. Place the mouse on its back to prevent bedding or debris from blocking the nares during the revovery.
Wait 90 min after the mouse recovers from anesthesia to allow alveolar macrophages to engulf influx of apoptotic cells after all the mice have awoken from anesthesia. Typically, awakening after anesthesia will take 1–2 min, which should not affect the outcome/timing of instillation.
4. Bronchoalveolar lavage fluid collection and processing (Day 2)
-
Euthanize each mouse per institutional guidelines 90 min after the mouse has woken up from anesthesia from dosing with apoptotic cells. Here, a lethal injection of ketamine and xylazine is used (90 mg/kg and 10 mg/kg, respectively) followed by excising the diaphragm.
NOTE: This time point allows sufficient time for alveolar macrophages to sense and engulf apoptotic cells38.
Weigh all mice (g) on a scale and record weights. Use the body weight to calculate BAL volume (26.25 mL/kg body weight).
Place mice on their backs and spray 70% ethanol to sterilize the chest and neck area.
Make a 2” longitudinal cut just below the sternum along the entire ventral side with surgical scissors, and while holding the sternum with forceps, nick the diaphragm to allow the lungs to fall back into the chest cavity.
Cut laterally along the sides of the rib cage to allow the lungs more room to expand when lavaging, then fold the chest cavity back with forceps.
Make a 1" vertical cut up along vasculature through the neck to expose the trachea.
Use two forceps to pull muscle and tissue off the trachea and expose it. Avoid additional potential bleeding and cutting the trachea, since it is surrounded by vasculature, longitudinal muscles, and connective tissue.
Use a needle to make a slit in the trachea (about one-quarter of the distance down from the head) and insert a cannula (18 G x 1.25") with a syringe pre-loaded with 1x PBS (26.25 mL/kg body weight, ~0.7-1.0 mL in an 8-10 week old female C57Bl/6J mouse) caudally into the trachea.
Push volume of PBS into the lungs slowly to allow the lungs to inflate then, pull the volume back out into the syringe. Repeat this process a total of 3 times.
Collect the pooled lavage fluid from each specific mouse in a 15 mL tube.
Centrifuge the bronchoalveolar lavage at 610 x g for 6 min at 4 °C and collect supernatant into a 1.5 mL tube and freeze at −80 °C. The pellet represents cells from the bronchoalveolar space.
Remove residual red blood cells in collected BAL fluid by adding 1 mL of ACK RBC lysis buffer to the cell pellet, then vortex well and lyse for 1 min on ice. Afterwards, add 4 mL of PBS to stop the lysis reaction.
Pellet cells by centrifugation at 610 x g for 6 min at 4 °C and aspirate the supernatant with a vacuum aspirator.
Resuspend cells in 1 mL of 1x PBS + 10% FBS to each BAL sample tube. Count cells on a hemocytometer for the quantification of total airspace cells from each sample (no trypan blue). Centrifuge 120 μL of each sample onto slides at 56 x g for 3 min, using medium acceleration and a cytocentrifuge. Dry the slides overnight.
5. Calculation of alveolar macrophage efferocytic index (Day 3)
Stain the slides with hematoxylin and eosin to allow for calculation of both efferocytic and differential cell counts, with at least 200 cells counted from each slide.
View slides under a bright-field setting on a biological microscope (a 20x or 40x objective will work best).
Calculate the efferocytic index based on the ratio of the number of alveolar macrophages that phagocytosed apoptotic Jurkat T cells to alveolar macrophages without apoptotic cell uptake out of a total 200 macrophages on a cell differential slide. Convert the ratio to a percentage for data input. Use the following equation:
Representative Results
O3 exposure is known to induce pulmonary inflammation and injury, and efferocytosis is required to maintain tissue homeostasis. C57BL/6J female mice were exposed to filtered air (FA) or 1 ppm O3 for 3 h and necropsied 24 h post-exposure to examine pulmonary inflammation and injury. O3-exposed mice displayed a significant increase in macrophages and neutrophils in the airspace compared to the FA control group (Figure 1A,B). Additionally, O3-exposed mice had a significant increase in BAL protein, a marker of alveolar epithelial barrier dysfunction 24 h post-exposure (Figure 1C).
Figure 1: O3 exposure induces pulmonary inflammation and injury.

C57BL/6J female mice were exposed to filtered air (FA) or 1 ppm O3 for 3 h. 24 h post-exposure, mice were necropsied to analyze pulmonary inflammation and injury (n = 6 per group). (A) Bronchoalveolar lavage (BAL) cell differentials were calculated, then epithelial (epi), eosinophils (eos), lymphocytes (lymph), macrophages (Mɸ), and neutrophils (PMN) were identified with at least 200 cells counted from each slide. (B) A representative image of cellular differentials. (C) Total protein in the BAL fluid. Data are expressed as ± SEM (**p < 0.01).
To determine if O3-induced pulmonary inflammation is associated with defects in alveolar macrophage efferocytosis in vivo, C57BL/6J female mice were instilled with apoptotic Jurkat T cells via oropharyngeal aspiration 24 h post-FA or post-O3 exposure. Apoptosis in Jurkat T cells was confirmed by flow cytometry prior to dosing, and there was a significant increase in early (annexin V+ PI− and late (annexin V+ and PI+) apoptotic cells (Figure 2A,B). The exposure level and incubation time resulted in repetitive results of ~75% apoptotic Jurkat T cells. A magnified image of what was identified as an efferocytic macrophage is shown in Figure 3A. Efferocytic macrophages were identified as macrophages that had engulfed a Jurkat T cell (indicated by black arrows), compared to regular alveolar macrophages (indicated by white arrows) (Figure 3B). When alveolar macrophage efferocytosis was assessed utilizing the protocol, there was a statistically significant decrease in the efferocytic index of the O3-exposed group compared to FA controls (Figure 3B,C). These data indicate that O3-induced pulmonary inflammation is associated with decreased clearance of apoptotic cells, which may prolong lung injury and inflammation.
Figure 2: Confirmation of UV induced apoptosis in Jurkat T cells.
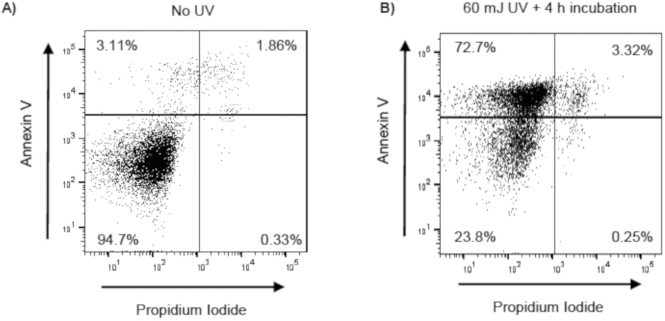
Jurkat T cells were exposed to UV (60 mJ/cm2) using a UV Crosslinker (Model 1800). Following UV exposure, Jurkat T cells were incubated at 37 °C with 5% CO2 for 4 h. Following incubation, Jurkat T cells were stained with annexin V and propidium iodide (PI), and apoptosis was evaluated by flow cytometry. Early apoptotic, late apoptotic, and necrotic cells are identified as annexin V+/PI−, annexin V+/PI+, annexin V−PI+, respectively. Representative flow cytometry scatter plots (with 10,000 events recorded) of (A) unexposed Jurkat T cells and (B) UV-exposed Jurkat T cells.
Figure 3: O3 exposure decreases alveolar macrophage efferocytosis.

C57BL/6J female mice were exposed to filtered air (FA) or 1 ppm O3for 3 h. 24 h post-exposure, mice were oropharyngeally instilled with approximately 5 x 106 apoptotic Jurkat T cells. 1.5 h after instillation, bronchoalveolar lavage (BAL) was performed, and the efferocytic index was calculated in BAL macrophages by light microscopy after counting 200 macrophages (n = 11 per group). (A) Representative image of an efferocytic macrophage. (B) Identification of alveolar macrophages (white arrows) and efferocytic macrophage (black arrows) after FA or O3 exposure. (C) Calculation of the efferocytic index after FA or O3 exposure (***p < 0.0001).
Discussion
Efferocytosis is an anti-inflammatory process in which macrophages clear apoptotic cells and debris as well as produce multiple anti-inflammatory mediators9,10,11,12,16,18. Multiple models of efferocytosis have provided insight into how the macrophage is a critical cell in the resolution of inflammation6,7. Recently, the progression of chronic lung diseases has been associated with defects in efferocytosis8,9,15,16,17. However, it is currently unclear whether exposure to air pollutants such as O3, results in defects in efferocytosis. This protocol enables the evaluation of alveolar macrophage efferocytosis after O3 exposure. It also quantifies efferocytosis in vivo using light microscopy and allows the measurement of efferocytosis in the context of the lung microenvironment, without ex vivo manipulations or expensive fluorescent dyes. Although this protocol is performed in the context of O3 exposure, multiple models of lung inflammation and injury can be used with this protocol to evaluate alveolar macrophage efferocytosis.
Advantages of this method over existing methods are its ability to analyze alveolar macrophages in the context of physiological environment. Ex vivo analysis of alveolar macrophages includes plating and incubation with apoptotic cells. Plating alveolar macrophages can induce both physiological and genomic changes that may alter efferocytosis28,29,30. Additionally, in the lung, alveolar macrophages exist in a microenvironment that contains surfactant and components of the lung lining fluid that are known to influences macrophage function31,32,33,34,35. Our method allows efferocytosis measurements in the lung with no ex vivo manipulations, which is more physiologically relevant. Future applications of this protocol can lead to more in-depth studies about how the lung microenvironment can alter alveolar macrophage efferocytosis.
A critical component of this protocol is the generation of apoptotic cells for evaluation of alveolar macrophage efferocytosis. This involves optimizing the correct UV exposure level to induce apoptosis, not necrosis. Our protocol uses the UV crosslinker with 254 nm wavelength emission bulbs and an exposure level of 60 mJ/cm2. The UV bulb choices are critical in producing apoptosis, not necrosis. 350 nm UV bulbs are excellent for protein membrane cross-linking and sterilization but fail to induce apoptosis35,36,37. An example dot plot of Jurkat T cells exposed to 60 mJ/cm2 with 350 nm bulbs is shown in Figure 4 with a significant increase in late apoptotic and necrotic cells. Additionally, the protocol uses a 4 h incubation post-UV exposure. To optimize this part of the protocol, we previously examined various incubation times and found that 1.5 and 2 h incubation post-exposure only yielded approximately 40% apoptosis, with an efferocytic index of less than 5% (data not shown). Based on current literature, ~70%-80% total apoptotic cells are sufficient for measuring efferocytosis38.
Figure 4: Suboptimal Jurkat T cell apoptosis using 350 nm frosted bulbs.
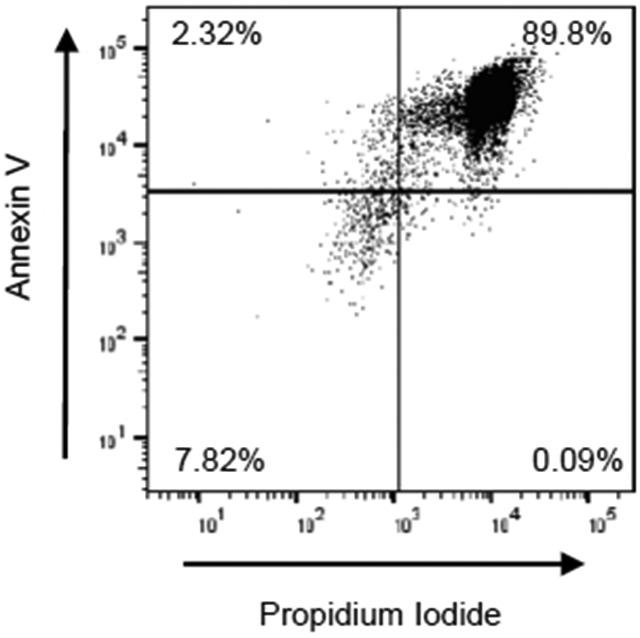
Jurkat T cells were irradiated using the UV Crosslinker for 10 min and incubated at 37 °C at 5% CO2 for 1 h. Following UV exposure, Jurkat T cells were incubated at 37 °C with 5% CO2 for 4 h. Following incubation, Jurkat T cells were stained with annexin V and propidium iodide (PI), then apoptosis was evaluated by flow cytometry. Early apoptotic, late apoptotic, and necrotic cells are identified as annexin V+/PI−, annexin V+/PI+, and annexin V−/PI+, respectively. Representative flow cytometry plots (with 10,000 events recorded) of UV-exposed Jurkat T cells with 350 nm bulbs are shown.
A limitation to this protocol is that it examines the efferocytic response of all macrophages in the airspace after FA or O3 exposure and does not distinguish tissue resident macrophages from recruited macrophages. The lung resident macrophage termed alveolar macrophage originate from the fetal liver, whereas recruited macrophages derive from a blood-borne embryonic origin. Upon injury, the lung can have a highly heterogeneous macrophage population with unique genetics and expression of cell surface markers28,29,30,31,32,33,34,35. It is known that the immunological response and function of these macrophage populations are different; however, recent studies have indicated that the tissue resident macrophage have a greater efferocytic response compared to recruited macrophages29,30,31. Determining the efferocytic response of tissue resident macrophages vs. recruited macrophages can be assessed with the current protocol; however, the macrophage populations need to be purified by FACS and plated on slides for analysis. Additionally, this protocol only assesses alveolar macrophage efferocytic function in one strain of inbred, commercially available mice. It has previously been reported that different strains of mice show different responses to O3 exposure, including pulmonary inflammation39,40. Therefore, there may be differences in alveolar macrophage efferocytosis based on the strain examined. This is a variable that should be considered when performing this in vivo assay.
In conclusion, the protocol described above allows the evaluation of alveolar macrophage efferocytosis in vivo. This protocol is cost-effective and simple, making it an assay that can be widely utilized. Moreover, this method can be applied to numerous models of lung injury and/or inflammation to increase the understanding of how various pulmonary insults can alter macrophage efferocytosis.
Supplementary Material
Acknowledgments
This study is funded by Health Effects Institute Walter A. Rosenblith Award and NIEHS R01ES028829 (to K. M. G). We would like to thank Dr. Dianne Walters (Department of Physiology, ECU) for her assistance with obtaining representative images of alveolar macrophages.
Footnotes
Disclosures
The authors declare no conflicts of interest.
Video Link
The video component of this article can be found at https://www.jove.com/video/60109/
References
- 1.Puttur F, Gregory LG, Lloyd CM Airway macrophages as the guardians of tissue repair in the lung. Immunology and Cell Biology. (2019). [DOI] [PubMed] [Google Scholar]
- 2.Gregoire M et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. European Respiratory Journal. 52 (2), (2018). [DOI] [PubMed] [Google Scholar]
- 3.Fan EKY, Fan J Regulation of alveolar macrophage death in acute lung inflammation. Respiratory Research. 19 (1), 50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michlewska S, McColl A, Rossi AG, Megson IL, Dransfield I Clearance of dying cells and autoimmunity. Autoimmunity. 40 (4), 267–273 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharya J, Westphalen K Macrophage-epithelial interactions in pulmonary alveoli. Seminars in Immunopathology. 38 (4), 461–469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donnelly LE, Barnes PJ Defective phagocytosis in airways disease. Chest. 141 (4), 1055–1062 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Morimoto K, Janssen WJ, Terada M Defective efferocytosis by alveolar macrophages in IPF patients. Respiratory Medicine. 106 (12), 1800–1803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vandivier RW et al. Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. American Journal of Physiology Lung Cellular and Molecular Physiology. 297 (4), L677–686 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grabiec AM et al. Diminished airway macrophage expression of the Axl receptor tyrosine kinase is associated with defective efferocytosis in asthma. The Journal of Allergy and Clinical Immunology. 140 (4), 1144–1146 e1144 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Chen W, Frank ME, Jin W, Wahl SM TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu, Immunity. 14 (6), 715–725 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Gao Y, Herndon JM, Zhang H, Griffith TS, Ferguson TA Antiinflammatory effects of CD95 ligand (FasL)-induced apoptosis. Journal of Experimental Medicine. 188 (5), 887–896 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Brien BA, Fieldus WE, Field CJ, Finegood DT Clearance of apoptotic beta-cells is reduced in neonatal autoimmune diabetes-prone rats. Cell Death and Differentiation. 9 (4), 457–464 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Shen ZX et al. Mineralocorticoid Receptor Deficiency in Macrophages Inhibits Atherosclerosis by Affecting Foam Cell Formation and Efferocytosis. Journal of Biological Chemistry. 292 (3), 925–935 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allard B, Panariti A, Martin JG Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Frontiers in Immunology. 9, 1777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamon R et al. Bushfire smoke is pro-inflammatory and suppresses macrophage phagocytic function. Science Reports. 8 (1), 13424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angsana J, Chen J, Liu L, Haller CA, Chaikof EL Efferocytosis as a regulator of macrophage chemokine receptor expression and polarization. European Journal of Immunology. 46 (7), 1592–1599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karaji N, Sattentau QJ Efferocytosis of Pathogen-Infected Cells. Frontiers in Immunology. 8, 1863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brouckaert G et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Molecular Biology of the Cell. 15 (3), 1089–1100 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez-Guevara E et al. Exposure to ozone induces a systemic inflammatory response: possible source of the neurological alterations induced by this gas. Inhalation Toxicology. 26 (8), 485–491 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Robertson S et al. CD36 mediates endothelial dysfunction downstream of circulating factors induced by O3 exposure. Toxicological Sciences. 134 (2), 304–311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilburg-Basnyat B et al. Specialized Pro-Resolving Lipid Mediators Regulate Ozone-Induced Pulmonary and Systemic Inflammation. Toxicological Sciences. 163 (2), 466–477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jakab GJ, Spannhake EW, Canning BJ, Kleeberger SR, Gilmour MI The effects of ozone on immune function. Environ Health Perspect. 103 Suppl 2, 77–89 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilmour MI, Hmieleski RR, Stafford EA, Jakab GJ Suppression and recovery of the alveolar macrophage phagocytic system during continuous exposure to 0.5 ppm ozone. Experimental Lung Research. 17 (3), 547–558 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Nayak DK, Mendez O, Bowen S, Mohanakumar T Isolation and In Vitro Culture of Murine and Human Alveolar Macrophages. Journal of Visualized Experiments. 10.3791/57287 (134) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao H et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. Journal of Lipid Research. 56 (8), 1449–1460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coe LM et al. FGF-23 is a negative regulator of prenatal and postnatal erythropoiesis. Journal of Biological Chemistry. 289 (14), 9795–9810 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yong WK, Abd Malek SN Xanthohumol induces growth inhibition and apoptosis in ca ski human cervical cancer cells. Evidence-Based Complementary and Alternative Medicine. 921306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van de Laar L et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity. 44 (4), 755–768 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Lavin Y et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 159 (6), 1312–1326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beattie L et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. Journal of Hepatology. 65 (4), 758–768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svedberg FRet al.The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nature Immunology. 10.1038/s41590-019-0352-y, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowther JE et al. Pulmonary surfactant protein a inhibits macrophage reactive oxygen intermediate production in response to stimuli by reducing NADPH oxidase activity. Journal of Immunology. 172 (11), 6866–6874 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Silveyra P, Floras J Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Frontiers in Bioscience. 17, 407–429 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schagat TL, Wofford JA, Wright JR Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. Journal of Immunology. 166 (4), 2727–2733 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Gomez Perdiguero E et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 518 (7540), 547–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nebbioso A et al. Time-resolved analysis of DNA-protein interactions in living cells by UV laser pulses. Scientific Reports. 7 (1), 11725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novak Z et al. Efficacy of different UV-emitting light sources in the induction of T-cell apoptosis. Photochemistry and Photobiology. 79 (5), 434–439 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Park YJ et al. PAI-1 inhibits neutrophil efferocytosis. Proceedings of the National Academy of Sciences of the United States of America. 105 (33), 11784–11789 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleeberger SR, Reddy S, Zhang LY, Jedlicka AE Genetic susceptibility to ozone-induced lung hyperpermeability: role of toll-like receptor 4. American Journal of Respiratory Cell and Molecular Biology. 22 (5), 620–627 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Wesselkamper SC, Chen LC, Kleeberger SR, Gordon T Genetic variability in the development of pulmonary tolerance to inhaled pollutants in inbred mice. American Journal of Physiology-Lung Cellular and Molecular Physiology. 281 (5), L1200–1209 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.