

Abstract
Liver repopulation after injury is a crucial feature of mammals which prevents immediate organ failure and death after exposure of environmental toxins. A deeper understanding of the changes in gene expression that occur during repopulation could help identify therapeutic targets to promote the restoration of liver function in the setting of injuries. Nonetheless, methods to isolate specifically the repopulating hepatocytes are inhibited by a lack of cell markers, limited cell numbers, and the fragility of these cells. The development of translating ribosome affinity purification (TRAP) technology in conjunction with the Fah−/− mouse model to recapitulate repopulation in the setting of liver injury allows gene expression profiling of the repopulating hepatocytes. With TRAP, cell type-specific translating mRNA is rapidly and efficiently isolated. We developed a method that utilizes TRAP with affinity-based isolation of translating mRNA from hepatocytes that selectively express the green fluorescent protein (GFP)-tagged ribosomal protein (RP), GFP:RPL10A. TRAP circumvents the long time period required for fluorescence-activated cell sorting that could change the gene expression profile. Furthermore, since only the repopulating hepatocytes express the GFP:RPL10A fusion protein, the isolated mRNA is devoid of contamination from the surrounding injured hepatocytes and other cell types in the liver. The affinity-purified mRNA is of high quality and allows downstream PCR- or high-throughput sequencing-based analysis of gene expression.
Keywords: liver regeneration, cell type-specific, TRAP, TRAP-seq, RNA-seq, immunoprecipitation, expression profiling, hepatocyte, hydrodynamic tail-vein injection
SUMMARY:
Translating ribosome affinity purification (TRAP) enables rapid and efficient isolation of cell type-specific translating mRNA. Here, we demonstrate a method that combines hydrodynamic tail-vein injection in a mouse model of liver repopulation and TRAP to examine the expression profile of repopulating hepatocytes.
INTRODUCTION:
As the main metabolic organ in vertebrates, the liver is responsible for glucose homeostasis, serum protein synthesis, bile acid secretion, and xenobiotic metabolism and detoxification. The liver possesses an extraordinary capacity to regenerate the injured parenchyma upon exposure to toxins to prevent immediate liver failure1. However, failure of regeneration can occur in the setting of acetaminophen or alcohol overconsumption, which can lead to acute liver failure2. Furthermore, chronic liver injury caused by viral hepatitis infection, fatty liver disease, and steatohepatitis can cause liver fibrosis, cirrhosis, and hepatocellular carcinoma3. The only available curative treatment for end-stage liver disease is transplantation but is limited by organ shortage, preventing efficient treatment for all patients4. A better understanding of the recovery process after toxic liver injury is therefore crucial for the development of treatments to stimulate regeneration sufficient to rescue function in the diseased organ.
The most broadly applied model system for the study of liver regeneration is partial hepatectomy in rodents, in which a large proportion of the liver is resected to stimulate rapid hepatocyte expansion5. However, partial hepatectomy does not recapitulate hepatocyte expansion following toxic liver injury due to the lack of immune cell infiltration and hepatocyte cell necrosis often observed in the setting of acute liver injury in humans6. A more suitable system to model this form of organ renewal is the Fah−/− mouse, which lacks functional fumarylacetoacetate hydrolase (FAH) required for proper tyrosine metabolism and develops severe liver damage leading to death7. These mice can be maintained in a healthy state indefinitely by treatment with the drug nitisinone in the drinking water. Alternatively, FAH expression can be restored by transgene delivery to a subset of hepatocytes, which will expand to repopulate the liver upon nitisinone removal8.
To profile the gene expression changes of repopulating hepatocytes, a tool to specifically isolate replicating hepatocytes in the Fah−/− mouse without contamination from the neighboring injured hepatocytes and other cell types is required. Unfortunately, fluorescence-assisted cell sorting (FACS) of hepatocytes is difficult since (1) the fragility of repopulating hepatocytes leads to poor recovery after liver perfusion, (2) replicating hepatocytes are highly variable in size, making isolation of a pure population by FACS difficult, and (3) the procedure time from liver perfusion to RNA isolation is greater than 2 hours, hence gene expression profiles may undergo substantial artificial changes before samples are acquired9.
Alternatively, the expression of epitope-tagged ribosomes specifically in repopulating hepatocytes allows for the rapid isolation of actively translating mRNA bound by ribosomes using affinity purification immediately after organ harvest, with bulk liver tissue lysates. Here, we describe a protocol to perform translating-ribosome affinity purification (TRAP)10 followed by high-throughput RNA-sequencing (TRAP-seq), to specifically isolate and profile mRNA in repopulating hepatocytes in the Fah−/− mouse9. Coexpression of green fluorescent protein-tagged ribosomal protein (GFP:RPL10A) with FAH allows affinity purification of translating mRNA bound by polysomes containing GFP:RPL10A. This method avoids any cell dissociation steps, such as liver perfusion to isolate fragile repopulating hepatocytes. Instead, it utilizes lysis of whole organ tissue and antibodies to rapidly extract the RNA specifically from the target cells. Finally, isolation of abundant, high-quality mRNA via TRAP-seq enables downstream applications such as sequencing analysis to profile the dynamic change of gene expression during the repopulation process.
PROTOCOL:
All methods that involve the use of mice are consistent with the guidelines provided by the IACUC of the Penn Office of Animal Welfare at the University of Pennsylvania.
- Reagent preparation
- 1.1 Cycloheximide
-
1.1.1. To make 500 μL of 0.1 g/mL cycloheximide, suspend 50 mg of cycloheximide in 500 μL of methanol.CAUTION: Cycloheximide is extremely toxic to the environment and can cause congenital malformation. All wastes and buffers containing cycloheximide should be collected for proper disposal.NOTE: Cycloheximide can be stored at 4 °C for up to 1 day. Cycloheximide inhibits translation.
- 1.2. Dithiothreitol (DTT)
-
1.2.1. To make 1 mL of 1 M DTT, suspend 0.15 g of DTT powder in RNase-free water.CAUTION: DTT can cause irritation to the skin, eye, and respiratory tract.NOTE: DTT is a detergent. DTT can be stored at −20 °C. It is recommended to store 1 M DTT in single-use aliquots of 50 μL.
- 1.3. Deoxycholate (DOC)
-
1.3.1. To make 10% DOC, suspend 1 g of DOC in a 50 mL conical tube and add RNase-free water up to 10 mL. Shake vigorously until the powder is dissolved.NOTE: The 10% DOC solution is slightly yellow and can be stored at room temperature (RT) for up to 1 year. DOC is used for nuclear lysis.
- 1.4. GFP antibodies
-
1.4.1. Aliquot GFP antibodies when using for the first time. Snap freeze the aliquots and store at −80 °C.NOTE: It is recommended to store 50 μg of GFP antibodies in single-use aliquots.
- 1.5. B biotinylated protein L
-
1.5.1. Resuspend biotinylated protein L in 1x phosphate-buffered saline (PBS) to make the final concentration 1 μg/μL.NOTE: Resuspended solution can be stored at −20 °C for up to 6 months.
- Buffer preparation
- 2.1. Bovine serum albumin (BSA) buffer
-
2.1.1. To make 50 mL of 3% BSA buffer, add 1.5 g of IgG- and protease-free BSA powder into 40 mL of PBS followed by vortex. After the BSA is dissolved, add PBS to a final volume of 50 mL.NOTE: The BSA buffer can be stored at 4 °C for up to six months.
- 2.2. Dissection buffer
-
2.2.1. To make 50 mL of dissection buffer stock, combine 5 mL of 10x Hank’s balanced salt solution (HBSS), 125 μL of 1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1,750 μL of 1 M glucose, and 200 μL of 1 M NaHCO3. Add RNase-free water to a final volume of 50 mL.NOTE: The dissection buffer stock can be stored at 4 °C for up to six months.
- 2.2.2. Immediately prior to use, add 100 μg/mL of 0.1 g/mL cycloheximide and keep on ice.
- 2.3. High-salt buffer
-
2.3.1. To make 50 mL of high-salt buffer stock, add 1 mL of 1 M HEPES, 8.75 mL of 2 M KCl, 500 μL of 1 M MgCl2, and 500 μL of 100% nonylphenyl polyethylene glycol (Table of Materials) to RNase-free water.NOTE: The high-salt buffer stock can be stored at 4 °C for up to six months.
- 2.3.2. Immediately prior to use, add 0.5 μL/mL of 1 M DTT and 1 μL/mL of 0.1 g/mL cycloheximide. Keep the fresh high-salt buffer on ice.
- 2.4. Low-salt buffer
-
2.4.1. To make 50 mL of low-salt buffer stock, add 1 mL of 1 M HEPES, 3.75 mL of 2 M KCl, 500 μL of 1 M MgCl2, and 500 μL of 100% nonylphenyl polyethylene glycol to 44.25 Ml of RNase-free water.NOTE: The low-salt buffer stock can be stored at 4 °C for up to six months.
- 2.4.2. Add 0.5 μL/mL of 1 M DTT and 1 μL/mL of 0.1 g/mL cycloheximide prior to use. Keep the fresh low-salt buffer on ice.
- 2.5. Tissue lysis buffer
-
2.5.1. To make 50 mL of tissue lysis buffer stock, combine 1 mL of 1 M HEPES, 3.75 mL of 2 M KCl, and 500 μL of 1 M MgCl2. Add RNase-free water to a final of 50 mL.NOTE: The dissection buffer stock can be stored at 4 °C for up to six months.
- 2.5.2. Add 1 tablet/10mL of EDTA-free protease inhibitor, 1 μL/mL of 0.1 g/mL cycloheximide, 10 μL/mL of RNase inhibitors each immediately prior to use. Keep the fresh tissue lysis buffer on ice.
- Conjugation of antibodies to magnetic beads
- 3.1. Antibodies
-
3.1.1. Calculate the amount of GFP antibodies required for all samples and prepare for one extra sample.NOTE: For each sample, 50 μg of each GFP antibody is required.
-
3.1.2. Thaw GFP antibodies on ice and spin at maximum speed (>13,000 x g) for 10 min at 4 °C and transfer supernatants to a new microcentrifuge tube.NOTE: The antibody preparation step can be performed prior to bead preparation and the thawed antibodies can be kept on ice. Alternatively, this part can be performed during incubation of the magnetic bead and biotinylated protein L.
- 3.2. Resuspend magnetic beads
- 3.2.1. Resuspend magnetic beads (Table of Materials) by gentle pipetting.
- 3.2.2. For each sample, use 150 μL of magnetic bead. Calculate the volume of magnetic bead required for all samples and prepare one extra. Transfer the resuspended magnetic beads to a 1.5 mL or 2 mL microcentrifuge tube. If more than 1 mL is required for an experiment, split the total amount into equal volumes.
- 3.2.3. Collect beads on a magnetic stand for >1 min and remove the supernatant. Remove the microcentrifuge tube from the magnetic stand and add 1 mL of PBS followed by pipetting up and down to wash the beads. Collect beads on a magnetic stand for >1 min and remove PBS.
- 3.3. Preparation of protein L-coated beads
- 3.3.1. For each sample, use 60 μL of biotinylated protein L. If protein L is previously resuspended and stored at −20 °C, thaw protein L on ice. If protein L is resuspended prior to use, take the amount required for all samples and prepare one extra.
-
3.3.2. Add the calculated volume of biotinylated protein L to the resuspended and washed magnetic beads. Add 1x PBS to make the final volume 1 mL if using a 1.5 mL microcentrifuge tube, or 1.5 mL if using a 2 mL microcentrifuge tube. Incubate magnetic beads with biotinylated protein L for 35 min at RT on a tube rotator.NOTE: Antibodies can be prepared at this step while the beads are incubating with biotinylated protein L.
- 3.3.3. Collect protein L-coated beads on a magnetic stand for >1 min and remove the supernatant. Remove the microcentrifuge tube from the magnetic stand and add 1 mL of 3% BSA buffer followed by gentle pipetting for at least 5 times to wash the protein L-coated beads.
- 3.3.4. Collect coated beads on a magnetic stand for >1 min and remove the supernatant. Repeat the washing steps with 3% BSA for another 4 times (a total of 5 times).
- 3.4. Antibody binding
-
3.4.1. Add the calculated amount of GFP antibodies into the protein L-coated beads and incubate for 1 h at 4 °C on a tube rotator.NOTE: After antibody incubation, take special care to not vortex the affinity matrix.
-
3.4.2. During incubation, prepare low-salt buffer by calculating the total volume required for all samples and add 0.5 μL/mL of 1 M DTT and 1 μL/mL of 0.1 g/mL cycloheximide to low-salt buffer stock prior to use.NOTE: 3 mL of low-salt buffer for washing each tube of GFP-conjugated beads and 200 μL/sample for resuspension of the GFP-conjugated beads is required. Fresh low-salt buffer can be kept on ice for a couple of hours.
- 3.4.3. Collect the affinity matrix on a magnetic stand for >1 min and remove the supernatant. Add 1 mL of low-salt buffer and gently pipette up and down to wash the affinity matrix. Collect the affinity matrix on a magnetic stand for >1 min and remove low-salt buffer. Repeat the washing steps with low-salt buffer for another 2 times (a total of 3 times).
-
3.4.4. Resuspend the beads in low-salt buffer so that each sample has 200 μL of affinity matrix.NOTE: The affinity matrix can be stored in 0.02% NaN3 at 4 °C for up to 2 weeks. The affinity matrix should be quickly washed in low-salt buffer 3 times and resuspended gently on a tube rotator at 4 °C for at least 10 min if the affinity matrix is prepared within 1 week or overnight if the affinity matrix is stored for over 1 week. The protocol can be paused after this step.CAUTION: Sodium azide is extremely toxic to the environment. Contact with acids produces toxic gas. All wastes should be collected for proper disposal.
- Liver tissue lysis
- 4.1. Buffer preparation and equipment setup
-
4.1.1. Calculate the number of microcentrifuge tubes required, label and chill on ice.NOTE: Usually, seven 1.5 mL microcentrifuge tubes are required for each sample: 1 for the remaining dissected liver, 4 for 4 mL of homogenized liver lysate, and 2 for transferring supernatants.
-
4.1.2. Prepare fresh dissection buffer by calculating the total volume required for all samples and add 1 μL/mL of 0.1 g/mL cycloheximide. Place the fresh dissection buffer on ice to keep cold throughout the experiment.NOTE: For each sample, 10 mL of dissection buffer is required.
-
4.1.3. Prepare fresh lysis buffer by calculating the total volume required for all samples and add 1 tablet/10mL of EDTA-free protease inhibitor, 1 μL/mL of 0.1 g/mL cycloheximide, and 10 μL/mL of RNase inhibitors each. Keep the lysis buffer on ice throughout the experiment.NOTE: For each sample, 4 mL of lysis buffer is required.
- 4.1.4. Set up the tissue grinder (Table of Materials) so that the polytetrafluoroethylene (PTFE)-glass tubes can be placed on ice during homogenization of liver pieces. Put 4 mL of cold lysis buffer in the PTFE-glass tubes.
- 4.2. Repopulating liver homogenization
- 4.2.1. Euthanize an 8–12-week-old Fah−/− mouse injected with the TRAP vector and repopulated for 1–4 weeks with anesthesia and cervical dislocation according to approved animal experimental guidelines.
- 4.2.2. Place the mouse on a dissection board and spray the abdomen with 70% ethanol. Tent the skin and peritoneum low in the abdomen using forceps and use scissors to make a transverse incision. Continue to cut with the scissors to make a wide U-shaped peritoneal flap, with care to not cut the viscera. Flip the peritoneal flap over the sternum to expose the liver.
-
4.2.3. Carefully remove the liver using fine scissors and forceps and quickly place the tissue in cold dissection buffer to rinse. To homogenize frozen tissues, quickly move the desired amount of liver tissue into PTFE-glass tubes with cold lysis buffer without thawing the tissue.NOTE: The dissected tissue can be flash-frozen and stored at −80 °C after it is washed with dissection buffer. The protocol can be paused after this step.
- 4.2.4. Weigh the liver on a Petri dish and Isolate 200–500 mg of liver pieces and move into the PTFE-glass tubes. Place the remaining liver tissue into a pre-chilled microcentrifuge tube and flash freeze.
- 4.2.5. Homogenize the samples in a motor-driven homogenizer starting at 300 rpm to dissociate hepatocytes from the liver structure for at least 5 strokes. Lower the glass tube each time but take care to not let the pestle rise above the solution to prevent aeration that could cause protein denaturation.
- 4.2.6. Raise the speed to 900 rpm to fully homogenize the liver tissues for at least 12 full strokes.
-
4.2.7. Transfer the lysate into the labeled and pre-chilled tubes, with no more than 1 mL of lysate per 1.5 mL microcentrifuge tubes. If 4 mL of lysis buffer is used, keep 1 tube and flash freeze the remaining 3 tubes.NOTE: The lysates can be kept on ice for up to 1 h while dissecting the next animal and preparing fresh lysates. The homogenized liver can be flash frozen after the lysis step and stored at −80 °C. There could be a 50% decrease in isolated RNA if frozen lysates are used. The protocol can be paused after this step.
- 4.3. Nuclear lysis
- 4.3.1. Centrifuge the liver lysate at 2,000 x g at 4 °C for 10 min and transfer the supernatant to a new, prechilled microcentrifuge tube on ice.
- 4.3.2. Add 1/9 of the supernatant volume of 10% nonylphenyl polyethylene glycol to make the final concentration 1% nonylphenyl polyethylene glycol and mix by gently inverting the microcentrifuge tubes.
- 4.3.3. Quickly spin down the microcentrifuge tubes and add 1/9 of the sample volume of 10% DOC to make the final concentration 1% DOC and mix by gently inverting the microcentrifuge tubes. Quickly spin down the microcentrifuge tubes and incubate on ice for 5 min.
-
4.3.4. Centrifuge the nuclear lysate at 20,000 x g at 4 °C for 10 min and transfer the supernatant to a new, prechilled microcentrifuge tube on ice.NOTE: The mitochondria-depleted supernatant can be placed on ice for a couple of hours while the remaining samples are being collected.
- Immunoprecipitation
- 5.1. For each tube, take out 1% of the total volume of the supernatant as a pre-immunoprecipitation control to compare target enrichment after incubation with the affinity matrix. Place pre-immunoprecipitation controls on a tube rotator at 4 °C overnight, the same way as the immunoprecipitated samples are processed.
-
5.2. Add 200 μL of affinity matrix to each sample. Take extra care to resuspend the beads by gentle pipetting prior to adding the affinity matrix to each sample. Incubate the lysates with affinity matrix at 4 °C overnight with gentle mixing on a tube rotator.NOTE: The protocol can be paused for up to a day after this step.
- RNA isolation
- 6.1. Buffer preparation and equipment setup
- 6.1.1. Place the magnetic rack at 4 °C for at least 30 min to pre-chill and keep the rack on ice throughout the experiment.
-
6.1.2. Calculate the number of microcentrifuge tubes required and pre-chill on ice or at 4 °C.NOTE: Usually, each sample requires 1 microcentrifuge tube for the final purified RNA.
-
6.1.3. Quickly spin down the tubes from step 5.2 and collect the beads by placing on the magnetic rack for at least 1 min. Collect or discard the supernatant in additional microcentrifuge tubes.NOTE: The collected supernatant that contains unbound fraction can be flash-frozen and stored at −80 °C to compare with the bound fraction for transcript enrichment after purification. The protocol can be paused after this step.
-
6.1.4. Prepare high-salt buffer by adding 0.5 μL/mL of 1 M DTT and 1 μL/mL of 0.1 g/mL cycloheximide to high-salt buffer stock.NOTE: For each sample, 5 mL of high-salt buffer is required.
- 6.2. RNA isolation
-
6.2.1. Add 1 mL of fresh high-salt buffer to each tube followed by gentle pipetting for at least 5 times without introducing bubbles.NOTE: Insufficient washing could introduce backgrounds of unbound transcripts while the introduction of bubbles could accelerate RNA degradation.
- 6.2.2. Collect beads on a magnetic stand for >1 min and remove the supernatant. Repeat the washing steps with high-salt buffer for another 4 times (a total of 5 times).
- 6.2.3. Remove remaining high-salt buffer and remove microcentrifuge tubes from the magnetic stand and place at RT for 5 min to warm up.
-
6.2.4. Resuspend the beads in 100 μL of lysis buffer (provided in the RNA isolation kit) with β-mercaptoethanol.NOTE: Any RNA isolation and purification kit that contains the denaturant guanidine thiocyanate can be used as a lysis buffer to release bound RNA from the affinity matrix. RNA extraction should be processed at RT since guanidine thiocyanate can crystallize at low temperatures.
- 6.2.5. Vortex the beads and buffer for at least 5 s at the highest speed, quickly spin down to collect the buffer on the side of the microcentrifuge tube and incubate the beads with the buffer at RT for 10 min to release the bead-bound RNA into the lysis buffer.
-
6.2.6. Collect beads on a magnetic stand for >1 min and collect the supernatant to proceed immediately to RNA cleanup according to the RNA purification protocol as specified in the kit (Table of Materials).NOTE: The supernatant containing the eluted RNA in lysis buffer can also be stored at −80 °C for up to 1 month before cleanup by warming up to RT upon thawing.
-
6.2.7. To achieve maximum quality of the isolated RNA, perform all optional steps including DNase digestion and all RNA elution steps. Heat up the elution buffer provided by the RNA isolation kit or RNase-free water to 60 °C for maximum RNA recovery.NOTE: The isolated RNA can be stored at −20 °C for up to 1 month or −80 °C for several years. The protocol can be paused after this step.
- Optional RNA quality analysis (recommended)
-
7.1. Assess RNA quality using a bioanalyzer11 and quantity with a spectrophotometer12 to determine if repeating the immunoprecipitation process is required to obtain ample and high-quality RNA.NOTE: The optimal RNA quality for high-throughput sequencing should follow protocols specified by library preparation kits and sequencing platforms.
-
-
Downstream applications
NOTE: Total RNA isolated by the TRAP protocol can be used in a number of standard downstream applications, including RNA-seq (TRAP-seq). Standard reverse transcription and quantitative PCR protocols can also be used following TRAP.
8.1. For TRAP-seq, perform RNA-seq library preparation according to standard methods13.
8.2. For RNA-seq, prepare cDNA sequencing libraries using commercial RNA-seq kits with oligo d(T)-based enrichment of polyadenylated poly(A) transcripts. Alternatively, if the total RNA quality is lower than recommended for poly(A) enrichment, use rRNA depletion modules. However, expect to see more rRNA alignment after sequencing.
Table of Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| Absolutely RNA Miniprep Kit | Agilent | 400800 | |
| Anti-GFP antibodies | Memorial Sloan-Kettering Antibody & Bioresource Core | GFP Ab #19C8 and GFP Ab #19F7 | |
| Bovine Serum Albumin, IgG-Free, Protease-Free | Jackson ImmunoResearch | 001-000-162 | |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 | |
| Cycloheximide | Millipore Sigma | C7698 | |
| D-Glucose, Dextrose | Fisher Scientific | D16 | |
| Deoxycholic acid, DOC | Millipore Sigma | D2510 | |
| DL-Dithiothreitol | Millipore Sigma | D9779 | |
| Dynabeads MyOne Streptavidin T1 | Thermo Fisher Scientific | 65602 | |
| HBSS (10X), calcium, magnesium, no phenol red | Thermo Fisher Scientific | 14065–056 | |
| HEPES, 1M Solution, pH 7.3, Molecular Biology Grade, Ultrapure, Thermo Scientific | Thermo Fisher Scientific | AAJ16924AE | |
| Magnesium chloride, MgCl2 | Millipore Sigma | M8266 | |
| Methanol | Fisher Scientific | A452 | |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England BioLabs | E7490S | |
| NEBNext Ultra RNA Library Prep Kit for Illumina | New England BioLabs | E7530S | |
| Nonylphenyl polyethylene glycol, NP-40. IGEPAL CA-630 | Millipore Sigma | I8896 | |
| Nuclease-Free Water, not DEPC-Treated | Ambion | AM9932 | |
| PBS Buffer (10X), pH 7.4 | Ambion | AM9625 | |
| Pierce Recombinant Protein L, Biotinylated | Thermo Fisher Scientific | 29997 | |
| Potassium chloride, KCl | Millipore Sigma | P4504 | |
| RNaseZap RNase Decontamination Solution | Invitrogen | AM9780 | |
| RNasin Ribonuclease Inhibitors | Promega | N2515 | |
| RNA 6000 Pico Kit & Reagents | Agilent | 5067–1513 | |
| Sodium azide, NaN3 | Millipore Sigma | S2002 | |
| Sodium bicarbonate, NaHCO3 | Millipore Sigma | S6297 | |
| SUPERase·In RNase Inhibitor | Invitrogen | AM2694 | |
| Overhead Stirrer | DWK Life Sciences (Wheaton) | 903475 | |
| 10 ml Tissue Grinder, Potter-Elv, Coated | DWK Life Sciences (Wheaton) | 358007 | |
REPRESENTATIVE RESULTS:
To profile gene expression in repopulating hepatocytes of the Fah−/− mouse, Gfp:Rpl10a fusion and Fah transgenes are co-delivered within a transposon-containing plasmid8 (TRAP vector) to livers by hydrodynamic injection (Figure 1A). The removal of nitisinone induces a toxic liver injury that creates a selection pressure for hepatocytes stably expressing FAH to repopulate the injured parenchyma9. Immunofluorescence staining confirms the co-expression of FAH and the GFP:RPL10A fusion protein in repopulating hepatocytes after two weeks of liver repopulation (Figure 1B).
Figure 1: Implementation of TRAP with Fah−/− to profile gene expression change of repopulating hepatocytes.
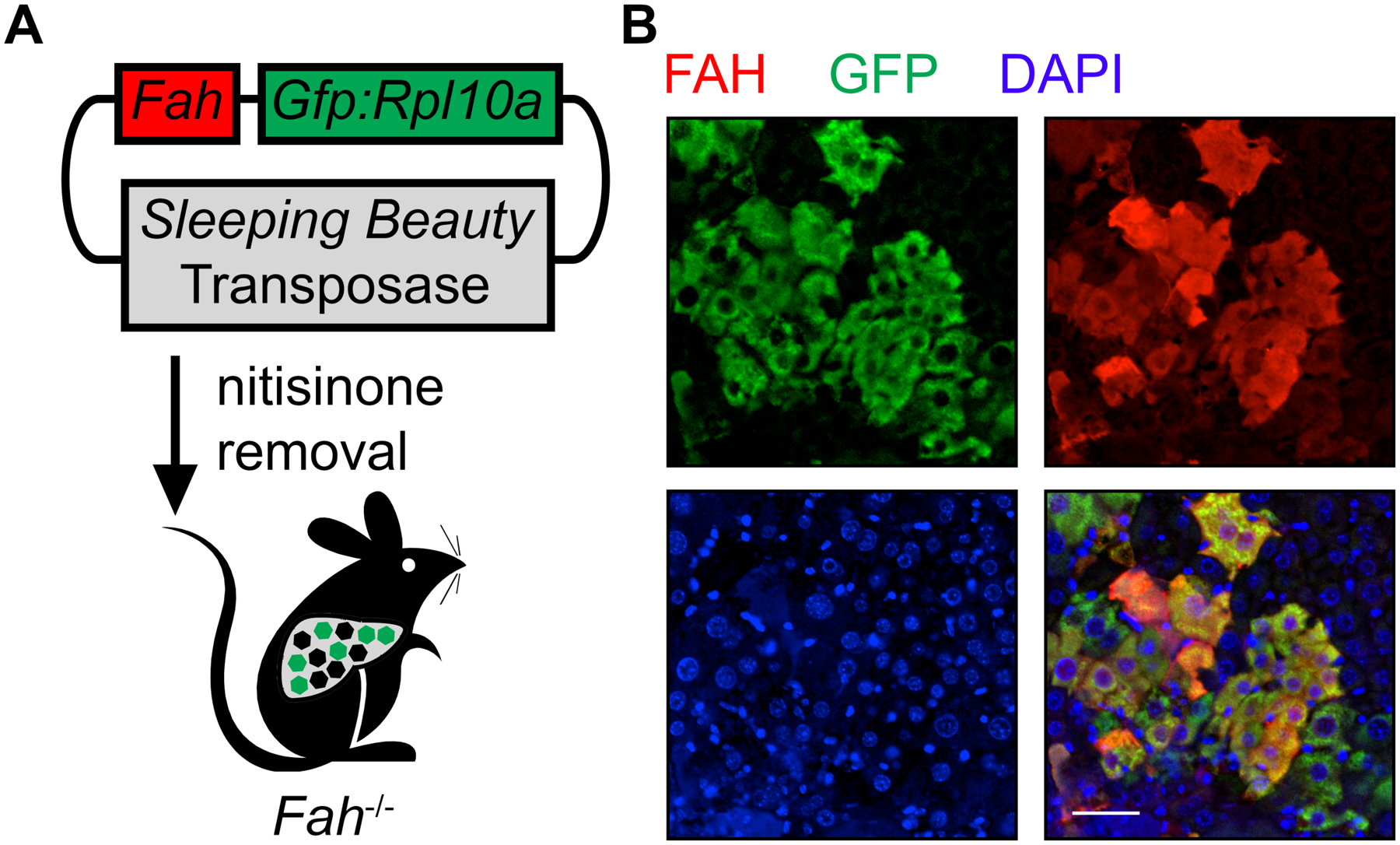
(A) Schematic of expressing the GFP:RPL10A fusion protein with FAH in the Sleeping Beauty transposon system followed by injection into the Fah−/− mouse. Green hexagons indicate repopulating hepatocytes with stable expression of FAH and GFP:RPL10A, whereas black hexagons represent injured, dying hepatocytes. (B) Representative immunofluorescence staining demonstrates coexpression of GFP-tagged ribosomal protein L10A (green) and FAH (red) in the repopulating hepatocytes. Scale bar = 50 μm.
In the following representative experiment, TRAP-seq was performed using quiescent and repopulating mouse hepatocytes. First, to obtain GFP-tagged ribosomes from quiescent hepatocytes, transgenic RosaLSL-GFP-L10A mice were injected with AAV8-TBG-Cre 7 days prior to sacrifice to induce GFP:RPL10A expression in all hepatocytes14. We also processed a liver sample collected from a wild type mouse as a negative control to ensure isolation of translating mRNA was specific, meaning RNA could only be extracted from mice expressing GFP:RPL10A. The concentration of isolated RNA correlated with the number of cells expressing the fusion protein, with the quiescent sample producing the highest yield since all hepatocytes express GFP:RPL10A after AAV8-TBG-Cre injection (Figure 2A). Conversely, barely any RNA was detectable in wild type controls that did not have the GFP:RPL10A transgene, indicating the TRAP procedure is highly specific and has a low background. When TRAP was used on liver tissue undergoing repopulation with GFP:RPL10A-transduced hepatocytes, abundant, high-quality RNA was obtained (Figure 2B). In contrast, no RNA trace was detected via bioanalyzer for the negative control sample.
Figure 2: TRAP allows cell type-specific isolation of high-quality RNA.
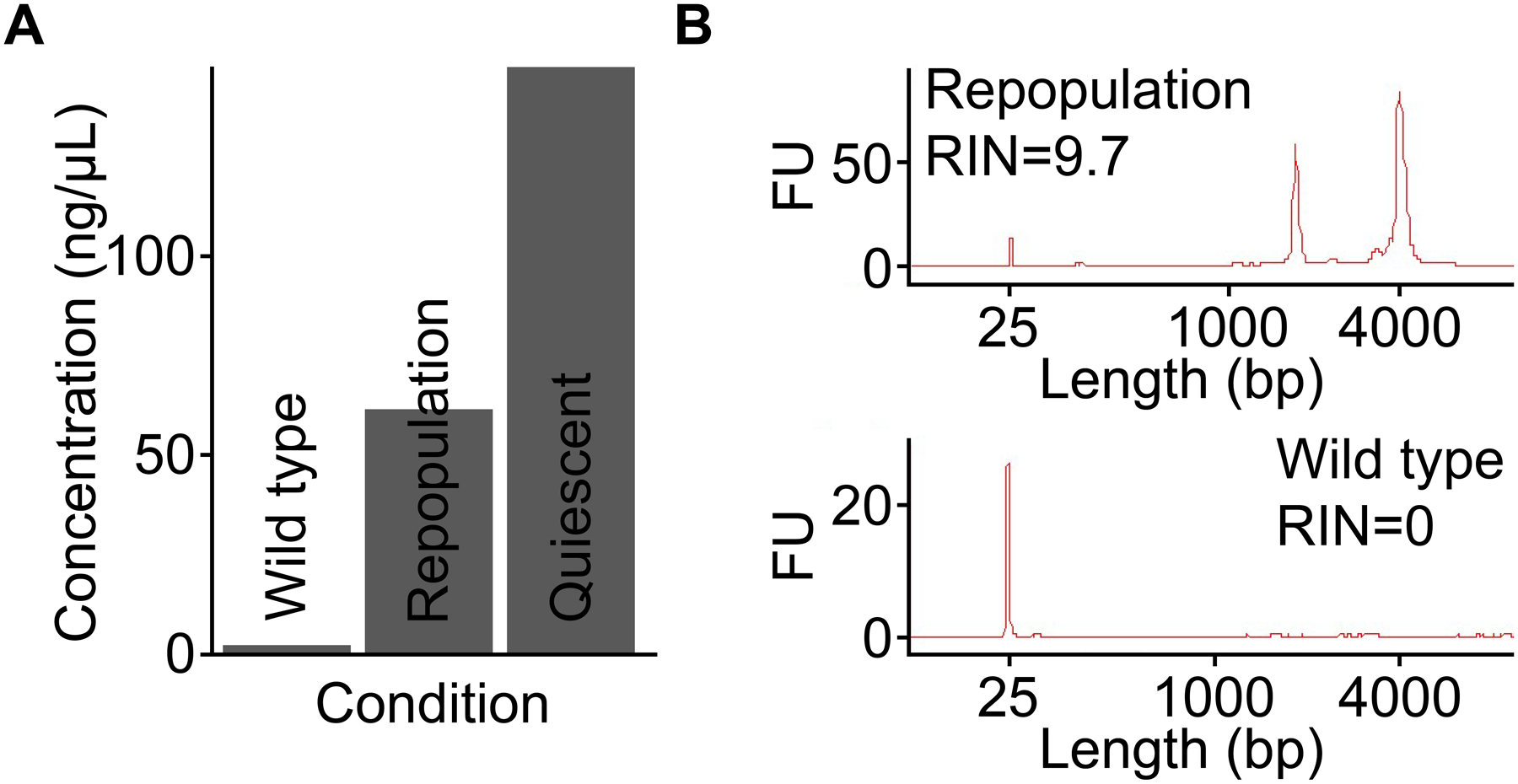
(A) The yield of RNA is positively correlated with the number of hepatocytes expressing GFP:RPL10A. The low yield of RNA from a wild type mouse demonstrates the specificity of TRAP from sources without the expression of GFP:RPL10A. (B) Bioanalyzer traces of total RNA isolated from repopulating livers expressing GFP:RPL10A and from wild type livers demonstrate the specificity of TRAP. Total RNA isolated from wild type liver tissue devoid of the GFP:RPL10A transgene shows that minimal RNA has been collected, whereas transgene-expressing tissue provides ample high-quality RNA. Note that ribosomal RNA peaks are present following successful TRAP10. FU, fluorescence unit. RIN, RNA integrity number.
Downstream gene expression analysis can be carried out via reverse transcription and quantitative PCR or RNA-seq on TRAP-isolated RNA. Gsta1 encodes glutathione S-transferase that plays an important role for the metabolism of glutathione, the main detoxifying peptide to protect the liver against oxidative stress damage15. Gsta1 expression is induced by over 10-fold in repopulating hepatocytes as compared to quiescent hepatocytes, while no threshold cycle (Ct) value was detected with TRAP-isolated RNA from the wild type mouse due to the lack of input RNA (Figure 3A). Note that RNA quality can greatly impact the gene expression analysis. In the case of RNA-seq experiments, assessment of RNA quality should be performed according the recommendations of the library preparation kit and the sequencing platform (Figure 3B). A bioanalyzer is often used to determine the RNA integrity number (RIN), with a high RIN correlating with a higher rate of mRNA alignments to the genome (Figure 3B, left), whereas a lower RIN leading to a higher rate of ribosomal reads, indicating mRNA degradation (Figure 3B, right). Figure 3C,D demonstrates that TRAP-seq can identify differential gene expression in quiescent and repopulating hepatocytes. For instance, Alb expression is inhibited and Afp expression is upregulated during liver repopulation, reflecting that the replicating hepatocytes assume a less differentiated state to inhibit liver metabolic functions during repopulation9,16.
Figure 3: TRAP-isolated RNA can be used for downstream gene expression analysis.
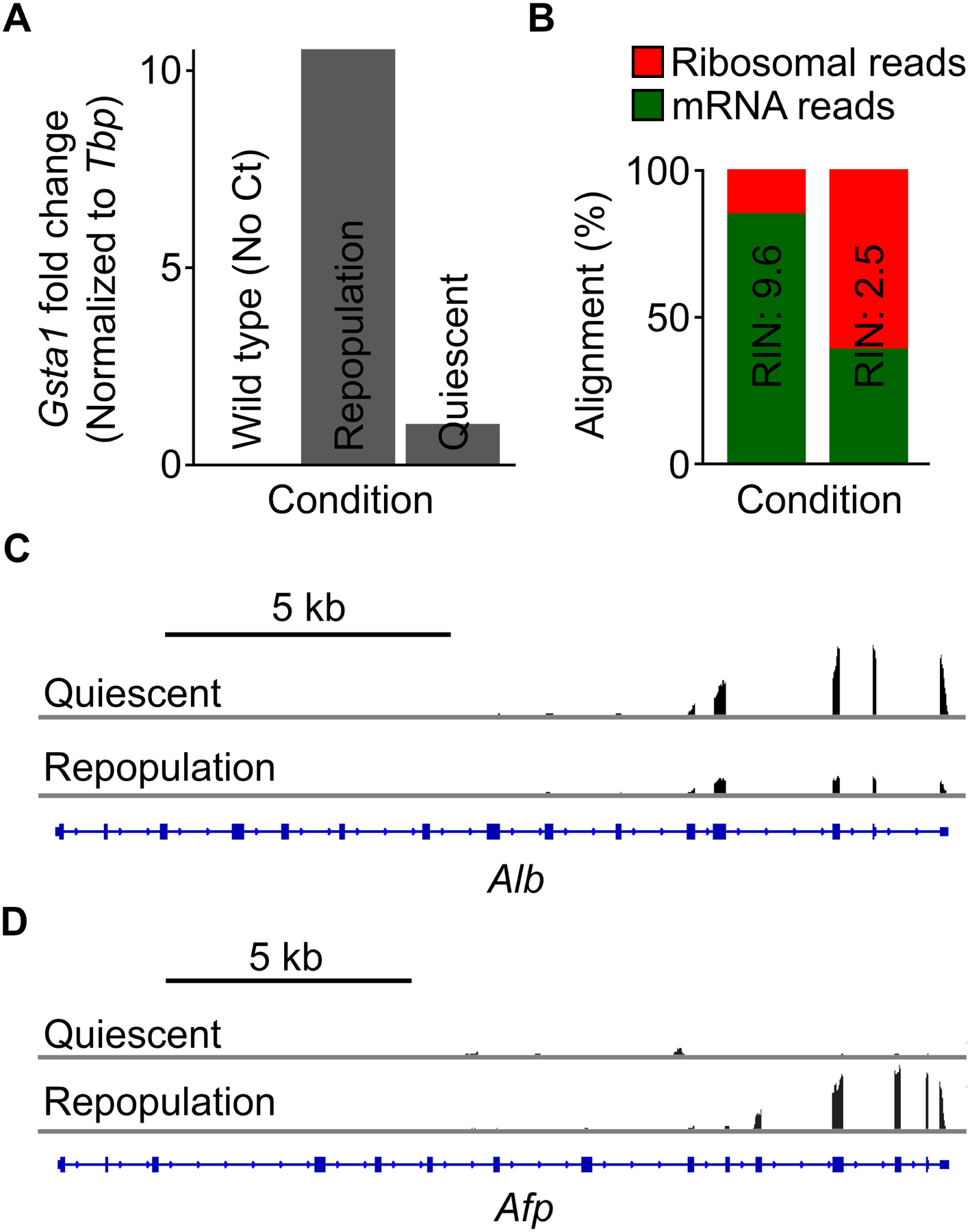
(A) Representative reverse transcription and quantitative PCR results of Gsta1 in quiescent and repopulating hepatocytes. No Ct value was detected with RNA isolated from wild type animals. (B) Alignment analysis of isolated RNA after high-throughput sequencing, demonstrating the importance of determining RNA integrity after isolation. High-quality RNA results in a higher percentage of mRNA reads (green), while low-quality RNA leads to a much higher percentage of ribosome reads (red), as most mRNA is degraded. RIN, RNA integrity number. (C,D) Integrative Genomics Viewer (IGV) tracks of RNA-seq reads of mRNA affinity-purified from quiescent and repopulating hepatocytes at the (C) Alb and (D) Afp loci. Note that the 3’ read bias is typical of a poly(A) selection pipeline.
DISCUSSION:
TRAP-seq is a technique for cell type-specific isolation of translating mRNA via epitope-tagged ribosomes and presents an alternative to FACS approaches, as it circumvents limitations such as time requirements of FACS9. Instead, TRAP allows rapid and efficient isolation of RNA directly from bulk tissues, helping to avoid any alterations in gene expression. TRAP-seq is especially well-suited for use in the repopulating Fah−/− mouse liver, as hepatocyte expansion following removal of nitisinone is cell autonomous and enables gene expression profiling of the subset of hepatocytes with integrated transgenes. The TRAP vector can also be coexpressed with gene-activating or -silencing molecules17, including cDNA, short-hairpin RNA, and guide RNA, to study the effects on global gene expression of activation or inhibition of a specific gene. Alternatively, the RosaLSL-GFP-L10A transgenic mouse provides the ability to profile gene expression in any cell with Cre recombinase activity. Since GFP:RPL10A can be specifically expressed in any cells that express Cre, the role of other cell types in the liver during liver injury and repopulation could be studied. For instance, crossing CK19-Cre mouse with the TRAP transgenic mouse could be used to express GFP:RPL10A in cholangiocytes followed by TRAP-seq to study the change of gene expression in the biliary epithelium during the repopulation process.
To ensure accurate profiling of gene expression, it is critical to prepare all buffers and the affinity matrix prior to tissue dissection. All steps should be performed on ice with cold buffers unless otherwise specified to ensure polysome stabilization10 and prevent RNA degradation. All buffers should be prepared with RNase-free reagents and the TRAP-seq protocol should be carried out in an RNase-free environment to prevent RNA degradation and low yield of immunoprecipitated RNA. The affinity matrix can be prepared up to 2 weeks prior to use with gentle resuspension on a tube rotator overnight. Special care should be taken to not vigorously shake the matrix to prevent disruption of the antibody-conjugated, protein L-coated magnetic beads. The methods to prepare the affinity matrix includes conjugation of magnetic beads to biotinylated protein L followed by incubation with anti-GFP antibodies. However, commercially available protein A/G magnetic beads can be substituted; if used, skip the initial conjugation step and proceed directly to antibody binding. Furthermore, alternative epitope tags are presumably feasible with the above protocol with appropriate modification.
There are various points in which the RNA isolation and purification step can be paused (see protocol above). However, once liver samples have been harvested, continuing to the immunoprecipitation is recommended, as the yield of isolated RNA could drop by ~50% with freezing at this step10. Tissues should be quickly rinsed with dissection buffer that contains cycloheximide to inhibit mRNA translation. Insufficient tissue lysis could also contribute to low RNA yield. It is critical to homogenize tissues on ice until no tissue chunks are visible with the motor homogenizer while ensuring minimal aeration10. Additionally, sufficient washing with high-salt buffer is crucial to ensure removal of nonspecific binding of ribosomal proteins to the affinity matrix. Including a wild type negative control helps to assess the specificity of the immunoprecipitation and the efficiency of the wash steps. Additionally, using a commercial RNA purification kit that includes RNase-free DNase treatment will increase RNA purity.
Moreover, it is recommended to verify the expression and abundance of the GFP:RPL10A fusion protein and assess the amount of tissue required to obtain ample RNA for downstream analysis. Tissue sections or lysates could be used for immuno-based detection methods to validate the expression of GFP:RPL10A. The amount of RNA isolated can vary by: (1) the number of cells expressing GFP:RPL10A, (2) the expression level of the transgene, and (3) the size and ploidy of the cells expressing the transgene. A pilot experiment using half and double the amount of the recommended amount of tissue could be useful in determining the optimal input lysate for TRAP-seq. In our hands, we could obtain ~150 ng of RNA with as little as 1–2% of hepatocytes expressing GFP:RPL10A from 200 mg of the repopulating Fah−/− liver, representing ~2×105 polyploid hepatocytes with transgene expression9.
The TRAP-seq methodology isolates ribosome-bound mRNA to profile a cell’s translating mRNA pool. The resulting sequencing reads therefore correspond to the ‘translatome’ rather than the transcriptome. Note that translating ribosome footprints will not be collected, as TRAP is performed on native rather than crosslinked complexes. If footprinting analyses are desired, the above protocol should be modified with relevant cross-linking followed by immunoprecipitation (CLIP) methodologies18. Another limitation of TRAP is the requirement of a sufficient number of cells expressing the GFP:RPL10A fusion protein. For experiments in which the cell type of interest is small, combining multiple biological samples may be required to isolate sufficient RNA to enable RNA-seq19. Furthermore, TRAP-seq requires the presence of GFP:RPL10A in the cell type of interest. This could pose a challenge if there is no specific delivery system to the cells or if a cell-type specific promoter to drive Cre expression is not available.
The recent development of single-cell RNA-seq (scRNA-seq) technology has allowed direct sequencing followed by in silico identification of various cell types, enabling sequencing without sorting for specific cell types of interests20–22. However, scRNA-seq still requires dissociation of cells from the organ. In the case of the Fah−/− repopulation model, liver perfusion and hepatocyte isolation is extremely difficult and inefficient due to the fragility of both the injured and replicating hepatocytes. In fact, we have not yet been able to isolate sufficient hepatocytes from Fah−/− mice undergoing repopulation after hydrodynamic injection of Fah plasmids. Additionally, in the time it takes to process tissues, gene expression levels could change. Protocols for liver perfusion take up to 30 minutes of warm ischemia time. Future methodologies to optimize liver perfusion to decrease processing time and increase isolation efficiency could allow scRNA-seq integration to the Fah−/− mouse model system and possibly to other injury and repopulation models. This would also enable the study of all liver cell types.
In conclusion, the integration of TRAP-seq with the Fah−/− mouse allows specific isolation and gene expression profiling of replicating hepatocytes to identify therapeutic targets that could promote liver repopulation. This method can be implemented to study other cell types in the liver and other organ systems for disease-specific identification of gene expression changes to identify potential drug targets or biomarkers. An analogous technique can be used to collect nuclei from repopulating hepatocytes using affinity purification, and then to perform an epigenetic analysis of these specific cells16.
ACKNOWLEDGMENTS:
This work was supported by the following grants: F31-DK113666 (AWW), K01-DK102868 (AMZ), K08-DK106478 (KJW), and P30-DK050306 (Pilot grant to KJW).
Footnotes
DISCLOSURES:
The authors have nothing to disclose.
REFERENCES:
- 1.Taub R Liver regeneration: from myth to mechanism. Nature Reviews Molecular Cell Biology. 5 (10), 836–847, 10.1038/nrm1489 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Lee WM Etiologies of acute liver failure. Seminars in Liver Disease. 28 (2), 142–152, 10.1055/s-2008-1073114 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Sanyal AJ, Yoon SK, Lencioni R The etiology of hepatocellular carcinoma and consequences for treatment. The Oncologist. 15 Suppl 4, 14–22, 10.1634/theoncologist.2010-S4-14 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Jadlowiec CC, Taner T Liver transplantation: Current status and challenges. World Journal of Gastroenterology. 22 (18), 4438–4445, 10.3748/wjg.v22.i18.4438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michalopoulos GK, DeFrances MC Liver regeneration. Science. 276 (5309), 60–66 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Michalopoulos GK Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. The American Journal of Pathology. 176 (1), 2–13, 10.2353/ajpath.2010.090675 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grompe M et al. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes & Development. 7 (12A), 2298–2307 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Wangensteen KJ et al. A facile method for somatic, lifelong manipulation of multiple genes in the mouse liver. Hepatology. 47 (5), 1714–1724, 10.1002/hep.22195 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang AW et al. TRAP-seq identifies cystine/glutamate antiporter as a driver of recovery from liver injury. The Journal of Clinical Investigation. 128 (6), 2297–2309, 10.1172/JCI95120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heiman M, Kulicke R, Fenster RJ, Greengard P, Heintz N Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nature Protocols. 9 (6), 1282–1291, 10.1038/nprot.2014.085 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agilent Technologies. Agilent RNA 6000 Pico: User Manual. Retrieved from https://www.agilent.com/cs/library/usermanuals/Public/G2938-90046_RNA600Pico_KG_EN.pdf (2016).
- 12.NEBNext. NEBNext Ultra RNA Library Prep Kit for Illumina: User Manual. Retrieved from https://international.neb.com/-/media/nebus/files/manuals/manuale7530.pdf?rev=e6b4aa14481a42b59232dce8359979da (2018).
- 13.ThermoScientific. NanoDrop 2000/2000c Spectrophotomerter: User Manual. Retrieved from https://assets.thermofisher.com/TFS-Assets/CAD/manuals/NanoDrop-2000-User-Manual-EN.pdf (2009).
- 14.Liu J et al. Cell-specific translational profiling in acute kidney injury. The Journal of Clinical Investigation. 124 (3), 1242–1254, 10.1172/JCI72126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu SC Regulation of glutathione synthesis. Molecular Aspects of Medicine. 30 (1–2), 42–59, 10.1016/j.mam.2008.05.005 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang AW et al. The dynamic chromatin architecture of the regenerating liver. bioRxiv. 10.1101/664862 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zahm AM et al. A high-content in vivo screen to identify microRNA epistasis in the repopulating mouse liver. bioRxiv. 10.1101/664847 (2019). [DOI] [Google Scholar]
- 18.Stork C, Zheng S Genome-Wide Profiling of RNA-Protein Interactions Using CLIP-Seq. Methods in Molecular Biology. 1421, 137–151, 10.1007/978-1-4939-3591-8_12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao X et al. Glutathione antioxidant pathway activity and reserve determine toxicity and specificity of the biliary toxin biliatresone in zebrafish. Hepatology. 64 (3), 894–907, 10.1002/hep.28603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halpern KB et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 542 (7641), 352–356, 10.1038/nature21065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camp JG et al. Multilineage communication regulates human liver bud development from pluripotency. Nature. 546 (7659), 533–538, 10.1038/nature22796 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Wang YJ, Kaestner KH Single-Cell RNA-Seq of the Pancreatic Islets--a Promise Not yet Fulfilled? Cell Metabolism. 29 (3), 539–544, 10.1016/j.cmet.2018.11.016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]