

Abstract
Our ability to engineer protein structure and function has grown dramatically over recent years. Perhaps the next level in protein design is to develop proteins whose function can be regulated in response to various stimuli, including ligand binding, pH changes, and light. Endeavors toward these goals have tested and expanded on our understanding of protein function and allosteric regulation. In this chapter, we provide examples from different methods for developing new allosterically regulated proteins. These methods range from whole insertion of regulatory domains into new host proteins, to covalent attachment of photoswitches to generate light-responsive proteins, and to targeted changes to specific amino acid residues, especially to residues identified to be important for relaying allosteric information across the protein framework. Many of the examples we discuss have already found practical use in medical and biotechnology applications.
Keywords: Allostery, Protein regulation, Protein engineering, Energy landscape, Amino acid network, Domain insertion, Covalent modification
15.1. Introduction
Allostery in biomacromolecules can be broadly defined as “action at a distance,” in which a perturbation at one site (e.g., ligand binding, amino acid change) has an effect on the structure or function of some other distant site. The ability to allosterically regulate biomacromolecules is essential for life itself, with examples in DNA replication [57, 116], gene regulation [21, 56, 66, 119, 129], and biosynthetic pathways [2, 38, 58]. There is renewed interest in the design of therapeutics that target allosteric sites in proteins involved in disease [18, 60, 64, 100, 106, 121, 132]. There have also been significant advances in the engineering of novel allosteric regulation mechanisms into proteins. Such proteins are of substantial interest in biotechnology and medical fields, including in the production of biofuels and pharmaceuticals [80, 102, 103].
We have only just begun to understand allostery, this “second secret of life” [53, 136]. While some details differ among allosteric proteins, it is often useful to develop more general models to both understand and then even design new allosteric proteins. Towards this end, we first briefly outline two of the most widely accepted modern frameworks for allostery, the free energy landscape and network models, and then present examples that leverage these models toward engineering allostery into proteins. It should be noted that our examples are representative of recent work, but are not exhaustive.
15.1.1. The Energy Landscape Model
The best-known early models for describing allostery are the Monod-Wyman-Changeux (MWC) and Koshland-Nemethy-Filmer (KNF) models, which focused on conformational changes in multimeric complexes [73]. While these models have proven extremely useful for quantifying conformational states for large complexes such as hemoglobin, they do not take into account non-rigid body dynamics. For example, there are now well-known examples of allosteric proteins that do not undergo substantial structural changes upon ligand binding [88, 94, 97], which could thus not be described by either the MWC or KNF type models. The free energy landscape model [25] can be viewed as a broader generalization of these early models, which can then also describe allostery in proteins that lack large conformational changes, and also expand our definition of allostery to include the effects of amino acid changes or changes to other physiochemical parameters (e.g., temperature, pressure). Within the free energy landscape model, proteins can fluctuate among many unique substates populated according to their relative free energies. Low-amplitude, high-frequency motions such as a methyl rotation or a change in a side chain dihedral angle are represented as small energy barriers between substates, while larger barriers represent lower-frequency, higher-amplitude motions such as exchange between major and minor conformational states. Molecular dynamics (MD) techniques such as Markov state models [93] and biophysical techniques such as nuclear magnetic resonance (NMR) spin relaxation [19] and relaxation dispersion [3], fluorescence resonance energy transfer (FRET) [78], and isothermal titration calorimetry (ITC) [92] are commonly used together to better understand these energy landscapes.
The free energy landscape of a protein can be highly malleable in that it changes in response to stimuli [76] such as ligand binding, mutagenesis, or even pressure [83, 139]. Efforts to engineer allostery in the context of the energy landscape model therefore focus on ways to restrict or increase conformational exchange under certain conditions, or to stabilize certain minor states correlated with protein function (Fig. 15.1a).
Fig. 15.1.
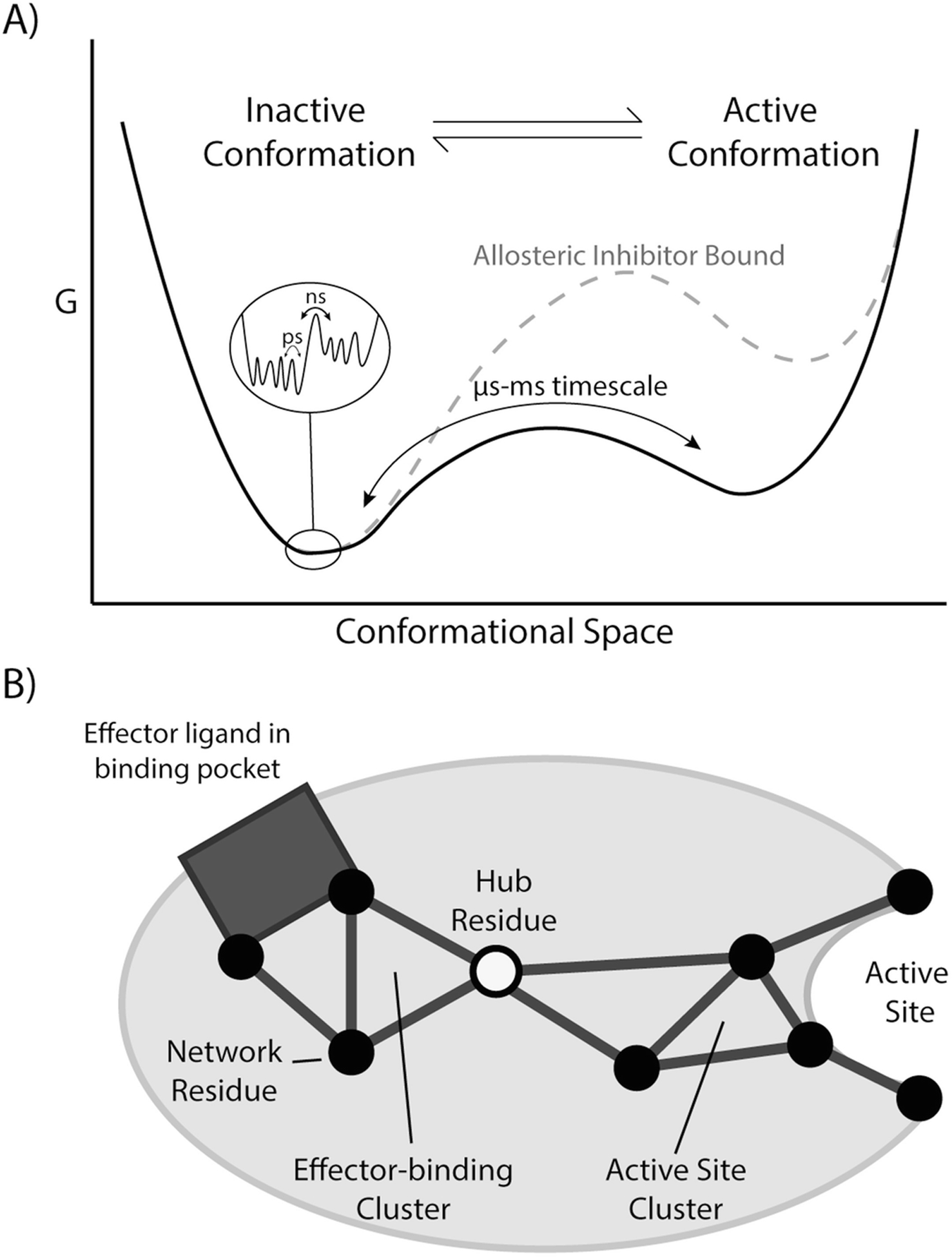
Frameworks for understanding allostery. (a) In the free energy landscape model, proteins are considered to be ensembles of different conformations. Local minima represent different conformations, with their free energies determining relative populations, and free energy barriers determining exchange rates between conformations. Allosteric inhibitors and activators can change the topology of the energy landscape by altering populations or the kinetics of exchange between conformations. (b) The network model places emphasis on correlated structural dynamics of amino acid residues being responsible for allosteric effects. A perturbation at one site can have an effect on a surrounding cluster of amino acids, which then can propagate through the amino acid residue interaction network to effect changes at a distant active site or other binding site. The network signals travel through highly connected hub residues, which may be important engineering points to change protein function and regulation
15.1.2. The Network Model
Another way to view a protein is to focus on the atomic-level noncovalent interactions that hold together its three-dimensional structure [89]. Within an amino acid interaction network, the nodes are usually full amino acid residues, and the edges contain information on which residues are interacting and the relative strength of those interactions (Fig. 15.1b). Residues with a large number of interactions are often considered to be important “hub” residues, and groups of interacting residues are sorted into cliques, communities, or clusters depending on the connectivity within the group [123]. Networks can be identified through a variety of techniques including bioinformatics analyses of multiple sequence alignments to identify evolutionarily coupled residues as performed by the statistical coupling analysis [63] (SCA) and direct coupling analysis [75] (DCA) algorithms. MD cross-correlation analysis [29] can identify energetically and dynamically coupled residues. NMR spectroscopy also offers a number of physics-based methods to identify networks, including the Chemical Shift Covariance Analysis (CHESCA) [11] and RASSMM [40] algorithms.
Ligand binding to an allosteric protein at one site may transmit a series of structural changes, and changes to the noncovalent interactions lead to structural and functional changes at a distant site. Efforts to engineer allostery in the context of the network model focus on changing network residues by mutagenesis or covalent modification to allosterically “tune” an enzyme’s activity or even give it new functionality by modifying these network connections [1, 6, 7].
In our view, the network model is compatible with the free energy landscape model; the network model focuses on paths and webs of physical interactions within the protein, while the free energy landscape attempts to assign these interactions to various conformational substates. Traversing over the free energy landscape from one conformational substate to another necessitates the breakage and formation of noncovalent interactions that might be identified as part of an important amino acid interaction network.
15.2. Engineering Protein Allostery Through Domain Insertion
Under favorable circumstances, new allosteric proteins can be created by inserting a regulatory domain from a known allosteric protein into a new host protein (Fig. 15.2 and 15.3). Upon allosteric effector binding, the regulatory domain undergoes a large conformational change, leading to substantial structural and functional changes in the host protein. The regulatory domain often changes the free energy landscape of the host protein by introducing new steric interactions between the regulatory domain and host protein. The goals of this section are to present select examples that highlight the different approaches for creating these chimeric proteins.
Fig. 15.2.
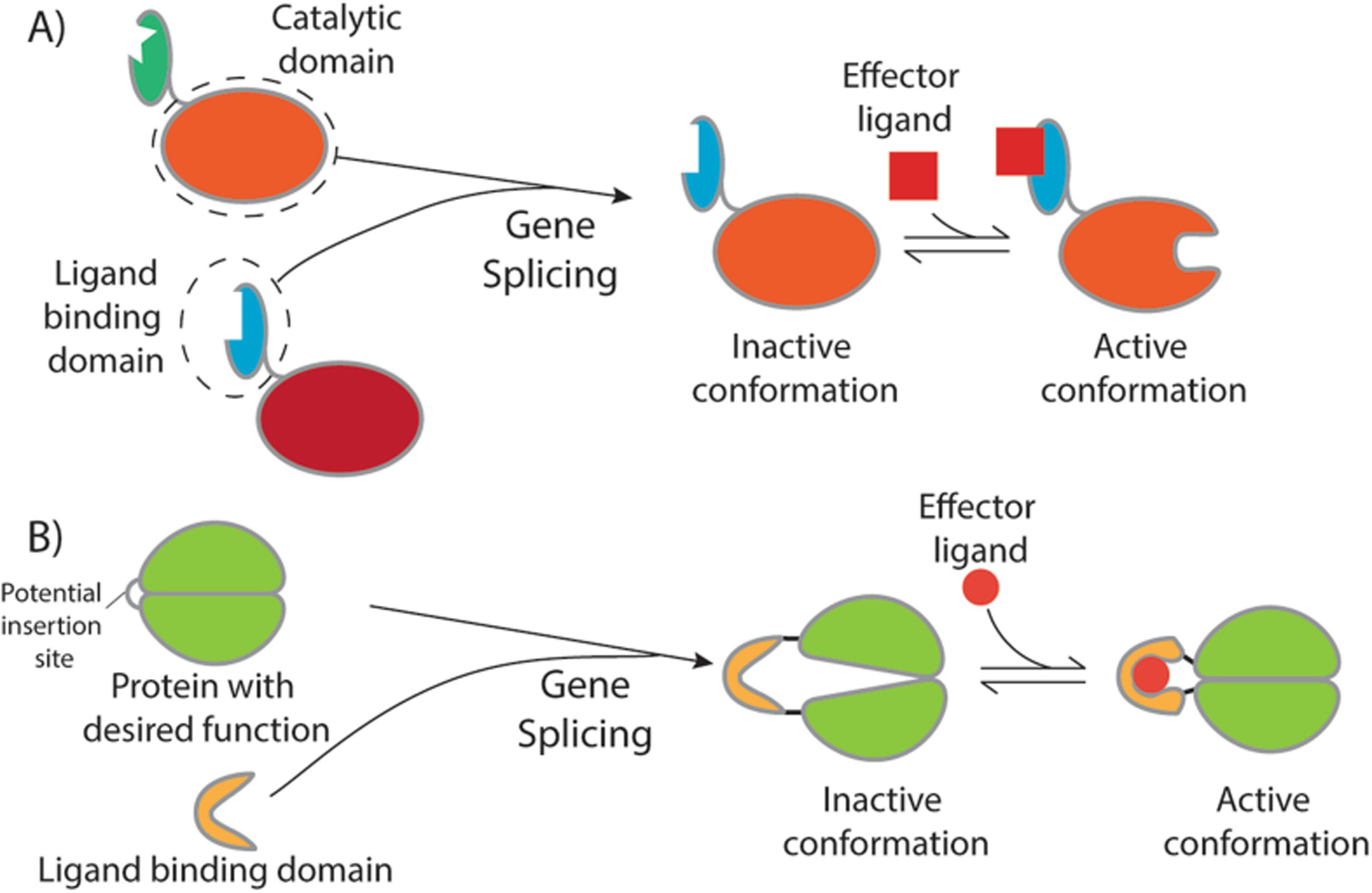
Methods of rational domain insertion. (a) Homologous proteins with catalytic domains are often structurally similar enough that regulatory domains sensitive to different effectors can be swapped or added to homologs without a regulatory domain, as in the above example where the blue regulatory domain is spliced onto the orange catalytic domain. (b) In cases where the protein with the desired function does not have any allosteric homologs, a conditionally disordered linker that becomes ordered in response to environmental changes or a regulatory domain can be inserted into the middle of a loop in the host protein sequence. In this example, the green enzymatic domain is inactive when the yellow regulatory domain is in its open apo conformation. Once the regulatory domain binds its effector ligand, it adopts a conformation that allows the enzymatic domain to adopt its active conformation
Fig. 15.3.
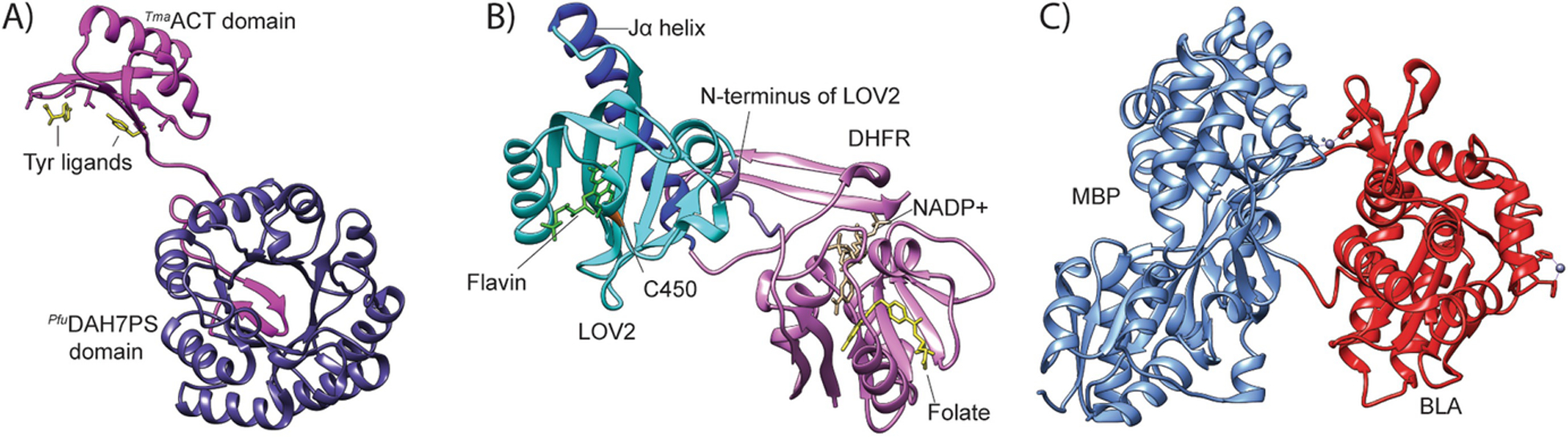
Examples of allosteric enzymes engineered through domain insertion. (A) PfuACTTmaDAH7PS with Tyr (yellow) bound to the ACT domain (magenta) with the DAH7PS domain in slate (PDB: 4GRS) [16]. PfuACTTmaDAH7PS is typically depicted as a homotetramer. It should be noted that only one subunit is shown here for clarity. (B) DHFR-LOV2 with LOV2 domain inserted at residue 120 of DHFR (PDBs: 1RX2, 2V0U) [59]. Upon irradiation with light, C450 covalently bonds to the flavin cofactor in the LOV2 domain. The changes in conformation and dynamics propagate through both domains. (C) The MBP-BLA construct RG13 generated by random domain insertion of BLA (red) into MBP (blue) (PDB: 4DXB) [48]
15.2.1. Rational Insertion of Regulatory Domains
The Parker lab has explored how allostery has evolved in nature, specifically in the amino acid biosynthesis pathways of various bacteria [4, 5, 16, 23, 43, 47, 58, 126]. Their method has primarily consisted of splicing ACT or CM regulatory domains from one (β/α)8 barrel enzyme onto another, similar to the schematic in Fig. 15.2a. The ACT domain is named for aspartate kinase, (AroH) chorismate mutase, and TyrA (i.e., TyrA is the bifunctional chorismate mutase/prephenate dehydratase in the Tyr biosynthetic pathway), all of which contain this domain. ACT domains consist of a βαββαβ fold, and typically occur when in contact with a second ACT domain to form a binding pocket for one or more small molecule effector ligands [58]. Ligand binding triggers a conformational change or a change in the oligomeric state of the enzyme that in turn modulates function of the catalytic domain. The AroQ chorismate mutase effector binding (CM) domain, in contrast, is an all-α-helical bundle that can turnover chorismic acid to prephenic acid, and binds prephenic acid when used as a regulatory binding domain [23]. Upon binding prephenic acid, the CM domain also undergoes a large conformational change. Cross et al. [16] spliced an ACT domain from Thermotoga maritima 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase (DAH7PS) onto the catalytic (β/α)8 barrel domain of the unregulated DAH7PS from Pyrococcus furiosus via a flexible linker on the N-terminus of P. furiosus DAH7PS (Fig. 15.3a). Surprisingly, the new chimeric protein was both catalytically active and allosterically inhibited by Tyr in a manner similar to that of T. maritima DAH7PS without any further modification. In a later publication, Fan et al. [23] swapped the regulatory domains of DAH7PS from T. maritima and Geobacillus sp. (TmaDAH7PS and GspDAH7PS, respectively). TmaDAH7PS and GspDAH7PS differ structurally, primarily in that TmaDAH7PS has an ACT regulatory domain and GspDAH7PS has an AroQ CM regulatory domain. In GspDAH7PS, the CM reduces DAH7PS activity in response to binding prephenic acid, and when transplanted onto the TmaDAH7PS (β/α)8 catalytic domain conferred that regulation. Similarly, the fusion of the TmaDAH7PS Tyr-binding ACT domain onto the (β/α)8 catalytic domain of GspDAH7PS conferred inhibition upon Tyr binding. These studies (and others48) were remarkable in how readily new allosteric mechanisms could be conferred onto these proteins, suggesting that the ability to allosterically regulate the catalytic domains may be intrinsic to these proteins, and perhaps to these types of protein folds.
For the creation of new allosteric chimeric proteins, it is common to insert the regulatory domain into the middle of the catalytic domain (Fig. 15.2b). Some of these new allosteric proteins have inserted an intrinsically disordered protein (IDP) domain that undergoes a transition between a high entropy disordered state to a low entropy ordered state, depending on the ligand-bound state, temperature, and/or pH. For example, Choi et al. [14] designed a maltose-dependent β-lactamase-maltose binding protein (BLA-MBP) chimeric protein in which the BLA domain was inserted in place of residues 317 and 318 of the MBP domain, and the amino acid sequence Asp-Lys-Thr was placed between the N-terminus of BLA and residue 319 of MBP. The construct, known as C4, was not allosterically regulated but did have BLA activity. Nonetheless, by changing the linker, they were able to develop allosterically regulated variants. For example, they added varying amounts of Gly residues to the linker resulting in temperature-dependent allosteric regulation, which was attributed to differences to entropic contributions with and without maltose. In another variant, a varying repeat Glu-Ala-Gln-Ala linker was substituted for Asp-Lys-Thr. Since Glu-Ala-Gln-Ala is more α-helical at low pH and more random coil at high pH, the proposal was to create variants that had a pH-dependent sensitivity to maltose. Indeed, experimental evidence showed that higher linker flexibility at high pH or high temperature resulted in greater conformational heterogeneity in the BLA domain, resulting in a higher population of less-active states until maltose was bound. Maltose binding under high pH or high temperature conditions reduced conformational entropy but increased the population of the active conformations, resulting in greater β-lactamase activity. The constructs lacked allosteric regulation when their respective linkers were α-helical at low pH or under low temperature conditions. These studies indicated that proteins can be engineered to respond simultaneously to ligand binding and changes to physiochemical parameters (i.e., pH and temperature), providing additional levels of control over protein function.
In a similar example, Meister et al. [71] took advantage of a previously identified insertion site in BLA [31–33] and inserted calmodulin (CaM) in such a way that BLA was split into two halves, BLA 24–194 and BLA 196–286, in a manner analogous to Fig. 15.2b. The resulting chimeric protein had β-lactamase activity in the absence of Ca2+ because the central helix of CaM was disordered under those conditions, allowing the two halves of the BLA domain to come together. Upon addition of Ca2+, the helix became rigid, which separated the two half BLA domains and abrogated catalytic activity. In this case, the high-entropy state was more active because it is likely thermodynamically favorable for the hydrophobic core of BLA to come back together and exclude solvent from those nonpolar residues. These sets of studies indicated that domain insertion and thoughtful linker engineering can lead to rational enthalpy and entropy changes to the underlying free energy landscapes of these proteins.
In a rather unique example, Guo et al. [34] created an allosteric Ca2+ electrochemical sensor. A calmodulin domain was inserted into the redox enzyme PQQ-glucose dehydrogenase with the goal of creating a biosensor that could quantify Ca2+ concentrations in biological fluids. PQQ-glucose dehydrogenase had been used as an electrochemical biosensor for glucose, but Guo et al. attempted to further engineer this enzyme through domain insertion of a calmodulin domain. The loop connecting strands A and B of β-sheet 3 of PQQ-glucose dehydrogenase was chosen as the insertion site since strand A is part of the catalytic domain involved in abstracting a proton from the glucose O1 atom. The thought was that the large conformational change in the CaM domain upon binding Ca2+ would result in a structural change that would activate the redox reaction catalyzed by PQQ-glucose dehydrogenase, resulting in an electrical impulse. Indeed, the chimeric protein could detect dilute Ca2+ in saliva. In a later refinement, the chimeric protein was placed in direct contact with conducting electrodes and a semiconductor by conjugating it to a graphene nanosheet associating ligand, PBSE [54].
In the above examples, new allosteric proteins were created that responded to ligand binding. There are also several examples of newly designed light-controlled proteins. The LOV2 domain from Avena sativa has been used to control the function of a variety of proteins via photoswitchable allosteric regulation [15, 26, 27, 39, 59, 65, 72, 74, 77, 84, 86, 105, 110, 114, 117, 125, 127, 130, 133, 137, 138]. A conformational change occurs upon absorption of blue light due to a conserved cysteine in the LOV2 domain forming a covalent bond with the flavin cofactor, altering the conformation of the Jα helix at the C-terminus and a helix at the N-terminus (Fig. 15.3b). The differences in structure and conformational dynamics between the “dark” and “light” states allow the LOV2 domain to be used as a photoswitch [35, 36]. Another strategy for rationally engineering light-controllable allostery is exemplified in the design of a photoswitchable mammalian pyruvate kinase by Gehrig et al. [27]. Through MD simulations, they found sites on the kinase that are highly surface-exposed and undergo conformational changes when an allosteric effector is bound. They inserted the LOV2 domain into these locations and demonstrated reversible control of the enzyme by light in mammalian cells.
Most of the aforementioned examples can be framed within the free energy landscape model, but in one notable example, Lee et al. fused a LOV2 domain and Escherichia coli dihydrofolate reductase (DHFR) at surface sites in each protein that are connected to allosteric amino acid interaction networks (Fig. 15.3b). By connecting the allosteric networks of the two proteins, they were able to control the catalytic activity of the DHFR enzyme with light [59].
15.2.2. Random Insertion of Regulatory Domains
While there are many elegant examples of well-designed chimeric allosteric proteins, rational domain insertion often requires prior knowledge of the regulatory domain and host proteins’ mechanisms and structures, which may not be generally available for proteins of interest. In these cases, random domain insertion may still generate new allosteric proteins, regardless of the depth of protein structure and function knowledge. For random domain insertion, an appropriate library of insertion variants must be created, and then a method for screening functional allosteric enzymes must be available. The Ostermeier lab’s methods for the creation of insertion libraries include random double-stranded breaks introduced by DNase I [17, 31, 32] or S1 nuclease [132], or by inverse PCR [91] into the insertion target within an expression vector (Fig. 15.4). The Ostermeier methods utilize random deletions and insertions of amino acids near the gene insertion site and random circular permutations of the insertion DNA to increase the diversity of the library. The resulting plasmid prior to insertion is blunt-ended linear DNA, with productive linear plasmids having the insert target split on either end. The blunt-ended DNA insert is then ligated into the plasmid, resulting in a circular expression vector for the chimeric protein. In contrast, the method of the Kim lab [95, 112, 113] involves the use of a Mu transposon for gene insertion (Fig. 15.4). A transposon is a double-stranded DNA sequence that can be randomly inserted into a target DNA sequence through the use of a transposase. This method also allows for random circular permutations. In both methods, the resulting libraries contain many variations on the insert sequence. The need for screening limits which chimeric proteins can be generated through random domain insertion. There must be an observable indication that either the insertion target or the insertion domain is active, where cell- or lysate-based assays are more amenable to high throughput screening.
Fig. 15.4.
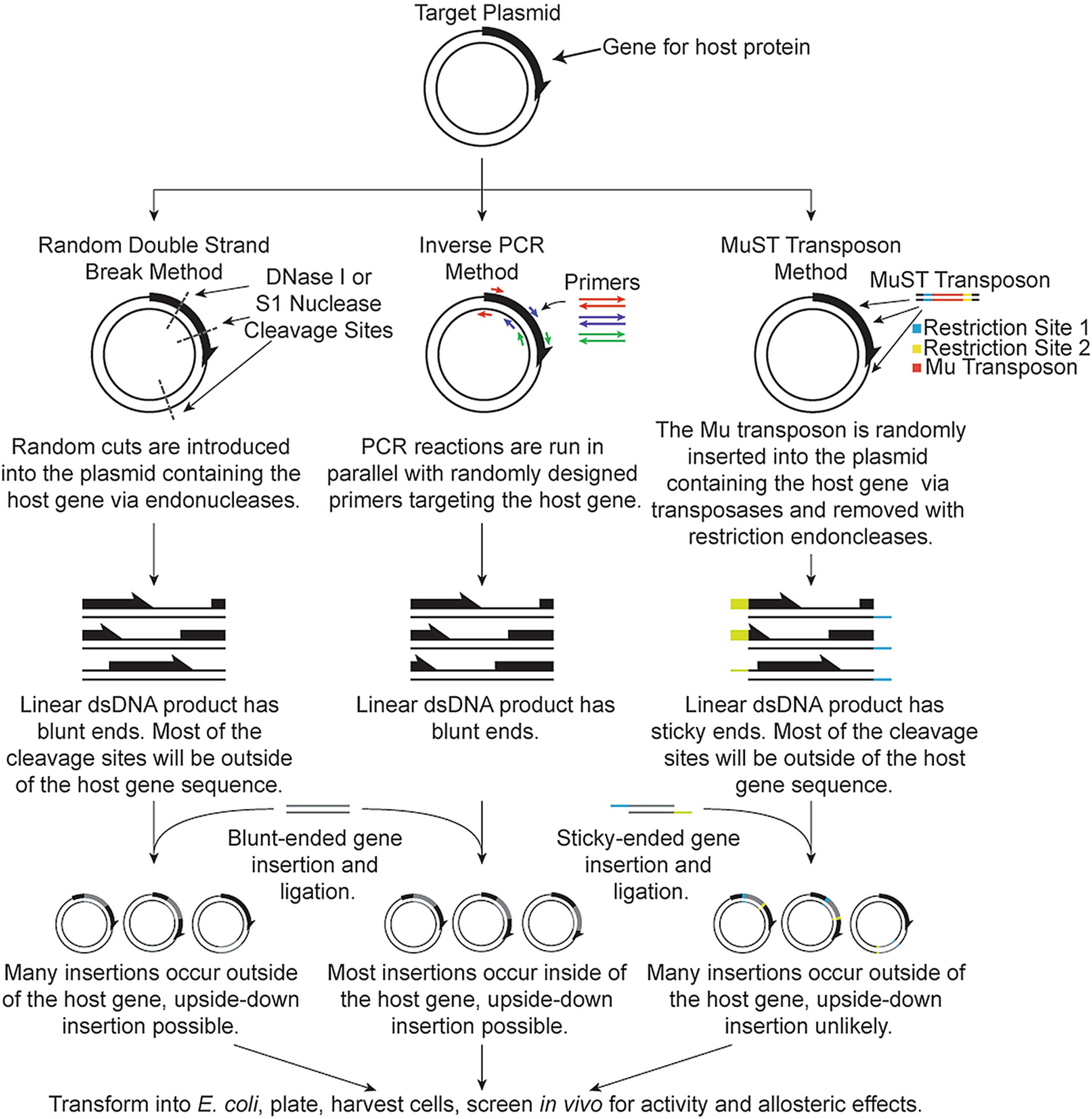
Methods of random domain insertion. Details on the major steps of creating gene insertion libraries are shown in brief
The work of Guntas et al. [31–33] provided an early template for random domain insertion for chimeric protein production with circular permutations and tandem deletions and repeats. The initial screening process for the target maltose-dependent β-lactamase consisted of transforming the vector library into E. coli and plating the cells on Luria-Bertani agar with ampicillin and maltose. Only cells that contained library members with functional β-lactamase activity were further tested for maltose-dependent β-lactamase activity. In the first iterations of this study [32], 70% of library members had β-lactamase successfully inserted into MBP, and 4% could also bind maltose. One member, RG13 (Fig. 15.3c), was shown to have a great amount of switching behavior, with very little β-lactamase activity in the absence of maltose and a 25-fold increase in the β-lactamase activity in the presence of maltose. Wright et al. [131] determined that the BLA domain of the chimeric protein primarily samples conformations where it is less active, and addition of maltose allows BLA to sample conformations closer to its native state in the full BLA protein.
Wright et al. [132] later applied the blunt-end random domain insertion method (Fig. 15.4) to create a prodrug-activating protein that selectively activates in the presence of the hypoxia-inducible factor 1α (HIF-1α), a marker for some types of cancer. This protein, Haps59, was a fusion of the HIF-1α-binding CH1 domain of the p300 protein and yeast cytosine deaminase (yCD), an enzyme that converts the prodrug 5-fluorocytosine (5FC) to the toxic 5-fluorouracil (5FU), a chemotherapeutic agent. The library was first transformed into GIA39 E. coli, a uracil auxotroph lacking cytosine deaminase. Since the target chimeric protein would convert 5FC into the toxin 5FU in the presence of HIF-1α, the first stage of screening consisted of negative selection where 5FC and uracil were present and cells possessing cytosine deaminase activity in the absence of HIF-1α did not grow due to 5FU toxicity. Surviving cells then had their plasmids isolated and transformed into GIA39 cells containing a glutathione-s-transferase-HIF-1a fusion (gstHIF-1α) plasmid with an arabinose promoter. The cells were plated on agar supplemented with cytosine and arabinose so that members that had cytosine deaminase activity in the presence of HIF-1α could grow but members without cytosine deaminase activity could not. The most promising chimeric protein, Haps59, was demonstrated to significantly decrease cell survival in human breast cancer and colorectal cancer cells in the presence of 5FC under hypoxic conditions similar to those in tumors, but not under normal oxygen conditions present outside of tumors.
Flow cytometry analysis fluorescent-assisted cell sorting (i.e., FACS) has become a popular method for screening libraries for desired functions. Ribeiro et al. [102, 103] used this selection method to create a more efficient endo-β-1,4-xylanase. Xylanase is an important enzyme for the conversion of plant biomass to fermentable sugars for biofuel production. Unfortunately, product inhibition from xylose is a significant limiting factor for xylanase function. The xylose binding domain from xylose binding protein (XBP) was selected as an insert domain since it undergoes a significant conformational change upon binding xylose. The resulting protein possessed increased xylanase activity even after large amounts of xylose product were formed. Because the goal was to create an improved version of an already active enzyme where the effector molecule was also the product, initial screening consisted of determining xylose-binding activity. E. coli cells lacking wild-type XBP were used since XBP is required for the efficient uptake of xylose. Cells were transformed with a pT7T3GFP_XBP plasmid, which contained a xylose promoter and would express GFP and the chimeric protein containing the XBP domain. FACS was used to sort for cells that displayed xylose binding, and these cells were further screened for xylanase activity. Cells with xylanase activity were determined by the formation of halos on agar xylan plates after staining with Congo Red, indicating xylan breakdown to xylose.
Nadler et al. [81] utilized the Mu transposon to insert GFP (green fluorescent protein) into a ligand-binding domain to demonstrate the creation of allosteric metabolite sensors. To demonstrate their technique, they created a single-fluorescent protein biosensor (SFPB) to quantify in-cell levels of trehalose. A binding event in the ligand binding domain was predicted to trigger an allosteric change in fluorescence in the inserted circularly permuted GFP domain. Oakes et al. [90] also utilized this technique with the goal of introducing allosteric regulation into the CRISPR-associated protein Cas9 by inserting the alpha estrogen receptor, which crystallographic evidence had shown undergoes a significant conformational change upon ligand binding. To identify sites where the estrogen receptor domain could be inserted, mouse PDZ-domain insertion sites were screened for several rounds in E. coli, with round one selecting for insertions that did not cause a frameshift and were not reversed. Round two screened for Cas9’s ability to suppress RFP (red fluorescent protein) expression with the insertion present at a random site to weed out non-functional Cas9 activity. Sites on Cas9 that best tolerated insertion were clustered around flexible loops, the ends of helices, and on the surface of the protein. Once functional insertion sites were identified, the protein variants were then screened for estrogen receptor insertion and assayed for Cas9 activity in a similar way to the PDZ screening in the presence and absence of a 4-hydroxytamoxifen ligand.
The rational design methods of regulatory domain insertion benefit from computation and structural knowledge to tune the thermodynamic and steric parameters of the chimeric protein to achieve the desired functional modulation. The random domain insertion methods take advantage of clever library development and screening assays, benefitting from but not necessitating structural knowledge of the regulatory domains and host proteins. These methods have brought new insights into the free energy landscapes underlying protein regulation, and have already resulted in new allosteric proteins finding practical use in biotechnology and medical applications.
15.3. Engineering Protein Allostery Through Covalent Modification
Allostery and long-range communication may be intrinsic to all protein folds. Instead of whole domain insertions, more subtle changes may likewise result in new allosteric regulation. Covalent modification of protein scaffolds represents another method for altering or creating allostery in proteins. Rather than adding a new domain to perturb a structural ensemble, covalent modification often either restricts the accessible conformations with a linker or perturbs an amino acid interaction network by conjugating a bulky molecule to a labile, surface-exposed amino acid.
Putri et al. used a spiropyrin photoswitch linker (Fig. 15.5a) to engineer a light-sensitive human serum albumin (HSA) [98]. The linker was conjugated to a surface-accessible cysteine via Michael addition in subdomain IA, which resulted in the regulation of ligand release at subdomain IB greater than 130 Å away. For example, the HSA ligands methylene orange and bromocresol green were released upon UV irradiation, and methyl orange binding affinity decreased by three-fold after UV irradiation. MD simulations confirmed that the subdomains were not directly interactions, indicating that changes to ligand affinities were due to longer-range network interactions.
Fig. 15.5.
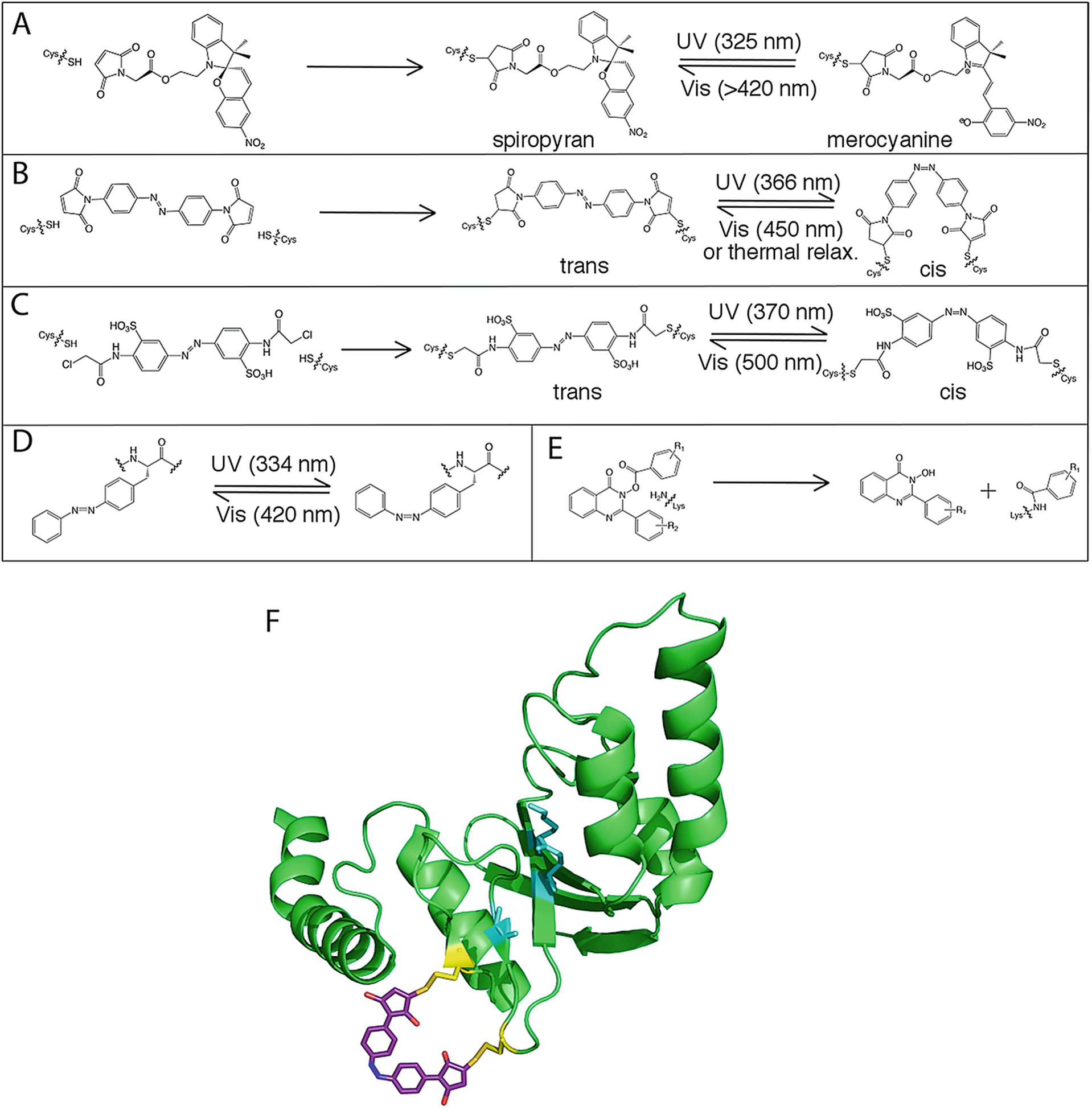
Covalent modification of proteins to control allostery. (a) Conjugation of spiropyran to a Cys residue and isomerization to the merocyanine form. (b) Conjugation of the bifunctional azobenzene derivative 4,4′-bis (maleimido)azobenzene to two cysteine residues and isomerization between trans and cis forms. (c) Conjugation of the bifunctional azobenzene derivative 3,3′-bis (sulfonato)-4,4′-bis (chloroacetamido)azobenzene (BSBCA) to two Cys residues and isomerization between trans and cis forms. (d) Isomerization of the genetically encoded azobenzene derivative AzoPhe. (e) Covalent modification of a Lys residue by quinazolin-4 (3H)-one hydroxamic ester. (f) Covalent modification of PvuII with azomal (PDB: 1NI0), adapted from Schierling et al. [108]. Active site residues (cyan) are disrupted upon isomerization of azomal to the trans form
One of the most common and useful covalent attachments is the azobenzene photoswitch, which reversibly isomerizes between cis and trans conformations [67, 98, 101]. Several strategies have been employed to integrate the azobenzene photoswitch: it can be directly incorporated into a peptide during peptide synthesis reactions [61, 122], incorporated into a protein via non-natural amino acids in vitro [79, 82] or in vivo [9], crosslinked to the protein to one or two solvent-exposed cysteines [79, 128, 135], and/or incorporated into a protein-binding ligand [96, 109, 124]. Generally, the isomerization of an azobenzene derivative between trans and cis conformations changes the relative free energies of the active and inactive protein conformations. In one example, Schierling et al. crosslinked an azobenzene derivative to the restriction enzyme PvuII, allowing allosteric inhibition to be reversibly controlled by light. Here, two cysteine residues were conjugated to the bifunctional azobenzene derivative 4,4′-bis (maleimido)azobenzene (Fig. 15.5b, f). The switch from cis to trans conformation disrupted the PvuII secondary structure, altered the active site structure, and reduced enzyme activity by 16-fold [108]. In another example, Ritterson et al. crosslinked two cysteine residues in cadherin, a cell-cell adhesion protein, using the bifunctional azobenzene derivative BSBCA (Fig. 15.5c). Upon irradiation with near-UV light, the apparent Ca2+ binding affinity decreased 18-fold. Because Ca2+ binding is linked to cadherin dimerization, the function of cadherin was controlled by light. The photoswitch was reversible up to at least three light/dark cycles. A genetically encoded azobenzene derivative has also been developed for use in E. coli allowing for the generation of azobenzene photoswitches in live cells [79] (Fig. 15.5d).
Key surface exposed residues might also be covalently labeled to control protein function. For example, Bongard et al. [8] discovered a class of small molecules, quinazolin-4 (3H)-one hydroxamic esters, that covalently modify Lys residues (Fig. 15.5e). They found that these molecules allosterically activate DegS, a bacterial serine protease, by modifying the conformation of a loop in the protease domain. By changing the Lys residues that were covalently modified, they were able to determine which modifications were responsible for allosteric activation. The Lys residue responsible for the activation was found to be part of an allosteric amino acid interaction network. This strategy of covalent modification can be applied to other proteins for allosteric regulation or to find noncatalytic residues that are part of allosteric amino acid interaction networks (see Fig. 15.1b).
15.4. Modifying Protein Allostery Through Mutagenesis
The ability to quickly create point mutations in a target protein has been a boon for molecular biology and biochemistry. With some prior knowledge about a protein’s structure and amino acid sequence, small modifications to the amino acid sequence can be made to create pH switchable proteins or identify and modify the amino acid interaction networks that allow a protein to function. These studies generally represent more subtle changes than whole domain insertions or covalent modification. This section will summarize select examples from the literature where mutagenesis was used as a tool to modify, study, or create new allostery.
15.4.1. Generating pH-Dependent Proteins Through Mutagenesis
Since a protein’s conformation is so dependent on its charge, there are many examples of pH-switchable proteins [44, 51, 68, 70, 87, 107, 115, 120]. Examples found in nature include the E. coli chaperone HdeA [41], β-ureidopropionase in the pyrimidine catabolic pathway [69], bovine β-lactoglobulin [99], human prolactin [49, 55], and nitrophorin 4 (NP4) [20]. The ability to engineer pH-dependent allosteric proteins has great medical potential. For example, it is well known that tumors often create an acidic environment, which may allow for pH-sensitive proteins to functionally target cancer cells. There are also stark pH differences within cells, potentially allowing for even more specific targeting in diseased states that affect membrane-bound organelles such as mitochondria or lysosomes.
NP4 is a protein used by the kissing bug Rhodnius prolixus to selectively release nitric oxide into its victim’s tissues via pH-dependent conformational changes. When deprotonated at the higher pH in the tissue, a hydrogen bond with Asp30 is broken, causing a conformational change that results in the release of bound nitric oxide (Fig. 15.6). Di Russo et al. used pH replica exchange molecular dynamics (pH-REMD) [46, 118] to describe the free energy landscape of NP4’s pH-dependent conformational change. By creating a variant that destabilized the open conformation but did not affect the pKa of Asp30 in either conformation, they were able to show that the observed pKa depends on both the equilibrium constant for the conformations and the pKa at each conformation as predicted by their model. In addition to changing the equilibrium constant between conformations, the pKa and local dynamics of buried ionizable residues in proteins can be altered by changing the distribution of surface charges, as demonstrated by Pey et al. using E. coli thioredoxin [104].
Fig. 15.6.
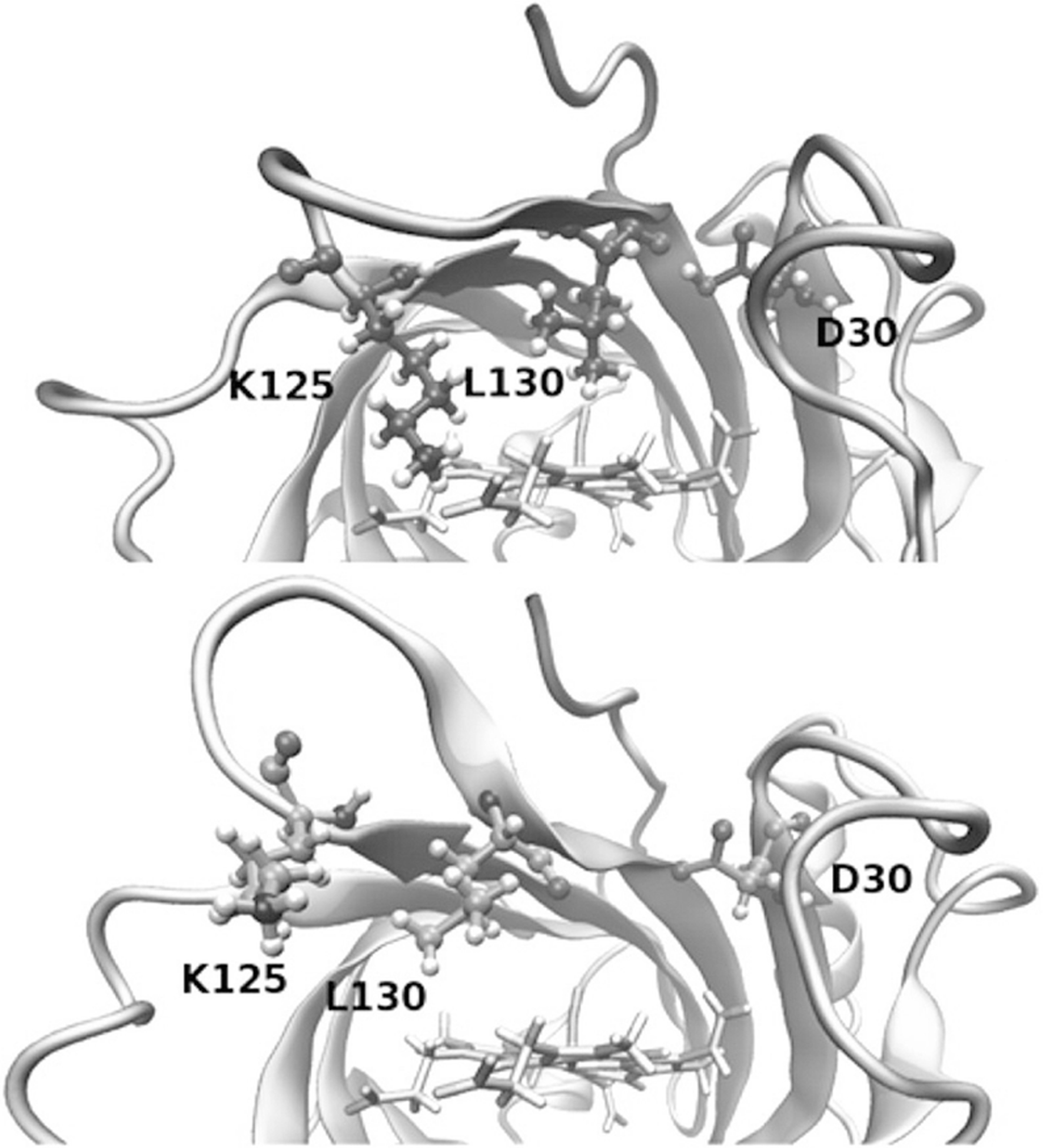
pH-dependent modulation of the NP4 protein. Closed (top) and open (bottom) conformations of NP4. Protonation of Asp30 stabilizes the closed conformation, while deprotonation stabilizes the open conformation. (Reprinted with permission from Di Russo, N. V.; Martí, M. A.; Roitberg, A. E. Underlying Thermodynamics of PH-Dependent Allostery. J. Phys. Chem. B 2014, 118 (45), 12,818–12,826. Copyright 2014 American Chemical Society)
In these studies, the coupling between protonation state equilibrium and conformational equilibrium must be considered in order to understand the thermodynamics of the pH-dependent conformational change. Exploring this further, Liu et al. studied the coupling of the protonation state to the conformation of the inner residues of staphylococcal nuclease by substituting ionizable residues into the interior of the protein and measuring the pKa values of these residues [62]. By taking into account the conformational equilibrium and the pKa of the residue of interest in each conformation there was excellent agreement with the experimentally observed pKa values and structural information for buried ionizable residues in the same protein [45].
There are now various examples of engineering proteins to be responsive to pH changes by substituting buried residues for ionizable residues. Zimenkov et al. [140] designed peptides that reversibly self-assemble into fibrils in response to pH changes. The self-assembly results from helix-coil transitions in the peptides when His residues in the helix are protonated. There are also biomedical applications to engineering pH-sensitive proteins. The fibronectin (Fn3) domain is a useful scaffold for engineering proteins that bind to proteins associated with cardiovascular disease and cancer. By engineering His residues into the hydrophobic core, Heinzelman et al. [37] engineered Fn3 domains that bind epidermal growth factor receptor (EGFR) with much lower affinity at low pH, and the effect was shown to be reversible. A similar strategy was used to engineer pH-responsive binding proteins for the Fc portion of human immunoglobulin (HIgG) derived from the hyperthermophilic Sso7d protein by Gera et al. [28]
Although most examples of engineered pH-dependent allostery involve substituting ionizable residues into the hydrophobic core of a protein, other residues can also be substituted to create pH-dependent allostery. Lysteriolysin O, a pore-forming protein secreted by Listeria cytogenes that destroys cholesterol-rich cell membranes, was engineered to be activated at slightly acidic pH via an allosteric mechanism. Kisovec et al. [50] substituted an Ala residue for a Tyr residue in the vicinity of ionizable residues previously identified to affect pH-dependent stability of the protein, changing the pKa values of the buried ionizable residues.
15.4.2. Perturbing Allosteric Amino Acid Interaction Networks with Mutagenesis
In the past decade, there have been several examples of mutational studies that identified and perturbed amino acid interaction networks. In several cases described here, changing network residues distal from the binding or active sites of proteins have wide implications for protein function and allostery. These studies have been of recent interest due to the rise of allosteric drugs and the hunt for druggable “cryptic” allosteric sites.
A long-standing problem in cellular signaling is tuning substrate and protein interaction specificity. For example, several PDZ domains have intersecting substrate specificity but play discrete roles in signaling pathways [24]. Gianni et al. investigated this problem using double mutant cycle analysis [42] on two specific PDZ domains that have computational evidence supporting distinct allosteric networks [52]. Double mutant cycle analysis helps to determine if two sites are allosterically and energetically coupled. Through these studies, they were able to determine the binding kinetics for 31 protein variants of each binding partner. These energetic constraints determined that the two binding partners had distinct allosteric mechanisms and that these allosteric networks were not conserved across the protein family. The authors emphasized the importance of amino acid composition to allosteric networks [30].
Introducing point mutations can also provide experimental evidence for the presence of amino acid interaction networks that play a critical role in allostery. Holliday and coworkers identified two dynamic amino acid clusters within cyclophilin A using RASSMM (relaxation techniques and single-site multiple mutations) [40]. RASSMM utilizes the high sensitivity of NMR methods to identify hot spots that are related to active site structural dynamics. Within the two coupled networks in cyclophilin A, two key “hot spot” residues were identified (Fig. 15.7). When these residues were perturbed, the active site dynamics and enzyme turnover were negatively impacted, despite both residues being greater than 15 Å from the active site. Similar methods can be applied to discover dynamically-coupled networks in other proteins.
Fig. 15.7.
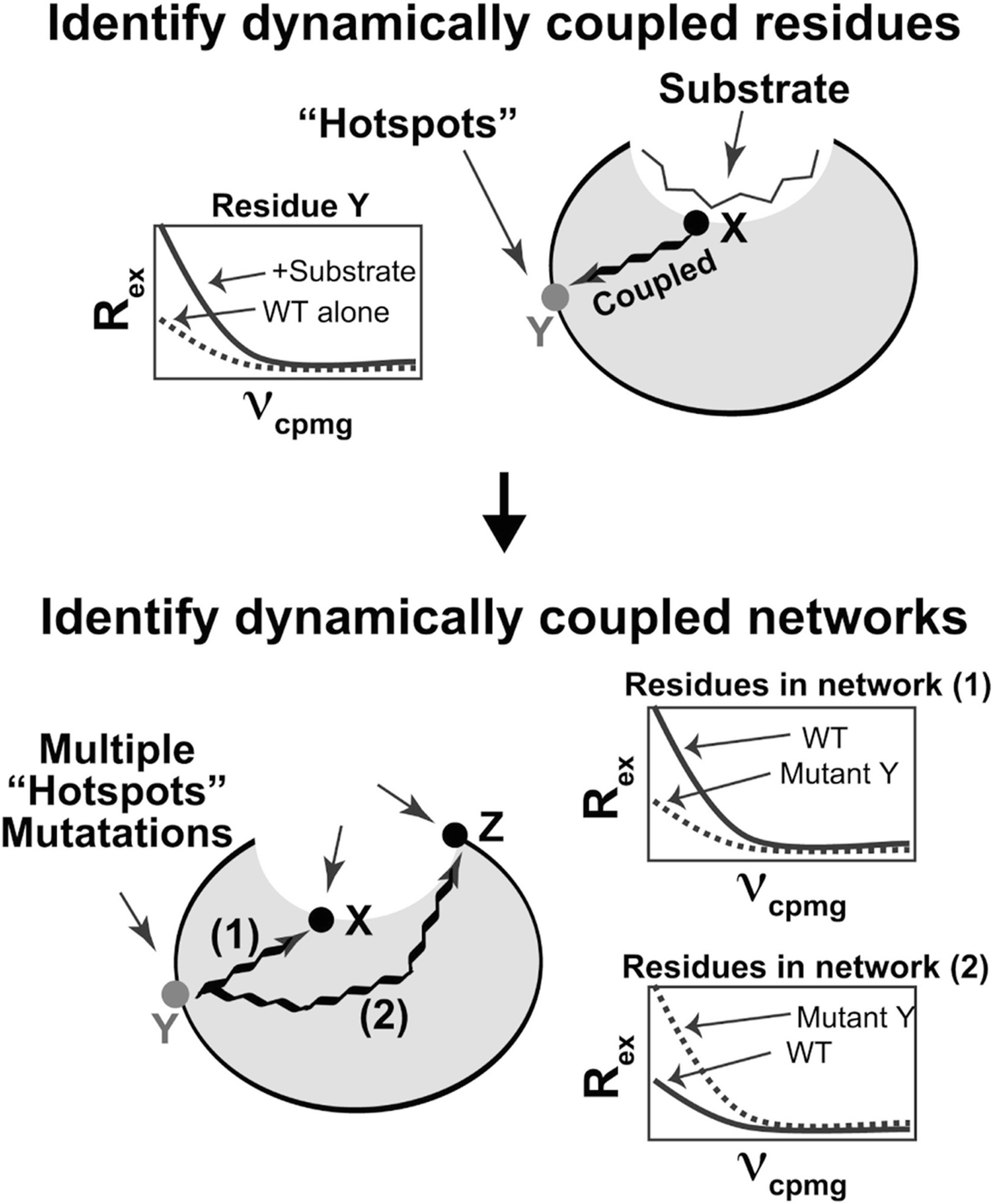
The RASMM method for identifying dynamically coupled amino acid interaction networks in proteins. Amino acids distal from the active site are identified as “hot spots” based on their R2 relaxation dispersion profiles in the apo and substrate-bound states. Residues that are linked allosterically to the active site are expected to show a measurable change in R2 relaxation dispersion behavior in the presence of substrate. To identify the global communications networks, single site mutations of “hot spot” residues are generated and global R2 relaxation dispersion profiles are examined. Reprinted from Structure, Volume 25 Issue 2, MJ Holliday, C Camilloni, GS Armstrong, M Vendruscolo, EZ Eisenmesser. Networks of Dynamic Allostery Regulate Enzyme Function, 276–286, Copyright (2017), with permission from Elsevier
There are several other NMR methods that can be used to identify and perturb allosteric networks in proteins. Among these is chemical shift covariance analysis (CHESCA) [111], which is similar to RASSMM in that it helps to elucidate allosteric mechanisms in proteins. CHESCA has been utilized to determine amino acid interaction networks in several proteins, including the alpha subunit of tryptophan synthase [2], a DNA repair protein (Rad50) [10], and phosophohexomutase [134]. In a different NMR approach to allostery, Cui and coworkers described a method that solely relies on total protein 1H, 15N chemical shift perturbations, or CSPs [17]. In order to probe the allosteric sites in tyrosine phosphatases, they introduced several Ala substitutions in an acidic loop region and then monitored chemical shift changes in residues distal from the active site. Using this method, they identified two separate amino acid interaction networks. When network residues were changed, a marked decrease in enzyme activity was noted, despite the distance of these network residues from the active site. Tyrosine phosphatases have been proposed to be potential drug targets for the treatment of diabetes, cancer, and obesity, and so the identification of these network residues helps to identify protein surfaces that might be targetable by allosteric drugs.
15.4.3. Engineering Allostery “Out Of” Proteins
All of the examples above have explored ways to generate new allosteric proteins. However, it is sometimes advantageous to render a protein unresponsive to allosteric regulation while maintaining other functions. For example, metabolic enzymes are often allosterically regulated, but such regulation might interfere in a particular synthetic scheme [22]. One great recent example is the engineering of tryptophan synthase (TS) for the synthesis of novel indole analogs. TS is composed of two subunits, the alpha subunit (αTS) that generates indole, which is then directly channeled through a hydrophobic tunnel to the beta subunit (βTS) that completes tryptophan biosynthesis. Only the catalytic activity of βTS was desired for the synthesis of indole analogs, but the large decrease in βTS activity in the absence of αTS made synthesis difficult [85]. The Arnold lab [13] sought to remedy this problem by using directed evolution to generate βTS that recapitulated the allosteric activation in the absence of αTS. The resulting βTS had greater catalytic activity than the native complex and also a more diversified substrate portfolio.
In further studies, Buller and coworkers sought to identify the mechanism by which their evolved βTS was able to fully function in the absence of αTS [12]. By monitoring the steady-state concentrations of βTS intermediates, they determined that their directed evolution variants progressively changed the rate-limiting step of the reaction. In addition, their variants shifted the steady-state distribution, favoring the tryptophan bound products (Fig. 15.8). They concluded that the amino acid substitutions stabilized a population in βTS that is typically only favored when bound to αTS – therefore recapitulating the complex activity in just the βTS subunit. The TS example provides an important counterexample for allostery engineering – instead of engineering in allostery, the goal was to engineer allostery out of βTS so the enzyme was fully capable of functioning alone.
Fig. 15.8.
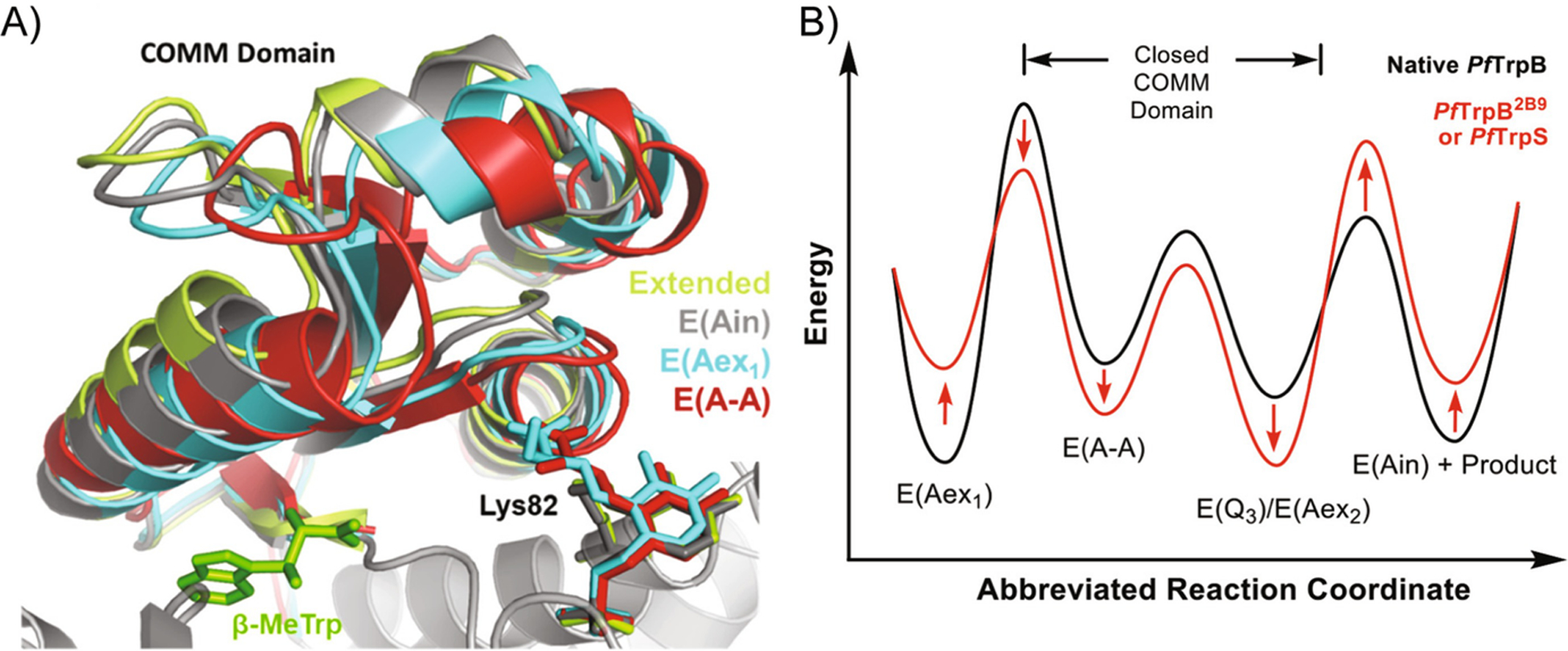
Engineering allostery “out of” the beta subunit of tryptophan synthase. (a) A crystal structure of the COMM domain of βTS representing the differences in the open/closed conformations through the catalytic cycle. The “extended” state represents the open conformation. As the nreaction proceeds, the COMM domain enters the fully closed conformation (E (A-A)). (b) A theoretical reaction coordinate illustrating the energy levels of the βTS intermediates. The engineered beta subunit (PfTrpB2B9) creates a decrease in energy of the product bound state. This results in a more efficient enzyme without the presence of the allosteric effector (αTS). Reprinted with permission from Buller, A. R. et al. Directed Evolution Mimics Allosteric Activation by Stepwise Tuning of the Conformational Ensemble. J. Am. Chem. Soc. 140, 7256–7266 (2018). Copyright 2018 American Chemical Society
15.5. Conclusion
The ability to control engineered proteins allosterically would revolutionize medicine and industry. Here, we have provided examples that illuminated the fundamentals of allostery, and have shown that there are newly designed allosteric proteins already finding practical use. Rational and random domain insertion methods appear to be the most prolific in creating novel allosteric proteins. The modular nature of certain types of catalytic and regulatory domains may be why these approaches are so successful. The ability to engineer photoswitchable proteins is especially exciting, which has been accomplished mainly through LOV2 domain insertion and/or insertion of the azobenzene photoswitch through covalent modification. Likewise, pH-switchable proteins could offer means to specifically target different cellular locations or activate only under certain disease conditions. The identification of amino acid interaction networks through a variety of biophysical and computational methods [1] offers not only a means to potentially engineer allostery and function, but these methods can also identify protein surfaces that may be amenable to small molecule targeting and allosteric regulation.
References
- 1.Axe JM, O’Rourke KF, Kerstetter NE, Yezdimer EM, Chan YM, Chasin A, Boehr DD (2015) Severing of a hydrogen bond disrupts amino acid networks in the catalytically active state of the alpha subunit of tryptophan synthase. Protein Sci 24(4):484–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Axe JM, Yezdimer EM, O’Rourke KF, Kerstetter NE, You W, Chang CE, Boehr DD (2014) Amino acid networks in a (beta/alpha) (8) barrel enzyme change during catalytic turnover. J Am Chem Soc 136(19):6818–6821 [DOI] [PubMed] [Google Scholar]
- 3.Berliner L (2015) Protein NMR: modern techniques and biomedical applications [Google Scholar]
- 4.Blackmore NJ, Nazmi AR, Hutton RD, Webby MN, Baker EN, Jameson GB, Parker EJ (2015) Complex formation between two biosynthetic enzymes modifies the allosteric regulatory properties of both: AN EXAMPLE OF MOLECULAR SYMBIOSIS. J Biol Chem 290(29):18187–18198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blackmore NJ, Reichau S, Jiao W, Hutton RD, Baker EN, Jameson GB, Parker EJ (2013) Three sites and you are out: ternary synergistic allostery controls aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J Mol Biol 425(9):1582–1592 [DOI] [PubMed] [Google Scholar]
- 6.Boehr DD, D’Amico RN, O’Rourke KF (2018) Engineered control of enzyme structural dynamics and function. Protein Sci 27(4):825–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boehr DD, Schnell JR, McElheny D, Bae SH, Duggan BM, Benkovic SJ, Dyson HJ, Wright PE. A distal mutation perturbs dynamic amino acid networks in dihydrofolate reductase. pp 1520–4995 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bongard J, Lorenz M, Vetter IR, Stege P, Porfetye AT, Schmitz AL, Kaschani F, Wolf A, Koch U, Nussbaumer P, Klebl B, Kaiser MA, Ehrmann MA. Identification of Noncatalytic Lysine Residues from Allosteric Circuits via Covalent Probes. pp 1554–8937 (Electronic) [DOI] [PubMed] [Google Scholar]
- 9.Bose M, Groff D, Xie J, Brustad E, Schultz PG (2006) The incorporation of a photoisomerizable amino acid into proteins in E. coli. J Am Chem Soc 128(2):388–389 [DOI] [PubMed] [Google Scholar]
- 10.Boswell ZK, Rahman S, Canny MD, Latham MP (2018) A dynamic allosteric pathway underlies Rad50 ABC ATPase function in DNA repair. Sci Rep 8(1):1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boulton S, Akimoto M, Selvaratnam R, Bashiri A, Melacini G (2014) A tool set to map allosteric networks through the NMR chemical shift covariance analysis. Sci Rep 4:7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buller AA-O, van Roye P, Cahn JKB, Scheele RA, Herger M, Arnold FA-OX. Directed Evolution Mimics Allosteric Activation by Stepwise Tuning of the Conformational Ensemble. pp 1520–5126 (Electronic)) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buller AR, Brinkmann-Chen S, Romney DK, Herger M, Murciano-Calles J, Arnold FH (2015) Directed evolution of the tryptophan synthase beta-subunit for stand-alone function recapitulates allosteric activation. Proc Natl Acad Sci U S A 112(47):14599–14604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi JH, Laurent AH, Hilser VJ, Ostermeier M (2015) Design of protein switches based on an ensemble model of allostery. Nat Commun 6:6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cristian C, Laura A, Sabrina G, Marco A, Edoardo R, Solei C, Paolo Z, Jan P, Monica B, van Etten JL (2015) Optogenetics. Engineering of a light-gated potassium channel. Science 348(6235):707. [DOI] [PubMed] [Google Scholar]
- 16.Cross PJ, Allison TM, Dobson RC, Jameson GB, Parker EJ. Engineering allosteric control to an unregulated enzyme by transfer of a regulatory domain. pp 1091–6490 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui DS, Beaumont V, Ginther PS, Lipchock JM, Loria JP (2017) Leveraging reciprocity to identify and characterize unknown allosteric sites in protein tyrosine phosphatases. J Mol Biol 429(15):2360–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies BR, Guan N, Logie A, Crafter C, Hanson L, Jacobs V, James N, Dudley P, Jacques K, Ladd B, D’Cruz CM, Zinda M, Lindemann J, Kodaira M, Tamura K, Jenkins EL (2015) Tumors with AKT1E17K mutations are rational targets for single agent or combination therapy with AKT inhibitors. Mol Cancer Ther 14(11):2441–2451 [DOI] [PubMed] [Google Scholar]
- 19.Dayie KT, Wagner G, Lefevre JF (1996) Theory and practice of nuclear spin relaxation in proteins. Annu Rev Phys Chem 47:243–282 [DOI] [PubMed] [Google Scholar]
- 20.Di Russo NV, Marti MA, Roitberg AE (2014) Underlying thermodynamics of pH-dependent allostery. J Phys ChemB 118(45):12818–12826 [DOI] [PubMed] [Google Scholar]
- 21.Djuranovic S, Nahvi A, Green R (2011) A parsimonious model for gene regulation by miRNAs. Science (New York, NY) 331(6017):550–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du L, Lou L (2010) PKS and NRPS release mechanisms. Nat Prod Rep 27(2):255–278 [DOI] [PubMed] [Google Scholar]
- 23.Fan Y, Cross PJ, Jameson GB, Parker EJ (2018) Exploring modular allostery via interchangeable regulatory domains. Proc Natl Acad Sci U S A 115(12):3006–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng W, Zhang M (2009) Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat Rev Neurosci 10(2):87–99 [DOI] [PubMed] [Google Scholar]
- 25.Frauenfelder H, Sligar SG, Wolynes PG (1991) The energy landscapes and motions of proteins. Science (New York, NY) 254(5038):1598–1603 [DOI] [PubMed] [Google Scholar]
- 26.Gautier A, Gauron C, Volovitch M, Bensimon D, Jullien L, Vriz S (2014) How to control proteins with light in living systems. Nat Chem Biol 10(7):533–541 [DOI] [PubMed] [Google Scholar]
- 27.Gehrig S, Macpherson JA, Driscoll PC, Symon A, Martin SR, MacRae JI, Kleinjung J, Fraternali F, Anastasiou D (2017) An engineered photoswitchable mammalian pyruvate kinase. FEBS J 284(18):2955–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gera N, Hill AB, White DP, Carbonell RG, Rao BM. Design of pH sensitive binding proteins from the hyperthermophilic Sso7d scaffold. pp 1932–6203 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh A, Vishveshwara S (2007) A study of communication pathways in methionyl- tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc Natl Acad Sci U S A 104(40):15711–15716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gianni S, Haq SR, Montemiglio LC, Jurgens MC, Engstrom A, Chi CN, Brunori M, Jemth P. Sequence-specific long range networks in PSD-95/discs large/ZO-1 (PDZ) domains tune their binding selectivity. pp 1083–351X (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guntas G, Mansell TJ, Kim JR, Ostermeier M (2005) Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc Natl Acad Sci U S A 102(32):11224–11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guntas G, Mitchell SF, Ostermeier M (2004) A molecular switch created by in vitro recombination of nonhomologous genes. Chem Biol 11(11):1483–1487 [DOI] [PubMed] [Google Scholar]
- 33.Guntas G, Ostermeier M (2004) Creation of an allosteric enzyme by domain insertion. J Mol Biol 336(1):263–273 [DOI] [PubMed] [Google Scholar]
- 34.Guo Z, Johnston WA, Stein V, Kalimuthu P, Perez-Alcala S, Bernhardt PV, Alexandrov K. Engineering PQQ-glucose dehydrogenase into an allosteric electrochemical Ca (2+) sensor. pp 1364–548X (Electronic) [DOI] [PubMed] [Google Scholar]
- 35.Halavaty AS, Moffat K (2007) N- and C-terminal flanking regions modulate light-induced signal transduction in the LOV2 domain of the blue light sensor phototropin 1 from Avena sativa. Biochemistry 46(49):14001–14009 [DOI] [PubMed] [Google Scholar]
- 36.Harper SM, Neil LC, Gardner KH (2003) Structural basis of a phototropin light switch. Science (New York, NY) 301(5639):1541–1544 [DOI] [PubMed] [Google Scholar]
- 37.Heinzelman P, Krais J, Ruben E, Pantazes R (2015) Engineering pH responsive fibronectin domains for biomedical applications. J Biol Eng 9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helmstaedt K, Krappmann S, Braus GH (2001) Allosteric regulation of catalytic activity: Escherichia coli aspartate transcarbamoylase versus yeast chorismate mutase. Microbiol Mol Biol R 65(3):404–421, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hisatomi O, Furuya K (2015) A light-regulated bZIP module, photozipper, induces the binding of fused proteins to the target DNA sequence in a blue light-dependent manner. Photochem Photobiol Sci: Off J Eur Photochem Assoc Eur Soc Photobiol 14(11):1998–2006 [DOI] [PubMed] [Google Scholar]
- 40.Holliday MJ, Camilloni C, Armstrong GS, Vendruscolo M, Eisenmesser EZ (2017) Networks of dynamic Allostery regulate enzyme function. Structure (London, England: 1993) 25(2):276–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong W, Jiao W, Hu J, Zhang J, Liu C, Fu X, Shen D, Xia B, Chang Z (2005) Periplasmic protein HdeA exhibits chaperone-like activity exclusively within stomach pH range by transforming into disordered conformation. The J Biol Chem 280(29):27029–27034 [DOI] [PubMed] [Google Scholar]
- 42.Horovitz A, Fersht AR (1990) Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol 214(3):613–617 [DOI] [PubMed] [Google Scholar]
- 43.Huisman FH, Koon N, Bulloch EM, Baker HM, Baker EN, Squire CJ, Parker EJ (2012) Removal of the C-terminal regulatory domain of alpha-isopropylmalate synthase disrupts functional substrate binding. Biochemistry 51(11):2289–2297 [DOI] [PubMed] [Google Scholar]
- 44.Idili A, Vallee-Belisle A, Ricci F (2014) Programmable pH-triggered DNA nanoswitches. J Am Chem Soc 136(16):5836–5839 [DOI] [PubMed] [Google Scholar]
- 45.Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno B. Large shifts in pKa values of lysine residues buried inside a protein. pp 1091–6490 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Itoh SG, Damjanovic A, Brooks BR (2011) pH replica-exchange method based on discrete protonation states. Proteins 79(12):3420–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiao W, Hutton RD, Cross PJ, Jameson GB, Parker EJ. Dynamic cross-talk among remote binding sites: the molecular basis for unusual synergistic allostery. pp 1089–8638 (Electronic) [DOI] [PubMed] [Google Scholar]
- 48.Ke W, Laurent AH, Armstrong MD, Chen Y, Smith WE, Liang J, Wright CM, Ostermeier M, van den Akker F (2012) Structure of an engineered beta-lactamase maltose binding protein fusion protein: insights into heterotropic allosteric regulation. PLoS One 7(6):e39168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keeler C, Jablonski EM, Albert YB, Taylor BD, Myszka DG, Clevenger CV, Hodsdon ME (2007) The kinetics of binding human prolactin, but not growth hormone, to the prolactin receptor vary over a physiologic pH range. Biochemistry 46(9):2398–2410 [DOI] [PubMed] [Google Scholar]
- 50.Kisovec M, Rezelj S, Knap P, Cajnko MM, Caserman S, Flasker A, Znidarsic N, Repic M, Mavri J, Ruan Y, Scheuring S, Podobnik M, Anderluh G. Engineering a pH responsive pore forming protein. pp 2045–2322 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohse S, Neubauer A, Pazidis A, Lochbrunner S, Kragl U. Photoswitching of enzyme activity by laser-induced pH-jump. pp 1520–5126 (Electronic) [DOI] [PubMed] [Google Scholar]
- 52.Kong Y, Karplus M (2009) Signaling pathways of PDZ2 domain: a molecular dynamics interaction correlation analysis. Proteins 74(1):145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koshland DE Jr, Nemethy G, Filmer D (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5(1):365–385 [DOI] [PubMed] [Google Scholar]
- 54.Koushanpour A, Gamella M, Guo Z, Honarvarfard E, Poghossian A, Schoning MJ, Alexandrov K, Katz EA. Ca (2+)-Switchable Glucose Dehydrogenase Associated with Electrochemical/Electronic Interfaces: Applications to Signal-Controlled Power Production and Biomolecular Release. pp 1520–5207 (Electronic) [DOI] [PubMed] [Google Scholar]
- 55.Kulkarni MV, Tettamanzi MC, Murphy JW, Keeler C, Myszka DG, Chayen NE, Lolis EJ, Hodsdon ME. Two independent histidines, one in human prolactin and one in its receptor, are critical for pH-dependent receptor recognition and activation. pp 1083–351X (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar R, McEwan IJ (2012) Allosteric modulators of steroid hormone receptors: structural dynamics and gene regulation. Endocr Rev 33(2):271–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lafrance-Vanasse J, Williams GJ, Tainer JA (2015) Envisioning the dynamics and flexibility of Mre11-Rad50-Nbs1 complex to decipher its roles in DNA replication and repair. Prog Biophys Mol Bio 117(2–3):182–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lang EJ, Cross PJ, Mittelstadt G, Jameson GB, Parker EJ (2014) Allosteric ACTion: the varied ACT domains regulating enzymes of amino-acid metabolism. Curr Opin Struct Biol 29:102–111 [DOI] [PubMed] [Google Scholar]
- 59.Lee J, Natarajan M, Nashine VC, Socolich M, Vo T, Russ WP, Benkovic SJ, Ranganathan R (2008) Surface sites for engineering allosteric control in proteins. Science (New York, NY) 322(5900):438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ling Y, Jing M, Wang XD (2015) Allosteric therapies for lung cancer. Cancer Metast Rev 34(2):303–312 [DOI] [PubMed] [Google Scholar]
- 61.Liu D, Karanicolas J, Yu C, Zhang Z, Woolley GAJB, Letters MC (1997) Site-specific incorporation of photoisomerizable azobenzene groups into ribonuclease S. Bioorganic Med Chem Lett 7(20):2677–2680 [Google Scholar]
- 62.Liu J, Swails J, Zhang JZH, He X, Roitberg AE (2018) A coupled ionization-conformational equilibrium is required to understand the properties of Ionizable residues in the hydrophobic interior of staphylococcal nuclease. J Am Chem Soc 140(5):1639–1648 [DOI] [PubMed] [Google Scholar]
- 63.Lockless SW, Ranganathan R (1999) Evolutionarily conserved pathways of energetic connectivity in protein families. Science (New York, NY) 286(5438):295–299 [DOI] [PubMed] [Google Scholar]
- 64.Lu S, Li S, Zhang J. Harnessing allostery: a novel approach to drug discovery. pp 1098–1128 (Electronic) [DOI] [PubMed] [Google Scholar]
- 65.Lungu OI, Hallett RA, Choi EJ, Aiken MJ, Hahn KM, Kuhlman B (2012) Designing photoswitchable peptides using the AsLOV2 domain. Chem Biol 19(4):507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mandal M, Breaker RR (2004) Gene regulation by riboswitches. Nat Rev Mol Cell Biol 5(6):451–463 [DOI] [PubMed] [Google Scholar]
- 67.Mart RJ, Allemann RK (2016) Azobenzene photocontrol of peptides and proteins. Chem Commun 52(83):12262–12277 [DOI] [PubMed] [Google Scholar]
- 68.Marttila AT, Hytonen VP, Laitinen OH, Bayer EA, Wilchek M, Kulomaa MS. Mutation of the important Tyr-33 residue of chicken avidin: functional and structural consequences. pp 0264–6021 (Print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maurer D, Lohkamp B, Krumpel M, Widersten M, Dobritzsch DA. Crystal structure and pH-dependent allosteric regulation of human beta-ureidopropionase, an enzyme involved in anticancer drug metabolism. pp 1470–8728 (Electronic) [DOI] [PubMed] [Google Scholar]
- 70.McConnell EM, Bolzon R, Mezin P, Frahm G, Johnston M, DeRosa MC (2016) pHAST (pH-driven aptamer switch for thrombin) catch-and-release of target protein. Bioconjug Chem 27(6):1493–1499 [DOI] [PubMed] [Google Scholar]
- 71.Meister GE, Joshi NS (2013) An engineered calmodulin-based allosteric switch for peptide biosensing. Chembiochem 14(12):1460–1467 [DOI] [PubMed] [Google Scholar]
- 72.Mills E, Chen X, Pham E, Wong S, Truong K (2012) Engineering a photoactivated caspase-7 for rapid induction of apoptosis. ACS Synth Biol 1(3):75–82 [DOI] [PubMed] [Google Scholar]
- 73.Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12:88–118 [DOI] [PubMed] [Google Scholar]
- 74.Mootz HD (2017) Split Inteins [Google Scholar]
- 75.Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, Weigt M (2011) Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci U S A 108(49):E1293–E1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Motlagh HN, Wrabl JO, Li J, Hilser VJ (2014) The ensemble nature of allostery. Nature 508(7496):331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Motta-Mena LB, Reade A, Mallory MJ, Glantz S, Weiner OD, Lynch KW, Gardner KH (2014) An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat Chem Biol 10(3):196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Munro JB, Vaiana A, Sanbonmatsu KY, Blanchard SC (2008) A new view of protein synthesis: mapping the free energy landscape of the ribosome using single-molecule FRET. Biopolymers 89(7):565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muranaka N, Hohsaka T, Sisido M (2002) Photoswitching of peroxidase activity by position-specific incorporation of a photoisomerizable non-natural amino acid into horseradish peroxidase. FEBS Lett 510(1–2):10–12 [DOI] [PubMed] [Google Scholar]
- 80.Murciano-Calles J, Romney DK, Brinkmann-Chen S, Buller AR, Arnold FH (2016) A panel of TrpB biocatalysts derived from tryptophan synthase through the transfer of mutations that mimic allosteric activation. Angew Chem Int Ed Engl 55(38):11577–11581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nadler DC, Morgan SA, Flamholz A, Kortright KE, Savage DF (2016) Rapid construction of metabolite biosensors using domain-insertion profiling. Nat Commun 7:12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakayama K, Endo M, Majima T (2004) Photochemical regulation of the activity of an endonuclease BamHI using an azobenzene moiety incorporated site-selectively into the dimer interface. Chem Commun 21:2386–2387 [DOI] [PubMed] [Google Scholar]
- 83.Nguyen LM, Roche J (2017) High-pressure NMR techniques for the study of protein dynamics, folding and aggregation. J Magnet Reson (San Diego, Calif: 1997) 277:179–185 [DOI] [PubMed] [Google Scholar]
- 84.Nihongaki Y, Kawano F, Nakajima T, Sato M (2015) Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol 33(7):755–760 [DOI] [PubMed] [Google Scholar]
- 85.Niks D, Hilario E, Dierkers A, Ngo H, Borchardt D, Neubauer TJ, Fan L, Mueller LJ, Dunn MF. Allostery and substrate channeling in the tryptophan synthase bienzyme complex: evidence for two subunit conformations and four quaternary states. pp 1520–4995 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Niopek D, Benzinger D, Roensch J, Draebing T, Wehler P, Eils R, Di Ventura B (2014) Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat Commun 5:4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nordlund HR, Hytonen VP, Laitinen OH, Uotila ST, Niskanen EA, Savolainen J, Porkka E, Kulomaa MS (2003) Introduction of histidine residues into avidin subunit interfaces allows pH-dependent regulation of quaternary structure and biotin binding. FEBS Lett 555(3):449–454 [DOI] [PubMed] [Google Scholar]
- 88.Nussinov R, Tsai CJ (2015) Allostery without a conformational change? Revisiting the paradigm. Curr Opin Struct Biol 30:17–24 [DOI] [PubMed] [Google Scholar]
- 89.O’Rourke KF, Gorman SD, Boehr DD. Biophysical and computational methods to analyze amino acid interaction networks in proteins. pp 2001–0370 (Print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oakes BL, Nadler DC, Flamholz A, Fellmann C, Staahl BT, Doudna JA, Savage DF (2016) Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol 34(6):646–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ochman H, Gerber AS, Hartl DL (1988) Genetic applications of an inverse polymerase chain reaction. Genetics 120(3):621–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Olsson U, Wolf-Watz M (2010) Overlap between folding and functional energy landscapes for adenylate kinase conformational change. Nat Commun 1:111. [DOI] [PubMed] [Google Scholar]
- 93.Pande VS, Beauchamp K, Bowman GR (2010) Everything you wanted to know about Markov State Models but were afraid to ask. Methods (San Diego, Calif) 52(1):99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL (2009) Hidden dynamic allostery in a PDZ domain. Proc Natl Acad Sci U S A 106(43):18249–18254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pierre B, Shah V, Xiao J, Kim JR. Construction of a random circular permutation library using an engineered transposon. pp 1096–0309 (Electronic) [DOI] [PubMed] [Google Scholar]
- 96.Pittolo S, Gomez-Santacana X, Eckelt K, Rovira X, Dalton J, Goudet C, Pin JP, Llobet A, Giraldo J, Llebaria A, Gorostiza P (2014) An allosteric modulator to control endogenous G protein-coupled receptors with light. Nat Chem Biol 10(10):813–815 [DOI] [PubMed] [Google Scholar]
- 97.Popovych N, Sun S, Ebright RH, Kalodimos CG (2006) Dynamically driven protein allostery. Nat Struct Mol Biol 13(9):831–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Putri RM, Zulfikri H, Fredy JW, Juan A, Tananchayakul P, Cornelissen J, Koay MST, Filippi C, Katsonis N (2018) Photoprogramming Allostery in human serum albumin. Bioconjug Chem 29(7):2215–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Qin BY, Bewley MC, Creamer LK, Baker HM, Baker EN, Jameson GB (1998) Structural basis of the Tanford transition of bovine beta-lactoglobulin. Biochemistry 37(40):14014–14023 [DOI] [PubMed] [Google Scholar]
- 100.Ramanathan RK, McDonough SL, Kennecke HF, Iqbal S, Baranda JC, Seery TE, Lim HJ, Hezel AF, Vaccaro GM, Blanke CD (2015) Phase 2 study of MK-2206, an allosteric inhibitor of AKT, as second-line therapy for advanced gastric and gastroesophageal junction cancer: a SWOG cooperative group trial (S1005). Cancer 121(13):2193–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Renner C, Moroder L (2006) Azobenzene as conformational switch in model peptides. Chembiochem 7(6):868–878 [DOI] [PubMed] [Google Scholar]
- 102.Ribeiro LF, Nicholes N, Tullman J, Ribeiro LF, Fuzo CA, Vieira DS, Furtado GP, Ostermeier M, Ward RJ (2015) Insertion of a xylanase in xylose binding protein results in a xylose-stimulated xylanase. Biotechnol Biofuels 8:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ribeiro LF, Tullman J, Nicholes N, Silva SR, Vieira DS, Ostermeier M, Ward RJ (2016) A xylose-stimulated xylanase-xylose binding protein chimera created by random nonhomologous recombination. Biotechnol Biofuels 9:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Richman DE, Majumdar A, Garcia-Moreno EB (2015) Conformational reorganization coupled to the ionization of internal Lys residues in proteins. Biochemistry 54(38):5888–5897 [DOI] [PubMed] [Google Scholar]
- 105.Richter F, Fonfara I, Bouazza B, Schumacher CH, Bratovic M, Charpentier E, Moglich A (2016) Engineering of temperature- and light-switchable Cas9 variants. Nucleic Acids Res 44(20):10003–10014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rockwell KLA, (24 November 2016) Allosterism in drug discovery [Google Scholar]
- 107.Sarkar CA, Lowenhaupt K, Horan T, Boone TC, Tidor B, Lauffenburger DA (2002) Rational cytokine design for increased lifetime and enhanced potency using pH-activated “histidine switching”. Nat Biotechnol 20(9):908–913 [DOI] [PubMed] [Google Scholar]
- 108.Schierling B, Noel AJ, Wende W, Hien LT, Volkov E, Kubareva E, Oretskaya T, Kokkinidis M, Rompp A, Spengler B, Pingoud A. Controlling the enzymatic activity of a restriction enzyme by light. pp 1091–6490 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schonberger M, Trauner D (2014) A photochromic agonist for mu-opioid receptors. Angew Chem Int Ed Engl 53(12):3264–3267 [DOI] [PubMed] [Google Scholar]
- 110.Seifert S, Brakmann S (2018) LOV Domains in the design of photoresponsive enzymes. ACS Chem Biol 13(8):1914–1920 [DOI] [PubMed] [Google Scholar]
- 111.Selvaratnam R, Chowdhury S Schouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. pp 1091–6490 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shah V, Kim JR (2016) Transposon for protein engineering. Mob Genet Elem 6(6):e1239601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shah V, Pierre B. Fau-Kim JR, Kim JR. Facile construction of a random protein domain insertion library using an engineered transposon. pp 1096–0309 (Electronic) [DOI] [PubMed] [Google Scholar]
- 114.Spiltoir JI, Strickland D, Glotzer M, Tucker CL (2016) Optical control of Peroxisomal trafficking. ACS Synth Biol 5(7):554–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Srivastava J, Barber DL, Jacobson MP (2007) Intracellular pH sensors: design principles and functional significance. Physiology (Bethesda, Md) 22:30–39 [DOI] [PubMed] [Google Scholar]
- 116.Stauffer ME, Chazin WJ (2004) Structural mechanisms of DNA replication, repair, and recombination. J Biol Chem 279(30):30915–30918 [DOI] [PubMed] [Google Scholar]
- 117.Strickland D, Moffat K, Sosnick TR (2008) Light-activated DNA binding in a designed allosteric protein. Proc Natl Acad Sci U S A 105(31):10709–10714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Swails JM, Roitberg AE. Enhancing conformation and protonation state sampling of Hen Egg White Lysozyme Using pH Replica Exchange Molecular Dynamics. pp 1549–9618 (Print) [DOI] [PubMed] [Google Scholar]
- 119.Swint-Kruse L, Matthews KS (2009) Allostery in the LacI/GalR family: variations on a theme. Curr Opin Microbiol 12(2):129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tillotson BJ, Goulatis LI, Parenti I, Duxbury E, Shusta EV (2015) Engineering an anti-transferrin receptor ScFv for pH-sensitive binding leads to increased intracellular accumulation. PLoS One 10(12):e0145820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tsai YT, Chuang MJ, Tang SH, Wu ST, Chen YC, Sun GH, Hsiao PW, Huang SM, Lee HJ, Yu CP, Ho JY, Lin HK, Chen MR, Lin CC, Chang SY, Lin VC, Yu DS, Cha TL (2015) Novel Cancer therapeutics with allosteric modulation of the mitochondrial C-Raf-DAPK complex by Raf inhibitor combination therapy. Cancer Res 75(17):3568–3582 [DOI] [PubMed] [Google Scholar]
- 122.Ueda T, Murayama K, Yamamoto T, Kimura S, Imanishi Y (1994) Photo-regulation of hydrolysis activity of semisynthetic mutant phospholipases A2 replaced by non-natural aromatic amino acids. J Chem Soc 2(2):225–230 [Google Scholar]
- 123.Vishveshwara S, Brinda K, Nkjjo T, Chemistry C (2002) Protein structure: insights from graph theory. J Theoret Comput Chem 1(01):187–211 [Google Scholar]
- 124.Volgraf M, Gorostiza P, Szobota S, Helix MR, Isacoff EY, Trauner D (2007) Reversibly caged glutamate: a photochromic agonist of ionotropic glutamate receptors. J Am Chem Soc 129(2):260–261 [DOI] [PubMed] [Google Scholar]
- 125.Wang H, Vilela M, Winkler A, Tarnawski M, Schlichting I, Yumerefendi H, Kuhlman B, Liu R, Danuser G, Hahn KM (2016) LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat Methods 13(9):755–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Webby CJ, Jiao W, Hutton RD, Blackmore NJ, Baker HM, Baker EN, Jameson GB, Parker EJ (2010) Synergistic allostery, a sophisticated regulatory network for the control of aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J Biol Chem 285(40):30567–30576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wehler P, Niopek D, Eils R, Di Ventura B (2016) Optogenetic control of nuclear protein import in living cells using light-inducible nuclear localization signals (LINuS). Curr Protoc Chem Biol 8(2):131–145 [DOI] [PubMed] [Google Scholar]
- 128.Willner I, Rubin S, Riklin A (1991) Photoregulation of papain activity through anchoring photochromic azo groups to the enzyme backbone, vol 113 [Google Scholar]
- 129.Wilson CJ, Zhan H, Swint-Kruse L, Matthews KS (2007) The lactose repressor system: paradigms for regulation, allosteric behavior and protein folding. Cell Mol Life Sci 64(1):3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wong S, Mosabbir AA, Truong K (2015) An engineered Split Intein for Photoactivated protein trans-splicing. PLoS One 10(8):e0135965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wright CM, Majumdar A, Tolman JR, Ostermeier M (2010) NMR characterization of an engineered domain fusion between maltose binding protein and TEM1 beta-lactamase provides insight into its structure and allosteric mechanism. Proteins 78(6):1423–1430 [DOI] [PubMed] [Google Scholar]
- 132.Wright CM, Wright RC, Eshleman JR, Ostermeier M (2011) A protein therapeutic modality founded on molecular regulation. Proc Natl Acad Sci U S A 108(39):16206–16211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM (2009) A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461(7260):104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu J, Sarma AVS, Wei Y, Beamer LJ, Van Doren SR (2017) Multiple ligand-bound states of a Phosphohexomutase revealed by principal component analysis of NMR peak shifts. Sci Rep 7(1):5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yamada MD, Nakajima Y, Maeda H, Maruta S (2007) Photocontrol of kinesin ATPase activity using an azobenzene derivative. J Biochem 142(6):691–698 [DOI] [PubMed] [Google Scholar]
- 136.Yates FE, Iberall ASJAOBE (1973) Chance and necessity: an essay on the natural philosophy of modern biology by Jacques Monod. Ann Biomed Eng 1(3):381–384 [Google Scholar]
- 137.Yi JJ, Wang H, Vilela M, Danuser G, Hahn KM (2014) Manipulation of endogenous kinase activity in living cells using photoswitchable inhibitory peptides. ACS Synth Biol 3(11):788–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yumerefendi H, Lerner AM, Zimmerman SP, Hahn K, Bear JE, Strahl BD, Kuhlman B. Lightinduced nuclear export reveals rapid dynamics of epigenetic modifications. pp 1552–4469 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang Y, Kitazawa S, Peran I, Stenzoski N, McCallum SA, Raleigh DP, Royer CA (2016) High pressure ZZ-exchange NMR reveals key features of protein folding transition states. J Am Chem Soc 138(46):15260–15266 [DOI] [PubMed] [Google Scholar]
- 140.Zimenkov Y, Dublin SN, Ni R, Tu RS, Breedveld V, Apkarian RP, Conticello VP (2006) Rational design of a reversible pH-responsive switch for peptide self-assembly. J Am Chem Soc 128(21):6770–6771 [DOI] [PubMed] [Google Scholar]