

Summary
Induction of protective mucosal T cell memory remains a formidable challenge to vaccinologists. Using a combination adjuvant strategy that elicits potent CD8 and CD4 T cell responses, we define the tenets of vaccine-induced pulmonary T cell immunity. An acrylic-acid-based adjuvant (ADJ), in combination with Toll-like receptor (TLR) agonists glucopyranosyl lipid adjuvant (GLA) or CpG, promotes mucosal imprinting but engages distinct transcription programs to drive different degrees of terminal differentiation and disparate polarization of TH1/TC1/TH17/TC17 effector/memory T cells. Combination of ADJ with GLA, but not CpG, dampens T cell receptor (TCR) signaling, mitigates terminal differentiation of effectors, and enhances the development of CD4 and CD8 TRM cells that protect against H1N1 and H5N1 influenza viruses. Mechanistically, vaccine-elicited CD4 T cells play a vital role in optimal programming of CD8 TRM and viral control. Taken together, these findings provide further insights into vaccine-induced multifaceted mucosal T cell immunity with implications in the development of vaccines against respiratorypathogens, including influenza virus and SARS-CoV-2.
Keywords: adjuvants, CD8, CD4, respiratory immunity, influenza virus, T1 and T17 programming, tissue-resident memory, heterosubtypic, mucosal subunit vaccines
Graphical Abstract
Highlights
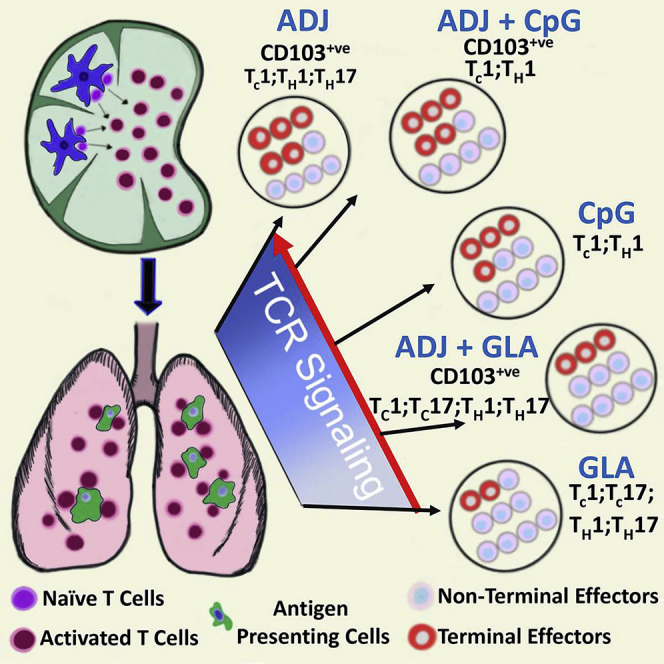
Combination adjuvants stimulate potent TRM cell immunity in the respiratory tract
Differentiation and functional programming depend on adjuvant and TCR signaling
Vaccine-induced T cell immunity to influenza requires CD4 and CD8 T cells
CD4 T cells regulate optimal positioning and programming of CD8 TRM in lungs
Marinaik et al. report that carbomer and TLR-based combination adjuvants elicit airway- and lung-resident memory and confer heterosubtypic immunity to influenza virus. Further, they show that combination adjuvants stimulate varying levels of TCR signaling and cytokines in lungs and drive disparate patterns of effector/memory differentiation, mucosal imprinting, and functional polarization.
Introduction
Viral mucosal infections, such as influenza, cause considerable morbidity and even mortality in very young and geriatric patients.1 Protection afforded against influenza A virus (IAV) by antibodies is typically virus type/subtype specific; however, T cells are believed to provide broad heterosubtypic immunity.2, 3, 4, 5 IAV infection elicits strong effector CD8 and CD4 T cell responses in the lungs, leading to the development of protective lung- and airway-resident memory T cells.3,6 However, influenza-specific mucosal memory T cells exhibit attrition and T-cell-based protection wanes in a span of 3–6 months.7,8 Therefore, unlike systemic viral infections that typically engender enduring immunity,9,10 mucosal viral infections fail to program durable T cell immunity in the respiratory tract (RT). Although engagement of multiple innate receptors early in the response might be key to long-lived immunological memory following systemic infections,11,12 there is a lack of understanding of why mucosal infections lead to shorter duration of cellular immunity. There is a general paucity of adjuvants that induce strong T cell responses, and we have limited knowledge of mucosal T cell responses to adjuvanted subunit vaccines, especially in the RT. These knowledge gaps pose daunting constraints in the development of immunization strategies targeted at the establishment of durable protective T cell immunity in the RT.12, 13, 14, 15
Adjuplex (ADJ) is a polyacrylic-acid (carbomer)-based adjuvant that is a component of some current veterinary vaccines and also known to induce neutralizing antibodies against HIV and malaria.16, 17, 18, 19 Here, we report that ADJ, in combination with Toll-like receptor (TLR) 4/9 agonists, elicits unexpectedly potent and functionally diverse CD8 and CD4 T cell responses to a subunit viral protein in the RT. Studies with this adjuvant system provided the means to differentially program distinct patterns of effector and memory T cell differentiation in the RT. Further, these studies provide a glimpse of the evolution of T cell responses to adjuvanted vaccines in the lungs to define the quantitative, phenotypic, and functional attributes of mucosal effector/memory CD8 and CD4 T cells that are associated with effective viral control in the lungs and protection against H1N1 and H5N1 influenza infections. Collectively, these findings provide insights into the immunological apparatus underlying the generation and establishment of protective and durable T cell immunity in the RT in response to adjuvanted subunit vaccines.
Results
Differential Programming and Mucosal Imprinting of Effector CD8 T Cells in the RT by Carbomer/TLR-Agonist-Based Combination Adjuvants
Differentiation of effector T cells in the RT has been extensively characterized following IAV infection.6 However, the extent to which adjuvanted subunit vaccines drive the expansion and differentiation of effector T cells in the RT is unknown. Here, we assessed whether ADJ, CpG, and glucopyranosyl lipid adjuvant (GLA) individually or in combination differed in terms of the magnitude and nature of the effector T cell response to intranasal vaccination with influenza virus nucleoprotein (NP). Mice were vaccinated with various adjuvants twice (prime and boost) at an interval of 3 weeks. Effector T cell responses were analyzed 8 days after the second (i.e., booster) vaccination. Because protein vaccination without an adjuvant failed to elicit detectable CD8 T cell responses,20 we did not include the NP only group in this study. At day 8 post-vaccination (PV), all adjuvants elicited surprisingly potent NP-specific CD8 T cell responses in the lungs and airways (Figure 1A). ADJ+GLA stimulated the strongest CD8 T cell response, and remarkably, 15%–40% of CD8 T cells were specific to the NP366 epitope in lungs or airways. ADJ+GLA also elicited systemic CD8 T cell responses in spleen (Figure S1A).
Figure 1.

Effector CD8 T Cell Response to Adjuvanted Vaccines
C57BL/6 mice were vaccinated intranasally (IN) twice (3 weeks apart) with influenza virus nucleoprotein (NP) formulated in the indicated adjuvants. At day 8 post-booster vaccination (PV), cells in the lungs and bronchoalveolar lavage (BAL) were stained with Db/NP366 tetramers along with antibodies to cell surface molecules, granzyme B, and transcription factors directly ex vivo.
(A, B, C, and E) Fluorescence-activated cell sorting (FACS) plots show percentages of gated tetramer-binding CD8 T cells in respective gates/quadrants.
(D) Median fluorescence intensities (MFIs) for transcription factors in NP366-specific CD8 T cells.
Data are pooled from two independent experiments or represent one of two independent experiments. Comparisons were made using one-way ANOVA test with Tukey-corrected multiple comparisons; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Elevated CX3CR1 and KLRG-1 expressions are associated with terminal differentiation of effector T cells.21, 22, 23, 24 Among single adjuvants, ADJ induced the highest level of CX3CR1 expression, followed by CpG and GLA (Figure 1B). ADJ and/or CpG promoted CX3CR1 expression, but the percentages of KLRG-1HI cells were comparable between the groups. Notably, the percentages of terminally differentiated CX3CR1HIKLRG-1HI NP366-specific CD8 T cells in lungs of ADJ, CpG, and ADJ+CpG groups were comparable but significantly higher (p < 0.05) than in the GLA group. Interestingly, comparison of ADJ, GLA, and ADJ+GLA groups suggested that GLA limited the development of CX3CR1HI CD8 T cells.
As another surrogate marker for effector differentiation, we quantified granzyme B levels in CD8 T cells directly ex vivo (Figure 1C). The percentages of granzyme BHI CD8 T cells among NP366-specific CD8 T cells in ADJ, CpG, and ADJ+CpG groups were significantly (p < 0.05) higher than in GLA or ADJ+GLA groups. Clearly, ADJ and CpG promoted granzyme B expression, but GLA antagonized the granzyme-B-enhancing effects of ADJ.
Studies to determine the transcriptional basis for the disparate differentiation of effector CD8 T cells in different adjuvant groups showed that the expressions of T-bet, interferon regulatory factor 4 (IRF-4), and basic leucine zipper ATF-like transcription factor (BATF) were substantially greater in ADJ and ADJ+CpG groups, compared to GLA and ADJ+GLA groups (Figure 1D). Although ADJ appeared to be the primary driver of T-bet, IRF-4, and BATF expression, GLA effectively negated this effect in ADJ+GLA mice (Figure 1D). The levels of EOMES did not differ between adjuvants, but analysis of T-bet and EOMES co-expression showed that a higher percentage of CD8 T cells co-expressed T-bet and EOMES (T-betHIEOMESHI) in the CpG and ADJ+CpG groups (Figure S1B). By contrast, a greater proportion of CD8 T cells in GLA and ADJ+GLA groups expressed EOMES, but not T-bet (T-betLOEOMESHI; Figure S1B). Taken together, terminal differentiation of effector CD8 T cells in ADJ and/or CpG was associated with high levels of T-bet, IRF-4, and BATF.
Next, we assessed expression of CD103 and CD69 to ask whether adjuvants affected mucosal imprinting of CD8 T cells in the RT. The majority of NP366-specific CD8 T cells in lungs and bronchoalveolar lavage (BAL) expressed CD69, but not CD103, in all groups. The percentages of CD103HICD69HI CD8 T cells in ADJ, ADJ+CpG, and ADJ+GLA groups were higher than in CpG and GLA groups, which suggested that ADJ was a potent inducer of CD103 (Figure 1E). Altogether, Figure 1 shows that ADJ and/or CpG promoted different facets of CD8 T cell terminal differentiation. Remarkably, however, when combined with ADJ, GLA antagonized ADJ-driven terminal differentiation program without affecting mucosal imprinting of CD8 T cells. Thus, ADJ-driven CD8 T cell differentiation program can be augmented or antagonized by TLR agonists CpG and GLA, respectively.
Adjuvants Regulate Differentiation and Mucosal Imprinting of Effector CD4 T Cells in the RT
Next, we characterized NP-specific CD4 T cell responses to various adjuvants following mucosal immunization. At day 8 PV, high percentages of NP311-specific CD4 T cells were detected in lungs and airways of all groups of mice (Figure 2A). The percentages and total numbers of NP311-specific CD4 T cells in lungs and airways were comparable between ADJ, CpG, GLA, and ADJ+CpG groups. However, the total numbers of NP311-specific CD4 T cells in the lungs and airways of ADJ+GLA group were significantly higher than in other groups (Figure 2A).
Figure 2.

Effector CD4 T Cell Response to Adjuvanted Vaccines
Groups of C57BL/6 mice were vaccinated IN, as in Figure 1. At day 8 PV, cells from lungs and BAL were stained with I-Ab/NP311 tetramers along with antibodies to cell surface molecules and transcription factors.
(A) FACS plots show the percentages of I-Ab/NP311 tetramer-binding cells among CD4 T cells.
(B) Percentages of the indicated cell population among NP311-specific, tetramer-binding CD4 T cells.
(C) FACS plots are gated on I-Ab/NP311 tetramer-binding cells, and the numbers in each quadrant are the percentages of cells among the gated population; MFIs for transcription factors in NP311-specific CD4 T cells are plotted in the adjoining graphs.
(D) FACS plots in (C) were used to quantify the percentages of T-betLOEOMESHI cells (quadrant 4) among NP311-specific CD4 T cells.
(E) Percentages of CD103HI and CD69HI cells among NP311-specific CD4 T cells.
Data are representative of two independent experiments. Comparisons were made using one-way ANOVA test with Tukey-corrected multiple comparisons; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Phenotypically, ADJ and CpG promoted the expression of terminal differentiation markers CX3CR1 and KLRG-1, respectively (Figure 2B). By contrast, expressions of CX3CR1 and KLRG-1 were lowest in the GLA group (Figure 2B) and GLA tempered ADJ-induced expression of CX3CR1 in ADJ+GLA group. NP311-specific CD4 T cells from ADJ and/or CpG groups contained greater levels of T-bet, as compared to other groups (Figure 2C), but EOMES levels were not different between groups. GLA with or without ADJ induced the lowest levels of T-bet, which resulted in greater percentages of T-betLOEOMESHI CD4 T cells in GLA and ADJ+GLA groups (Figure 2D). Thus, ADJ and CpG might have promoted terminal differentiation of CD4 T cells by inducing T-bet expression, as compared to GLA or ADJ+GLA groups. Analysis of mucosal imprinting markers CD103 and CD69 showed that ADJ-containing adjuvants elicited higher percentages of CD103HI and CD103HICD69HI CD4 T cells in lungs (Figure 2E). Thus, in contrast to ADJ and CpG, combining ADJ with GLA promoted the development of less-differentiated mucosally imprinted CD4 T cells in the lungs and airways.
Distinct Functional Programming of Mucosal Effector CD8 and CD4 T Cells by Combination Adjuvants
We then asked whether adjuvants regulated functional programming of effector CD8 and CD4 T cells into TC1/TC17 or TH1/TH17 subsets, respectively, in lungs. NP366-specific interferon (IFN)-γ-producing TC1 CD8 T cells were induced in all groups, and the percentages of such cells among CD8 T cells were generally higher in the ADJ+GLA group (Figure 3A). Interestingly, however, interleukin-17 (IL-17)-producing NP366-specific TC17 CD8 T cells were strongly induced only in the GLA and ADJ+GLA groups. To further elucidate the relative dominance of TC1 versus TC17 in different adjuvant groups, we calculated the relative proportions of these cells among total cytokine-producing (IL-17+IFN-γ-producing cells), peptide-stimulated, NP366-specific CD8 T cells (Figure 3B); ∼80%–88% of NP366-specific, cytokine-producing CD8 T cells produced IFN-γ in the CpG and ADJ+CpG groups, and only 65%, 56%, and 36% of such cells produced IFN-γ in ADJ, GLA, and ADJ+GLA groups, respectively. Reciprocally, although only a relatively small fraction (12%–20%) of NP366-specific, cytokine-producing CD8 T cells produced IL-17 or IL-17+IFN-γ in CpG and ADJ+CpG groups, 40%–57% of NP366-specific CD8 T cells produced IL-17 or IL-17+IFN-γ in GLA and ADJ+GLA groups. Thus, CpG and ADJ+CpG promoted functional polarization of TC1 cells, and ADJ, GLA, and ADJ+GLA drove a balanced differentiation of TC1 and TC17 cells. Evaluation of the ability of NP366-specific CD8 T cells to co-produce IFN-γ, tumor necrosis factor alpha (TNF-α), and IL-2 (Figure 3C) showed that all adjuvants induced polyfunctional CD8 T cells, but a significantly higher percentage of NP366-specific CD8 T cells in the GLA group were polyfunctional, as compared to other groups (Figure 3C).
Figure 3.

Functional Polarization of Effector CD8 and CD4 T Cells
C57BL/6 mice were vaccinated as in Figure 1. On the 8th day PV, lung cells were stimulated with NP366 or NP311 peptides for 5 h. The percentages of NP366-specific CD8 T cells (A–C) or NP311-specific CD4 T cells (D–F) that produced IFN-γ, IL-17, TNF-α, and IL-2 were quantified by intracellular cytokine staining.
(A) Percentages of cytokine-producing cells among the gated CD8 T cells.
(B) To demonstrate relative dominance of TC1 versus TC17 in different groups, we calculated the relative proportions of these cells among total cytokine-producing CD8 or CD4 T cells (IL-17+IFN-γ-producing cells following stimulation with NP366 peptide).
(C) Plots are gated on IFN-γ-producing CD8 T cells, and the numbers are the percentages among the gated cells.
(D) Percentages of cytokine-producing cells among the gated CD4 T cells.
(E) Calculated percentages of IFN-γ and/or IL-17-producing CD4 T cells among NP311-specific, cytokine-producing CD4 T cells.
(F) Plots are gated on IFN-γ-producing CD4 T cells.
Data are representative of two independent experiments. Comparisons were made using one-way ANOVA test with Tukey-corrected multiple comparisons; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
NP311-specific TH1 and TH17 CD4 T cells were induced to varying levels by different adjuvants (Figure 3D). ADJ promoted TH17 polarization of effector CD4 T cells, but CpG promoted TH1 differentiation and negated the TH17 skewing effects of ADJ in the ADJ+CpG group. TH17 differentiation dominated over the TH1 development in GLA and ADJ+GLA groups (Figure 3E). In summary, although CpG and ADJ+CpG promoted the development of TH1 effector cells, ADJ, GLA, and ADJ+GLA favored the differentiation of TH17 cells (Figure 3E). Polyfunctionality among NP311-specific CD4 T cells was largely comparable between groups (Figure 3F).
Role of TCR Signaling and Inflammation in Regulating the Differentiation of Vaccine-Elicited Effector T Cells
Antigenic stimulation and the inflammatory milieu govern effector differentiation during infections.24, 25, 26 In order to determine whether adjuvants differed in terms of antigenic stimulation in draining lymph nodes (DLNs) and lungs, early after vaccination (days 2 and 5), we adoptively transferred 5 × 104 T cell receptor (TCR) transgenic OT-I CD8 T cells that express GFP under the control of Nur77 promoter; Nur77 expression faithfully reports specific TCR signaling in T cells.27 Subsequently, mice were vaccinated with chicken ovalbumin (OVA) mixed with different adjuvants, and GFP expression by OT-I CD8 T cells was assessed at days 2 and 5 PV. OT-I CD8 T cells expressed readily detectable levels of GFP in DLNs and lungs at different days PV (Figure 4A). Overall, GFP levels were not significantly different for OT-I CD8 T cells (p < 0.05) in DLNs between various groups (except between GLA and ADJ+GLA) at day 5 PV (Figure 4A). OT-I CD8 T cells were not detectable in lungs until day 5 PV; at day 5 PV, significantly (p < 0.05) higher levels of GFP were detected in OT-I CD8 T cells from the lungs of ADJ mice, compared to CpG, GLA, and ADJ+GLA groups (Figure 4A). Adoptive transfer of 5 × 104 TCR transgenic CD8 T cells was technically essential to assess T cell signaling early after vaccination (Figure 4A), but transfer of such high numbers of T cells might affect their differentiation.28 Therefore, for assessment of TCR signaling at day 8 PV, we transferred 103 Nur77-GFP OT-I TCR transgenic CD8 T cells prior to vaccination. The pattern of GFP fluorescence in donor OT-I CD8 T cells in DLNs and lungs of vaccinated mice at day 8 PV is shown in Figure S2. On the 8th day PV, OT-I CD8 T cells in the DLNs of ADJ mice expressed higher levels of GFP, compared to other groups, but the differences did not reach statistical significance. By contrast, on day 8 PV, GFP levels in OT-I CD8 T cells from lungs of ADJ mice were significantly higher (p < 0.05) than in OT-I CD8 T cells from lungs of CpG, GLA, and ADJ+GLA mice (Figure 4A). Collectively, a greater percentage of OT-I CD8 T cells in the lungs of ADJ group showed evidence of active TCR signaling in the lungs at days 5 and 8 after vaccination, and notably, this effect of ADJ was dampened by GLA, but not CpG. Enhanced TCR signaling in ADJ group (and to a lesser extent in CpG group) was consistent with elevation of IRF-4 and BATF (Figure 1D).29
Figure 4.

Regulation of the Effector T Cell Response by Antigen Receptor Signaling
(A) Ly5.1 Nur77-GFP OT-I CD8 T cells were adoptively transferred into congenic Ly5.2 C57BL/6 mice and vaccinated IN next day with OVA protein formulated in the indicated adjuvants. At days 2, 5, or 8 PV, cells from lymph nodes and lungs were stained with Kb/SIINFEKL tetramers, anti-Ly5.1, anti-Ly5.2, anti-CD8, and anti-CD44 antibodies. The GFP MFIs in donor Ly5.1+ve OT-I CD8 T cells were quantified by flow cytometry.
(B) Wild-type non-transgenic (WT) and transgenic KLF2-GFP mice were vaccinated with NP protein formulated in adjuvants, as in (A). At day 8 PV, lung cells were stained with anti-CD8, anti-CD44, and Db/NP366 tetramers. The overlay histogram shows GFP fluorescence (MFI) for the gated tetramer-binding CD8 T cells from WT (black) and KLF2-GFP transgenic (red) mice.
(C) B6 mice were vaccinated with NP protein formulated in various adjuvants, as in (A). At day 8, PV lung cells were stained with anti-CD8, anti-CD44, anti-PD-1, and Db/NP366 tetramers. Plots show the percentages of PD-1+ve cells among the gated Db/NP366 tetramer-binding CD8 T cells.
(D) Statistical correlation analysis between the percentages of PD-1+ve CD8 T cells and the percentages of tetramer+ve CD8 T cells at day 8 PV.
Data are pooled from two independent experiments or represent one of two independent experiments. Comparisons were made using one-way ANOVA test with Tukey-corrected multiple comparisons. For (D), we used two-way ANOVA, Student’s t test, and simple regression analysis; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Transcription factor KLF2, which regulates T cell trafficking, is downregulated by TCR signaling.30,31 Using KLF2-GFP reporter mice,31 we assessed whether high TCR signaling in ADJ-vaccinated mice led to KLF2 downregulation in polyclonal NP366-specific CD8 T cells in the DLNs and lungs at day 8 PV. In all groups, NP366-specific CD8 T cells downregulated KLF2 expression in lungs, relative to KLF2 levels in their respective DLNs (Figure 4B). In lungs of ADJ, CpG, and ADJ+CpG groups, NP366-specific CD8 T cells expressed lower levels of KLF2 than in CD8 T cells from GLA and ADJ+GLA groups (Figure 4B). These data suggested that ADJ and/or CpG might enhance TCR-signaling-induced KLF2 downregulation in lungs, as compared to ADJ+GLA.
During influenza virus infection in mice, TCR signaling drives PD-1 expression in lungs.32 Therefore, we investigated whether PD-1 expression was linked to varying levels of TCR signaling induced by different adjuvants. At day 8 PV, higher percentages of NP366-specific CD8 T cells in ADJ mice expressed PD-1, as compared to those in CpG and GLA mice (Figure 4C). Interestingly, addition of GLA, but not CpG, to ADJ significantly reduced ADJ-driven PD-1 expression on NP366-specific CD8 T cells (Figure 4C). To elucidate the possible relationship between the frequency of NP366-specific CD8 T cells and their PD-1 expression levels in the lungs, we calculated correlation co-efficient between the two parameters (Figure 4D). Strikingly, there was a significant linear inverse correlation between PD-1 expression and the frequency of NP366-specific CD8 T cells in lungs of mice vaccinated with ADJ, CpG, and GLA adjuvants. These findings suggested that TCR-signaling-induced PD-1 expression might limit the accumulation of CD8 T cells (clonal burst size) in the lungs. In summary (Figures 1 and 4), terminal differentiation of effector CD8 T cells in ADJ and ADJ+CpG groups was associated with enhanced TCR signaling in the lungs. Reciprocally, GLA might protect effector CD8 T cells from ADJ-driven terminal differentiation by limiting TCR signaling in the lungs.
To explore whether TCR signaling in CD8 T cells in ADJ+GLA mice is governed by the abundance of antigen-presenting cells in lungs, first we quantified innate immune cells, including dendritic cells (DCs), in lungs at days 5 and 8 PV (Figure S3A). ADJ+GLA and ADJ+CpG increased the infiltration of neutrophils in lungs at days 5 and 8, respectively. Only at day 5, but not at day 8, PV, lungs of ADJ, ADJ+CpG, and ADJ+GLA contained higher numbers of monocytes and monocyte-derived DCs than in CpG and GLA mice. There were no differences between the groups in the numbers of CD103+ve DCs or alveolar macrophages on either day after vaccination. We determined the abundance and type of antigen-processing cells in lungs by vaccinating mice with DQ-OVA, which emits green/red fluorescence upon degradation by proteases (Figure S3B). As compared to CpG and GLA groups, lungs of ADJ and ADJ+CpG (and ADJ+GLA to a slightly lesser degree) contained higher numbers of DQ-OVA-bearing monocyte-derived DCs, monocytes, and CD103+ve DCs at day 5 PV, but not at day 8 PV. These data suggested that dampened TCR signaling in ADJ+GLA group, as compared to augmented signaling in ADJ and ADJ+CpG groups (Figures 4A–4D), cannot be simply explained by reduced abundance of specific antigen-bearing cells in the lungs.
To determine whether early inflammatory response influenced the phenotypic and functional differentiation of effector T cells, we quantified cytokine expression in the lungs. At 24 (Figure S4) and 48 h (Figure S5) PV, the levels of cytokines/chemokines IL-1α, IL-1β, IL-6, KC, RANTES, G-CSF, and GM-CSF were higher in lungs of GLA and/or ADJ+GLA mice. However, the levels of IFN-β, IFN-λ, IL-10, IL-12p40, IL-12p70, MIP1α, MIP1β, MCP, transforming growth factor β1 (TGF-β1), and TNF-α in lungs were largely comparable between groups, except for the ADJ group (Figures S4 and S5). Thus, terminal differentiation of effector CD8 T cells in ADJ, CpG, and ADJ+CpG mice was not associated with excessive inflammation in the lungs, relative to other groups. Notably, however, TC17 and TH17 cell development in GLA and ADJ+GLA groups was associated with elevated IL-1α in the lungs. Further, development of TH1 effectors and enhanced T-bet induction (Figures 1 and 2) in CpG and ADJ+CpG groups was not associated with elevated levels of IL-12p70 in the lungs. Thus, the differences in accumulation and terminal differentiation of effector T cells in the lungs of vaccinated mice cannot be explained by the degree of early inflammation.
Mucosal CD8 and CD4 T Cell Memory in Vaccinated Mice
At 100 days PV, we quantified NP366-specific memory CD8 T cells in lungs, airways, and spleen. All adjuvants elicited robust CD8 T cell memory in the RT (Figure 5). Notably, both frequencies and total numbers of NP366-specific memory CD8 T cells in lungs and airways of ADJ+GLA group were significantly (p < 0.05) higher than in other groups (Figure 5A); total numbers were not different in spleen. Intravascular staining showed that 60%–80% of NP366-specific memory CD8 T cells in the lungs localized to the non-vascular compartment in ADJ, CpG, GLA, and ADJ+GLA groups; the percentages of non-vascular memory CD8 T cells were slightly reduced in the ADJ+CpG group (Figure 5B). The percentages of CD103+veCD69+ve lung resident memory (TRM) cells among NP366-specific CD8 T cells were comparable for various adjuvants (Figure 5C). However, lungs of ADJ+GLA group contained significantly (p < 0.05) greater numbers of both non-vascular and vascular CD103+ve NP366-specific CD8 T cells, as compared to other groups (Figure 5D). Thus, ADJ+GLA was the most effective adjuvant that elicited high numbers of CD103+ve TRM CD8 T cells in the airways and the non-vascular compartment of the lungs.
Figure 5.

Mucosal CD8 and CD4 T Cell Memory in Vaccinated Mice
At 100 days after booster vaccination, NP366-specific memory CD8 T cells (A–D) and NP311-specific CD4 T cells (E–H) were characterized in lungs, airways (BAL), and spleen. To stain for vascular cells, mice were injected intravenously with fluorescent-labeled anti-CD45.2 antibodies, 3 min prior to euthanasia. Cells from lungs and BAL were stained with Db/NP366 tetramers, I-Ab/NP311 tetramers, and anti-CD4, anti-CD8, anti-CD44, anti-CD103, and anti-CD69 antibodies.
(A) Percentages and total numbers of NP366-specific CD8 T cells in lungs, BAL, and spleen.
(B) FACS plots are gated on NP366-specific, tetramer-binding CD8 T cells; numbers are the percentages of vascular and non-vascular cells in the gated population.
(C) Percentages of CD69+veCD103+ve TRM cells among NP366-specific CD8 T cells.
(D) Total numbers of vascular and non-vascular CD103+ve NP366-specific CD8 T cells in lungs.
(E) Percentages and total numbers of NP311-specific CD4 T cells in lungs.
(F) Percentages of vascular and non-vascular cells among NP311-specific CD4 T cells in lungs.
(G) Percentages of IFN-γ- or IL-17-producing cells among CD4 T cells.
(H) Calculated percentages of IFN-γ- and/or IL-17-producing CD4 T cells among total NP311-specific, cytokine-producing (IFN-γ + IL-17) peptide-stimulated CD4 T cells.
Data are pooled from two independent experiments. Comparisons were made using one-way ANOVA test with Tukey-corrected multiple comparisons; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
At 100 days PV, all adjuvants induced strong CD4 T cell memory and the percentages of NP311-specific memory CD4 T cells ranged from 1.5% to 4% in the lungs (Figure 5E). The percentages of memory CD4 T cells in lungs of ADJ+GLA group were consistently higher than in other groups (Figure 5E). Regardless of adjuvants, 60%–80% of memory CD4 T cells localized to the non-vascular compartment in the lungs (Figure 5F). Likewise, the percentages (15%–20%) of lung CD69+ve TRM-like CD4 T cells were comparable for various adjuvants.
We determined whether polarization of TH1 versus TH17 was maintained in memory CD4 T cells of vaccinated mice. At 100 days PV, IFN-γ and/or IL-17-producing NP-specific memory CD4 T cells were detectable in the lungs of vaccinated mice (Figure 5G). Figure 5H illustrates that the percentages of NP-specific, cytokine-producing CD4 T cells that produce IFN-γ and/or IL-17 differed among various groups. IFN-γ-producing CD4 T cells were only dominant (∼60%) in the CpG group, but IL-17-producing CD4 T cells formed the dominant subset (∼75%) in the GLA and ADJ+GLA groups. About 50%–60% of NP-specific memory CD4 T cells produced IL-17 in the ADJ and ADJ+CpG groups. Therefore, functional programming in effector cells is largely preserved in memory T cells.
Adjuvants Regulate Recall T Cell Responses and Protective Heterosubtypic Immunity to Influenza A Virus in Mice
Mice were vaccinated twice with NP protein formulated in various adjuvants. At day 100 PV, we investigated whether NP-specific T cell memory protected against respiratory challenge with the virulent PR8/H1N1 IAV. On the 6th day after challenge, viral burden was high in lungs of mice that were unvaccinated or vaccinated with NP alone (without adjuvants; Figure 6A). Compared to the unvaccinated and NP-only groups, other groups exhibited varying degrees of protection. The ADJ+GLA vaccine provided the most effective protection, followed by GLA and ADJ+CpG vaccines (Figures 6A and S6A). Although relatively less effective, ADJ and CpG vaccines still reduced viral titers by >90%. Kinetically, at 100 days PV, viral burden was reduced in the lungs of all vaccinated mice within 2–4 days after PR8/H1N1 challenge (Figure S6B), but clear differences in viral control among adjuvants emerged between days 4 and 6 postchallenge (Figures 6A and S6B). Protection against IAV afforded by various adjuvant groups was maintained for at least until day 180 PV (Figure S6C).
Figure 6.

Vaccine-Induced Protective Immunity to H1N1 and H5N1 Influenza Viruses
(A–C) Groups of C57BL/6 mice were vaccinated twice IN, as in Figure 1. At 100 days after the booster vaccination, mice were challenged IN with H1N1/PR8 strain of influenza A virus. Viral tiers and virus-specific T cell responses in lungs were quantified on the 6th day after virus challenge.
(A) Viral titers in the lungs on the 6th day after virus challenge.
(B) Percentages of NP366-specific, IFN-γ- and IL-17-producing cells among CD8 T cells (bar graphs) and calculated proportions of IFN-γ- and/or IL-17-producing cells among total IFN-γ+IL-7-producing, peptide-stimulated, NP366-specific T cells (pie charts).
(C) Percentages of NP311-specific, IFN-γ- and IL-17-producing cells among CD4 T cells and calculated proportions of IFN-γ- and/or IL-17-producing cells among total IFN-γ+IL-7-producing, peptide-stimulated, NP311-specific T cells.
(D) C57BL/6 mice were vaccinated with NP+ADJ+GLA twice at an interval of 3 weeks. 180 days after the last vaccination, mice were challenged IN with H1N1/PR8 strain of influenza A virus; unvaccinated mice were challenged with virus as controls. Cohorts of vaccinated virus-challenged mice were treated with isotype control immunoglobulin G (IgG) or anti-IL-17A antibodies (intravenously [i.v.] and IN) at −1, 0, 1, 3, and 5 days relative to virus challenge. On the 6th day after viral challenge, viral titers and virus-specific T cell responses were quantified in lungs.
(E) Groups of C57BL/6 mice were vaccinated twice, as above. 50 days after booster vaccination, vaccinated and unvaccinated mice were challenged IN with the highly pathogenic H5N1 avian influenza A virus; weight loss and survival were monitored until day 14.
Data are pooled from 2 independent experiments or representative of two independent experiments. In (E), we used non-linear regression for analyzing weight loss data. For the rest, we used one-way ANOVA test with Tukey-corrected multiple comparisons; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To elucidate correlates of protection afforded by various adjuvanted vaccines, we quantified recall CD8 and CD4 T cell responses in the lungs at day 6 after PR8/H1N1 challenge. Interestingly, despite varying levels of protection afforded by various vaccines (Figure 6A), the numbers and extra-vascular localization of NP366-specific CD8 T cells and NP311-specific CD4 T cells in the lungs were comparable between the groups (Figure S6D). The percentages of NP366-specific, IFN-γ-producing CD8 T cells were also comparable for all groups of mice (Figure 6B). In striking contrast, percentages of NP366-specific, IL-17-producing TC17 cells were considerably higher in the lungs of ADJ+GLA and GLA groups (Figure 6B). The percentages of NP311-specific, IFN-γ-producing CD4 T cells in the CpG and ADJ+CpG groups were significantly higher than in other groups (Figure 6C). In addition, lungs of GLA and ADJ+GLA mice contained higher percentages of IL-17-producing, NP311-specific TH17 CD4 T cells than in other groups. In this adjuvant system, all adjuvants afforded considerable protection. However, differences in viral control between groups appeared to associate with disparate levels of TC17 and/or TH17 cells, but not TC1 or TH1 cells. For example, better viral control by GLA and ADJ+GLA groups was associated with increased percentages of IL-17-producing, NP366-specific TC17 and NP311-specific TH17 cells (Figures 6B and 6C). CpG and ADJ+CpG groups also differed in the percentages of NP311-specific TH17 cells, but not TH1 or TC1 cells. These data suggest that stimulation of TC17/TH17 cells in parallel with TC1/TH1 cells might constitute a correlate of enhanced immunity conferred by ADJ and GLA, as compared to ADJ and CpG groups. To test this inference, we assessed the importance of IL-17A in mediating protective immunity to IAV in mice vaccinated with NP formulated in ADJ+GLA. At 180 days after vaccination, ADJ+GLA-vaccinated mice were treated with isotype control antibodies or anti-IL-17A antibodies, just prior to viral challenge. Data in Figure 6D show that treatment with anti-IL-17A antibodies did not affect the accumulation of NP366-specific CD8 T cells or NP311-specific CD4 T cells in lungs following viral challenge. Lung viral titers in isotype control antibody and anti-IL-17A-treated mice were not significantly different (Figure 6D), which suggested that blockade of IL-17A did not affect viral control. Although IL-17 production is known to be protective against certain fungal and bacterial infections, it is also linked to immune pathology.33,34 In order to evaluate whether protective immunity in ADJ+GLA mice was associated with lung pathology, we analyzed histopathological changes in lungs after viral challenge (Figure S7). With the exception of the ADJ group, moderate necrotizing bronchiolitis was present in all mice and was most severe in the CPG, where it progressed to early-stage bronchiolitis obliterans and organizing pneumonia. Very mild extension to the surrounding alveoli was present in the GLA and AJ GLA group. Thus, we did not find evidence of augmented lung pathology in ADJ+GLA mice following viral challenge.
Next, we assessed whether NP-based adjuvanted vaccines conferred heterosubtypic immunity against a highly lethal infection with H5N1 avian influenza virus at 50 days PV. In the unvaccinated and NP-vaccinated group, 100% of mice lost significant weight and succumbed to H5N1 infection (Figure 6E). By contrast, 100% of ADJ+GLA and ADJ+CpG mice lost little weight and survived H5N1 challenge, although other groups showed excellent protection ranging from 70% to 90% (Figure 6E).
Role of CD4 T Cells in Programming Vaccine-Induced CD8 T Cell Memory and T-Cell-Based Protective Immunity to Influenza
In order to determine whether CD4 T cells regulate the quality of CD8 T cell memory and protective immunity induced by the ADJ+GLA vaccine, we depleted CD4 T cells only at the time of prime and boost vaccination. At 80 days PV, we examined CD4 and CD8 T cell memory in the RT (Figure 7). NP311-specific memory CD4 T cells were only detected in lungs and airways of non-depleted mice (Figure 7A). CD4 T cell depletion had no adverse effect on the numbers of NP366-specific memory CD8 T cells in the RT (Figure 7B). Among TRM markers, only the expression of CD103, but not CD69 or CD49a, was significantly reduced by CD4 T cell depletion (Figure 7C). Coincident with impaired CD103 expression, memory CD8 T cells in CD4 T-cell-depleted mice poorly localized to the lung parenchyma (Figure 7D). Functionally, the percentages of NP366-specific, IFN-γ-producing CD8 T cells were significantly (p < 0.05) increased in the lungs of CD4 T-cell-depleted mice, with no effect on IL-17-producing CD8 T cells (Figures 7E and 7F). In summary, loss of CD4 T cells reduced CD103 expression and extra-vascular localization of memory CD8 T cells but increased the percentages of NP366-specific memory TC1 cells in the lungs.
Figure 7.

Regulation of Vaccine-Induced CD8 T Cell Memory and Protective Immunity by CD4 T Cells
Groups of C57BL/6 mice were vaccinated with NP+ADJ+GLA, as in Figure 1, and treated with isotype control antibodies (non-depleted) or anti-CD4 antibodies (CD4-depleted) i.v. and IN on days −1, 0, and 1 relative to prime and boost vaccination. T cell memory in lungs (A–F) and protective immunity to influenza A virus (G–P) was determined at 80 days PV.
(A–F) T cell memory in lungs at day 80 PV. To stain for vascular cells, mice were injected i.v. with anti-CD45.2 antibodies, 3 min prior to euthanasia. Lung cells were stained directly ex vivo with Db/NP366 or I-Ab/NP311 tetramers along with the indicated antibodies. For cytokine analysis, lung cells were stimulated with NP366 or NP311 peptide for 5 h before intracellular staining.
(A) FACS plots are gated on CD4 T cells and show NP311-specific, tetramer-binding memory CD4 T cells only in non-depleted mice.
(B) NP366-specific tetramer-binding memory CD8 T cells in lungs of non-depleted and CD4 T-cell-depleted mice.
(C) Expression of tissue residency markers on NP366-specific, tetramer-binding memory CD8 T cells in lungs.
(D) Percentage of vascular (CD45.2+ve) and non-vascular (CD45.2−ve) cells among NP366-specific, tetramer-binding memory CD8 T cells in lungs.
(E) Percentages of IFN-γ- and IL-17-producing, NP366-specific cells among CD8 T cells in lungs.
(F) Calculated proportions of IFN-γ- and/or IL-17-producing cells among cytokine-producing, peptide-stimulated, IFN-γ+IL-17 NP366-specific CD8 T cells.
(G–P) At day 80 after booster vaccination, non-depleted and CD4 T-cell-depleted mice were challenged IN with PR8/H1N1 influenza A virus; recall virus-specific CD8/CD4 T cell responses and viral load in lungs were assessed at day 6 after challenge.
(G) Percentages of NP366-specific, tetramer-binding cells among CD8 T cells in lungs.
(H) Percentages of NP366-specific, tetramer-binding CD8 T cells in vascular and non-vascular lung compartment.
(I) Percentages of NP311-specific, tetramer-binding cells among CD4 T cells in lungs.
(J) Expression of tissue residency markers on NP366-specific, tetramer-binding CD8 T cells.
(K) Chemokine receptor and transcription factor expression in NP366-specific CD8 T cells in lungs.
(L) Granzyme B expression by NP366-specific CD8 T cells directly ex vivo.
(M) Percentages of IFN-γ- and IL-17-producing, NP366-specific CD8 T cells.
(N) Relative proportions of IFN-γ- and/or IL-17-producing cells among total IFN-γ plus IL-17-producing, peptide-stimulated, NP366-specific CD8 T cells.
(O) Viral titers in lungs at day 6 after challenge.
(P) Body weight measured as a percentage of starting body weight prior to challenge.
Data are pooled from two independent experiments. Comparisons were made using one-way ANOVA test with Tukey-corrected multiple comparisons; ∗p < 0.1, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To assess whether depletion of CD4 T cells affected protective immunity, we challenged undepleted and CD4 T-cell-depleted vaccinated mice with the PR8/H1N1 virus. On the 6th day after challenge, we assessed recall CD8 T cell responses and viral control in the lungs. The percentages of NP366-specific CD8 T cells in lungs of CD4 T-cell-depleted mice were higher than in undepleted mice (Figure 7G), and the majority of these effector cells localized to the non-vascular compartment (Figure 7H). NP311-specific CD4 T cells were only detected in the lungs of undepleted mice (Figure 7I). CD4 T cell depletion had no effect on the percentages of CD69+ve CD8 T cells, but the percentages of CD49a+ve cells were significantly (p < 0.05) increased in CD4 T-cell-depleted mice. A small percentage of NP366-specific CD8 T cells in the lungs of undepleted mice expressed CD103, and this fraction was significantly (p < 0.05) reduced by CD4 T cell depletion (Figure 7J). In the CD4 T-cell-depleted group, the percentages of CXCR3+ve NP366-specific CD8 T cells were significantly reduced, but the percentages of NP366-specific CD8 T cells that expressed elevated levels of CX3CR1, T-bet, and EOMES were higher in CD4 T-cell-depleted group than in undepleted group (Figure 7K). Increased accumulation of CX3CR1Hi cells and reduced expression of CXCR3 on CD8 T cells in CD4 T-cell-depleted mice is likely linked to elevated expression of T-bet.22,35 Significantly, the percentages of IFN-γ-producing and granzyme B+ve CD8 T cells were higher, but there was a concurrent reduction in IL-17-producing CD8 T cells in the lungs of CD4 T-cell-depleted mice (Figures 7L and 7M). Strikingly, >90% of NP366-specific CD8 T cells produced IFN-γ in the CD4 T-cell-depleted group as opposed to ∼56% in undepleted mice (Figure 7N). Undepleted mice controlled viral replication in the lungs (Figure 7O; >99% reduction in lung viral titers). Thus, surprisingly, despite markedly increased development of IFN-γ-producing granzyme B+ve TC1 CD8 T cells, CD4 T-cell-depleted mice showed poor control of influenza virus in the lungs (Figure 7O) and also exhibited exaggerated weight loss (Figure 7P). Taken together, data in Figure 7 demonstrated that CD4 T cells play an essential role in (1) optimal programming of protective CD8 T cell memory, (2) restraining the induction of T-bet and terminal differentiation of effector CD8 T cells, (3) promoting recall responses of TC17 cells, and (4) orchestrating protective immunity to influenza virus.
In order to dissect whether impaired viral control in CD4 T-cell-depleted mice was due to defective programming of CD8 T cells and/or due to loss of CD4 T-cell-dependent viral control, we depleted CD4 or CD8 T cells just prior to influenza virus challenge (Figure S8). As shown in Figure S8A, treatment with anti-CD4 or anti-CD8 antibodies reduced NP-specific CD4 and CD8 T cells, respectively. Unlike vaccinated mice treated with isotype control antibodies, vaccinated mice depleted of CD4 or CD8 T cells failed to effectively control viral load in lungs. These data suggested a role for both CD4 and CD8 T cells in vaccine-induced protective immunity to influenza A virus in ADJ+GLA-vaccinated mice.
Discussion
Mucosal viral infections, such as influenza, fail to induce durable T cell immunity in the RT.7,35 Here, we report an adjuvant system composed of a polyacrylic-acid-based adjuvant ADJ and TLR agonists that elicits strong, durable, and functionally diverse mucosal T cell immunity to disparate strains of IAV. As a mucosal adjuvant, ADJ afforded good protection against IAV.20 In this study, we define ways to broaden T cell immunity and enhance the protective efficacy of ADJ by combining with TLR4 and TLR9 agonists, GLA and CpG, respectively. This adjuvant system’s ability to elicit high numbers of antigen-specific CD8 and CD4 T cells enabled us to perform in-depth characterization of vaccine-elicited effector and memory T cells directly ex vivo, without the need for tetramer enrichment. Following IAV infection, activated CD8 T cells migrate from DLNs to the lungs, undergo another round of antigenic stimulation, and differentiate into effector cells.6 Likewise, antigen-specific CD8 T cells in all adjuvant groups experienced varying levels of TCR signaling in the lungs. Significantly, adjuvants differed in terms of the degree of effector differentiation, for both CD8 and CD4 T cells. ADJ- and/or CpG-adjuvanted vaccines drove terminal differentiation into CX3CR1HIKLRG-1HI effector cells; as in IAV-infected mice,3,36 the pathway to terminal differentiation is attributed at least in part to higher TCR signaling in the lungs and induction of transcription factors T-bet, IRF-4, and BATF.29,37,38 Notably, high TCR signaling also induced PD-1 expression in ADJ, CpG, and ADJ+CpG groups and likely limited the accumulation of CD8 T cells in lungs. PD-1 might restrain RT inflammation,32 but it would be worthwhile determining whether PD-1 limits vaccine-induced memory and protective immunity. It is noteworthy that, despite the presence of similar numbers of antigen-bearing cells in lungs of ADJ, ADJ+CpG, and ADJ+GLA mice, effector CD8 T cells in ADJ+GLA mice displayed substantially lower levels of TCR signaling in lungs. It is possible that GLA-induced TLR4 stimulation antagonized antigen-triggered TCR signaling in ADJ+GLA mice.39 By dampening TCR signaling, GLA might have mitigated terminal differentiation of effectors and promoted the development of TRMs in ADJ+GLA mice. High levels of inflammation and IL-12 have been linked to T-bet induction and terminal differentiation of CD8 T cells in spleen,24,26 but the rules that govern T cell differentiation in lungs versus spleen are likely different and worthy of further exploration.
We find that ADJ enhances CD103 expression in responding CD4 and CD8 T cells. TCR signaling, IL-10, and exposure to TGF-β promote CD103 expression and mucosal imprinting in T cells.3,40 However, we find that, at 24 and 48 h after vaccination, the levels of TGF-β1 or IL-10 in lungs did not explain differences in CD103 expression. ADJ promotes cross-presentation of antigen to CD8 T cells,20 and hence, ADJ-induced increase in the number of antigen-bearing cells in lungs likely enhances TCR signaling and CD103 expression on effector CD8 T cells. Interestingly, GLA inhibited TCR signaling in ADJ+GLA mice without abrogating the CD103-inducing effects of ADJ. It is possible that the residual TCR signaling in ADJ+GLA mice is sufficient to induce CD103 or other mechanisms, including IFN-γ production by CD4 T cells, might have contributed to CD103 expression on CD8 T cells.41 In summary, we infer that the magnitude of TCR signaling in lungs is a key factor that controls accumulation, mucosal imprinting, and effector/memory differentiation.
A salient feature of ADJ-based adjuvants is the diverse functional programming of effector and memory T cells. For CD8 T cells, all adjuvants induced comparable levels of IL-12 and elicited a strong TC1 response. However, GLA, by virtue of its ability to induce IL-1 and IL-6, also enabled a significant TC17/TH17 response, and induction of TH17 cells by GLA is consistent with published work.42 Importantly, from a vaccination perspective, here, we are reporting ways to tailor an adjuvant based on pathogen-specific correlates of protection. For example, ADJ formulated with CpG elicits strong TC1/TH1 memory, which protects against viruses and protozoan pathogens (e.g., leishmania). Alternatively, ADJ formulated with GLA stimulates balanced differentiation of TC1/TH1 and TH17 cells, which is protective against fungi, tuberculosis, and other bacterial pathogens.43,44
Effective T-cell-based protection against IAV requires a critical number of TRMs in the airways and the lung parenchyma.3,45 In this study, all adjuvants elicited readily detectable CD8 and CD4 TRMs in the RT. ADJ+GLA induced the largest number of TRMs and vascular memory CD8/CD4 T cells in the lungs, which is likely a sequel to less terminal differentiation and larger clonal burst size during the effector phase.46 TRMs are known to reside primarily in the tissue parenchyma and in the DLNs, but not as circulating cells.47 We find that lungs of ADJ+GLA mice contained CD103+ve memory CD8 T cells in the vasculature, which are similar to circulating skin-resident CD103+ve memory T cells in humans.48 Parabiosis studies are needed to elucidate whether vascular CD103+ve memory CD8 T cells in ADJ+GLA mice are circulating cells or lung-vasculature-resident memory T cells. The numbers of memory T cells in lungs of other adjuvant groups were comparable, but the differential polarity (TH1 versus TH17) programmed by each during the effector phase was preserved in memory T cells; CpG and ADJ+CpG displayed TH1 dominance, and ADJ, GLA, and ADJ+GLA showed skewed TH17 differentiation. Upon challenge with the PR8/H1N1 IAV, all vaccinated groups afforded considerable protection in the lungs. Interestingly, the extent of protection varied between the groups; ADJ+GLA provided the most effective protection, and the descending order of adjuvants in terms of protection is GLA ≥ ADJ+CpG > CpG ≥ ADJ. Upon challenge, all vaccinated groups mounted a strong recall response, and the accumulations of NP366-specific CD8 T cells and NP311-specific CD4 T cells in lungs were comparable. The percentages of IFN-γ-producing, NP366-specific CD8 T cells were similar between the groups, and the percentages of IFN-γ-producing, NP311-specific CD4 T cells showed no correlation with viral control. However, interestingly, differences in viral control tend to associate with the combined percentages of IL-17-producing CD8 and CD4 T cells. However, blocking IL-17A did not significantly affect IAV control in mice vaccinated with ADJ+GLA. In addition to IL-17A, TC17/TH17 cells also produce cytokines, such as IL-17F, IL-22, and GM-CSF, whose role in vaccine-induced control of IAV needs further investigation. It is also possible that TC17/TH17 programming, and not IL-17-mediated antiviral functions per se, might be important in protective immunity, because TH17 programming is associated with stem-cell-like functionally plastic memory T cells.49 It is likely that a battery of redundant mechanisms, including but limited to IL-17, IFN-γ, and major histocompatibility complex class I (MHC-class-I)/MHC-class-II-restricted cytotoxicity, orchestrates vaccine-induced protective immunity to IAV.50, 51, 52, 53
Our investigations into the CD4 T cells’ role in programming vaccinal immunity to IAV provided further insights into the mechanisms of protection in ADJ+GLA-vaccinated mice. Depletion of CD4 T cells during vaccination precluded priming of NP311-specific CD4 T cells but had no adverse effect on IFN-γ- or IL-17-producing, NP366-specific memory CD8 T cells in lungs. Importantly, however, CD4 T cell depletion reduced CD103 expression and the number of non-vascular CD8 TRMs in the lungs, as reported before.41 Upon IAV challenge, despite mounting a highly potent IFN-γ-producing TC1 recall response, CD4 T-cell-depleted mice exhibit considerable morbidity and high viral burden, which might be attributed to aberrant CD8 T cell response and/or a lack of CD4 T-cell-dependent viral control. Depletion of CD4 T cells or CD8 T cells just prior to virus challenge also impair viral control in lungs, which suggest that both CD4 T cells and CD8 T cells mediate IAV control in vaccinated mice. It remains to be determined whether CD4 T cells exert direct antiviral activity and/or orchestrate functions of other cell types, such as CD8 T cells. Further, antibodies against IAV NP are less likely to play a role in viral neutralization, but they could promote antigen uptake by FcR-dependent mechanisms, leading to enhanced antigen presentation to CD8 and CD4 T cells, and protect by antibody-dependent cell-mediated cytotoxicity (ADCC) or other mechanisms.54, 55, 56 Nonetheless, it is clear from our studies that CD4 T cells have a dual role in vaccine-induced protective immunity: appropriate programming of protective CD8 TRMs and orchestrating IAV control.
Findings reported in the manuscript provide fundamental insights into T-cell-based, vaccine-induced protective immunity in the RT. First, we document how combination adjuvants stimulate different degrees of terminal differentiation in effector cells, and their subsequent differentiation into memory T cells, by controlling TCR signaling in the lungs. Second, by inducing distinct sets of T-cell-polarizing cytokines, combination adjuvants differentially program TC1/TH1 and/or TC17/TH17 differentiation in lungs. Third, combining adjuvants enables us to selectively harness the most desirable properties and at the same time mitigate less-desirable effects of individual adjuvants. For example, by combining ADJ and GLA, we can harness the (1) mucosal imprinting and TC1/TH1 driving properties of ADJ, (2) TC17/TH17-polarizing effects of GLA, and (3) ability of GLA to dampen TCR signaling and mitigate ADJ-driven terminal differentiation of effector cells. Taken together, findings presented in this manuscript might have implications in the development of safe and effective subunit vaccines against mucosal pathogens.
Limitations of Study
There are several limitations to this study: (1) validation with other adjuvants will be needed to assess the general applicability of the findings on the effect of adjuvant on the differentiation of effector/memory T cells and development of protective immunity; (2) although we demonstrate a role for CD4 and CD8 T cells in vaccine-induced protective immunity to influenza virus, the precise mechanisms are unknown; (3) we do not know whether results from studies in specific-pathogen-free inbred mice can be recapitulated in pre-clinical models of outbred mice or non-human primates; and (4) this work did not determine whether vaccinations work in animals with pre-existing immunity.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Hamster anti-CD11c-BV786-conjugated (N418) | BD PharMingen | Cat# 563735 |
| Rat anti-CD11b-BV711-conjugated (M1/70) | BD PharMingen | Cat# 563168 |
| Rat anti-I-A/I-E-BV650-conjugated (M5/114.15.2) | BD PharMingen | Cat# 563415 |
| Rat anti-Siglec-F-Alexa Fluor 647-conjugated (E50-2440) | BD PharMingen | Cat# 562680 |
| Rat anti-Ly-6G-BUV 395-conjugated (1A8) | BD PharMingen | Cat# 563978 |
| Rat anti-Ly6C-PE-Cy7-conjugated (AL21) | BD PharMingen | Cat# 560593 |
| Rat Anti-CD4-BUV496-conjugated (GK1.5) | BD PharMingen | Cat# 564667 |
| Rat Anti-CD8-BUV395-conjugated (53-6.7) | BD PharMingen | Cat# 563786 |
| Rat Anti-CD44-BV510-conjugated (IM7) | BD PharMingen | Cat# 563114 |
| Hamster Anti-CD49a-BV605-conjugated (Ha31/8) | BD PharMingen | Cat# 740375 |
| Hamster Anti-KLRG1-BV711-conjugated (2F1) | BD PharMingen | Cat# 564014 |
| Rat Anti-CD62L-Alexa 700-conjugated (MEL-14) | BD PharMingen | Cat# 560517 |
| Rat Anti-IL-2-PE-CF594-conjugated (JES6-5H4) | BD PharMingen | Cat# 562483 |
| Rat Anti-TNF-BV 421-conjugated (MP6-XT22) | BD PharMingen | Cat# 563387 |
| Rat Anti-IFN-γ-APC-conjugated (XMG 1.2) | BD PharMingen | Cat# 554413 |
| Hamster Anti-CD279 (PD-1)-BV 650-conjugated (J43) | BD PharMingen | Cat# 744546 |
| Hamster Anti-Mouse CD69-PE-Cy7-conjugated (H1.2F3) | BD PharMingen | Cat# 552879 |
| Rat Anti- CD4-APC-conjugated | BD PharMingen | Cat# 553051 |
| Hamster Anti- CD103 Antibody-FITC-conjugated (2E7) | eBioscience | Cat# 11-1031-85 |
| Mouse anti-CD45.2 Monoclonal Antibody (104) | eBioscience | Cat# 12-0454-82 |
| Rat InVivoMAb anti-mouse CD4 | BioXcell | Cat# BE0003-1 |
| Rat InVivoMAb anti-mouse CD8α (53-6.7) | BioXcell | Cat# BE0004-1 |
| Rat InVivoMAb anti-mouse IL-17A (17F3) | BioXcell | Cat# BP0173 |
| Rat anti-EOMES-PE-eFluor 610-conjugated (Dan11mag) | eBioscience | Cat# 61-4875-82 |
| Mouse anti-BATF-PerCP-eFluor 710-conjugated (MBM7C7) | eBioscience | Cat# 46-9860-42 |
| Mouse anti-T-Bet-PerCP-Cy5.5-conjugated (4B10) | eBioscience | Cat# 45-5825-82 |
| Rat anti-IRF4-PE-Cyanine7-conjugated (3E4) | eBioscience | Cat# 25-9858-82 |
| Rat anti-IRF4-FITC-conjugated (3E4) | eBioscience | Cat# 11-9858-82 |
| Hamster anti-CD279 (PD-1)-PE-conjugated (J43) | eBioscience | Cat# 12-9985-82 |
| Mouse anti-Granzyme B-PE-conjugated (GB12) | Thermo Fisher Scientific | Cat# MHGB04 |
| Rat anti-CD127-PerCP-Cy5.5-conjugated (A7R34) | Biolegend | Cat# 135022 |
| Rat anti-CD127-BV650-conjugated (A7R34) | Biolegend | Cat# 135043 |
| Hamster anti-CD103-BV605-conjugated (2E7) | Biolegend | Cat# 121433 |
| Mouse anti-CX3CR1-BV785-conjugated (SA011F11) | Biolegend | Cat#149029 |
| Hamster anti-CXCR3-BV650-conjugated (CXCR3-173) | Biolegend | Cat# 126531 |
| Rat anti-IL-17A-FITC conjugated (TC11-18H10.1) | Biolegend | Cat# 506908 |
| APC-conjugated H2-Kb tetramers bearing the ovalbumin peptide SIINFEKL | NIH Tetramer Core Facility at Emory University | N/A |
| BV421-conjugated I-Ab tetramers bearing the NP peptide NP311 (QVYSLIRPNENPAHK) | NIH Tetramer Core Facility at Emory University | N/A |
| APC-conjugated-H2-Db tetramers bearing the NP peptide NP366 (ASNENMDTM) | NIH Tetramer Core Facility at Emory University | N/A |
| Mouse anti-CD64 (FcγRI)-PerCP-Cy5.5-conjugated (X54-5/7.1) | Biolegend | Cat# 139308 |
| Anti-CD16 + CD32 Rat Monoclonal Antibody [clone: 2.4G2]; FC block | Tonbo Biosciences | Cat# 70-0161-U500 |
| Bacterial and Virus Strains | ||
| A/PR8/8/1934 (H1N1) | Dr. Robert Webster (St. Judes Childrens Hospital) | N/A |
| A/Vietnam/1203/2004 (H5N1) | Virus was derived by reverse genetics in the laboratory of Dr. Yoshihiro Kawaoka | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Adjuplex (endotoxin-free) | Provided by Advanced Bioadjuvants | N/A |
| Recombinant nucleoprotein (NP) of the PR8/H1N1 influenza virus strain | Sino Biologicals | Cat# 11675-V08B |
| MPLA (PHAD), Monophosphoryl Lipid A (Synthetic); Glucopyranosyl Lipid Adjuvant (GLA) | Avanti Polar Lipids | Cat# 699800 |
| Hen egg white ovalbumin grade V (OVA) from chicken egg white | MilliporeSigma | Cat# A5503 |
| CpG ODN 1826 (CpG) oligonucleotide | InvivoGen | Cat# vac-1826-1 |
| DQ Ovalbumin | Thermo Fisher Scientific | Cat# D12053 |
| T-PER Tissue Protein Extraction Reagent | Thermo Fisher Scientific | Cat# 78510 |
| LiveDead eFlour 780 stain | eBioscience | Cat# A10628 |
| 16% Paraformaldehyde | Electron Microscopy Sciences | Cat# 15710-S |
| 96-well Clear Round Bottom TC-treated Microplate | Corning | Cat# 3799 |
| 96-well Clear Flat Bottom TC-treated Microplate | Corning | Cat# 3595 |
| 24 Well Cell Culture Plate, flat, TC, sterile, Bulk | Nest Scientific | Cat# 702002 |
| OVA 257-264 peptide | Thinkpeptides | Custom-made |
| NP311 peptide | Thinkpeptides | Custom-made |
| NP366 peptide | Thinkpeptides | Custom-made |
| GolgiPlug (Protein Transport Inhibitor (Containing Brefeldin A)) | BD Biosciences | Cat# 555029 |
| Recombinant Human IL-2 protein | BD Biosciences | Cat# 554603 |
| InvivoPure pH 8.0 Dilution Buffer | Bio X Cell | IPT080 |
| InvivoPure pH 7.0 Dilution Buffer | Bio X Cell | Cat# IP0070 |
| InvivoPure pH 6.5 Dilution Buffer | Bio X Cell | Cat# IP0065 |
| Critical Commercial Assays | ||
| Cytofix/Cytoperm fixation/permeabilization | BD Biosciences | Cat# 555028 |
| Foxp3 /Transcription Factor Staining Kit | Thermo Fisher Scientific | Cat# 00-5523-00 |
| Bio-Plex Pro Mouse Cytokine 23-plex Assay (Bio-rad) | Bio-Rad | Cat# M60009RDPD |
| Mouse IL-28B (IFN lambda 3) Uncoated ELISA Kit | Thermo Fisher Scientific | Cat# 88-7284-22 |
| Mouse IFN-β ELISA Kit | R&D Systems | Cat# DY8234-05 |
| Bio-Plex Pro TGF-β Assays | Bio-Rad | Cat# 171W4001M |
| Experimental Models: Cell Lines | ||
| Madin Darby Canine Kidney Cell Line (MDCK) | ATCC | Cat# PTA-6500 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | Jackson Labs | Cat# 000664 |
| B6. KLF2-GFP reporter mice | Dr. Stephen Jameson (University of Minnesota) | Cat# Weinreich et al.57 |
| B6. Nur77-GFP OT-1 mice | Dr. Ross Kedl (University of Colorado) | Cat# Moran et al.27 |
| Software and Algorithms | ||
| FlowJo v10 | TreeStar | N/A |
| Prism V 7.0 | Graphpad Prism | N/A |
| Bioplex Manager 6.1.1 | Bio-Rad | N/A |
| Other | ||
| RPMI-1640 Media | Lonza | Cat# 12-702F/12 |
| ACK buffer | Lonza | Cat# 10-0548E |
| Collagenase B | Sigma-Aldrich | Cat# 11088831001 |
| TPCK Treated Trypsin | Worthington Biochemical | Cat# LS003740 |
| Pennicillin/Streptomcyin | Lonza | Cat# 17-602F |
| L-Glutamine | Lonza | Cat# 12-702F/12 |
| RPMI 1640 without L-Gln or Phenol Red | Lonza | Cat# 12-918F |
| PBS | Lonza | Cat# 17-516F/24 |
| Fetal Bovine Serum (FBS) - premium | Atlanta Biologicals | Cat# S11150 |
| Bovine Serum Albumin | Fisher Scientific | Cat# BP1600-1 |
| SeaPlaque Agarose | Lonza | Cat# 50101 |
| PBS Powder | Bio Basic | Cat# PD0100 |
| GentleMacs C Tubes | Milltenyi Biotech | Cat# 130-096-334 |
| Non-sterile CellTrics® Filters | Sysmex | Cat# 04-0042-2317 |
| Brilliant Staining Buffer | BD Biosciences | Cat# 566385 |
| Bioplex 200 Systems | Bio-rad | Cat# 171000201 |
| GentleMACS Dissociator | Miltenyi Biotech | Cat# 130-093-235 |
| BD LSRFortessa | BD | Cat# N/A |
| Spectramax i3x Multi-mode Microplate Reader | Molecular Devices | Cat# I3X |
Resource Availbility
Lead Contact
Request for further information, resources and reagents should be directed to the Lead Contact: Dr. M. Suresh (sureshm@vetmed.wisc.edu).
Materials Availability
All materials or reagents used in this manuscript are available commercially or were obtained from other researchers. This study did not develop new or unique reagents.
Data and Code Availability
All data generated in this study are presented in Figures or in Supplemental Information. No codes were generated in this study.
Experimental Model and Subject Details
Mice
Six-to-twelve-week-old C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in specific-pathogen-free (SPF) conditions. The OT-I/Nur77-eGFP mice were maintained under SPF conditions at University of Colorado. KLF2-GFP reporter mice were provided by Dr. Jameson (University of Minnesota, Minneapolis, MN). Both male and female mice were used in all experiments, and we did not see sex-specific differences in response to vaccination or infection. All animal experiments were performed in accordance with the protocol (Protocol number V5308 and V5564) approved by the University of Wisconsin School of Veterinary Medicine Institutional Animal Care and Use Committee (IACUC). The animal committee mandates that institutions and individuals using animals for research, teaching, and/or testing much acknowledge and accept both legal and ethical responsibility for the animals under their care, as specified in the Animal Welfare Act (AWA) and associated Animal Welfare Regulations (AWRs) and Public Health Service (PHS) Policy.
Vaccination
All vaccinations were administered intranasally to anesthetized mice in 50 μL saline with 10 μg NP or OVA or DQ-OVA protein alone or with the following adjuvants: 10% Adjuplex (ADJ); 10 μg CpG (CpG); 10 μg GLA (GLA); 10% ADJ + 5 μg CpG (ADJ+CpG); 5%–7.5% ADJ + 5 μg GLA (ADJ+GLA). Mice were vaccinated twice (at an interval of 3 weeks) and analyzed for effector and memory T cell responses at day 8 and 100 after the booster vaccination, respectively.
Virus Challenge Studies and Viral Titration
For viral challenge studies, vaccinated mice were inoculated intranasally with 500 plaque forming units (PFU) of A/PR8/8/1934 (H1N1) or 10 MLD50 of A/Vietnam/1203/2004 (H5N1) strains of influenza A virus in 50 μL media. Lungs were collected from mice on the 6th day after viral challenge for viral titration by a plaque assay using Madin Derby Canine Kidney Cells (MDCK) cells.
Treatment of Mice with Antibodies
To assess the role of CD4 T cells in programming CD8 T cell memory, mice were depleted of CD4 T cells by administration of 200-250 μg of anti-CD4 antibodies (Bio X Cell, Clone: GK1.5) intraperotoneally and intranasally on days −1, 0 and 1 relative to vaccinations. To assess the role of CD4 T cells and CD8 T cells in protective immunity, mice were administered with 200 μg of anti-CD4 (Bio X Cell, Clone: GK1.5) or CD8 T cells (Bio X Cell; Clone 2.43) intravenously and intranasally at days −5. −3, −1 and 1 relative to challenge with influenza A virus. To determine whether IL-17A was involved in vaccine-induced protection against influenza virus, vaccinated mice were treated with 200 μg of isotype control antibodies or anti-IL-17A antibodies (Bio X Cell; intravenously and intranasally) at days -3, −1 and 1, 3 amd 5 relative to challenge with influenza A virus.
Adoptive transfer of Nur77-eGFP/OT-I CD8 T Cells
Single-cell suspensions of spleens and lymph nodes (LNs) from Nur77GFP OT-I (CD-45.1+) mice containing 103 or 5x104 of transgenic CD8+ T cells were injected intravenously into sex-matched congenic CD45.2 C57BL/6 mice, and 24 hours later, mice were intranasally vaccinated with OVA formulated with various adjuvants. At days 2, 5, and 8 PV, LNs and lungs were harvested and GFP expression by live OT-I CD8 T cells was quantified directly ex vivo by flow cytometry.
Method Details
Flow cytometry
Vascular staining of T cells was performed by intravenous administration of 3 μg of fluorochrome-labeled anti-CD45.2, three minutes prior to euthanasia. Single-cell suspensions from lymph nodes and spleen were prepared by mechanical disruption on stainless steel meshes using a syringe plunger. To extract cells from lungs, lung tissue was minced and processed using the gentleMACS Dissociator (Miltenyi Biotech) in 5 mL media containing 2mg/ml collagenase B, as per manufacturer’s instructions. Samples were incubated for 30 minutes at 37C, rehomogenized and resuspended in media containing 1% FBS. Subsequently, cells were spun down, resuspended in 10% RPMI and counted in a hemocytometer. Single cell suspensions of cells (107/ml) prepared from various tissues were stained for viability with Dye eFluor 780 (eBiosciences, San Diego, CA), and incubated with fluorochrome-labeled antibodies of MHC I tetramers (1:150 dilution) at 4C for 1 hour. For staining with the I-Ab/NP311 tetramer (1:150 dilution), cells were incubated with tetramer at 37C for 60 minutes, followed by staining with antibodies for cell surface molecules at 4C for 30 minutes. Following staining, cells were washed three times with FACS buffer (2% BSA in PBS). Stained cells were fixed with 2% paraformaldehyde for 20 minutes, then transferred to FACS buffer (2% BSA in PBS). When analyzed for GFP expression, as in KLF2-eGFP mice, cells were stained with MHC I/II tetramers and antibodies; live unfixed cells were acquired on a flow cytometer. In most cases, we acquired a total of 1-2x106 events/sample on the flow cytometer. Data from live single cells were analyzed with FlowJo software (TreeStar, Ashland, OR).
Intracellular Staining for Cytokines and Transcription Factors
For intracellular cytokine staining, 1x106 cells were stimulated for 5 hours at 37C in the presence of human recombinant IL-2 (10 U/well), and brefeldin A (1 μl/ml, GolgiPlug, BD Biosciences), with one of the following peptides: OVA257, NP366 or NP311 (thinkpeptides®, ProImmune Ltd. Oxford, UK) at 0.1ug/ml. After stimulation, cells were stained for 30 minutes with antibodies to cell surface markers, washed 3 times, fix/permeabilzed and stained for intracellular molecules with Cytofix/Cytoperm kit (BD Biosciences, Franklin Lakes, NJ). To stain for transcription factors, cells were first stained with antibodies for cell surface molecules for 30 minutes at 4C, fixed, permeabilized and subsequently stained for transcription factors using the transcription factors staining kit (eBioscience). Samples containing up to 2x106 cells were acquired on a LSRFortessa (BD Biosciences) flow cytometer; Upto 2x106 events/sample were acquired in a flow cytometer. Data from live single cells were analyzed with FlowJo software (TreeStar, Ashland, OR).
Cytokine production in lungs
At 24 and 48 hours after vaccination with OVA and various adjuvants, lungs were harvested for cytokine analysis Five hundred microliter of the mammalian tissue protein extraction reagent (T-PER) (Thermo Scientific) containing protease inhibitor cocktails (Roche, Indianapolis, IN), was added to the lung tissue and homogenized in a tissue homogenizer. Subsequently, tissue lystate was centrifuged at 10,000 x g for 5 minutes at 4C. Supernatants were collected and protein concentrations in extracts were quantified using BCA method and 1mg of total lysate was added for each well in a 96-well plate. Cytokines were quantified using Bio-Plex Pro Mouse Cytokine 23-plex and Bio-Plex Pro TGF-β Assays (Bio-Rad), IFN beta Mouse ProcartaPlex Simplex Kit and IL-28B/(IFN lambda 3) Mouse ELISA kit (eBioscience), as recommended by the manufacturer. The samples were acquired and analyzed using Bioplex-200 with the Bioplex Manager 6.1.1. The data were normalized by (the amount of cytokine/ml) ∗ (extraction volume) divided by the weight of the lung tissue (mg).
Virus Titration in Lungs
Madin-Darby canine kidney (MDCK) cells were obtained from ATCC (ATCC; Manassas, VA, USA) and propagated in growth media containing Modified Eagle’s Medium (MEM) with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), 2 mM L-glutamine, 1.5 g/l sodium bicarbonate, non-essential amino acids, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and incubated at 37°C in 5% CO2. 16-24 hours before the assay, 24-well plates were seeded with 500 ul of MDCK cells suspended at a concentration of 5x104cells/mL Lung tissues were homogenized in a bullet blender, and spun for 10 minutes at 2,000 RPM in a refrigerated centrifuge. Supernatants were collected and and 10-fold serial dilutions of the supernatant were used to infect MDCK cells grown to 90% confluency in 24-well plates, and incubated for 1hr at 37C; each sample was tested in duplicate wells. Cells were then washed with warm PBS and incubated in media containing 1% SeaPlaque agarose (Lonza, Basel, Switzerland) and TPCK-treated trypsin (final concentration of 10ug/ml). After 48hr incubation, cells were fixed in 10% neutral buffered formalin (NBF) for 1 hour, agarose plugs removed, and distinct plaques were counted at a given dilution to determine the plaque forming units (PFU) of virus per sample.
Quantification and Statistical Analysis
Statistical analyses were performed using GraphPad software (La Jolla, CA). Statistical tests used are outlined in figure legends. Unless otherwise noted, all comparisons were made using an one-way ANOVA test with Tukey corrected multiple comparisons or Students t test where p < 0.05 = ∗, p < 0.001 = ∗∗, p < 0.0001 = ∗∗∗ were considered significantly different among groups. In some experiments (Figure 4), we used one-way ANOVA, Students t test and simple regression analysis. In Figure 6, we used non-linear regression for analyzing weight loss data. Data are presented as mean ± SEM for biological replicates. Viral titers were log transformed prior to analysis. No data or outliers were excluded from analyses. For all statistical tests, normality of data was checked and Brown-Forsythe test was used to determine if standard deviations were equal, and if not a Brown-Forsythe test was used to correct for unequal SD.
Acknowledgments
We thank Dr. Jameson for providing the KLF2-GFP mice and Amulya Suresh for designing and drawing the graphical abstract. We acknowledge the efforts of the veterinary and animal care staff at UW-Madison. This study was supported by a PHS grant from the National Institutes of Health (U01AI124299 and R21 AI149793) and funds from the John E. Butler Professorship to M.S. W.L. was supported by an American Heart Association pre-doctoral fellowship (18PRE34080150). D.J.G.’s contribution was supported by NIH training grant (T32OD010423).
Author Contributions
C.B.M. planned and performed experiments, analyzed data, and wrote the paper. B.K.-B. and W.L. performed experiments, analyzed data, and wrote the paper. M.S., A.L., B.N., and M.H. performed experiments and analyzed data. D.J.G. evaluated tissue pathology. R.M.K. provided critical reagents and edited the manuscript. Y.K. provided critical reagents. M.S. was responsible for concept development and experimental planning and also performed data analysis and wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: September 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100095.
Supplemental Information
References
- 1.Gounder A.P., Boon A.C.M. Influenza pathogenesis: the effect of host factors on severity of disease. J. Immunol. 2019;202:341–350. doi: 10.4049/jimmunol.1801010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMaster S.R., Wilson J.J., Wang H., Kohlmeier J.E. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-γ production. J. Immunol. 2015;195:203–209. doi: 10.4049/jimmunol.1402975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takamura S., Kohlmeier J.E. Establishment and maintenance of conventional and circulation-driven lung-resident memory CD8+ T cells following respiratory virus infections. Front. Immunol. 2019;10:733. doi: 10.3389/fimmu.2019.00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sant A.J. The way forward: potentiating protective immunity to novel and pandemic influenza through engagement of memory CD4 T cells. J. Infect. Dis. 2019;219(Suppl_1):S30–S37. doi: 10.1093/infdis/jiy666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sridhar S. Heterosubtypic T-cell immunity to influenza in humans: challenges for universal T-cell influenza vaccines. Front. Immunol. 2016;7:195. doi: 10.3389/fimmu.2016.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hufford M.M., Kim T.S., Sun J., Braciale T.J. The effector T cell response to influenza infection. Curr. Top. Microbiol. Immunol. 2015;386:423–455. doi: 10.1007/82_2014_397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slütter B., Van Braeckel-Budimir N., Abboud G., Varga S.M., Salek-Ardakani S., Harty J.T. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol. 2017;2:eaag2031. doi: 10.1126/sciimmunol.aag2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Braeckel-Budimir N., Harty J.T. Influenza-induced lung Trm: not all memories last forever. Immunol. Cell Biol. 2017;95:651–655. doi: 10.1038/icb.2017.32. [DOI] [PubMed] [Google Scholar]
- 9.Kalia V., Sarkar S., Ahmed R. CD8 T-cell memory differentiation during acute and chronic viral infections. Adv. Exp. Med. Biol. 2010;684:79–95. doi: 10.1007/978-1-4419-6451-9_7. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed R., Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 11.Pulendran B., Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 12.Coffman R.L., Sher A., Seder R.A. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pulendran B., Oh J.Z., Nakaya H.I., Ravindran R., Kazmin D.A. Immunity to viruses: learning from successful human vaccines. Immunol. Rev. 2013;255:243–255. doi: 10.1111/imr.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foged C., Hansen J., Agger E.M. License to kill: formulation requirements for optimal priming of CD8(+) CTL responses with particulate vaccine delivery systems. Eur. J. Pharm. Sci. 2012;45:482–491. doi: 10.1016/j.ejps.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 15.Koff W.C., Burton D.R., Johnson P.R., Walker B.D., King C.R., Nabel G.J., Ahmed R., Bhan M.K., Plotkin S.A. Accelerating next-generation vaccine development for global disease prevention. Science. 2013;340:1232910. doi: 10.1126/science.1232910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta P.K., Mukherjee P., Dhawan S., Pandey A.K., Mazumdar S., Gaur D., Jain S.K., Chauhan V.S. Production and preclinical evaluation of Plasmodium falciparum MSP-119 and MSP-311 chimeric protein, PfMSP-Fu24. Clin. Vaccine Immunol. 2014;21:886–897. doi: 10.1128/CVI.00179-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakrabarti B.K., Feng Y., Sharma S.K., McKee K., Karlsson Hedestam G.B., Labranche C.C., Montefiori D.C., Mascola J.R., Wyatt R.T. Robust neutralizing antibodies elicited by HIV-1 JRFL envelope glycoprotein trimers in nonhuman primates. J. Virol. 2013;87:13239–13251. doi: 10.1128/JVI.01247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gualandi G.L., Losio N.M., Muratori G., Foni E. The ability by different preparations of porcine parvovirus to enhance humoral immunity in swine and guinea pigs. Microbiologica. 1988;11:363–369. [PubMed] [Google Scholar]
- 19.Mumford J.A., Wilson H., Hannant D., Jessett D.M. Antigenicity and immunogenicity of equine influenza vaccines containing a Carbomer adjuvant. Epidemiol. Infect. 1994;112:421–437. doi: 10.1017/s0950268800057848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gasper D.J., Neldner B., Plisch E.H., Rustom H., Carrow E., Imai H., Kawaoka Y., Suresh M. Effective respiratory CD8 T-cell immunity to influenza virus induced by intranasal carbomer-lecithin-adjuvanted non-replicating vaccines. PLoS Pathog. 2016;12:e1006064. doi: 10.1371/journal.ppat.1006064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerlach C., Moseman E.A., Loughhead S.M., Alvarez D., Zwijnenburg A.J., Waanders L., Garg R., de la Torre J.C., von Andrian U.H. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity. 2016;45:1270–1284. doi: 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sallin M.A., Sakai S., Kauffman K.D., Young H.A., Zhu J., Barber D.L. Th1 differentiation drives the accumulation of intravascular, non-protective CD4 T cells during tuberculosis. Cell Rep. 2017;18:3091–3104. doi: 10.1016/j.celrep.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarkar S., Kalia V., Haining W.N., Konieczny B.T., Subramaniam S., Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi N.S., Cui W., Chandele A., Lee H.K., Urso D.R., Hagman J., Gapin L., Kaech S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jameson S.C., Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity. 2009;31:859–871. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haring J.S., Badovinac V.P., Harty J.T. Inflaming the CD8+ T cell response. Immunity. 2006;25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Moran A.E., Holzapfel K.L., Xing Y., Cunningham N.R., Maltzman J.S., Punt J., Hogquist K.A. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badovinac V.P., Haring J.S., Harty J.T. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwata A., Durai V., Tussiwand R., Briseño C.G., Wu X., Grajales-Reyes G.E., Egawa T., Murphy T.L., Murphy K.M. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol. 2017;18:563–572. doi: 10.1038/ni.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo C.T., Veselits M.L., Leiden J.M. LKLF: a transcriptional regulator of single-positive T cell quiescence and survival. Science. 1997;277:1986–1990. doi: 10.1126/science.277.5334.1986. [DOI] [PubMed] [Google Scholar]
- 31.Skon C.N., Lee J.Y., Anderson K.G., Masopust D., Hogquist K.A., Jameson S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z., Wang S., Goplen N.P., Li C., Cheon I.S., Dai Q., Huang S., Shan J., Ma C., Ye Z. PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci. Immunol. 2019;4:eaaw1217. doi: 10.1126/sciimmunol.aaw1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eyerich K., Dimartino V., Cavani A. IL-17 and IL-22 in immunity: driving protection and pathology. Eur. J. Immunol. 2017;47:607–614. doi: 10.1002/eji.201646723. [DOI] [PubMed] [Google Scholar]
- 34.Wu X., Tian J., Wang S. Insight into non-pathogenic Th17 cells in autoimmune diseases. Front. Immunol. 2018;9:1112. doi: 10.3389/fimmu.2018.01112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson K.G., Masopust D. Editorial: Pulmonary resident memory CD8 T cells: here today, gone tomorrow. J. Leukoc. Biol. 2014;95:199–201. doi: 10.1189/jlb.0913493. [DOI] [PubMed] [Google Scholar]
- 36.McGill J., Legge K.L. Cutting edge: contribution of lung-resident T cell proliferation to the overall magnitude of the antigen-specific CD8 T cell response in the lungs following murine influenza virus infection. J. Immunol. 2009;183:4177–4181. doi: 10.4049/jimmunol.0901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nayar R., Schutten E., Bautista B., Daniels K., Prince A.L., Enos M., Brehm M.A., Swain S.L., Welsh R.M., Berg L.J. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J. Immunol. 2014;192:5881–5893. doi: 10.4049/jimmunol.1303187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nayar R., Enos M., Prince A., Shin H., Hemmers S., Jiang J.K., Klein U., Thomas C.J., Berg L.J. TCR signaling via Tec kinase ITK and interferon regulatory factor 4 (IRF4) regulates CD8+ T-cell differentiation. Proc. Natl. Acad. Sci. USA. 2012;109:E2794–E2802. doi: 10.1073/pnas.1205742109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.González-Navajas J.M., Fine S., Law J., Datta S.K., Nguyen K.P., Yu M., Corr M., Katakura K., Eckman L., Lee J., Raz E. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J. Clin. Invest. 2010;120:570–581. doi: 10.1172/JCI40055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson E.A., Darrah P.A., Foulds K.E., Hoffer E., Caffrey-Carr A., Norenstedt S., Perbeck L., Seder R.A., Kedl R.M., Loré K. Monocytes acquire the ability to prime tissue-resident T cells via IL-10-mediated TGF-β release. Cell Rep. 2019;28:1127–1135.e4. doi: 10.1016/j.celrep.2019.06.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laidlaw B.J., Zhang N., Marshall H.D., Staron M.M., Guan T., Hu Y., Cauley L.S., Craft J., Kaech S.M. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity. 2014;41:633–645. doi: 10.1016/j.immuni.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orr M.T., Beebe E.A., Hudson T.E., Argilla D., Huang P.W., Reese V.A., Fox C.B., Reed S.G., Coler R.N. Mucosal delivery switches the response to an adjuvanted tuberculosis vaccine from systemic TH1 to tissue-resident TH17 responses without impacting the protective efficacy. Vaccine. 2015;33:6570–6578. doi: 10.1016/j.vaccine.2015.10.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Dis E., Sogi K.M., Rae C.S., Sivick K.E., Surh N.H., Leong M.L., Kanne D.B., Metchette K., Leong J.J., Bruml J.R. STING-activating adjuvants elicit a Th17 immune response and protect against Mycobacterium tuberculosis infection. Cell Rep. 2018;23:1435–1447. doi: 10.1016/j.celrep.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDermott A.J., Klein B.S. Helper T-cell responses and pulmonary fungal infections. Immunology. 2018;155:155–163. doi: 10.1111/imm.12953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slütter B., Pewe L.L., Kaech S.M., Harty J.T. Lung airway-surveilling CXCR3(hi) memory CD8(+) T cells are critical for protection against influenza A virus. Immunity. 2013;39:939–948. doi: 10.1016/j.immuni.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hou S., Hyland L., Ryan K.W., Portner A., Doherty P.C. Virus-specific CD8+ T-cell memory determined by clonal burst size. Nature. 1994;369:652–654. doi: 10.1038/369652a0. [DOI] [PubMed] [Google Scholar]
- 47.Beura L.K., Wijeyesinghe S., Thompson E.A., Macchietto M.G., Rosato P.C., Pierson M.J., Schenkel J.M., Mitchell J.S., Vezys V., Fife B.T. T cells in non-lymphoid tissues give rise to lymph node resident memory T cells. Immunity. 2018;48:327–338.e5. doi: 10.1016/j.immuni.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klicznik M.M., Morawski P.A., Höllbacher B., Varkhande S.R., Motley S.J., Kuri-Cervantes L., Goodwin E., Rosenblum M.D., Long S.A., Brachtl G. Human CD4+CD103+ cutaneous resident memory T cells are found in the circulation of healthy individuals. Sci. Immunol. 2019;4:eaav8995. doi: 10.1126/sciimmunol.aav8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muranski P., Borman Z.A., Kerkar S.P., Klebanoff C.A., Ji Y., Sanchez-Perez L., Sukumar M., Reger R.N., Yu Z., Kern S.J. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Devarajan P., Bautista B., Vong A.M., McKinstry K.K., Strutt T.M., Swain S.L. New insights into the generation of CD4 memory may shape future vaccine strategies for influenza. Front. Immunol. 2016;7:136. doi: 10.3389/fimmu.2016.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamada H., Bassity E., Flies A., Strutt T.M., de Luz Garcia-Hernandez M., McKinstry K.K., Zou T., Swain S.L., Dutton R.W. Multiple redundant effector mechanisms of CD8+ T cells protect against influenza infection. J. Immunol. 2013;190:296–306. doi: 10.4049/jimmunol.1200571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hamada H., Garcia-Hernandez Mde.L., Reome J.B., Misra S.K., Strutt T.M., McKinstry K.K., Cooper A.M., Swain S.L., Dutton R.W. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J. Immunol. 2009;182:3469–3481. doi: 10.4049/jimmunol.0801814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKinstry K.K., Strutt T.M., Kuang Y., Brown D.M., Sell S., Dutton R.W., Swain S.L. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J. Clin. Invest. 2012;122:2847–2856. doi: 10.1172/JCI63689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.León B., Ballesteros-Tato A., Randall T.D., Lund F.E. Prolonged antigen presentation by immune complex-binding dendritic cells programs the proliferative capacity of memory CD8 T cells. J. Exp. Med. 2014;211:1637–1655. doi: 10.1084/jem.20131692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng B., Zhang Y., He H., Marinova E., Switzer K., Wansley D., Mbawuike I., Han S. Rectification of age-associated deficiency in cytotoxic T cell response to influenza A virus by immunization with immune complexes. J. Immunol. 2007;179:6153–6159. doi: 10.4049/jimmunol.179.9.6153. [DOI] [PubMed] [Google Scholar]
- 56.Padilla-Quirarte H.O., Lopez-Guerrero D.V., Gutierrez-Xicotencatl L., Esquivel-Guadarrama F. Protective antibodies against influenza proteins. Front. Immunol. 2019;10:1677. doi: 10.3389/fimmu.2019.01677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weinreich M.A., Takada K., Skon C., Reiner S.L., Jameson S.C., Hogquist K.A. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity. 2009;31:122–130. doi: 10.1016/j.immuni.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in this study are presented in Figures or in Supplemental Information. No codes were generated in this study.