

Abstract
The green fluorescent protein GFP from Aequorea victoria has been engineered extensively in the past to generate variants suitable for protein tagging. Early efforts produced the enhanced variant EGFP and its monomeric derivative mEGFP, which have useful photophysical properties, as well as superfolder GFP, which folds efficiently under adverse conditions. We previously generated msGFP, a monomeric superfolder derivative of EGFP. Unfortunately, compared to EGFP, msGFP and other superfolder GFP variants show faster photobleaching. We now describe msGFP2, which retains monomeric superfolder properties while being as photostable as EGFP. msGFP2 contains modified N- and C-terminal peptides that are expected to reduce nonspecific interactions. Compared to EGFP and mEGFP, msGFP2 is less prone to disturbing the functions of certain partner proteins. For general-purpose protein tagging, msGFP2 may be the best available derivative of A. victoria GFP.
Keywords: fluorescent protein, monomeric, superfolder, GFP, mCherry, mScarlet, photobleaching, cytotoxicity, Traffic, Intracellular Transport
1 |. INTRODUCTION
The green fluorescent protein (GFP) from Aequorea victoria was the first fluorescent protein (FP) to be used as a reporter and as a tool for labeling proteins, cells, and tissues.1 Multiple rounds of protein engineering have yielded improved GFP variants. Initial efforts focused on photophysical properties, and led to creation of the enhanced variant EGFP.1 Compared to wild-type GFP, EGFP shows faster chromophore maturation as well as a red-shifted excitation spectrum that allows fluorescence to be excited with blue light. EGFP is bright and photostable, and has been the most widely used GFP variant.
Yet EGFP and other first-generation GFP variants have limitations. Most notably, GFP has a weak tendency to dimerize, and this property can induce artifactual clustering of tagged proteins in the crowded environment of a cell.2,3 The self-association of GFP variants is effectively eliminated by point mutations that disrupt the dimerization interface.4 For example, an A206K mutant of EGFP yields the monomeric variant mEGFP. A more subtle issue is that GFP can misfold to a nonfluorescent state. For example, when EGFP is fused to bacterial proteins that form inclusion bodies, the EGFP tag fails to become fluorescent. This observation was the basis for creating a “superfolder” GFP that reaches the fluorescent state even in inclusion bodies.5 Superfolder GFP can outperform other GFP variants, most notably in oxidizing environments such as the lumen of the endoplasmic reticulum or the periplasm of gram-negative bacteria.6,7 The original superfolder GFP contained an A206V mutation5, but superfolder behavior is also obtained with the monomerizing A206K mutation3, thereby capturing the monomeric and superfolder properties in a single variant. We previously used a monomeric superfolder GFP called msGFP as a model protein to track secretion in yeast.8
A disadvantage of currently available superfolder GFP variants is that they photobleach faster than EGFP or mEGFP.5 This concern limits the utility of superfolder GFP variants for live-cell imaging. Our goal was to create a new monomeric superfolder GFP, termed msGFP2, that would retain the desirable photophysical properties of EGFP.
As part of this engineering effort, we also explored an issue that has received little attention, namely, the peptides at the N- and C-termini of FPs. Our interest in this topic was prompted by earlier work to optimize DsRed, a tetrameric red FP. DsRed undergoes higher-order aggregation that was suppressed by mutating residues near the N-terminus.9–11 Furthermore, mutations near the N-terminus of DsRed enhanced bacterial expression.10 A different approach was taken for generating the monomeric DsRed variant mCherry, which contains N- and C-terminal peptides derived from EGFP.12 The rationale was that EGFP is a well-behaved FP, so its N- and C-terminal peptides were expected to confer similarly good behavior on other FPs. This practice of incorporating the terminal peptides of EGFP has become widespread when developing new FPs. However, we report here that the terminal peptides from EGFP can cause bacterial cytotoxicity when present in FPs such as mCherry. To avoid this problem, we incorporated alternative N- and C-terminal peptides into msGFP2.
The enhanced utility of msGFP2 as a fluorescent tag will be most apparent with a subset of partner proteins. We demonstrate that msGFP2 can outperform EGFP or mEGFP in certain protein fusions.
2 |. RESULTS
2.1 |. Introduction of point mutations into EGFP
We previously modified EGFP using the following approach.8 First, we made the eight superfolder mutations S30R, Y39N, F99T, N105T, Y145F, M153T, V163A, and I171V. Those mutations are identical to the ones in the original superfolder GFP5, except that F99T was chosen instead of F99S because in the closely related GFP homolog phiYFP13, threonine is present at the position corresponding to F99. Second, instead of the A206V mutation in the original superfolder GFP, we made the monomerizing mutation A206K.3,4 Third, to revert the accidental introduction of leucine at position 231 during the creation of EGFP1, we made an L231H mutation. Collectively, these changes generated a monomeric superfolder variant of GFP.
2.2 |. Replacement of the terminal peptides in mCherry and mScarlet-I
The next set of changes focused on the N- and C-terminal peptides. This effort was motivated by our analysis of mCherry, which contains N- and C-terminal peptides derived from EGFP.12 We found that expression of mCherry in the E. coli strain DH10B results in cytotoxicity, presumably because mCherry associates with itself or with other cellular components. Screens of mCherry mutant libraries implicated the termini of the protein in cytotoxicity (data not shown). Therefore, to optimize N- and C-terminal peptides for use in an improved GFP variant, we employed mCherry as a test protein.
The six N-terminal amino acids of wild-type GFP are MSKGEE. In EGFP, immediately after the start codon, a valine codon (V1a) was inserted to create a Kozak sequence for efficient mammalian expression1, so the N-terminal peptides of EGFP and mCherry are MVSKGEE. We replaced the N-terminal peptide of mCherry with MDSTES, which incorporates the DST tripeptide that was found to enhance the solubility and bacterial expression of DsRed derivatives.10 The V1a codon was omitted because the D2 codon creates a Kozak sequence.
The six C-terminal amino acids of wild-type GFP and EGFP are MDELYK. This peptide can potentially undergo electrostatic and hydrophobic interactions, so we replaced the C-terminal six residues of mCherry with the linker sequence GSSGSS. At the same time, the previous three amino acids were changed from TGG to GSQ to match the sequence in another monomeric DsRed variant termed DsRed-Monomer.14 The resulting variant with modified N- and C-termini was termed mCherry2B.15 Subsequently, to match the more standard practice of using glycine-rich linkers16, we switched to using GGSGGS for the C-terminal peptide. A variant containing MDSTES at the N-terminus and GGSGGS at the C-terminus was termed mCherry2C.
DH10B cells were transformed to express mCherry, or mCherry2C, or an mCherry variant containing either the N-terminal MDSTES replacement alone or the C-terminal GSSGSS replacement alone. Compared to the colonies expressing mCherry, the colonies expressing mCherry2C were substantially larger (Figure S1 and Figure 1). Similar results were obtained with mCherry2B (data not shown). The N-terminal replacement evidently made the major contribution to reducing cytotoxicity (Figure S1A). This effect is probably not due to lower expression because the MDSTES coding sequence is expected to increase bacterial expression.10 We conclude that replacing the terminal peptides of mCherry can reduce cytotoxicity.
FIGURE 1.
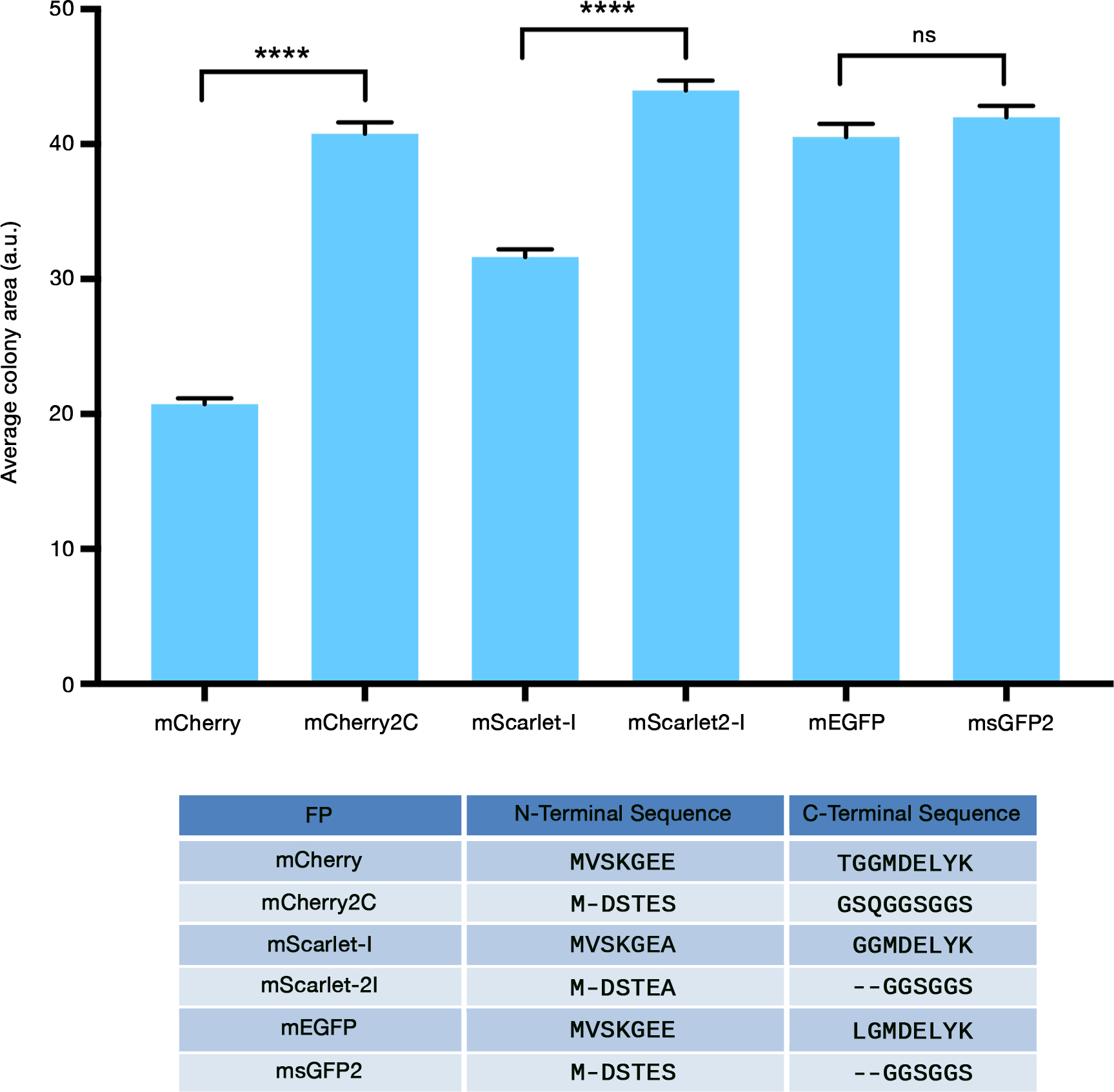
FP cytotoxicity in bacteria can be reduced by modifying the protein termini. E. coli cells of strain DH10B were transformed with the indicated FP expression constructs, and colony areas were measured. Average colony areas are plotted in arbitrary units (a.u.). Bars indicate SEM. Statistical significance was measured using Welch’s t-test. Four asterisks indicate significance at P < 0.0001, and “ns” indicates no significant difference. The table at the bottom lists the N- and C-terminal peptides of the FPs, with the dashes indicating amino acids that are missing from one of the two FPs in each pair.
To determine whether this improvement extends to another FP, we examined mScarlet-I, a synthetic monomeric red fluorescent protein with N- and C-terminal peptides from EGFP.17 The N-terminal peptide of mScarlet-I was replaced with MDSTEA because we found that MDSTES diminished the fluorescence (data not shown), and the C-terminal peptide was replaced with GGSGGS, yielding a variant that we termed mScarlet2-I. Although the difference between mScarlet-I and mScarlet-2I was less pronounced than the difference between mCherry and mCherry2C, the DH10B colonies expressing mSclarlet-2I were larger on average than those expressing mScarlet-I (Figure S1B,C and Figure 1). The combined results indicate that the terminal peptides of EGFP can render some FPs cytotoxic.
2.3 |. Replacement of the terminal peptides in monomeric superfolder GFP variants
To generate the original msGFP, we modified our monomeric superfolder GFP variant to contain the N-terminal peptide MDSTES and the C-terminal peptide SSGSSG.8 The MDSTES sequence preserves a single glutamate, which is needed at position 5 or 6 of GFP to obtain fluorescence.18 During further engineering to generate msGFP2, the C-terminal eight residues were replaced with the glycine-rich peptide GGSGGS, and an additional photostabilizing mutation was introduced as described below.
When expressed in DH10B cells, mEGFP (which contains the EGFP terminal peptides) yielded colonies as large as those obtained with msGFP2 (Figure S1B,C and Figure 1). Thus, for unknown reasons, cytotoxicity due to the EGFP terminal peptides was apparent with two monomeric red FPs but not with a monomeric GFP. We nevertheless included the modified N- and C-terminal peptides in msGFP2, based on the concern that even for a GFP variant, the EGFP terminal peptides might have deleterious effects under some circumstances.
2.4 |. Generation of the photostable msGFP2 variant
Superfolder GFP variants have been reported to photobleach faster than EGFP5,19, and we observed this effect as well. When purified msGFP and EGFP were illuminated with blue light on a microscope stage (Figure 2A), msGFP (blue curve) was substantially less photostable than EGFP (green curve).
FIGURE 2.
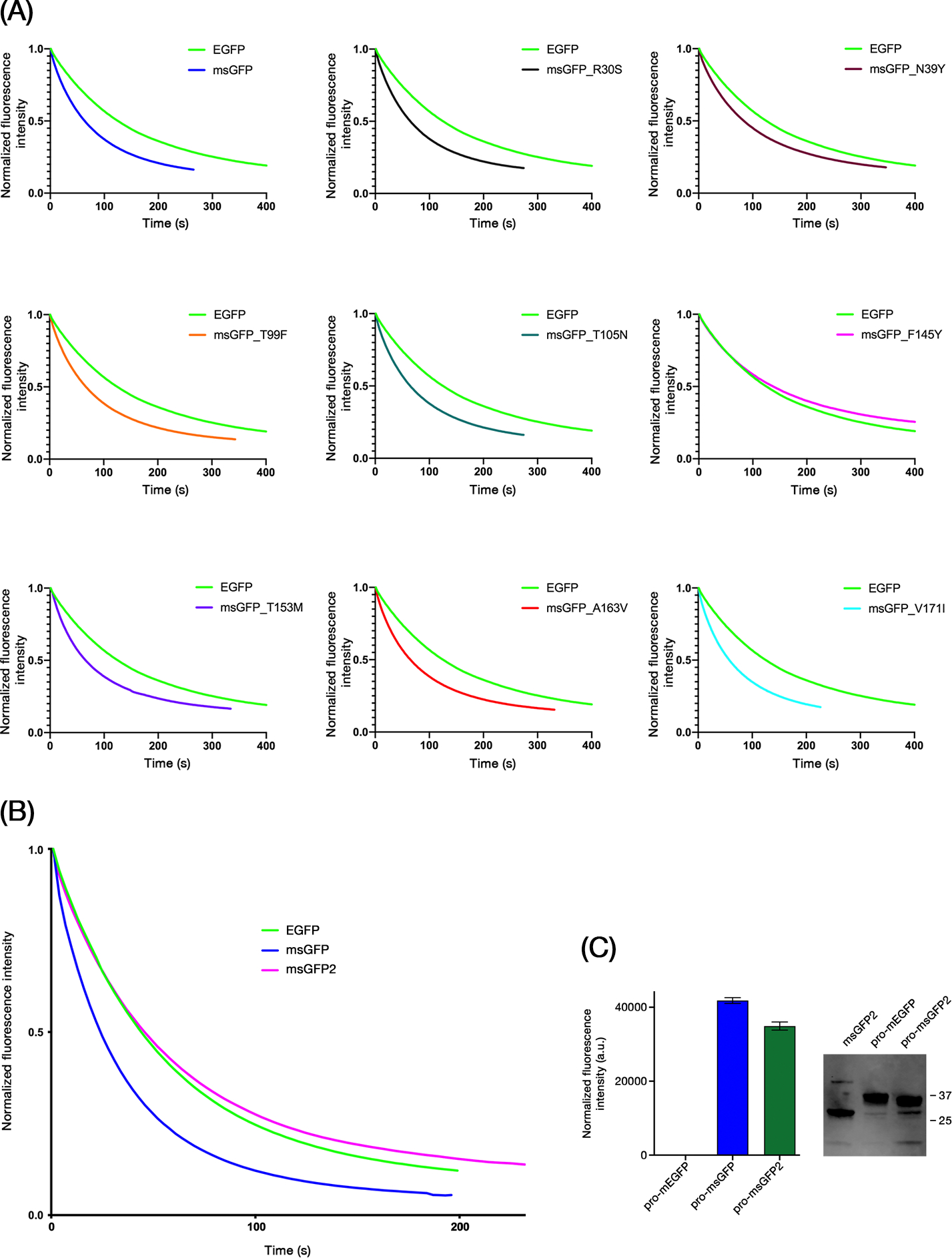
The F145Y reversion in GFP restores photostability without compromising superfolder activity. (A) EGFP, msGFP, and the indicated point revertants of msGFP were purified and subjected to photobleaching with blue light illumination on a microscope stage. Data points were collected every 3 s. Each signal was normalized to a starting value of 1.0. (B) Photobleaching of purified EGFP, msGFP, and msGFP2 was analyzed as in (A), except that the blue light illumination was stronger, and the curves were obtained by averaging the results from 3 experiments. (C) The indicated proinsulin fusion constructs were expressed in E. coli, and the green fluorescence signals were measured. Averaged signals from samples analyzed in triplicate are plotted in arbitrary units (a.u.) with SEM. To the right of the plot is an immunoblot with an anti-GFP antibody showing the levels of proinsulin-mEGFP and proinsulin-msGFP2 in extracts from the bacterial cells, with unfused msGFP2 as a control. The numbers indicate the sizes in kDa of molecular weight markers. The unfused msGFP2 construct yielded a fluorescence signal of about 57,000 arbitrary units, indicating that the efficiency of folding to the fluorescent state for proinsulin-msGFP and proinsulin-msGFP2 was greater than 50%.
We reasoned that one of the eight superfolder mutations in msGFP might be responsible for the reduced photostability. Those mutations were reverted individually, and the resulting purified FPs were examined. Strikingly, the F145Y reversion restored photostability to a level at least as high as that of EGFP (Figure 2A, magenta curve). None of the other reversions had a comparable effect. The F145Y reversion was therefore incorporated into msGFP2, which was much more photostable than msGFP and slightly more photostable than EGFP (Figure 2B). msGFP2 was similar to EGFP with regard to fluorescence spectra and brightness (Figure S2 and Table 1).
TABLE 1.
Photophysical properties of msGFP2 compared to EGFP.
| FP | Absorbance Maximum | Emission Maximum | Quantum Yield | Extinction Coefficient |
|---|---|---|---|---|
| msGFP2 | 489 nm | 509 nm | 0.59 | 52,200 |
| EGFP | 488 nm | 510 nm | 0.60 | 52,900 |
To confirm that the F145Y reversion did not abolish superfolder activity, we generated fusions of different GFP variants to mammalian proinsulin, which normally has disulfide bonds and is expected to misfold in the bacterial cytoplasm to form inclusion bodies.20 Expression of a proinsulin-mEGFP fusion in bacteria yielded no detectable fluorescence, presumably because mEGFP cannot mature to the fluorescent state in inclusion bodies.5 By contrast, expression of proinsulin-msGFP yielded strong fluorescence (Figure 2C). With proinsulin-msGFP2, the signal was somewhat lower but still strong (Figure 2C). As judged by immunoblot analysis of the bacterial cell extracts, the proinsulin-mEGFP and proinsulin-msGFP2 constructs were expressed at similar levels, indicating that the lack of fluorescence with proinsulin-mEGFP reflected misfolding (Figure 2C). These results show that msGFP2 retains superfolder activity.
In sum, compared to EGFP, the superfolder variant msGFP2 has similar photophysical properties but contains the following changes:
The monomerizing A206K mutation
The seven superfolding mutations S30R, Y39N, F99T, N105T, M153T, V163A, and I171V (but not Y145F)
An N-terminal MDSTES peptide in place of MVSKGEE
A C-terminal GGSGGS peptide in place of LGMDELYK
2.5 |. Evaluation of msGFP2 photostability in live yeast cells
To verify that msGFP2 showed improved photostability in live cells, we fused mEGFP, msGFP, or msGFP2 to yeast Sec7, a marker for late Golgi cisternae.21 This marker is used routinely in our laboratory to track Golgi cisternal maturation by time-lapse 3D fluorescence microscopy.22,23 Because the resulting movies are generated from many individual images, photobleaching is a concern. We captured movies of each yeast strain using both widefield and confocal fluorescence microscopes, with the illumination intensities set to cause at least 50% photobleaching of the original signal within 2 min. Widefield imaging (Figure 3A) and confocal imaging (Figure 3B) gave similar results: the msGFP2 signal was slightly more photostable than the EGFP signal and substantially more photostable than the msGFP signal. As a control, all three FPs labeled Sec7 with similar brightness values prior to photobleaching (Figure 3C).
FIGURE 3.
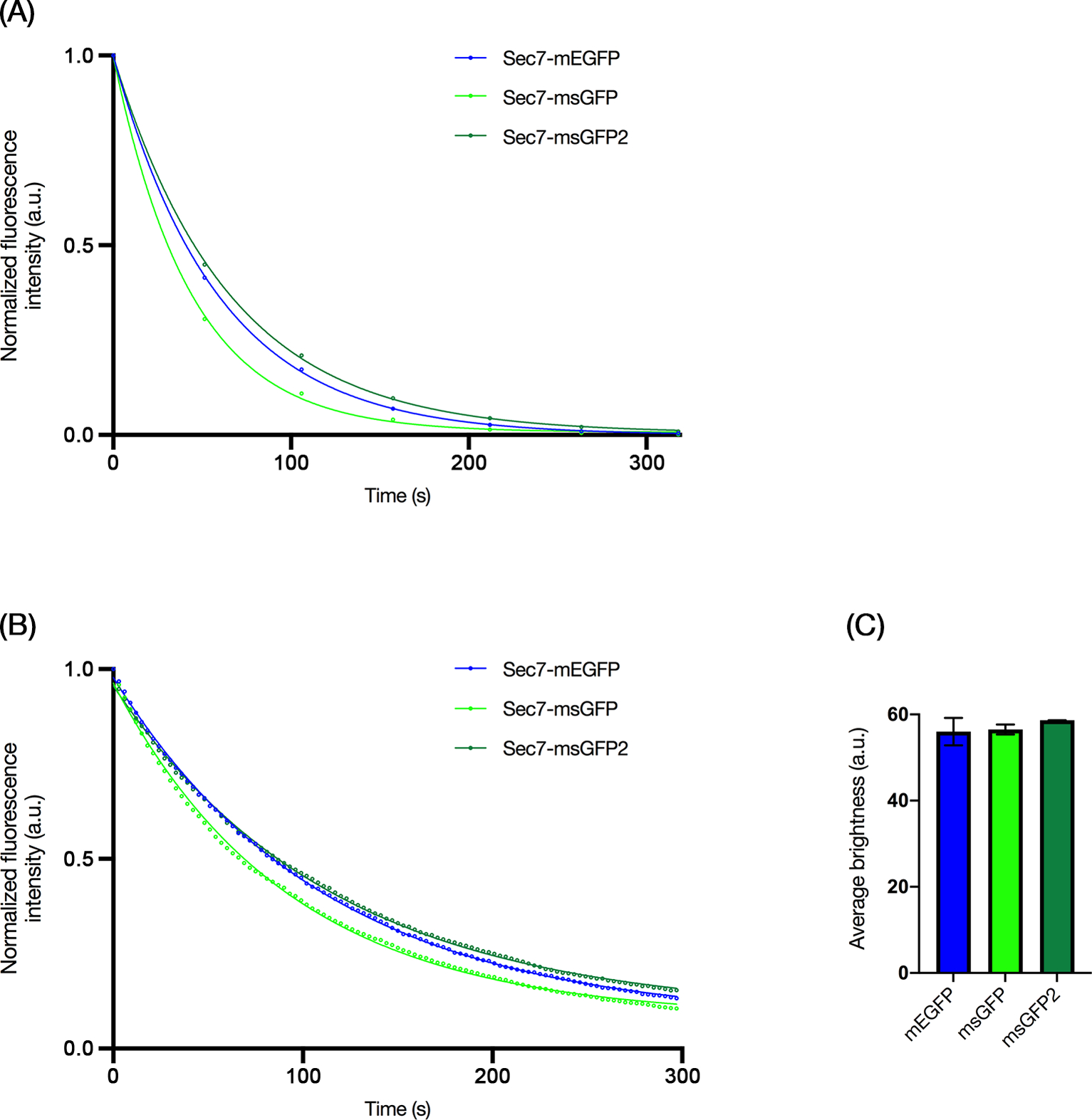
msGFP2 shows superior photostability in live cell imaging. Yeast Sec7 was tagged with mEGFP, msGFP, or msGFP2 by gene replacement, and the fluorescent late Golgi cisternae were imaged by time-lapse 3D microscopy. Shown are the fluorescence values from the projected Z-stacks in arbitrary units (a.u.) as well as best-fit exponential decay curves. (A) Photobleaching of tagged Sec7 was tracked by widefield microscopy, with Z-stacks collected every 54 s. The data points show the average values from 8 experiments. Typical SEM values were 2–5% of the average values. (B) Photobleaching of tagged Sec7 was tracked by confocal microscopy, with Z-stacks collected every 3 s. The data points show the average values from 6 experiments. Typical SEM values were 1–6% of the average values. (C) Brightness values were measured for Sec7-containing compartments labeled with the indicated FP tags. The time zero frames from the confocal movies in (B) were quantified to determine the average brightness values of late Golgi cisternae in arbitrary units (a.u.). Bars represent SEM.
When the photobleaching data for widefield imaging were fit to exponential decay curves as shown in Figure 3A, the time constant k for fluorescence decay of msGFP2 was 0.90 times the k value for mEGFP and 0.67 times the k value for msGFP. When the photobleaching data for confocal imaging were fit to exponential decay curves as shown in Figure 3B, the time constant k for fluorescence decay of msGFP2 was 0.97 times the k value for mEGFP and 0.81 times the k value for msGFP. We expect that these numbers will vary under different imaging conditions because photobleaching of FPs is a complex phenomenon24, but the results support the conclusion that msGFP2 is a uniquely photostable superfolder GFP for live cell microscopy.
2.6 |. Functional tests of msGFP2 in yeast
In the oxidizing environments of the ER lumen and the bacterial periplasm, superfolder GFP variants perform better than traditional GFP variants, which undergo aberrant disulfide bonding.6,25 We therefore tested whether msGFP2 was less disruptive than mEGFP as a tag for Saccharomyces cerevisiae Kar2, the yeast homolog of the ER luminal chaperone BiP.26 Chromosomal gene replacement was used to create Kar2-mEGFP and Kar2-msGFP2 fusions, with the FPs inserted immediately before the C-terminal HDEL signal that mediates retrieval to the ER.27 At 23°C or 30°C, Kar2-msGFP2 exhibited a typical yeast ER pattern consisting of a prominent nuclear envelope ring plus cortical labeling (Figure 4A).28 By contrast, Kar2-mEGFP produced abnormal fluorescent ER patterns at both 23°C and 30°C (Figure 4A). When growth was examined at 30°C, the strain expressing Kar2-msGFP2 grew similarly to a strain expressing wild-type Kar2, but the strain expressing Kar2-mEGFP grew more slowly (Figure 4B). These results fit with a previous study that employed a superfolder GFP to tag Kar229, but msGFP2 exhibits photostability that should make it a preferred option for live cell imaging.
FIGURE 4.
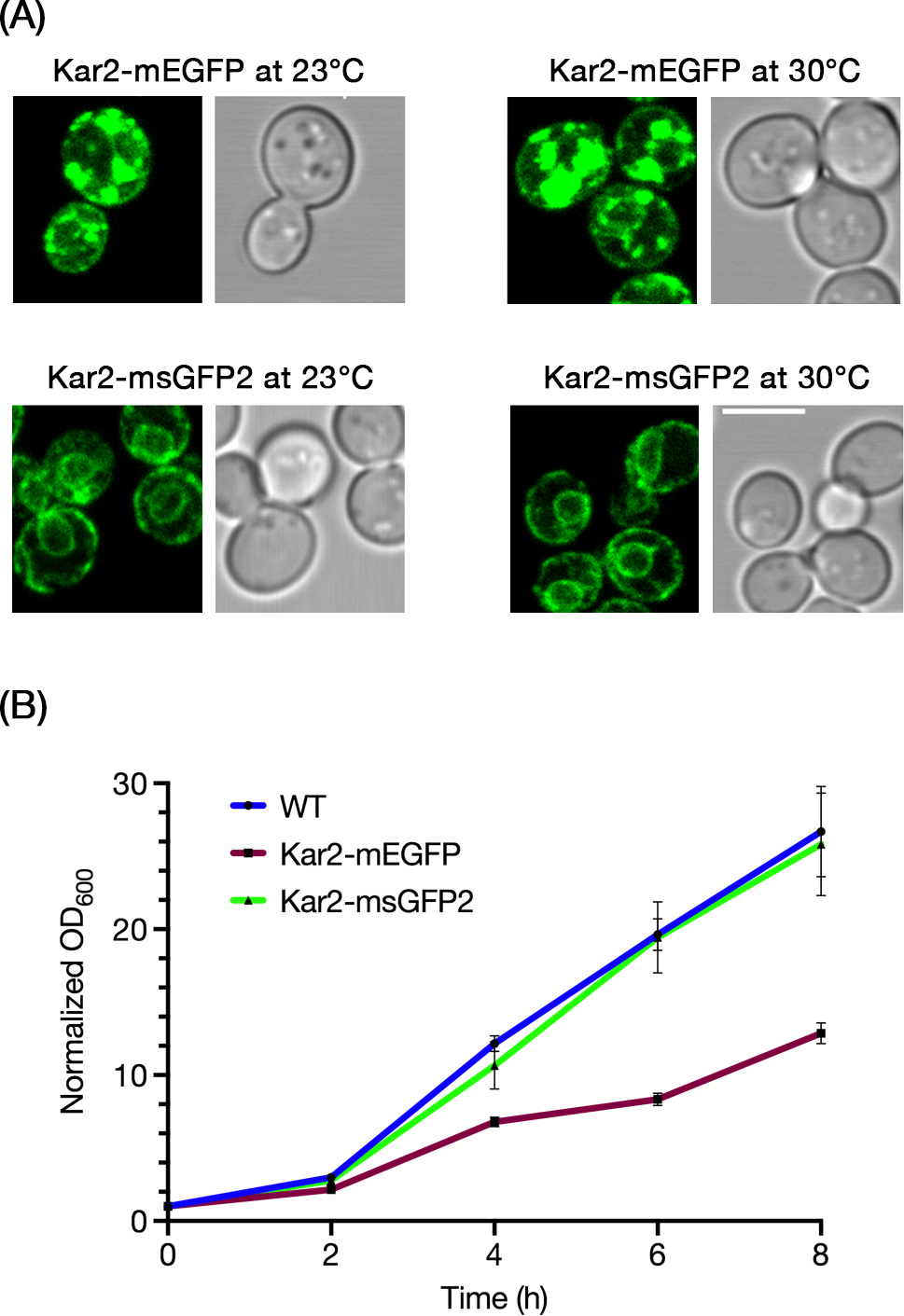
msGFP2 outperforms mEGFP as a fusion tag for the S. cerevisiae ER protein Kar2. (A) The endogenous Kar2 protein was tagged by gene replacement with mEGFP or msGFP2. Cells grown in minimal medium at either 23°C or 30°C were imaged by confocal microscopy. Optical sections corresponding to approximately the central half of each cell were projected. Scale bar, 5 μm. (B) Cells expressing wild-type Kar2, or Kar2-mEGFP, or Kar2-msGFP2 were grown overnight at 30°C in rich medium, then diluted to an OD600 of approximately 0.2 in the same medium. The diluted cultures were grown further at 30°C, and OD600 values were measured at 2-h intervals. OD600 values for each culture were normalized to the starting value. The graph represents averaged values for 3 independent experiments, with bars indicating SEM.
Are the benefits of msGFP2 also apparent when tagging a cytosolic protein? To address this question, we built on the finding that in the yeast Pichia pastoris, tagging the COPII vesicle coat protein Sec13 with GFP results in lethality at 36.5°C when combined with the P1092L point mutation in Sec16.30 The implication is that the Sec13-GFP fusion protein is partially defective. This effect was seen by plating yeast cells in a dilution series (Figure 5A). At 23°C, sec16-P1092L mutant cells grew well when Sec13 was tagged at the endogenous locus with either EGFP, or mEGFP, or msGFP2. By contrast, at 36.5°C, the strain expressing Sec13-msGFP2 grew much better than the strains expressing Sec13-EGFP or Sec13-mEGFP (Figure 5A). This effect was quantified by measuring growth in liquid culture at the semi-permissive temperature of 34°C. While none of the tagged Sec13 constructs conferred wild-type growth rates, the cells expressing Sec13-msGFP2 grew substantially better than those expressing Sec13-EGFP or Sec13-mEGFP (Figure 5B). When the ER exit sites were examined by fluorescence microscopy30,31 after 8 h at 34°C, fragmented ER exit sites were sometimes seen in cells expressing Sec13-EGFP or Sec13-mEGFP but not in cells expressing Sec13-msGFP2 (Figure 5C). All of the strains showed qualitatively similar fluorescence levels (Figure 5C), suggesting that the benefit of the superfolder property may have been due to the conformational stability of the GFP tag rather than the cellular abundance of Sec13. We conclude that Sec13 function is sensitive to tagging and that msGFP2 is less perturbing in this context than standard GFP variants.
FIGURE 5.
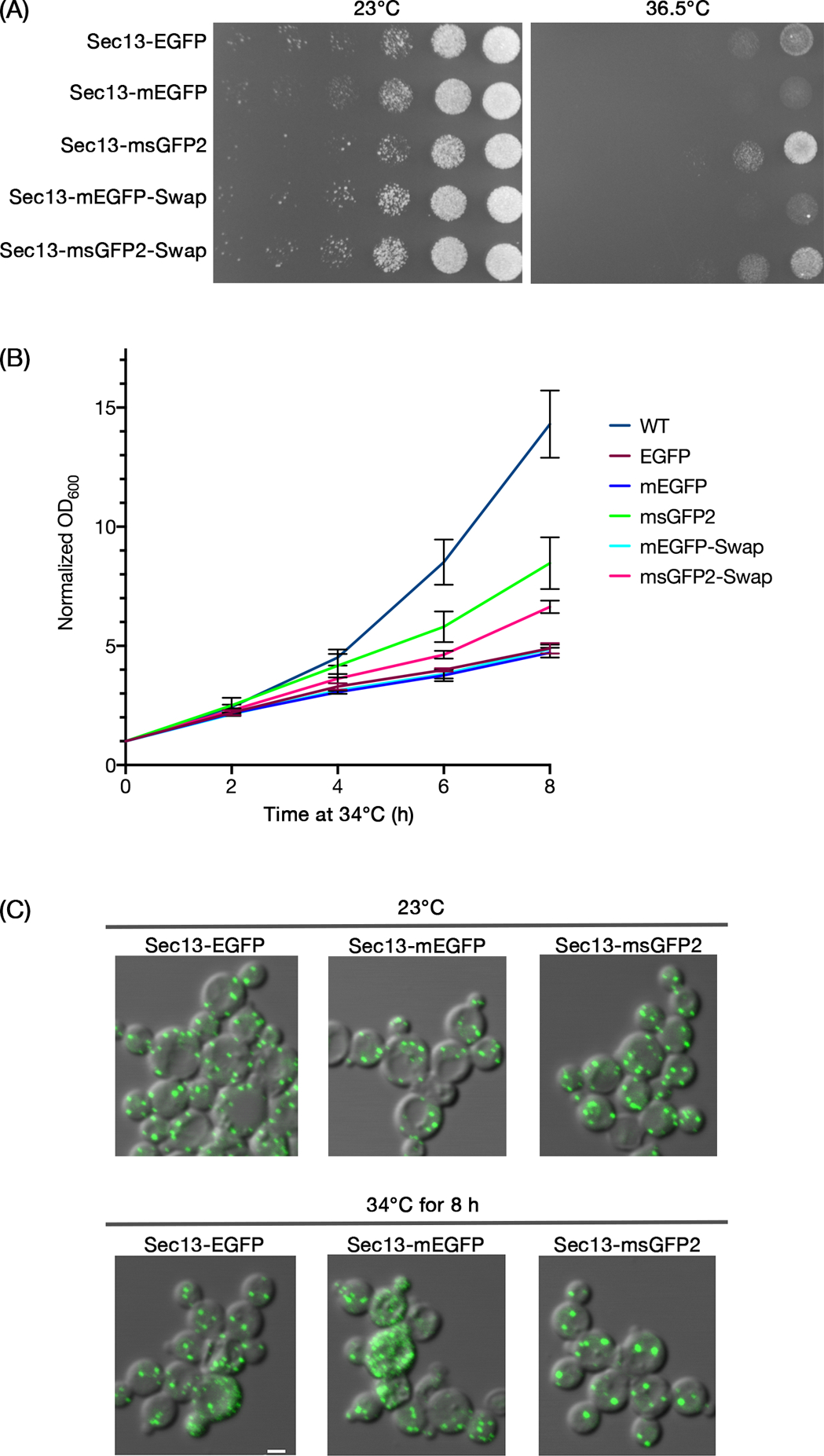
The superfolder property of msGFP2 enhances functionality in a fusion with the P. pastoris COPII coat protein Sec13. (A) P. pastoris strains with the indicated tags on Sec13 were grown in rich medium to mid-log phase, then diluted to an OD600 of 0.25. Serial 10x dilutions were spotted onto two identical minimal medium plates that selected for the integrated tag. The plates were then incubated for 3 days at either 23°C or 36.5°C. (B) P. pastoris strains with the indicated tags on Sec13 were grown overnight at 23°C in rich medium to early log phase. Then the cultures were diluted in rich medium to OD600 values of about 0.3, and incubated at 34°C for 8 h. OD600 measurements were taken every 2 h for 3 independent cultures per construct, normalized to the initial OD600 values, and averaged. Bars represent SEM. (C) Aliquots of the cells from (B) were imaged by fluorescence microscopy after initial growth at 23°C, and then again after incubation for 8 h at 34°C. As indicated by these representative images, after the 34°C incubation, a fraction of the cells expressing Sec13-EGFP and Sec13-mEGFP showed ER exit site fragmentation that presumably reflected toxic effects of the tags. Scale bar, 2 μm.
The stronger growth of the Sec13-msGFP2 strain could reflect either the superfolder property of msGFP2, or the modified N- and C-terminal peptides, or both. To test these possibilities, we created a construct in which Sec13 was fused either to mEGFP containing the msGFP2 terminal peptides (Sec13-mEGFP-Swap), or to msGFP2 containing the mEGFP terminal peptides (Sec13-msGFP2-Swap). As judged by plating yeast cells in a dilution series, only the Sec13-msGFP2-Swap construct yielded significant growth at 36.5°C (Figure 5A). This growth was reproducibly weaker than that obtained with Sec13-msGFP2, suggesting that the terminal peptides of msGFP2 made a difference. Similar results were obtained by comparing growth of the Sec13-msGFP2 and Sec13-msGFP2-Swap strains in liquid culture at 34°C (Figure 5B). In both assays, the superfolder property was necessary for improved growth, and the msGFP2 terminal peptides yielded a further improvement.
3 |. DISCUSSION
Variants of A. victoria GFP are typically among the best performing FPs for use as protein tags. Yet some GFP-tagged proteins show mislocalization and/or reduced functionality, suggesting that there is room for further improvement. We describe a variant called msGFP2 that has two enhancements.
The first enhancement in msGFP2 is the F145Y reversion, which restores photostability to the level seen with EGFP. We note that during the engineering of superfolder GFP, Y145F was the only mutation that altered the interior of the protein5, so it is not surprising that this mutation changed the photophysical properties. Fortunately, Y145F had only a minor effect on folding5, and we found that this mutation could be reverted with only a slight reduction in the superfolder property. Interestingly, the Y145F mutation was found to be detrimental and was omitted during the recent engineering of a cyan FP termed superfolder mTurquoise2.32 It is likely that other superfolder GFP variants could also benefit from preserving the original tyrosine at position 145.
The second enhancement in msGFP2 is replacement of the N- and C-terminal peptides. When these terminal peptides are incorporated into other FPs, they can cause cytotoxicity in certain E. coli strains. In particular, the widely used red FP mCherry contains the N- and C-terminal peptides of EGFP12, and we find that the N-terminal peptide of EGFP can cause cytotoxicity of mCherry during bacterial expression. Better results are seen with an mCherry variant that incorporates an N-terminal peptide related to the one we generated when optimizing tetrameric DsRed.10 This improvement may reflect lower interactivity of the new N-terminal peptide. We also replaced the mCherry C-terminal peptide, which is MDELYK, with the sequence GGSGGS, based on the expectation that the GFP-derived MDELYK peptide might be prone to undesirable electrostatic and hydrophobic interactions. Similar N- and C-terminal peptide replacements also reduced bacterial cytotoxicity of mScarlet-I17, a synthetic red FP. We suggest that the common practice of incorporating the EGFP terminal peptides into engineered FPs is unwise, and that the terminal peptides described here are a good alternative. Because effects of the type seen with red FPs might also occur with GFP in some situations, we incorporated the new N- and C-terminal peptides into msGFP2.
Compared to mEGFP, which has been a favored choice for protein tagging, msGFP2 is expected to be comparable and sometimes superior. The major advantage of msGFP2 is that it offers superfolder behavior without loss of photostability. One example of an environment in which the superfolder property is beneficial is the lumen of the ER. Standard GFP variants can accumulate in the ER in a nonfluorescent state, apparently because cysteines that are normally in the interior of folded GFP can form disulfides that inhibit folding.6,33 Indeed, we find that when the yeast ER luminal chaperone Kar2 (BiP) is tagged with mEGFP or msGFP2, only the msGFP2 fusion yields an unperturbed ER pattern. Expression of Kar2-mEGFP results in fluorescence, but the cells grow slowly and have abnormal ER structures that may indicate ER stress. Thus, Kar2-msGFP2 behaves much better than Kar2-mEGFP.
The superfolder property can also be beneficial in the cytosol, especially when a tagged protein is in a challenging local environment such as bacterial inclusion bodies.5 Similar challenges may arise whenever a GFP-tagged protein is highly concentrated at a particular cellular location. We explored a test case involving the polymeric coat of COPII vesicles. Our previous work had revealed that when the COPII subunit Sec13 was tagged with EGFP in the yeast P. pastoris, the Sec13-EGFP allele rendered the cells thermosensitive for growth when combined with a point mutation in the COPII-associated Sec16 protein.30 The same effect is seen with Sec13-mEGFP. By contrast, Sec13-msGFP2 allows significantly faster growth at elevated temperatures in a strain expressing mutated Sec16. The reason for the better behavior of Sec13-msGFP2 is not obvious, because the fluorescence signals at COPII-containing ER exit sites are similar for Sec13-mEGFP and Sec13-msGFP2. One possibility is that the cells expressing fluorescent Sec13-mEGFP also contain nonfluorescent Sec13-mEGFP molecules, which actively interfere with COPII function. A more intriguing possibility is that conformational stability is important even after a GFP variant has folded to the fluorescent state, with fluorescent Sec13-msGFP2 molecules being more conformationally stable and hence more functional than fluorescent Sec13-mEGFP molecules. Additional work will be needed to clarify how superfolder GFP variants reduce perturbation of tagged proteins.
Diverse applications can be envisioned for derivatives of msGFP2, including variants with altered spectra1 or with other specialized properties.34 We have obtained good results using a yeast codon-optimized msGFP2 (data not shown). This type of engineering is helping to unlock the full potential of FPs as fusion tags.
4 |. METHODS
4.1 |. DNA constructs and bacterial growth assay
DNA constructs were propagated in E. coli strain XL1-Blue, which suppresses cytotoxicity due to low constitutive expression of FPs. A supplemental folder contains SnapGene files for the constructs used in this study. Relevant constructs will be archived with Addgene.
To determine the effects of FP expression on bacterial growth, E. coli cells of strain DH10B were transformed with pQE-60NA constructs35 encoding the various FP variants. DH10B was chosen because this strain exhibited more pronounced cytotoxicity effects than the other E. coli strains that we tested. A transformed culture was diluted 1:30, then spread on an LB + ampicillin plate and grown overnight at 37°C for 19–20 h. The resulting colonies were photographed with a Bio-Rad FX Pro Plus gel imager, using ultraviolet light illumination to detect fluorescent clones with the exposure time adjusted based on FP brightness. These images were processed using ImageJ36 as follows: images were changed to 8-bit format, inverted to display black colonies on a white background, and subjected to thresholding to remove background spots and nonfluorescent colonies. Colony areas were automatically measured using the “Analyze Particles” tool. For each FP construct, the average colony areas were determined for at least 200 colonies derived from 3 separate experiments.
4.2 |. FP expression and purification
Hexahistidine-tagged FPs were expressed using a pQE-81-based vector.37 For modification of msGFP, the parental construct was subjected to QuikChange mutagenesis to generate single-codon revertants. Constructs were transformed into E. coli XL1-Blue cells, and clones were grown overnight in LB + ampicillin. Each culture was then diluted 1:50 in LB + ampicillin, grown to mid-log phase, induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), grown overnight, and centrifuged. The pelleted cells were lysed using B-PER II bacterial protein extraction reagent (Thermo Fisher) for 15 min, and centrifuged for 15 min at 27,000xg. The supernatant was adjusted to 300 mM NaCl, vortex mixed, and centrifuged for 1 min at 17,000xg. The soluble extract was incubated with 0.2 mL of Ni2+-NTA-agarose slurry (Macherey-Nagel) per 50 mL of starting culture, and mixed end-over-end for 1 h at 23°C. Then the beads were washed three times with 300 mM NaCl, 20 mM imidazole-HCl, pH 7.4, 0.5% Triton X-100, using 2 mL per 50 mL of culture, followed by three more washes with the same buffer lacking Triton X-100. Each FP was eluted by mixing the beads with 0.5 mL of 300 mM imidazole-HCl, pH 7.4 per 50 mL of starting culture for 20 min at 23°C. The final eluate was adjusted to 0.5 mM EDTA and stored in the dark at 4°C.
4.3 |. Photobleaching assay for purified FPs
Each purified 6xHis-tagged FP was adjusted to a protein concentration of 60 μM. In parallel, 1% low-melt agarose in 50 mM Na+-PIPES, pH 7.0, 100 mM NaCl, 0.02% sodium azide was melted in a microwave and then placed at 50°C, and 500 μL of the melted agarose was supplemented with 15 μL of 2-μm polystyrene beads (Polysciences). Twenty μL each of the FP and agarose/beads solutions were mixed thoroughly, and 2 μL of this mixture was placed on a preheated glass slide and compressed with a preheated coverslip, aiming for a complete and even spread. The polystyrene beads created a layer of uniform thickness. Melted wax was used to seal the edges of the coverslip. After solidification of the agarose, the sample was imaged. To induce photobleaching, the slide was exposed to continuous illumination with blue light using a Zeiss Axioplan 2 microscope with a GFP filter cube. Images of a single focal plane were taken every 3 s until the field of view was almost completely dark. Fluorescence intensity values were obtained from each image series, blank subtracted, normalized, and plotted as a function of time.
4.4 |. Superfolder activity assay
Vectors based on pQE60-NA were used to express human proinsulin with C-terminally fused FPs. The unfused msGFP2 construct contained the first four codons of proinsulin to minimize variations in bacterial expression due to the sequence near the start codon10. These constructs were transformed into E. coli XL1-Blue cells, and transformants were grown on LB + ampicillin plates. For each transformant, 3 clones were picked, inoculated in 5 mL LB + ampicillin medium, and grown at 37°C to an OD600 of 0.5. Cultures were diluted 4-fold in the same medium, and 5 mL were placed in a 50-mL baffled flask and grown with shaking at 37°C to an OD600 of 0.5. Three aliquots from each culture were transferred to a black 96-well plate to measure the background fluorescence from uninduced cells. The cultures were then induced by adding IPTG to 1 mM, and were incubated for 1.5 h. Aliquots from each induced culture were adjusted to an OD600 of 0.5 and transferred to a black 96-well plate to measure the cellular fluorescence. After subtracting the background values, the fluorescence values were averaged.
4.5 |. Spectral measurements
Purified 6xHis-tagged FPs were serially diluted in 300 mM imidazole-HCl, pH 7.4, and the absorbance and emission spectra of each dilution were measured using an Agilent 8453 UV-Visible spectrophotometer and a Horiba Fluorolog-3 fluorimeter, respectively. The emission spectrum values were integrated and plotted as a function of the peak absorbance values, and the slopes of the lines were used to calculate the quantum yields relative to EGFP, which was assumed to have a quantum yield of 0.60.1
4.6 |. Yeast methods
The parental yeast strains were S. cerevisiae JK9–3da38 and P. pastoris PPY12.39 Yeast proteins were C-terminally tagged with FPs using the pop-in/pop-out method for S. cerevisiae40 or integrative gene tagging for P. pastoris.31 In the case of Kar2, the FPs were inserted upstream of the C-terminal HDEL signal that mediates retrieval to the ER. Details of the constructs can be found in the supplemental SnapGene files. Cells were grown in baffled flasks in rich YPD medium or nonfluorescent minimal NSD medium.41
For fluorescence imaging of strains with tagged Kar2 or Sec13, strains were grown in liquid NSD to mid-log phase at 23°C. Cells were either attached to the bottom of a coverslip-bottom dish (MatTek Corporation) coated with concanavalin A42 for Kar2, or compressed beneath a coverslip and imaged on a glass slide for Sec13. Images were captured with a Leica SP5 confocal microscope using a 1.4 NA/63x oil immersion objective, 15% laser power for the 488-nm line at 70% intensity, image fields of 41 × 41 μm, a zoom factor of 6, line averaging of 16, and pinhole size of 1.0 AU, a Z-step interval of 0.25 μm, and either 27–30 optical sections for Kar2 or 17–20 optical sections for Sec13. The Z-stacks were imported into ImageJ for range adjustment and average projection. In the case of Kar2, to highlight the cortical and nuclear ER patterns, only the 10 middle slices of each Z-stack were average projected.
For photobleaching analysis of strains with tagged Sec7, cells were grown in NSD with shaking at 23°C to mid-log phase, and were then attached to a coverslip-bottom dish coated with concanavalin A. One set of experiments employed widefield imaging with a Zeiss Axio Observer Z1 microscope using a 1.4 NA/63x oil immersion objective, an exposure time of 460 ms (100% lamp intensity) for the green channel (488-nm excitation wavelength), a Z-step interval of 0.5 μm, and 16 optical sections (7.5 μm total depth) collected every 54 s for a total collection time of 320 s. The image fields were 113 × 71 μm, with an average of about 210 cells per image. This analysis was repeated 4 times for 2 independent clones, giving a total of 8 replicates, and the results were averaged. A second set of experiments employed confocal imaging with a Leica SP5 microscope using a 1.4 NA/63x oil immersion objective, 10% laser power for the 488-nm line at 100% intensity, a zoom factor of 4, line averaging of 6, a pinhole size of 1.2 AU, a Z-step interval of 0.25 μm, and 27 optical sections (6.5 μm total depth) collected every 3 s for a total collection time of 300 s. The image fields were 70×18 μm, with an average of about 45 cells per image. This analysis was repeated 3 times for 2 independent clones, for a total of 6 replicates, and the results were averaged. For both widefield and confocal movies, deconvolution and average projection of Z-stacks were performed as previously described.42 To quantify the brightness value of each FP tag when fused to Sec7, the time zero projections for the 6 confocal movies were processed with ImageJ as follows. After thresholding to exclude background in the fluorescence channel, the Analyze Particles tool was used to identify spots that presumably represented individual Golgi cisternae, with areas in the range of 0.1–0.6 μm2. The integrated density values for those spots were measured and averaged.
For the growth test with S. cerevisiae strains expressing tagged Kar2, cells were grown in YPD with shaking at 23°C to mid-log phase, then diluted to an OD600 of 0.2 and grown with shaking at 30°C for 8 h. OD600 values were measured every 2 h.
For the growth test with P. pastoris strains expressing tagged Sec13, cells were grown in YPD with shaking at 23°C to an OD600 of about 0.5. Aliquots of each culture were used to capture pre-treatment images. Then the cultures were diluted in YPD to an OD600 of 0.3, and were grown with shaking at 34°C for 8 h. OD600 values were measured every 2 h. At the end of the incubation, aliquots of each culture were used to capture post-treatment images.
4.7 |. Immunoblotting
Bacterial cells expressing GFP variants, either unfused or fused to proinsulin, were harvested and processed for SDS-PAGE as previously described.10 Immunoblotting was then performed8 with a 1:2500 dilution of a rabbit anti-GFP polyclonal antibody (Abcam, catalog number ab6556) followed by a 1:3000 dilution of an Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (Thermo Fisher, catalog number A-11008). Signals were detected using a LI-COR Odyssey CLx system.
4.8 |. Software analysis
DNA constructs were simulated and recorded using SnapGene (Insightful Science). Statistical analysis was performed using GraphPad Prism (Insightful Science). Figures were assembled using ImageJ together with Photoshop (Adobe). Deconvolution was performed using Huygens Essential (SVI).
Supplementary Material
FIGURE S1 Terminal peptides influence bacterial cytotoxicity for two red fluorescent proteins. (A) E. coli DH10B cells were transformed with constructs encoding mCherry, or mCherry2C, or mCherry with its N-terminal peptide replaced with MDSTES, or mCherry with its C-terminal peptide replaced with GSSGSS. Transformants were plated on LB + ampicillin, and the plate was photographed after overnight growth. (B) The experiment was performed as in (A), except that three separate plates were used for pairwise comparisons of transformants expressing the indicated FPs. (C) For transformants expressing the indicated FPs, a scatter plot shows a representative distribution of colony sizes in arbitrary units (a.u.). Averaged data from 3 such experiments are plotted as a bar graph in Figure 1.
FIGURE S2 EGFP and msGFP2 have similar fluorescence spectra. Absorbance and fluorescence emission spectra were measured for purified 6xHis-tagged FPs diluted to an absorbance at 488 nm of 0.1. Fluorescence is plotted in arbitrary units (a.u.).
Synopsis.
Superfolder GFP variants are suitable for imaging in challenging cellular environments, but they suffer from rapid photobleaching. Here we describe a photostable monomeric superfolder GFP called msGFP2. In addition to its enhanced photophysical properties, msGFP2 contains N- and C-terminal peptides derived from an mCherry variant that has reduced cytotoxicity. These improved fluorescent proteins should be of general utility.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 GM104010. F.M.V. was supported by NIH training grant T32 GM007183. Thanks for assistance with fluorescence microscopy to Vytas Bindokas and Christine Labno at the Integrated Microscopy Core Facility, which is supported by the NIH-funded Cancer Center Support Grant P30 CA014599. Thanks for assistance with the collection of spectra to Elena Solomaha at the Biophysics Core Facility. Additional thanks to the Glick lab for constructive feedback.
REFERENCES
- 1.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. [DOI] [PubMed] [Google Scholar]
- 2.Yang F, Moss LG, Phillips GN Jr. The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14(10):1246–1251. [DOI] [PubMed] [Google Scholar]
- 3.Costantini LM, Fossati M, Francolini M, Snapp EL. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 2012;13(5):643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. [DOI] [PubMed] [Google Scholar]
- 5.Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. [DOI] [PubMed] [Google Scholar]
- 6.Aronson DE, Costantini LM, Snapp EL. Superfolder GFP is fluorescent in oxidizing environments when targeted via the Sec translocon. Traffic. 2011;12:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dinh T, Bernhardt TG. Using superfolder green fluorescent protein for periplasmic protein localization studies. J Bacteriol. 2011;193(18):4984–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzgerald I, Glick BS. Secretion of a foreign protein from budding yeasts is enhanced by cotranslational translocation and by suppression of vacuolar targeting. Microb Cell Fact. 2014;13:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bevis BJ, Glick BS. Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed). Nat Biotechnol. 2002;20:83–87. [DOI] [PubMed] [Google Scholar]
- 10.Strack RL, Strongin DE, Bhattacharyya D, et al. A noncytotoxic DsRed variant for whole-cell labeling. Nat Methods. 2008;5:955–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yanushevich YG, Staroverov DB, Savitsky AP, et al. A strategy for the generation of non-aggregating mutants of Anthozoa fluorescent proteins. FEBS Lett. 2002;511:11–14. [DOI] [PubMed] [Google Scholar]
- 12.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. [DOI] [PubMed] [Google Scholar]
- 13.Shagin DA, Barsova EV, Yanushevich YG, et al. GFP-like proteins as ubiquitous metazoan superfamily: evolution of functional features and structural complexity. Mol Biol Evol. 2004;21(5):841–850. [DOI] [PubMed] [Google Scholar]
- 14.Strongin DE, Bevis B, Khuong N, et al. Structural rearrangements near the chromophore influence the maturation speed and brightness of DsRed variants. Protein Eng Des Sel. 2007;20:525–534. [DOI] [PubMed] [Google Scholar]
- 15.Day KJ, Casler JC, Glick BS. Budding yeast has a minimal endomembrane system. Dev Cell. 2018;44(1):56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bindels DS, Haarbosch L, van Weeren L, et al. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods. 2017;14(1):53–56. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Zhang G, Ngo N, Zhao X, Kain SR, Huang CC. Deletions of the Aequorea victoria green fluorescent protein define the minimal domain required for fluorescence. J Biol Chem. 1997;272(45):28545–28549. [DOI] [PubMed] [Google Scholar]
- 19.Lee S, Lim WA, Thorn KS. Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS One. 2013;8(7):e67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cowley DJ, Mackin RB. Expression, purification and characterization of recombinant human proinsulin. FEBS Lett. 1997;402(2–3):124–130. [DOI] [PubMed] [Google Scholar]
- 21.Rossanese OW, Reinke CA, Bevis BJ, et al. A role for actin, Cdc1p and Myo2p in the inheritance of late Golgi elements in Saccharomyces cerevisiae. J Cell Biol. 2001;153(1):47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casler JC, Papanikou E, Barrero JJ, Glick BS. Maturation-driven transport and AP-1–dependent recycling of a secretory cargo in the Golgi. J Cell Biol. 2019;218(5):1582–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Losev E, Reinke CA, Jellen J, Strongin DE, Bevis BJ, Glick BS. Golgi maturation visualized in living yeast. Nature. 2006;441(22 June):1002–1006. [DOI] [PubMed] [Google Scholar]
- 24.Diaspro A, Chirico G, Usai C, Ramoino P, Dobrucki J. Photobleaching In: Pawley JB, ed. Handbook of Biological Confocal Microscopy, 3rd Edition Springer; 2006:690–702. [Google Scholar]
- 25.Jain RK, Joyce PB, Molinete M, Halban PA, Gorr SU. Oligomerization of green fluorescent protein in the secretory pathway of endocrine cells. Biochem J. 2001;360(Pt 3):645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rose MD, Misra LM, Vogel JP. KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell. 1989;57:1211–1221. [DOI] [PubMed] [Google Scholar]
- 27.Pelham HR. The retention signal for soluble proteins of the endoplasmic reticulum. Trends Biochem Sci. 1990;15(12):483–486. [DOI] [PubMed] [Google Scholar]
- 28.Preuss D, Mulholland J, Kaiser CA, et al. Structure of the yeast endoplasmic reticulum: localization of ER proteins using immunofluorescence and immunoelectron microscopy. Yeast. 1991;7:891–911. [DOI] [PubMed] [Google Scholar]
- 29.Lajoie P, Moir RD, Willis IM, Snapp EL. Kar2p availability defines distinct forms of endoplasmic reticulum stress in living cells. Mol Biol Cell. 2012;23(5):955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connerly PL, Esaki M, Montegna EA, et al. Sec16 is a determinant of transitional ER organization. Curr Biol. 2005;15(16):1439–1447. [DOI] [PubMed] [Google Scholar]
- 31.Rossanese OW, Soderholm J, Bevis BJ, et al. Golgi structure correlates with transitional endoplasmic reticulum organization in Pichia pastoris and Saccharomyces cerevisiae. J Cell Biol. 1999;145:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meiresonne NY, Consoli E, Mertens LMY, Chertkova AO, Goedhart J, den Blaauwen T. Superfolder mTurquoise2ox optimized for the bacterial periplasm allows high efficiency in vivo FRET of cell division antibiotic targets. Mol Microbiol. 2019;111(4):1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jokitalo E, Cabrera-Poch N, Warren G, Shima DT. Golgi clusters and vesicles mediate mitotic inheritance independently of the endoplasmic reticulum. J Cell Biol. 2001;154:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reifenrath M, Boles E. A superfolder variant of pH-sensitive pHluorin for in vivo pH measurements in the endoplasmic reticulum. Sci Rep. 2018;8(1):11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strack RL, Keenan RJ, Glick BS. Noncytotoxic DsRed derivatives for whole-cell labeling. Methods Mol Biol. 2011;699:355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strack RL, Strongin DE, Mets L, Glick BS, Keenan RJ. Chromophore formation in DsRed occurs by a branched pathway. J Am Chem Soc. 2010;132:8496–8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunz J, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73:585–596. [DOI] [PubMed] [Google Scholar]
- 39.Gould SJ, McCollum D, Spong AP, Heyman JA, Subramani S. Development of the yeast Pichia pastoris as a model organism for a genetic and molecular analysis of peroxisome assembly. Yeast. 1992;8:613–628. [DOI] [PubMed] [Google Scholar]
- 40.Targeting Rothstein R., disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. [DOI] [PubMed] [Google Scholar]
- 41.Bevis BJ, Hammond AT, Reinke CA, Glick BS. De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nat Cell Biol. 2002;4(10):750–756. [DOI] [PubMed] [Google Scholar]
- 42.Johnson N, Glick BS. 4D microscopy of yeast. J Vis Exp. 2019(146). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Terminal peptides influence bacterial cytotoxicity for two red fluorescent proteins. (A) E. coli DH10B cells were transformed with constructs encoding mCherry, or mCherry2C, or mCherry with its N-terminal peptide replaced with MDSTES, or mCherry with its C-terminal peptide replaced with GSSGSS. Transformants were plated on LB + ampicillin, and the plate was photographed after overnight growth. (B) The experiment was performed as in (A), except that three separate plates were used for pairwise comparisons of transformants expressing the indicated FPs. (C) For transformants expressing the indicated FPs, a scatter plot shows a representative distribution of colony sizes in arbitrary units (a.u.). Averaged data from 3 such experiments are plotted as a bar graph in Figure 1.
FIGURE S2 EGFP and msGFP2 have similar fluorescence spectra. Absorbance and fluorescence emission spectra were measured for purified 6xHis-tagged FPs diluted to an absorbance at 488 nm of 0.1. Fluorescence is plotted in arbitrary units (a.u.).