

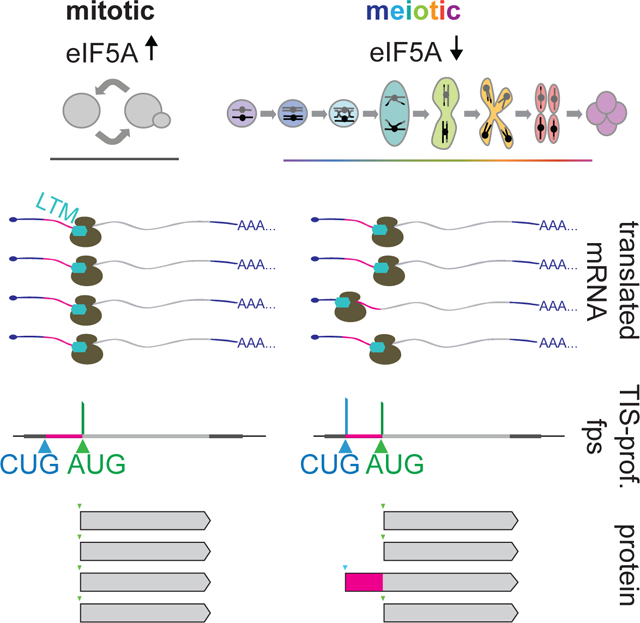
SUMMARY
Genomic analyses in budding yeast have helped define the foundational principles of eukaryotic gene expression. However, in the absence of empirical methods for defining coding regions, these analyses have historically excluded specific classes of possible coding regions, such as those initiating at non-AUG start codons. Here, we applied an experimental approach to globally annotate translation initiation sites in yeast and identified 149 genes with alternative N-terminally extended protein isoforms initiating from near-cognate codons upstream of annotated AUG start codons. These isoforms are produced in concert with canonical isoforms and translated with high specificity, resulting from initiation at only a small subset of possible start codons. The non-AUG initiation driving their production is enriched during meiosis and induced by low eIF5A, which is seen in this context. These findings reveal widespread production of non-canonical protein isoforms and unexpected complexity to the rules by which even a simple eukaryotic genome is decoded.
Graphical Abstract
eTOC blurb
Eisenberg et al. identify translation initiation sites genome-wide in budding yeast. They define a class of 149 genes with alternate extended isoforms that initiate at non-AUG start codons. These isoforms are produced in concert with their corresponding canonical isoforms, but typically at lower abundance, and are selectively induced during meiosis.
INTRODUCTION
Our understanding of cell function has been advanced by genome annotations that comprehensively predict the repertoire of protein products within the cell. Genes were historically annotated computationally based on a set of rules that were informed by existing knowledge of the mechanism of translation and the features shared by most well-studied genes (Brent, 2005). Open reading frames (ORFs), for example, have been defined as starting at an AUG and stopping at the next in-frame stop codon because this reflects characterized properties of translation of an mRNA by the ribosome (reviewed in Aitken and Lorsch, 2012). Development of experimental approaches to globally define translated regions has now made it possible to determine the prevalence of translated ORFs that do not follow these rules. Additionally, such approaches enable identification of condition-specific changes in ORF identity, such as during stress or developmental progression, which cannot be predicted from sequence-based annotation alone.
Ribosome profiling was the first method to allow genome-wide experimental identification of translated regions in vivo. This method involves isolating and sequencing the short (~30nt) regions of mRNA that are protected from nuclease digestion by translating ribosomes (Ingolia et al., 2009). We previously used ribosome profiling to assess changes in translation as yeast cells progress through meiosis (Brar et al., 2012), the highly conserved cellular differentiation program that leads to gamete formation. We observed pervasive and condition-specific non-canonical translation, including spans of translation that initiated at near-cognate start codons (which differ from AUG by one nucleotide) and translation of uORFs (upstream ORFs) in 5’ leader regions. However, the prevalence of overlapping translated ORFs in 5’ leader regions in meiotic cells made it challenging to unambiguously assign ribosome footprints, complicating our goal of achieving high-confidence annotations of all translated ORFs.
A modified ribosome profiling strategy, in which cells are pre-treated with drugs that inhibit post-initiation ribosomes, yields footprint reads that map primarily to translation initiation sites (TISs), aiding in the detection and annotation of ORFs (Ingolia et al., 2011; Lee et al., 2012). Global TIS mapping has been performed under several conditions (Fields et al., 2015; Fritsch et al., 2012; Ingolia et al., 2011; Lee et al., 2012; Machkovech et al., 2019; Sapkota et al., 2019; Stern-Ginossar et al., 2012), but thus far only in mammals and viruses, which have complex gene structures. Budding yeast (Saccharomyces cerevisiae) has relatively simple transcript architectures with far fewer known cases of complexity, such as from alternative splicing, despite extensive analyses of its transcriptome (Davis et al., 2000; Hossain et al., 2011; Juneau et al., 2009; Kim Guisbert et al., 2012; Yassour et al., 2009). This simple architecture allows for investigation of TISs to be more directly informative, as identification of the start codon alone can generally be used to define an ORF.
We developed a TIS identification approach for budding yeast, both in vegetative and meiotic conditions, with the goal of characterizing ORF types that were previously challenging to identify systematically by standard ribosome profiling. The class of ORFs that we were most interested in assessing, due to their potential to modulate the function of well-characterized genes, were those encoding alternate protein isoforms that result from translation initiation at non-AUG codons upstream of the characterized start codon (see Table S1 for a summary of prior studies of this class of proteins). Several individual examples of N-terminally extended proteins isoforms have been identified in an ad hoc manner using classical approaches (K.-J. Chang and Wang, 2004; Heublein et al., 2019; Kearse and Wilusz, 2017; Kritsiligkou et al., 2017; Monteuuis et al., 2019; Suomi et al., 2014; Tang et al., 2004; Touriol et al., 2003) and a recent computational study predicted the existence of many additional cases (Monteuuis et al., 2019). However, it was not previously possible to directly experimentally evaluate the prevalence of this class of translation products comprehensively in yeast. Our approach allowed us to determine that condition-specific translation of non-AUG-initiated protein isoforms is common, reflecting regulated induction of a pool of alternative proteins that is facilitated by low eIF5A levels. More broadly, this study revealed surprising complexity to translation-even at characterized loci-in this widely studied organism.
RESULTS
TIS-profiling in yeast globally defines translation initiation sites
We sought to perform TIS identification in yeast by using ribosome profiling following pre-treatment with harringtonine or lactimidomycin (LTM), two established drugs that preferentially inhibit post-initiation ribosomes but allow elongating ribosomes to run off, resulting in ribosome footprint enrichment at TISs (Figure 1A; Fresno et al., 1977; Ingolia et al., 2011; Lee et al., 2012; Sugawara et al., 1992). Initial testing of both drugs under the conditions used for this purpose in mammalian contexts was unsuccessful in yeast. Even treatment with extremely high concentrations of harringtonine (10-fold higher than used in mammalian cells; Ingolia et al., 2011) did not result in a growth defect, suggesting that this drug does not effectively inhibit translation in yeast. Harringtonine treatment did inhibit the growth of a yeast strain that lacks ABC transporter efflux pumps, pointing to active drug efflux as the mechanism of harringtonine resistance in wild-type yeast (Figure S1A; Suzuki et al., 2011). However, this strain could not efficiently undergo meiosis, precluding its use for our experiments (data not shown).
Figure 1: Translation initiation site ribosome profiling in mitotic and meiotic yeast cells.
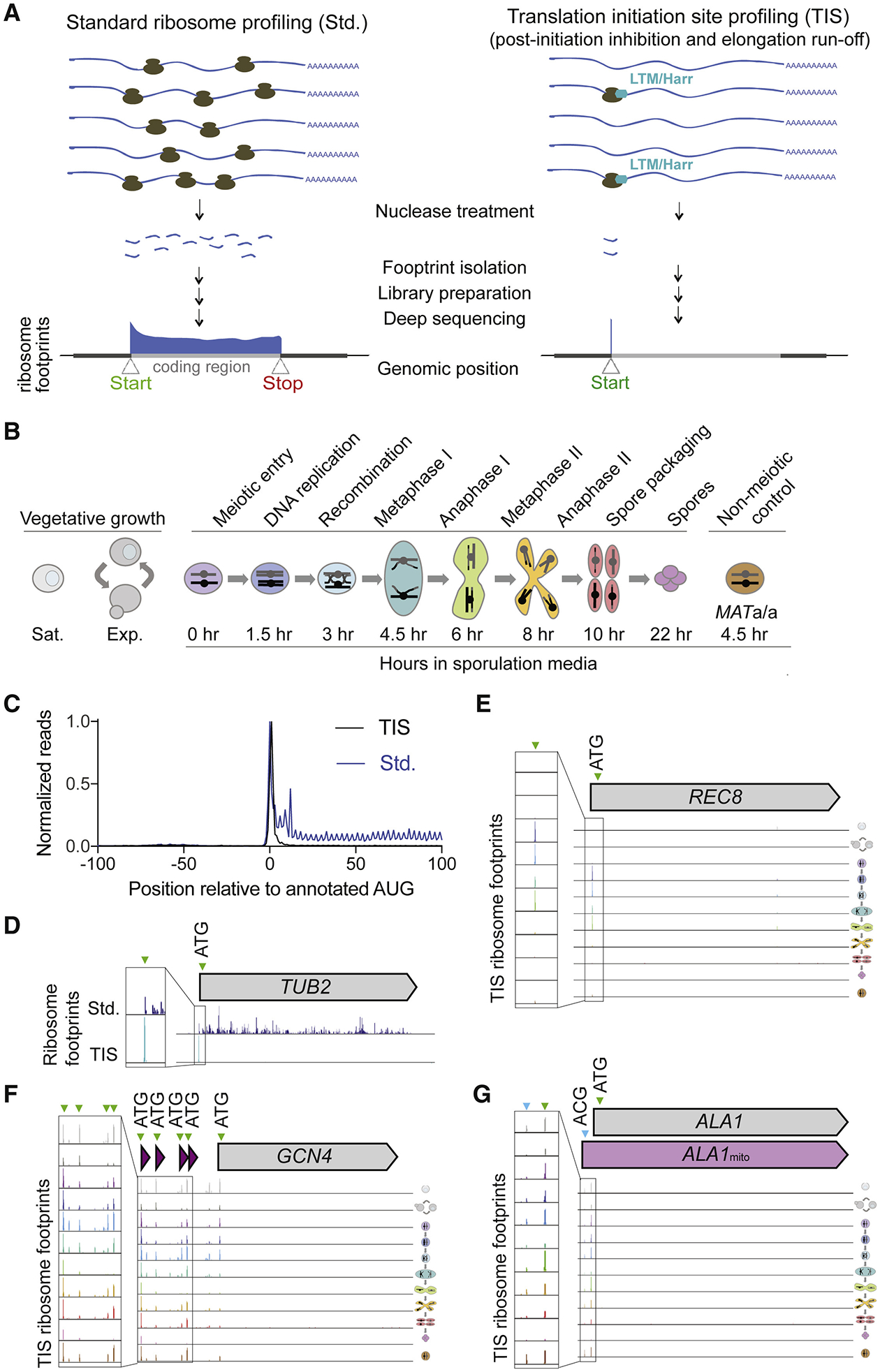
(A) Cartoon comparing standard (Std., left) and translation initiation site (TIS, right) ribosome profiling, with representative ribosome footprint profiles for a typical ORF.
(B) Schematic of yeast cell stages and samples collected for TIS-profiling, including vegetative saturated (sat.), vegetative exponential (exp.), 0 hr, 1.5 hr, 3 hr, 4.5 hr, 6 hr, 8 hr, 10 hr, and 22 hr after addition to sporulation media, and a MATa/a non-meiotic control taken at 4.5 hr in sporulation media.
(C) Metagene plot of normalized reads from standard ribosome profiling (blue) and TIS-profiling (black), 100 nucleotides upstream and downstream of annotated AUG start codons. Values are normalized to the peak at position zero.
(D) Comparison of standard and TIS-profiling for TUB2, a representative gene, from all timepoints combined. Green arrowheads indicate peaks at ATGs, and inset shows close-up view of region around predicted initiation site.
(E-G) TIS-profiling of REC8 (E), GCN4 (F) and ALA1 (G), showing ribosome footprints at the time points indicated in Figure 1B. Green arrowheads indicate peaks at ATGs, and blue arrowheads indicate non-ATG peaks.
Testing of previously used LTM treatment conditions resulted in ribosome profiling reads throughout ORFs in yeast, consistent with LTM inhibiting both post-initiation and elongating ribosomes at high concentrations (Figure S1B; Schneider-Poetsch et al., 2010). LTM concentrations 25-fold less than those used for TIS mapping in mammalian cells (Lee et al., 2012) still caused a growth defect in yeast (Figure S1C) and resulted in strong TIS enrichment of ribosome footprints (Figure S1D). This suggests that post-initiation ribosomes are more sensitive to LTM-based inhibition than elongating ribosomes. We selected an LTM concentration of 3 μM and a 20 minute incubation prior to harvesting to allow sufficient run-off time for elongating ribosomes. We performed translation initiation site profiling (TIS-profiling) for eight meiotic time points to assess translation initiation globally during meiosis (Figure 1B). For comparison, we also included samples from vegetative cells during either exponential growth or stationary phase, as well as diploid cells that cannot undergo meiosis grown in media matched to meiotic samples (MATa/a). Metagene analysis of the regions surrounding annotated start codons revealed a strong peak at the TIS and a low level of background reads in ORF bodies, suggesting that TISs were indeed being highly efficiently captured by our approach (Figure 1C). This is in contrast to the expected distribution of ribosome footprint reads across the entirety of the ORF seen for standard ribosome profiling, which is also seen for a representative gene, TUB2 (Figure 1C, 1D).
We confirmed that our data accurately reported the expected positions and condition-specificity of both canonical and non-canonical start codons through analysis of several well-studied genes. For example, at the locus of a meiotic gene, REC8, a single abundant peak was observed at the known TIS during time points when Rec8 is normally expressed (Figure 1E). TIS-profiling also revealed peaks at known non-canonical TISs, including the four AUG-initiated uORFs known to regulate GCN4 (Figure 1F). Finally, peaks at near-cognate codons were detected in our dataset, consistent with mammalian experiments using LTM or harringtonine (Ingolia et al., 2011; Lee et al., 2012). One of the few characterized examples of productive near-cognate translation initiation in yeast is for the tRNA synthetase gene ALA1, which encodes two functionally characterized protein isoforms (Tang et al., 2004). Translation of the canonical isoform initiates at an AUG, while translation of an N-terminally extended isoform initiates from an ACG in the 5’ leader. This upstream initiation event appends a mitochondrial targeting sequence to the canonical protein, which localizes this isoform to the mitochondria. We observed strong and specific peaks for both the upstream near-cognate start codon as well as the annotated AUG for ALA1 in our dataset (Figure 1G) and concluded that our TIS-profiling protocol could capture both known canonical and non-canonical TISs.
TIS-profiling reveals thousands of non-canonical ORFs
To systematically annotate translation products, including those that were challenging to assess by traditional ribosome profiling, like alternate protein isoforms, we used ORF-RATER, a linear regression algorithm (Fields et al., 2015). ORF-RATER integrates both standard and TIS-profiling data to evaluate read patterns over ORFs within annotated transcripts. It then assigns scores to detected peaks based on the similarity of their read patterns to annotated ORFs, with scores closest to 1 being the most similar. This method was particularly well suited to our goal of identifying uORFs and ORFs that overlap annotated ORFs, which were the most difficult to annotate from standard ribosome profiling data since they are often obscured by signal from elongating ribosomes.
ORF-RATER successfully called most previously annotated canonical coding regions using the TIS-profiling dataset and a timepoint-matched standard ribosome profiling dataset (Cheng et al., 2018). Of annotated ORFs in our yeast reference dataset, ORF-RATER identified 67% at a high score cutoff (>0.5; Figure 2A, Table S2). Of those that were not called by ORF-RATER, 45.8% are expressed at low abundance under the conditions tested (fewer than 5 mean reads per kilobase million, RPKM; Figure S2A, S2B). An interesting category of uncalled annotated ORFs includes cases of apparent misannotation, such as PEX32 and RSB1, for which the likely predominant initiation site based on TIS-profiling and ORF-RATER analysis is upstream or downstream of the annotated TIS (Figures 2B and 2C). In these cases, the previously annotated TIS does not show evidence of initiation in our dataset, indicating that the alternate TIS that is called is likely to be the correct one for these genes. This category represents approximately 39% of “uncalled” annotated ORFs, as these are instead erroneously called as extensions or truncations. This includes cases for which the previous annotation was based on the assumption that the predominant TIS is the one that produces the longest possible ORF at a given locus, and also includes cases in which the original reference genome annotation for the ORF was incorrect based on sequencing errors or sequence differences between yeast strains. An example of the latter is DEP1, which has a stop codon upstream of the annotated stop codon in our strain background (SK1; Figure S2C). Finally, we estimate that approximately 15% of uncalled canonical annotated ORFs (representing 5% of total annotated ORFs) are false negatives, like RIM11, for which ORF-RATER did not call an ORF despite an observable peak at the annotated start codon in the TIS-profiling data (Figure S2D).
Figure 2: ORF-RATER-based annotations of TIS-profiling data.
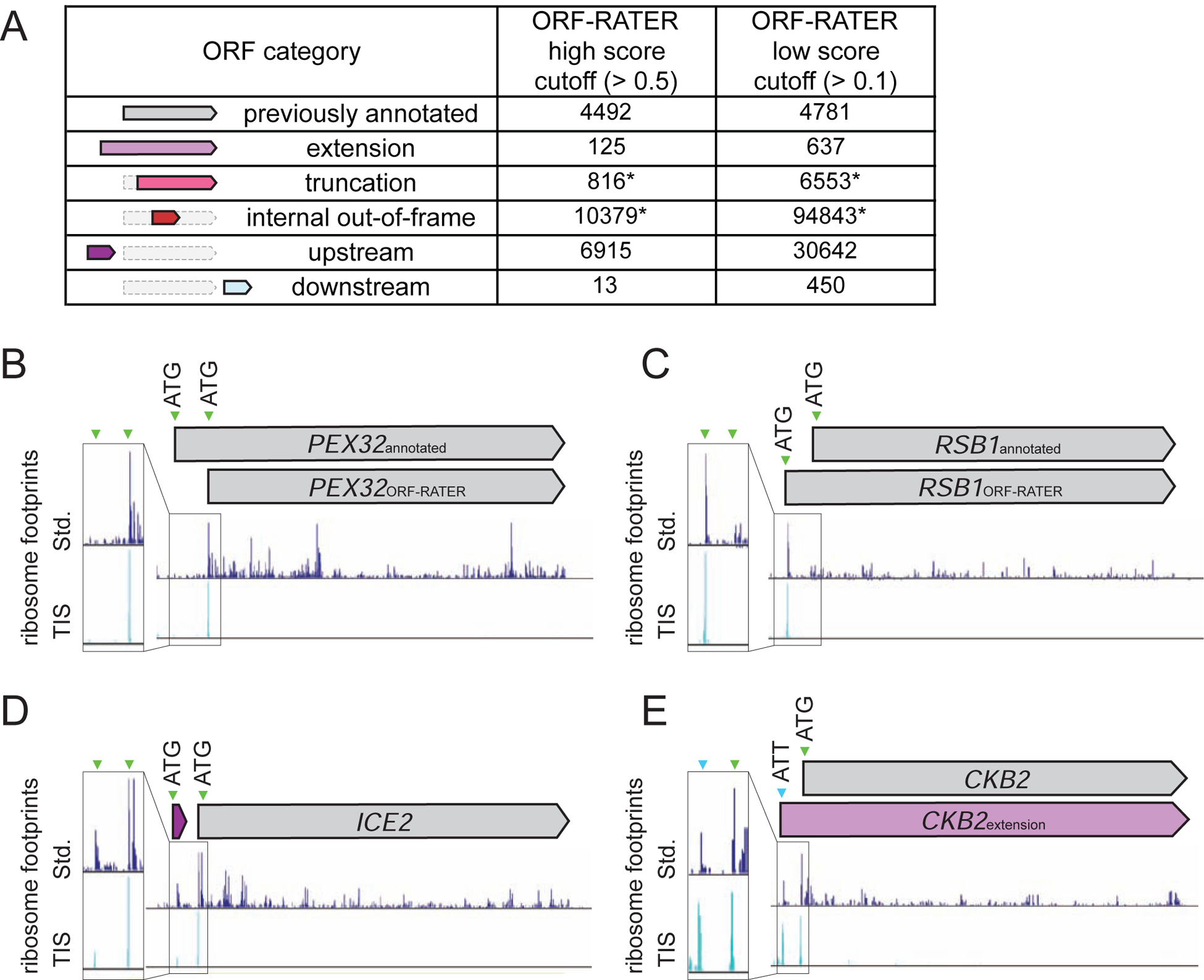
(A) Numbers of different types of ORFs called by ORF-RATER at two different score cutoffs - a high score cutoff (> 0.5) and a low score cutoff (> 0.1). Truncation and internal out-of-frame numbers are likely high overestimates due to high rates of false positives, indicated with a *.
(B-E) Comparison of standard and TIS-profiling for (B) PEX32, which has a likely incorrect TIS annotation. The likely correct (downstream) TIS was called by ORF-RATER, while the previously annotated site was not called. (C) RSB1, for which the likely correct TIS is upstream of the previously annotated site. (D) ICE2, which has a previously uncalled uORF identified by ORF-RATER. (E) CKB2, which has a previously uncalled 5’ extended ORF with a non-AUG TIS identified by ORF-RATER.
It is not surprising that ORF-RATER was generally successful at calling annotated canonical ORFs because the approach trains on this set. To assess its success in identifying unconventional translation products from our dataset, we examined ORF-RATER calls for the few previously well-characterized non-canonical ORFs, which includes 17 AUG-initiated uORFs, 6 near-cognate initiated extensions, and 6 AUG-initiated alternate isoforms (Table S3). Among this set, the high score cutoff (>0.5) was sufficiently sensitive to detect 71% (12/17) of the known AUG-initiated uORFs and 67% (4/6) of AUG-initiated alternate ORF isoforms but failed to detect 3 of the 6 (50%) known near-cognate initiated 5’ extended ORFs. We could detect all but one of these cases (83%) when using a lower ORF-RATER score cutoff (>0.1), which also slightly increased the detection of known AUG-initiated uORFs to 77% and AUG-initiated alternate ORFs to 83%. To increase the likelihood of detection of non-canonical ORFs, we used the lower score cutoff for further analyses, which resulted in the provisional annotation of 133,125 non-canonical ORFs in several classes (Figure 2A). This number was much higher than we expected to represent true translated regions, and so we investigated each class in more detail.
Case-by-case investigation of read patterns in the TIS-profiling and standard ribosome profiling data revealed substantial variability in apparent false positive calls between different ORF categories. A very high proportion of newly called internal ORFs (both truncations and out-of-frame; Figure 2A) are likely to be false positives, based on visual analysis of the LTM data (such as for SIN3 and CDC15; Figure S2E, S2F), and the fact that there were a median of 16 internal ORFs called per annotated gene (score > 0.1; Figure S2G). This high rate of apparent false positives is likely due to residual translation elongation inhibition at the concentration of LTM used in our method, resulting in background ribosome footprints within translated ORFs that erroneously result in internal TIS calls. While real internal initiation sites are expected to exist within these calls, the experimental and detection conditions here were not able to systematically separate true from false positives. In contrast to internally-initiated ORFs, manual visual analysis of the data for extensions and downstream ORFs called by ORF-RATER suggested that ORF-RATER calls of these classes of non-canonical ORFs are highly specific. We concluded that our analytical conditions are suitable to detect both canonical and non-canonical ORFs, with the exception of internal ORFs. We therefore excluded both out-of-frame internal ORFs and in-frame internally-initiated truncations from further analyses, and the ORF-RATER calls from these categories should be interpreted cautiously.
The remaining non-canonical ORFs that were confidently called at the low score cutoff included 637 N-terminal extensions (akin to ALA1; Figure 1G), 30,642 uORFs, and 450 downstream ORFs in which translation initiates within predicted 3’UTR regions (Figure 2A). Traditional ribosome profiling had previously predicted translation from some of these unannotated ORFs, but as expected, some were sensitively detected only with analysis incorporating the TIS-profiling data. Newly identified non-canonical ORFs included uORFs (for example, ICE2; Figure 2D), N-terminal extensions (for example, CKB2; Figure 2E), and downstream ORFs. We further refined the N-terminal extension class based on length. A cutoff of greater than 10 amino acids was chosen based on the minimum length predicted for function, such as for a targeting signal or binding domains (Figure S3A, Almagro Armenteros et al., 2019; Fukasawa et al., 2015). Excluding AUG-initiated extensions, many of which are likely to represent misannotations (as for RSB1; Figure 2C), left 231 extensions, representing 160 unique genes, as some genes contained multiple predicted extensions (Figure S3B, Table S4; this number was ultimately adjusted to 149 based on misannotations discovered through conservation analysis).
Translation of uORFs and 5’ extended ORFs is enriched in meiosis
Increased ribosome footprints within 5’ leader regions were previously observed in meiosis in yeast (Brar et al., 2012). To determine whether TIS-profiling detected increased meiotic translation initiation within 5’ leaders, we compared metagene profiles surrounding annotated start codons for vegetative exponentially growing cells to a representative mid-meiotic time point (4.5 h). This indeed revealed a meiosis-specific increase in translation initiation 5’ of annotated start codons (Figure 3A) but no difference between the vegetative and meiotic LTM-based ribosome footprints in regions surrounding annotated stop codons (Figure 3B). The increased read density in 5’ leaders during meiosis could reflect an increase in translation of either uORFs or 5’ extended ORFs. To investigate this, we compared the types of ORFs called in the vegetative exponential time point to the mid-meiotic time point. The calls for both uORFs and 5’ extensions are increased in meiosis, while the number of annotated and downstream ORFs are similar between the two conditions (Figure 3C). Although annotated ORFs all begin with an AUG start codon, extensions and uORFs initiate at near-cognate start codons in 93.6% and 73.3% of cases, respectively (Figure 3D). The translation of both uORFs and N-terminally extended proteins results from increased translation initiation within 5’ leaders, but the consequences of these two classes of non-canonical translation are fundamentally different. Translation initiation at the start codon of a uORF may regulate the translation of the downstream canonical ORF or produce a small peptide, whereas translation initiation at the start codon for an N-terminal extension generates a modified protein product with potentially distinct function (Hood et al., 2009; Morris and Geballe, 2000). For example, the extended isoform of Ala1 is targeted to the mitochondria rather than the cytosol, providing alanyl-charged tRNAs for mitochondrial translation (Tang et al., 2004). Our TIS-profiling data identified translation of the known extensions at the ALA1, YMR31/KGD4, HYR1/GPX3, TRZ1 and HFA1 loci, as well as 155 other genes, which we proceeded to evaluate in more detail (Table S4; Heublein et al., 2019; Kritsiligkou et al., 2017; Monteuuis et al., 2019; Suomi et al., 2014; Tang et al., 2004).
Figure 3: Specificity of uORF and N-terminal extension translation is influenced by condition and start codon identity.
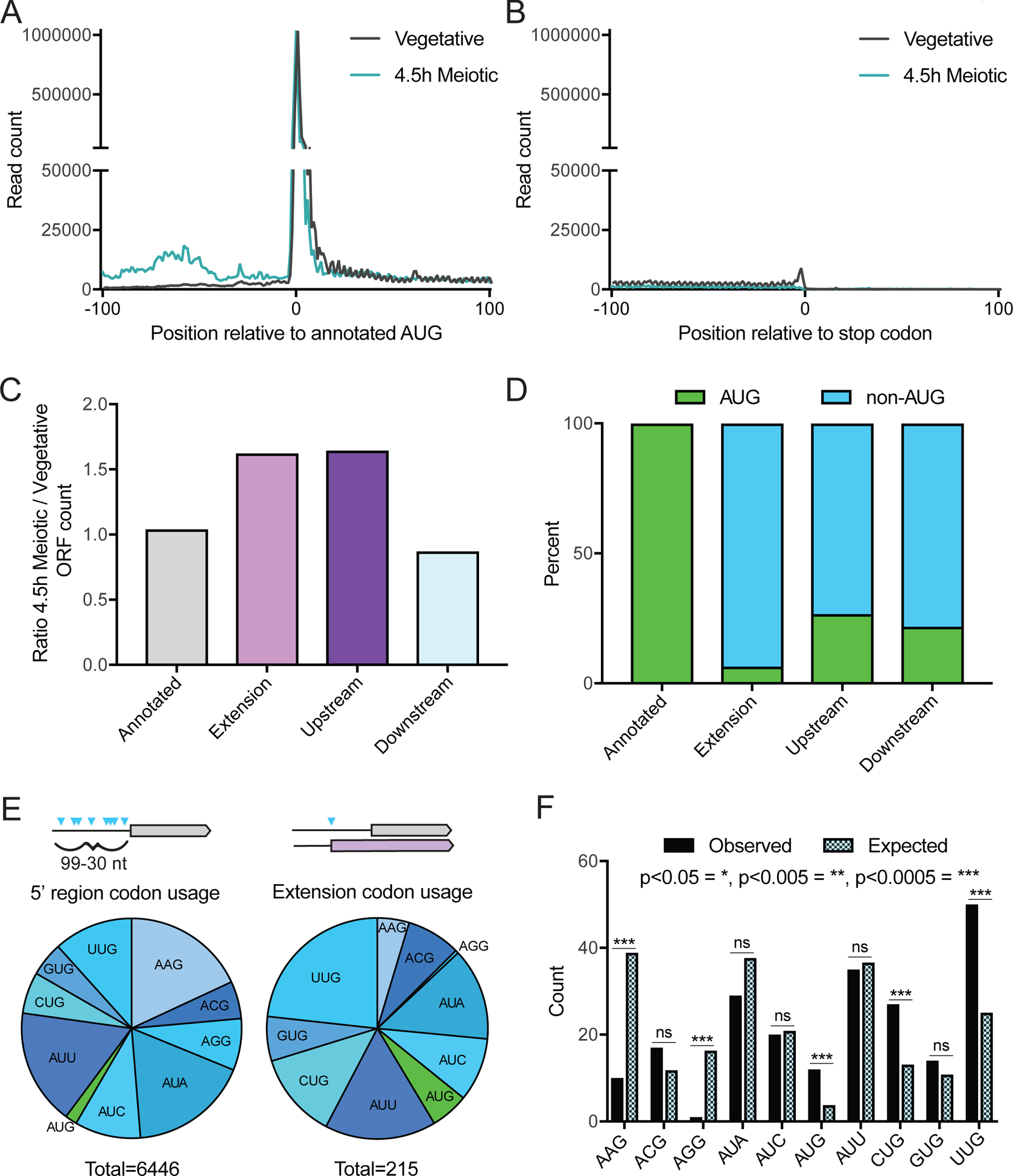
(A) Metagene plot of read counts from vegetative exponential and 4.5 hr time points, 100 nucleotides upstream and downstream of annotated AUG start codons. Reads are normalized to aligned reads for that timepoint. Increased read density is observed for the meiotic time point upstream of annotated start codons, but not after.
(B) Metagene plot of read counts from vegetative exponential and 4.5 h time points, 100 nucleotides upstream and downstream of annotated stop codons. Reads are normalized to aligned reads for that timepoint.
(C) Relative numbers of ORFs from different ORF categories, comparing the 4.5 h meiotic time point to vegetative exponential. More 5’ extensions and upstream ORFs are called in the meiotic time point, while annotated and downstream ORFs are similar between the two conditions.
(D) Percent of AUG versus non-AUG TISs for different ORF types. Annotated ORFs all have AUG start codons, while 5’ extensions, upstream, and downstream ORFs have primarily non-AUG TISs.
(E) Distribution of AUG and non-AUG start codon usage 99–30 nucleotides (nt) upstream of annotated AUG start codons for all possible TISs (left) and called 5’ extensions (right). Of the 6,446 sites possible in 5’ regions, 215 are observed to initiate translation of 5’ extensions called by ORF-RATER.
(F) Near-cognate codon usage for called extensions (observed) compared to relative abundance of all possible near-cognate codons within upstream regions (expected). Expected distribution is derived from counts of all possible TISs in the 99–30 nt upstream of annotated AUG start codons. P-values calculated by Fisher’s exact test, with p<0.05 = *, p<0.005 = **, p<0.0005 = ***, and ns = not significant.
Non-AUG-initiated isoform translation is specific and does not preclude canonical isoform translation
The low number of AUG-initiated N-terminal extensions identified here (Figure 3D) likely reflects the fact that traditional genome annotations selected the longest AUG-initiated ORF at a locus as the one most likely to be translated. We wondered whether these extended ORFs generally represented an additional translated ORF or whether these were the sole translated ORF at these loci. Consistent with the former, 85% (136/160) of genes encoding extended ORFs had a corresponding annotated ORF that was called by ORF-RATER. Of the 24 that were not called, 17 show evidence of translation initiation at the annotated AUG-initiation site in our TIS-profiling data but were not called by ORF-RATER (Table S4). Four of the remaining seven are misannotations, similar to RIM11 (Figure S2D), and one (YPL034W) includes a likely frameshifting event (see note in Table S4). This leaves only 2 cases in which the near-cognate-initiated extension is the sole or predominant translation product: HFA1, which is indeed the only characterized gene in yeast in which a non-AUG-initiated product is thought to be the primary translation product (Suomi et al., 2014) and YNL187W, a poorly characterized gene. We concluded from these analyses that loci that encode near-cognate-initiated extended protein isoforms generally express them in concert with the canonical AUG-initiated isoform.
Given the prevalence of translation initiation within 5’ leaders in meiosis, most of which is at near-cognate start codons, we wondered if generally less stringent TIS selection in meiotic conditions might produce 5’ extended ORFs non-specifically. To estimate the number of theoretically possible N-terminal extensions based on non-specific “sloppy” initiation, we calculated the number of in-frame cognate and near-cognate start codons that fall between 99–30 nucleotides upstream of annotated start codons and do not have an in-frame stop codon before the canonical start codon. We chose this region to account for the average length of yeast 5’ UTRs and to include only the potential ORF extensions that would be expected to be long enough to confer new biological function (>10 additional amino acids; David et al., 2006; Nagalakshmi et al., 2008). We found 6446 possible sites, only 3.3% of which have evidence of being used to initiate translation in our TIS-profiling dataset. This indicates highly stringent selection of certain near-cognate TISs to produce N-terminal extensions.
Some of this specificity resulted from preferential initiation at certain near-cognate codons (Figure 3E, 3F). The codons that we found to be enriched for initiation of 5’ extended ORFs, including CUG and UUG, have been previously shown through in vitro assays to be the most efficiently initiated near-cognate codons (Kolitz et al., 2009). The preference for specific near-cognate codons alone could not explain the small percentage of potential start codons in 5’ leaders used to translate extended ORFs, so we also searched for evidence that start codon context influenced the set of used versus theoretically possible TISs. We found only weak enrichment for the optimal (Kozak-like) motif found around annotated AUG-initiated ORFs (Figure S4A; Kozak, 2002; 1999; 1984; 1978), which is consistent with previous reports of differences between optimal contexts around near-cognate and AUG start codons (C.-P. Chang et al., 2010). We were unable to identify any simple context cues that were enriched specifically in the translated near-cognate TISs (data not shown), suggesting that other, yet-to-be-determined features define the specific start codons used for translation initiation of extended isoforms.
Predicted N-terminal extensions can be detected by mass spectrometry
To determine whether the identified N-terminally extended protein isoforms are abundant in meiosis, we re-analyzed a previously generated quantitative mass spectrometry dataset, searching for peptides that uniquely arise from the N-terminally extended regions (Cheng et al., 2018). Our search set contained all extensions with an ORF-RATER score of 0.1 or higher, an extension length greater than ten amino acids, and initiation at a near-cognate start codon (Figure S3A). Of the 160 unique genes searched in this way, seven showed at least one peptide originating from the extension. Three of the seven had ORF-RATER scores well below the high score cutoff of 0.5 (Figure 4A), suggesting that our choice of the lower cutoff to define extended isoforms is appropriate. For the majority (69%), the annotated isoform was quantifiable, but we detected extension-derived peptides for only 6.25% of those searched (average extension length of 25 amino acids). By comparison, a parallel search for peptides within the first 25 amino acids of annotated proteins identified 43.2% of cases. The high degree of discrepancy in detection between these two classes, and the fact that we only identified two of the six established extensions (HYR1 and YMR31), suggests that near-cognate-initiated extended proteins, as a class, may be lowly expressed relative to canonical proteins.
Figure 4: The abundance of near-cognate-initiated isoforms is not reflective of TIS-profiling peak height.
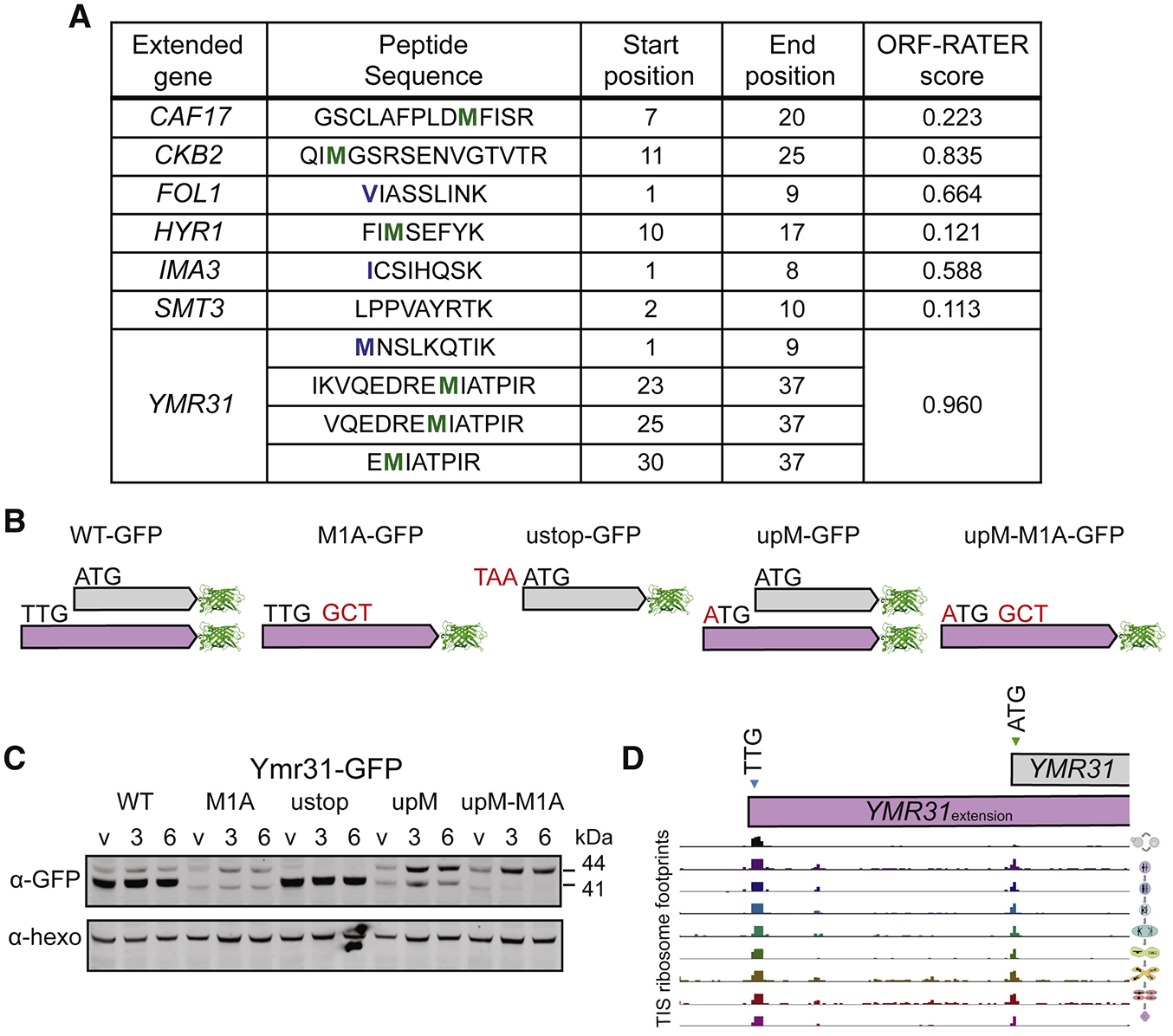
(A) Extensions with peptides identified that match to the extension-specific region of the protein from a meiotic mass spectrometry dataset. The annotated methionine is highlighted in green and the extension start codon is highlighted in blue where relevant.
(B) Cartoon of tagging and mutagenesis strategy for validation of extensions. All constructs include a C-terminal GFP tag. Mutations include: M1A to mutate the annotated methionine to alanine, ustop to mutate the codon upstream of the annotated start codon to a stop codon, and upM to mutate the extension’s upstream non-AUG start codon to a methionine.
(C) Western blot of Ymr31-GFP showing the WT construct with two bands corresponding to translation of the extension (44 kDa) and annotated ORF (41 kDa). M1A and ustop constructs show translation of the extension and annotated ORF individually, respectively. upM and upM-M1A constructs show an increase in the extension isoform. Samples were taken in vegetative exponentially growing cells (v), and at 3h and 6h after addition to sporulation media. Anti-hexokinase (α-hexo) is a loading control. The band around 40 kDa visible in the M1A construct is of unknown identity, and may represent translation from a downstream AUG.
(D) TIS-profiling of YMR31, showing ribosome footprints at the time points indicated in Figure 1B, with the extension (TTG) and annotated (ATG) start-codon-encoding sites indicated.
Extended protein isoform levels are lower than expected based on TIS-profiling peak height
To probe the relative levels of near-cognate initiated and canonical protein isoforms, we characterized in more detail the expression of Ymr31, a subunit of the mitochondrial alpha-ketoglutarate dehydrogenase recently found to be produced from both a canonical AUG and upstream UUG start codon (Heublein et al., 2019). We chose Ymr31 for this analysis for three reasons. First, mass spectrometry had detected multiple peptides from this extension, indicating that the extended protein isoform was likely to be abundant in our conditions. Second, it was the highest scoring extension called by ORF-RATER. Lastly, the discrepancy in size between the GFP-tagged small canonical protein (41 kDa) and the relatively large extended protein (44 kDa) made the two isoforms readily distinguishable by western blot. This last property, which was rare among genes with extended isoforms, was especially valuable in enabling in vivo analyses of isoform regulation.
To evaluate relative expression levels of the two YMR31-encoded isoforms, a C-terminally GFP tagged version of this protein was expressed with either the wild-type (WT) start codon, the annotated ATG start-codon-encoding site mutated to an alanine-encoding codon (M1A), or a stop codon inserted directly upstream of this ATG (ustop). In M1A cells, the extension is expected to be the only isoform translated, and cells carrying the ustop construct are expected to only produce the canonical AUG-initiated isoform (Figure 4B). Samples were collected in vegetative cells, and at 3h and 6h after inducing meiosis. In YMR31-M1A and YMR31-ustop cells, only the extended or canonical forms were observed, respectively, confirming our predicted YMR31 ORF annotations (Figure 4C, S5A). The extended form of Ymr31 was ten times lower in abundance than the canonical form in WT cells by western blot analysis (Figure S5A), which is in marked contrast with the TIS-profiling data showing over eight times higher ribosome footprint read density at the near-cognate initiation site than at the canonical start codon (Figure 4D, S6A).
Mutation of the near-cognate-encoding initiation codon to ATG resulted in higher levels of the N-terminally extended Ymr31 isoform, either with (upM-M1A) or without (upM) mutation of the canonical start codon (Figure 4C). This suggested that the native near-cognate TIS is used inefficiently for translation initiation relative to AUG, consistent with in vitro and mutagenesis experiments comparing AUG and near-cognate initiation (Chen et al., 2008; Kolitz et al., 2009). This result also suggested that the peak height observed by TIS-profiling at near-cognate and AUG codons may not be comparable. This may be due to differences in the ability of LTM to inhibit the two different types of post-initiation ribosome complexes or in their timespan of initiation. We also considered the possibility that near-cognate-initiated proteins might be subject to proteasome-mediated degradation, but at least for Ymr31, we did not observe an increase in the alternate isoform in cells in which proteasome activity was inhibited by MG132 (Figure S6B, S6C).
We further investigated whether the discrepancy between protein levels and TIS peak height indicated that TIS-profiling peaks were not quantitatively predictive of translation levels. This was not generally true, at least for AUG-initiated ORFs, as the height of TIS peaks appeared to reflect known regulation patterns during meiosis for characterized genes. Across annotated ORFs, there was a positive association between the read count at the TIS for TIS-profiling and the density of ribosome footprints over ORFs for standard ribosome profiling (Figure S6D, S6E). This was seen by comparisons of individual time points (Figure S6E), as well as by calculating correlation scores for each gene across all time points (Figure S6D). Individual examples, such as Rec8 (Figure 1E), showed a strong correlation between TIS-profiling peaks and standard profiling reads (Pearson correlation coefficient = 0.833), and correlations were significantly enriched for positive values compared to a random distribution of genes (Figure S6D). This is consistent with a study using a similar approach in mammalian cells that suggested ribosome footprint peaks at AUG start codons following LTM treatment quantitatively reflect translation initiation levels (Lee et al., 2012). We concluded that our TIS-profiling protocol reports at least weakly quantitative values for translation initiation levels at AUG start codons but that TIS-profiling peak heights at near-cognate start codons are much higher than expected based on our poor detection of near-cognate-initiated peptides by mass spectrometry, as well as the inferred translation levels from western blotting analysis of the two Ymr31 isoforms.
5’ extensions are poorly conserved as a class
To probe the likelihood that the N-terminally extended protein isoforms have conserved functionality within Saccharomyces, we analyzed the evolutionary protein coding potential of the extensions using PhyloCSF, which reports a score indicating whether the local alignment of a region is more likely under coding or non-coding models of evolution (Lin et al., 2011). Positive scores are more likely in conserved coding regions (Figure 5A). We noted that among the highest scoring cases were 11 in which the putative extension was a misannotation resulting from sequencing errors or strain-specific stop codons or indels, leaving 149 genes with apparent true near-cognate initiated extensions (Table S4). Alignments of individual true extensions illustrate the degree of conservation, which for Ymr31 is high, reflected in its high PhyloCSF score (Figure 5B). We further evaluated two true extensions with high PhyloCSF scores, for the genes HYR1 and YML020W (Figures 5C and 5D). In these cases, as well as for nearly every other extension-encoding gene we examined, the size difference between the extended and canonical isoform was too small to detect by western blot for the WT construct, making the M1A construct critical in confirming the expression of the extended isoform. For HYR1, using the tagging strategy previously described, we observed a lowly expressed band corresponding to the extended isoform in extract from cells carrying the HYR1-M1A mutant construct (Figure 5E, S5B). Similarly, we detect an N-terminally extended isoform of Yml020w in cells carrying the YML020W-M1A construct (Figure 5F, S5C).
Figure 5: Most 5’ ORF extensions are poorly conserved.
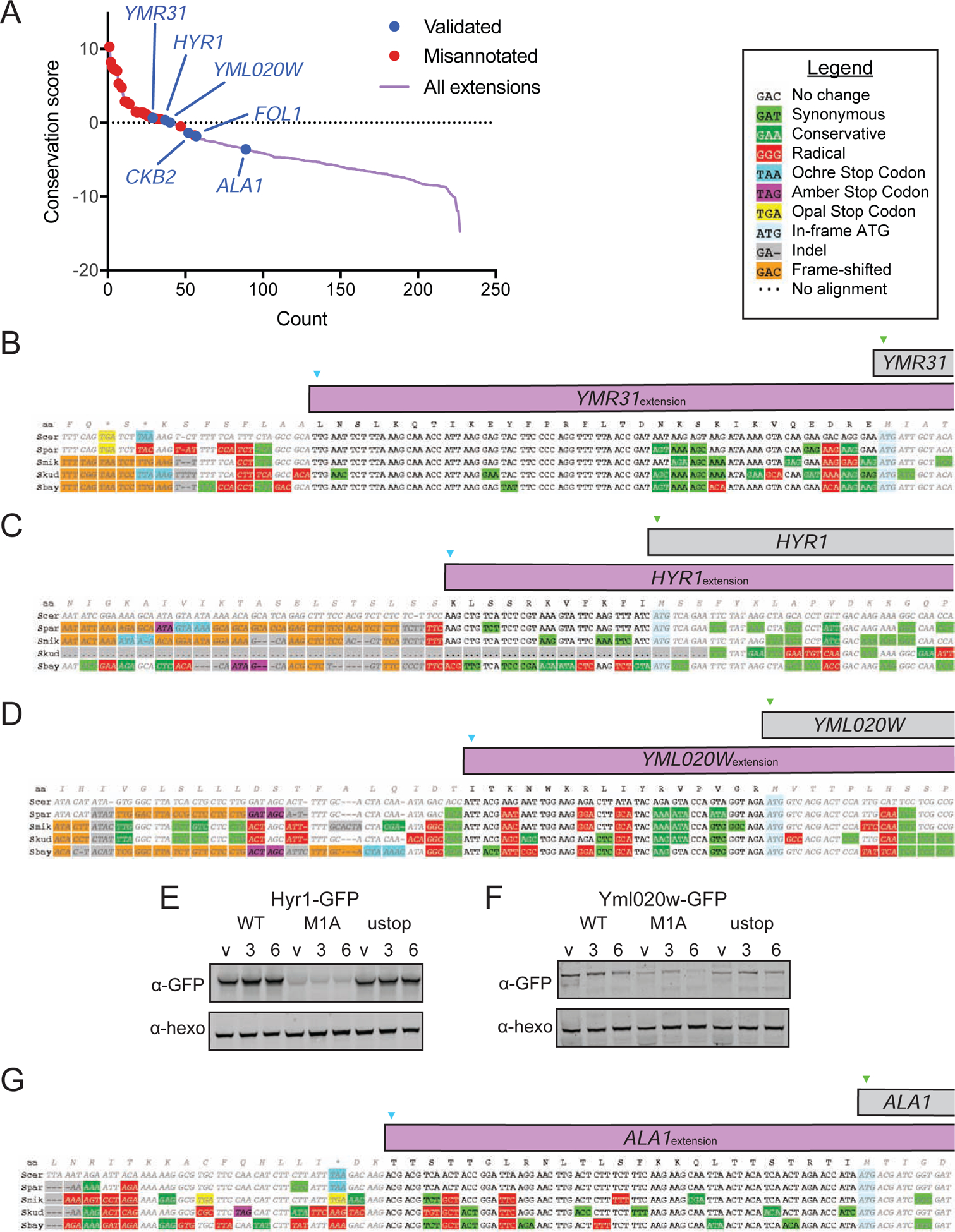
(A) Plot of PhyloCSF conservation scores for 5’ extended ORFs. Misannotated extensions are shown with red dots, and validated extensions are shown with blue dots, including three previously validated extensions (YMR31, HYR1 and ALA1). The additional “validated” extensions (YML020W, CKB2 and FOL1) were validated in this study.
(B-D) Alignments showing level of conservation for YMR31 (B), HYR1 (C) and YML020W (D), all of which have positive conservation scores.
(E) Western blot of Hyr1-GFP including WT, M1A and ustop constructs. Samples were taken in vegetative exponentially growing cells (v), and at 3h and 6h after addition to sporulation media.
(F) Western blot of Yml020w-GFP including WT, M1A and ustop constructs. Samples were taken in vegetative exponentially growing cells (v), and at 3h and 6h after addition to sporulation media.
(G) Alignment showing level of conservation for ALA1, which has a negative conservation score.
The majority of extensions analyzed had scores below zero, suggesting a lack of conserved functionality (Figure 5A). In some cases, however, the extension might have conserved function but nonetheless have a negative PhyloCSF score because the amino acid sequence is under only weak purifying selection or is subject to an atypical constraint. An example of the latter is ALA1, where the ACG start codon and the reading frame are conserved in five species but the extension itself had a negative PhyloCSF score (−3.587; Figure 5A, 5G). A possible explanation is that the mitochondrial targeting function of the extension is present in the other species but imposes a constraint that PhyloCSF is not able to detect.
Transcripts with canonical start codon mutations are NMD targets
The length of the extended Ala1 protein relative to the canonical isoform was too small to allow both versions to be detected by western blotting and, because the start codon at the endogenous locus could not be manipulated to isolate production of the extended isoform without affecting cell fitness, GFP reporters (ALA1GFP) were constructed to further investigate translation from this gene (Figure 6A). When the canonical start codon was present in the reporter (ALA1GFP-WT), both Ala1 reporter isoforms were observed (Figure 6B, 6C, S5D). The canonical Ala1 reporter isoform could be detected alone in extract from cells carrying the ALA1GFP-ustop construct (Figure 6B, 6C). Surprisingly, in cells carrying the ALA1GFP-M1A construct, however, we could not detect production of either protein isoform (Figure 6B, 6C). The dramatic difference in production of the extended reporter with and without the canonical start codon mutation cannot be explained by inefficient near-cognate usage alone. The difference we observed exceeded even the ~10–100 fold decrease we would expect based on inefficient near-cognate usage (Chen et al., 2008; Clements et al., 1988; Kolitz et al., 2009). We further found that the mRNA levels of GFP from the ALA1GFP-M1A construct were dramatically decreased relative to the ALA1GFP-WT construct (Figure 6D). This led us to explore the possibility that the nonsense-mediated decay (NMD) pathway degrades transcripts from mutated constructs lacking the canonical in-frame start codon, likely due to efficient translation initiation at a downstream out-of-frame AUG that results in early translation termination (Figure S7A). Consistent with this hypothesis, we observed that both mRNA and protein levels of the ALA1GFP-M1A reporter construct increased in an NMD-deficient mutant background (upf1Δ), although not to the level of the extended isoform in the ALA1GFP-WT reporter construct (Figure 6B–D).
Figure 6: Extended ORF-encoding transcripts lacking canonical AUG start codons are degraded by NMD.
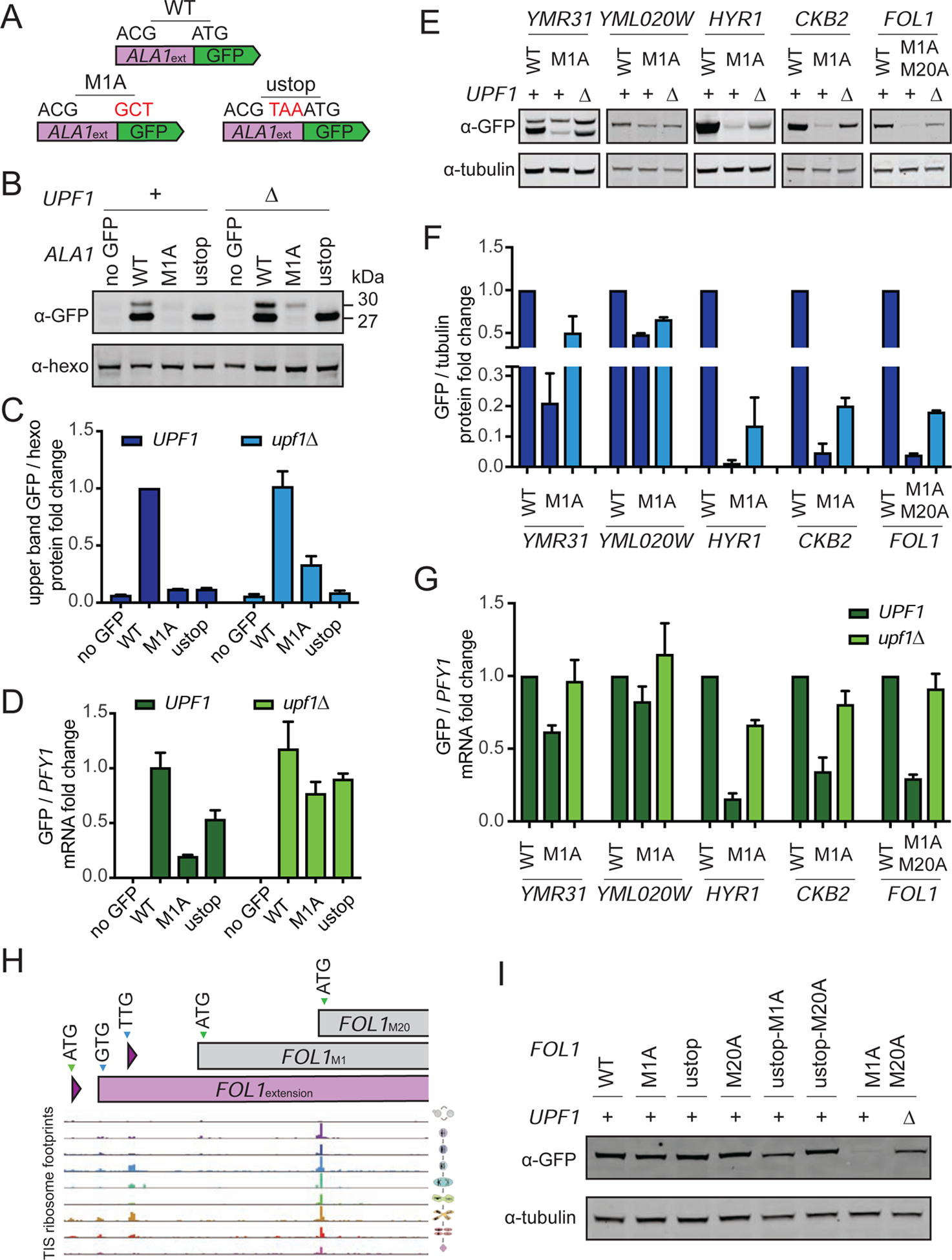
(A) Schematic for ALA1 tagging strategy, using a reporter including the region upstream of the ATG, and either including (WT) or not including (M1A) the in-frame ATG in front of the GFP, and a mutant with a stop codon upstream of the in-frame ATG (ustop).
(B) Western blot for Ala1GFP reporters in UPF1 and upf1Δ vegetative cells. The band corresponding to the extension (30 kDa) can be seen in the WT construct, but is not seen in the M1A construct in UPF1 cells. In a upf1Δ background, the M1A construct now shows the extended proten.
(C) Western blot quantification of Ala1-GFP upper band intensity from Figure 6B normalized to hexokinase, for 3 replicates.
(D) qPCR fold change of ALA1-GFP transcript relative to PFY1, for 3 replicates. The level of the M1A mRNA in UPF1 cells is low due to NMD acting on this transcript, and this effect is lessened in the upf1Δ background.
(E) Western blot analysis of Ymr31-GFP, Hyr1-GFP, Fol1-GFP, Ckb2-GFP and Yml020w-GFP for the WT and M1A constructs in UPF1 cells and the M1A construct in upf1Δ cells at 4.5 hours in meiosis.
(F) Western blot quantification of GFP tagged proteins from Figure 6E normalized to tubulin, for 3 replicates.
(G) qPCR fold change of GFP transcripts relative to PFY1 for 3 replicates from strains from Figure 6E.
(H) TIS-profiling of FOL1, showing ribosome footprints at the time points indicated in Figure 1B, with the positions of the extension (GTG), M1 (ATG) and M20 (ATG) start-codon-encoding sites indicated.
(I) Western blot analysis of Fol1-GFP for constructs including mutations at the annotated methionine (M1) as well as a methionine at position 20 (M20), indicating that translation can begin at three in-frame start codons.
In addition to the ALA1 reporters, several other M1A constructs showed little to no tagged protein in otherwise wild-type cells. This was consistent with our findings for the extended isoform of Hyr1, which was detected in our mass spec dataset (Figure 4A) but was detected at extremely low levels in cells carrying the HYR1-M1A construct (Figure 5E; Kritsiligkou et al., 2017). Analysis of the HYR1-M1A construct in upf1Δ cells revealed increased levels of the N-terminally extended protein and HYR1 mRNA (Figures 6E–6G, and S5E), consistent with NMD targeting of the mutant transcript. Analyses in the upf1Δ background allowed validation of additional N-terminally extended isoforms predicted by TIS-profiling-based annotation. These include CKB2, encoding the casein kinase beta subunit, and FOL1, which encodes a folic acid synthesis pathway enzyme. For these genes, like ALA1 and HYR1, the mutant construct lacking the AUG start codon(s) (M1A for CKB2; M1A M20A for FOL1, see below) was not detected with UPF1 present, but was in upf1Δ cells (Figures 6E–6G).
For the two examples that were robustly detected in a WT background, Ymr31 and Yml020w, little increase in protein levels from M1A constructs in upf1Δ cells was seen for the extended versions (Figure 4C, 5F). Consistently, YMR31-M1A and YML020W-M1A mRNA levels were not dramatically decreased in WT cells relative to unmutated constructs (Figure 6G). The difference between cases like CKB2, FOL1, ALA1 and HYR1, in which mutation of the canonical start codon leads to high mRNA degradation by NMD, and YMR31 and YML020W, in which it does not, is intriguing, as all loci produce the extended proteins at lower levels than the canonical protein, and all M1A constructs are expected to result in translation of a short out-of-frame ORF that should trigger NMD. Among this group, there is no correlation between the distance from the new presumptive out-of-frame stop codon to the end of the transcript and the strength of NMD, as measured by the percent abundance of M1A relative to WT mRNA (Figure S7A–C), although this distance is thought to be a key factor in specifying yeast NMD substrates (Reviewed in Hug et al., 2016). We did, however, observe a moderately positive association between the distance of the transcription start site to the location of the first downstream AUG (which is out-of-frame) in the M1A constructs and the degree of NMD (Figure S7B).
The FOL1 locus encodes three protein isoforms
Among the 149 genes identified as having alternate N-terminally extended isoforms by our TIS-profiling analysis, several cases appeared to have more than two alternate TISs. At the FOL1 locus, for example, our data reveals translation initiation at two uORF start codons, an upstream in-frame GUG start codon (producing an N-terminally extended isoform), the annotated AUG start codon and an AUG 19 codons downstream of the annotated AUG (Figure 6H). The relative usage of these start codons, as gauged by TIS-profiling peak height, differed among the conditions that we assayed. The three GFP tagged Fol1 isoforms predicted based on these data could not be resolved by western blotting, but high Fol1 protein levels were observed in cells carrying either a ustop-M1A or ustop-M20A construct, confirming protein production from the downstream AUG (M20) alone and the canonical AUG (M1) alone, respectively (Figure 6I). FOL1-M1A-M20A cells showed a drastic decrease in FOL1 mRNA and protein levels that were partially rescued in upf1Δ cells, confirming translation from the upstream GUG identified by TIS-profiling (Figure 6H, 6I, S5F). Such coding complexity is surprising to find in a eukaryote as simple as budding yeast and would not have been readily identifiable without TIS-profiling data.
eIF5A levels alter non-AUG TIS usage in yeast meiosis
The preferential translation of non-AUG-initiated ORFs in meiotic cells (Figure 3C), and the increase in TIS-profiling reads in 5’ leader regions in meiotic time points relative to vegetative cells suggests condition-specific modulation of translation initiation (Figure 7A). To identify candidates for this regulation, we performed quantitative mass spectrometry of 40S and 60S ribosomal subunits isolated by sucrose gradient centrifugation of cell extract from meiotic and vegetative cells. We found that eIF5A (encoded by HYP2 in yeast) was strongly and reproducibly disenriched in meiotic relative to vegetative samples, indicating decreased ribosome association of this factor in meiotic cells (Figure 7B). Many of the initiation factors found to associate with the 40S and 60S subunits have lower overall levels in meiotic cells, but the disenrichment of ribosome association seen for eIF5A is greater than could be explained by its overall decrease in abundance relative to vegetative cells (Figure S8). eIF5A has recently been shown to influence translation elongation and termination (Gregio et al., 2009; Henderson and Hershey, 2011; Saini et al., 2009; Schuller et al., 2017), but was initially identified for activity in promoting a late stage of translation initiation in vitro (Benne and Hershey, 1978; Kemper et al., 1976; Lopo et al., 1986; Schreier et al., 1977). A CRISPRi screen in human cell lines identified eIF5A as a factor that enhanced translation of the CUG-initiated, N-terminally extended isoform of MYC when transcriptionally repressed (Manjunath et al., 2019). In this context, low eIF5A levels are thought to impair translation elongation, leading to ribosome queuing, which contributes to increased initiation at upstream near-cognate sites (Ivanov et al., 2018; Manjunath et al., 2019).
Figure 7: eIF5A levels regulate pervasive non-AUG-initiated translation.
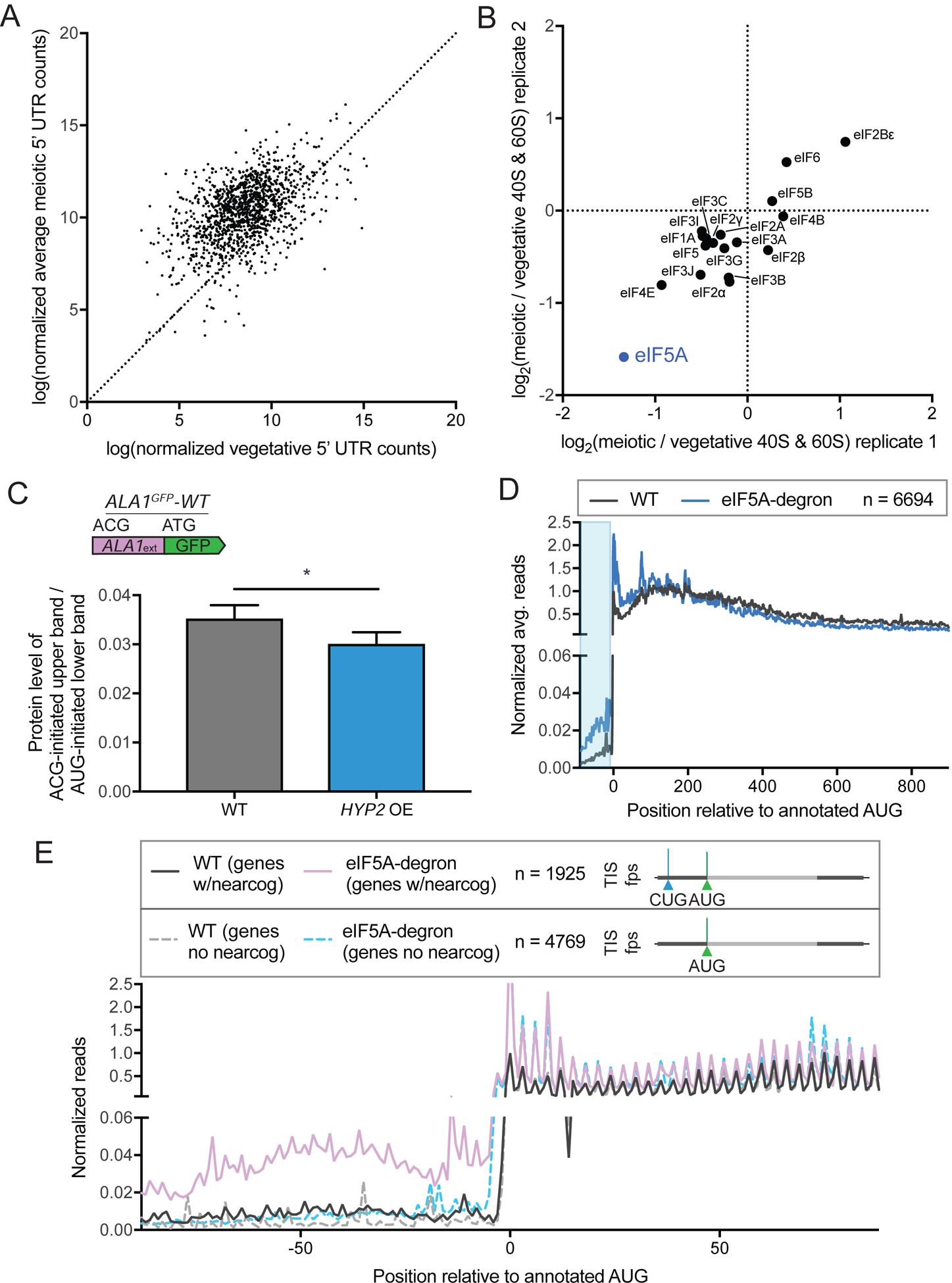
(A) Comparison of vegetative and average meiotic 5’ read density measurements.
(B) Enrichment of translation factors, comparing meiotic and vegetative samples for two replicates, determined by quantitative mass spectrometry of 40S and 60S ribosomal subunits isolated by sucrose gradient centrifugation of cell extract from meiotic and vegetative cells.
(C) Western blot quantification of ALA1GFP-WT reporter in meiosis with copper induction in strains containing or lacking a copper-inducible overexpression (OE) HYP2 allele. Non-AUG-initiated GFP upper band protein is normalized to AUG-initiated GFP lower band protein, which runs as a doublet. Both bands were used for quantification. The decrease seen with HYP2 OE is significant (p < 0.0146, 4 replicates).
(D) Metagene plot of normalized average reads from WT (black) and eIF5A-degron (blue) samples, 100 nt upstream and 900 nt downstream of annotated AUG start codons for all genes (n = 6694). Reads are normalized to WT at position zero and averaged across three nucleotides. Ribosome profiling data was re-analyzed from a previous study (Schuller et al., 2017). The area boxed in blue highlights the increased reads seen for the eIF5A-degron relative to WT in 5’ leader regions.
(E) Metagene plot around annotated start codons comparing genes with 5’ near-cognate-initiated ORFs annotated by ORF-RATER (n=1925, WT (genes w/nearcog): solid black and eIF5A-degron (genes w/nearcog): solid purple) and genes that do not contain 5’ near-cognate-initiated ORFs (n=4769, WT (genes no nearcog): dotted gray and eIF5A-degron (genes no nearcog): dotted light blue). Increased reads in the 5’ region are seen only in the eIF5A-degron samples for genes containing ORFs translated from near-cognate start codons in 5’ regions, based on TIS-profiling.
To test whether increased expression of eIF5A might alter the high near-cognate TIS selection that we observe in meiosis, we placed HYP2 under a copper-inducible promoter and quantified the change in the non-AUG-initiated form of ALA1GFP-WT in meiotic cells upon Hyp2 induction. We see a small but significant decrease in non-AUG-initiated translation, dependent on increased levels of HYP2 (Figure 7C, S5G, S5H), suggesting that lower eIF5A is at least partly responsible for the increased translation from near-cognate codons seen in meiotic cells. The small effect seen here is not surprising, as simply overexpressing eIF5A may not increase the relevant functional pool of this factor, which not only has multiple characterized roles as noted above, but is also regulated by hypusine modification (Hershey et al., 1990). Indeed, mass spectrometry data show that Lia1, one of the enzymes responsible for Hyp2 hypusination, is dramatically decreased in meiotic cells, which would be expected to lead to low Hyp2 activity (Figure S8). Moreover, our data suggests that meiotic ribosomes show changes in association with multiple translation initiation factors relative to vegetative cells, some of which are known to be involved in TIS selection (Figure 7B; reviewed in Hinnebusch, 2011; Kearse and Wilusz, 2017). It may be that multiple changes in concert mediate the large increase in near-cognate initiation seen during meiosis.
A previously published vegetative ribosome profiling dataset (Schuller et al., 2017) was examined for evidence that the loss of eIF5A in a non-meiotic context mimicked the high near-cognate initiation we observe in meiosis. Metagene analysis of ribosome footprint reads over all genes was consistent with the elongation defect previously reported within ORFs (Schuller et al., 2017) and also revealed enrichment in 5’ leader reads in cells depleted for eIF5A relative to WT controls, supporting the reported role for this factor in repressing translation from 5’ leader TISs (Figure 7D; Manjunath et al., 2019). When the set of genes we identified as having near-cognate initiated translation in 5’ leaders in our TIS-profiling data was separated from the set that do not, a dramatic difference was evident. The set that we identified as having near-cognate initiation in 5’ leaders in meiosis (n=1925) are enriched for ribosome footprint reads upstream of canonical start codons in eIF5A-depleted mitotic cells, but there was no difference seen for the set that we did not identify as having near-cognate initiation in 5’ leaders (n=4769), relative to WT cells (Figure 7E). This shows that low eIF5A levels alone can lead to selective enhanced near-cognate-initiated translation in the specific subset of genes with this non-canonical type of initiation in meiosis. Together, our data point to eIF5A as a factor that contributes to the condition-specific unmasking of near-cognate-initiated alternate protein isoforms in meiosis.
DISCUSSION
Here we report the first method for globally mapping translation initiation sites, and thus defining translated ORFs, in budding yeast. Traditional ribosome profiling has allowed detection of translated regions genome-wide, but the combined signal of initiating and elongating ribosomes makes identification of alternative and overlapping ORFs challenging. Ribosome profiling following treatment with a post-initiation translation inhibitor, first applied in mammalian cells, overcomes this issue by isolating sites of translation initiation. This type of approach has not been widely used, likely because of the difficulty of identifying drug treatment conditions that are highly specific to inhibition of initiating ribosomes and the challenges of data analysis in organisms with complex transcript architectures.
Our application of this method in vegetative and meiotic budding yeast cells indicates that genome decoding in this simple eukaryote is much more complex than previously appreciated. The many newly identified ORFs from our analyses indicate the need for substantial revision to genome annotations. We identified, for example, the second case (to our knowledge) in which a yeast locus encodes three distinct proteins (Martin and Hopper, 1994). Whereas decades of study have resulted in the validation of only a handful of non-canonical translation products, our systematic experimental approach defined many cases, including 149 near-cognate-initiated N-terminally extended proteins. This is complementary to previous studies (Table S1) and adds direct experimental evidence for widespread translation initiation at near-cognate codons in budding yeast, especially during meiosis. We also found that protein levels resulting from near-cognate initiation, for N-terminal extensions, are not proportional to peak heights observed by TIS-profiling (as exemplified by Ymr31, compare Figure 4C and 4D). Rather, we detect much lower levels than expected, suggesting fundamental differences between AUG- and near-cognate-initiated translation. Both protein synthesis and degradation could contribute to the low steady-state protein levels, but blocking proteasome degradation did not appear to increase the level of the extended isoform (Figure S6C). We favor a model in which near-cognate-initiating ribosomes pause longer at TISs and are thus captured there more efficiently by ribosome profiling. It is also possible that ribosomes initiating at near-cognate and AUG TISs differ in their susceptibility to LTM-based inhibition, leading to preferential capture of reads at near-cognate sites by TIS-profiling.
Although previous studies have identified individual cases of extensions or predicted potential extensions computationally, it has not been possible to experimentally determine the pervasiveness of alternate protein isoforms beginning at non-AUG codons. This has become a recent area of interest, with three of the six established cases in yeast identified in just the last three years (Heublein et al., 2019; Kritsiligkou et al., 2017; Monteuuis et al., 2019). One of these studies predicted this class of proteins to be common, based largely on elegant computational analyses (Monteuuis et al., 2019). Our data are consistent with their general prediction, providing the first direct and comprehensive evidence for translation of a large set of N-terminally extended proteins in budding yeast. We also report these proteins to be conditionally unmasked, with their translation enriched in the context of meiosis.
The few known loci that encode extended proteins have been studied either by mutating the upstream near-cognate codon to encode an AUG, or by using a strong promoter to increase production of the extended protein, presumably by necessity due to low natural expression of extended isoforms (Kritsiligkou et al., 2017; Monteuuis et al., 2019). Conservation and mass spectrometry analyses of N-terminally extended proteins provided evidence for function and stability of only a small subset of the proteins resulting from the alternate isoforms that our TIS-profiling predicted. Because the detection efficiency of both approaches has length-dependence, however, it is not surprising that this class of short protein extensions are generally poorly detected. Moreover, the low abundance of these isoforms, as a class, might explain their especially poor detection by mass spectrometry. The lack of PhyloCSF signal for this class of coding regions may also suggest species-specific translation or unusual constraints on the amino acid sequence. For example, the extended portion of the alanyl tRNA synthetase Ala1 did not show evidence of conserved coding potential despite its critical role in mitochondrial translation. This extension was also not detected by mass spectrometry analysis, highlighting the challenges in using existing global approaches to comprehensively identify this class of alternative protein isoforms.
The large class of non-AUG-initiated 5’ extended ORFs defined in this study reveals trends that could not be determined from the few such cases previously confirmed in vivo. Our study also highlights the challenges of studying near-cognate-initiated extended protein isoforms by classical approaches, and the reasons that few have been confirmed to date. First, as noted above, the protein levels for extended proteins appear low relative to the canonical isoform, making it difficult to study their localization or activity compared to the canonical form, or even to detect their presence in many cases. The efficiency of initiation at near-cognate codons has been reported at between 1% and 10% that of AUG initiation (Chen et al., 2008; Kearse and Wilusz, 2017; Kolitz et al., 2009), and a model in which many fewer ribosomes initiate at the near-cognate TIS relative to the canonical AUG is consistent with our data. Second, the length of the extension relative to the rest of the protein is small (with a median of 21 amino acids in our set), making it difficult to resolve the two isoforms by western blotting. Of the extensions validated by western blot here, only Ymr31 had a large enough size difference to discriminate the two isoforms, while all others necessitated mutating the canonical start codon (M1A constructs) to confirm production of the extended isoform. However, we also found that isolated production of the extended isoforms from the M1A construct can result in low mRNA levels due to NMD, presumably caused by downstream initiation at an out-of-frame AUG (Celik et al., 2017). The degree to which such transcripts are targets of NMD varied greatly and these differences did not seem to correlate with the distance from the newly used out-of-frame stop codon to the end of the transcript, a distance proposed to affect NMD (Hug et al., 2016). Interestingly, however, a moderate positive association was seen with the distance from the beginning of the transcript to the downstream out-of-frame AUG. It is currently unclear how or if this observation might inform the mechanism of NMD for these transcripts, but it is intriguing in light of our incomplete understanding of what defines an NMD target in budding yeast.
Are near-cognate-initiated alternate protein isoforms translated from the same transcripts as canonical isoforms or from distinct transcript isoforms? Our TIS-profiling data cannot distinguish between these possibilities, but we favor the former model for several reasons. First, as discussed above, ribosomes frequently bypassing the near-cognate TIS in favor of initiating at the canonical AUG TIS would make translation of the two isoform types in concert possible from one transcript. Second, 5’RACE analysis of two genes with near-cognate-initiated extensions showed the vast majority (33/34) of transcription start sites to be upstream of the extension’s TIS (Figure S9A, S9B). Finally, the data for genes in which the canonical AUG start is mutated (M1A, Figures 4C, 5E, 5F, 6B, and 6E) supports both isoforms being translated from the same pool of transcripts. Otherwise, we would not expect ATG mutation to result in dramatic downregulation of extended isoform production and deletion of UPF1 (and the resultant NMD deficiency) to rescue it. Finally, in the case of previously-studied extensions encoded by ALA1 and HFA1, the transcription start sites identified by 5’RACE were all upstream of the near-cognate TIS (Suomi et al., 2014; Tang et al., 2004).
Although we identified 149 genes for which translation initiation from a 5’ leader-positioned near-cognate codon produces an alternate extended isoform of a characterized protein, this represents only ~3% of possible in-frame TISs upstream of annotated ORFs. It is unclear which cis factors contribute to this strong specificity, although a bias for the usage of some near-cognate codons over others appears to be a factor. The preferential usage of these codons, including prominently CUG and UUG, is consistent with previous studies of near-cognate translation initiation (Chen et al., 2008; Diaz de Arce et al., 2018; Kolitz et al., 2009). The basis for the additional specificity beyond near-cognate codon identity cannot be explained by optimal context cues used to define the set of AUG start codons used for translation of traditional ORFs. Our attempts to identify simple shared context motifs around the near-cognate codons used to translate alternate isoforms did not reveal signal beyond the preference for a central U in the start codon itself (data not shown). Identifying the context cues that underlie the strong specificity that we observe is an interesting future area of study that may illuminate differences in the mechanism of translation initiation at AUG and near-cognate codons. It is possible that the case of HFA1 is informative in this respect, as it is one of only two genes encoding extended isoforms for which we do not see translation initiation at the annotated downstream AUG. This is suggestive of very efficient initiation at the upstream near-cognate codon that prevents leaky downstream scanning of initiation complexes. The sequence downstream of the near-cognate (AUU) start codon for HFA1 has very high nucleotide-level conservation in yeast, with many positions intolerant to even synonymous mutations (Figure S9C). Such constraint typically indicates function beyond protein coding, such as RNA structure. Consistently, a conserved, stable RNA structure is predicted downstream of the AUU by RNAz analysis, (Figure S9C), which may contribute to the high initiation efficiency at this site (Kozak, 1990).
We found that eIF5A is a trans-factor that contributes to translation of near-cognate-initiated protein isoforms in meiotic cells. eIF5A is known to associate with 60S ribosomal subunits and has been reported to affect multiple aspects of translation (Gregio et al., 2009; Melnikov et al., 2016; Schuller et al., 2017). We found low eIF5A association with ribosomal subunits in meiosis, leading us to investigate its role in meiotic cells. Inducing higher levels of eIF5A decreased translation of a reporter for near-cognate-initiated translation, and re-analysis of published data for eIF5A depletion in mitotic cells showed higher translation within 5’ leaders generally (consistent with Manjunath et al., 2019; Schuller et al., 2017). Strikingly, the subset of genes that we identified as having near-cognate-initiated translation in 5’ leaders during meiosis were the same genes that were responsible for the higher 5’ leader ribosome occupancy in eIF5A-depleted cells, suggesting that the specific near-cognate TISs that we report here are coordinately and selectively “unmasked” by low eIF5A levels. A possible mechanism for this enhanced near-cognate initiation is elongation stalling at specific motifs in eIF5A-deficient cells, leading to ribosome queuing and increased opportunity to initiate at upstream near-cognate sites (Gutierrez et al., 2013; Ivanov et al., 2018; Manjunath et al., 2019; Schuller et al., 2017). The recent finding that low eIF5A enhances CUG-initiated MYC translation in mammals, as well, suggests a conserved mechanism in the regulation of near-cognate-initiated protein isoforms (Manjunath et al., 2019).
An especially intriguing outstanding question raised by this study is the potential function of the many new protein extensions that were identified. Their generally low conservation suggests that they could expand the function of conserved proteins in a species-specific manner. All six known cases of near-cognate initiated alternate protein isoforms result in mitochondrial targeting of the extended protein and dual mitochondrial and cytoplasmic targeting has been suggested as a general role for this type of alternate isoform (Pujol et al., 2007; Yogev and Pines, 2011). However, mitochondrial localization signals are not significantly enriched in the full set of such extensions that we identify (Figure S9D), leaving investigation of their function (or range of functions) an important area of future study. It remains unclear whether most extensions mediate key cellular roles, akin to the case for Ala1, or whether they might represent noisy expression that provides a selective advantage to cells only under specific new or stressful conditions. Because one third of random DNA sequences can mediate organellar protein localization, modified protein localization is an attractive hypothesis for the function of these extended isoforms that could drive the evolution of new roles for existing protein products (Kaiser and Botstein, 1990). That these alternative protein isoforms can be induced in concert, potentially by a decrease in the stringency of start codon selection during translation initiation, points to a simple strategy for cells to modulate the features of a discrete subset of the proteome in response to a change in condition.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gloria Brar (gabrar@berkeley.edu).
Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gloria Brar (gabrar@berkeley.edu).
Data and Code Availability
The accession number for the sequencing data reported in this paper is GEO: GSE150375.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strain construction
All yeast strains used were Saccharomyces cerevisiae of the SK1 background. Strains used in this study are listed in Table S5. GFP-tagged strains were created using single-integration plasmids constructed by Gibson assembly of PCR-amplified genomic regions including 5’ leader regions and PCR-amplified single-integration vector pÜB731/pNH604 (which contains a TRP1 selection marker, yEGFP tag and ADH1 terminator; described in Zalatan et al., 2012). Plasmids were mutated using the Q5 Site Directed Mutagenesis kit. M1A constructs were generated by mutating the annotated ATG to a GCT, and for genes where the next downstream ATG was in-frame, this ATG was also mutated to a GCT. Ustop constructs were generated by mutating the codon prior to the annotated ATG to a stop codon. Deletion strains were created using pÜB1/pFA6A-KanMX (described in Longtine et al., 1998), and overexpression strains were created using pÜB189/pFA6A-KanMX-pCUP1.
Yeast growth and sporulation
Vegetative cells were grown in YEPD at 30°C, with exponentially growing cells grown from an OD600 of 0.2 to an OD600 of 1, and saturated cells to an OD600 >10. For meiotic time courses, strains were inoculated in YEPD for 24 hours, then diluted to an OD600 of 0.2 in buffered YTA and grown for 16 hours at 30°C. Cells were washed once in water, resuspended in sporulation media (SPO), and grown at 30°C. Time points were taken at times indicated in figures.
METHOD DETAILS
TIS-profiling
Cells were treated with 3 μM LTM (Millipore) for 20 minutes, then harvested by filtration and flash freezing in liquid nitrogen. Samples were lysed by mixermilling at 15 Hz for 6 rounds of 3 minutes each. Samples were thawed at 30°C and spun down at 3000 rcf for 5 minutes at 4°C. The supernatant was removed and cleared at 20,000 rcf for 10 minutes at 4°C, and 200 μL aliquots of cleared supernatant were flash frozen. Ribosome profiling library preparation was as in (Brar et al., 2012). In brief, samples were treated with RNaseI (Ambion), then monosome peaks were collected from sucrose gradients. RNA was extracted, size selected, dephosphorylated, polyA-tailed, subjected to rRNA subtraction, RT-PCR, circularization, and PCR amplification. Samples were sequenced on an Illumina HiSeq 2500, 50SRR, with multiplexing, at the UC-Berkeley Vincent Coates QB3 Sequencing facility.
Polysome gradient analysis
Extract from mixermilling flash-frozen cells was subjected to polysome gradient analysis as described in (Ingolia et al., 2009). In short, 200 μl extract was loaded on 10–50% sucrose gradients with or without RNAseI treatment, depending on if sample would be used for ribosome profiling or 40S/60S isolation, respectively. Samples were centrifuged in a Beckman XL-70 Ultracentrifuge, using a Sw-Ti41 rotor for 3 hours at 35,000 rpm at 4°C. Tube was loaded on a Bio-Comp Gradient Station and analyzed for absorbance at 260 nm. For mass spectrometry of 40S/60S fraction, sucrose fraction was collected and flash frozen prior to precipitation and mass spectrometry.
Mass-spectrometry-based protein identification of the 40S/60S peaks by iTRAQ-labeling
Proteins from the collected 40S/60S fractions were precipitated by adding −20°C cold acetone to the lysate (acetone to eluate ratio 10:1) and overnight incubation at −20°C. The proteins were pelleted by centrifugation at 20,000 g for 15 min at 4°C. The supernatant was discarded and the pellet was left to dry by evaporation. The protein pellet was reconstituted in 100 μl urea buffer (8 M Urea, 75 mM NaCl, 50 mM Tris/HCl pH 8.0, 1 mM EDTA) and protein concentrations were determined by BCA assay (Pierce). 10 μg of total protein per sample (with the exception of the “Master spike-in Total Extract” where we used 20 μg - see below) were processed further. Disulfide bonds were reduced with 5 mM dithiothreitol and cysteines were subsequently alkylated with 10 mM iodoacetamide. Samples were diluted 1:4 with 50 mM Tris/HCl (pH 8.0) and sequencing grade modified trypsin (Promega) was added in an enzyme-to-substrate ratio of 1:50. After 16 hours of digestion, samples were acidified with 1% formic acid (final concentration). Tryptic peptides were desalted on C18 StageTips according to (Rappsilber et al., 2007) and evaporated to dryness in a vacuum concentrator. Desalted peptides were labeled with the iTRAQ reagent according to the manufacturer’s instructions (AB Sciex) and as previously described (Mertins et al., 2013). Briefly, replicate 1 and replicate 2 were each measured in their own iTRAQ mix. In addition, each mix had the same two “Master spike-in” samples added. The “Master spike-in Total Lysate” contained an equal mix of total protein extract from vegetative, meiotic cells and spores. The “Master spike-in Polysomes” contained an equal mix of proteins from all polysome fractions from vegetative, meiotic cells and spores. Briefly, 0.33 units of iTRAQ reagent were used per IP. Peptides were dissolved in 10 μl of 0.5 M TEAB pH 8.5 solution and the iTRAQ reagent was added in 23 μl of ethanol. After 1 h incubation the reaction was stopped with 50 mM Tris/HCl (pH 8.0). Differentially labeled peptides were mixed and subsequently desalted on C18 StageTips (Rappsilber et al., 2007) and evaporated to dryness in a vacuum concentrator. Peptides were reconstituted in 50 μl 3% MeCN/0.1% formic acid. LC-MS/MS analysis was performed as previously described (Mertins et al., 2013).
| Mix 1 | ||
|---|---|---|
| Sample | iTRAQ label | Peptides labeled (μg) |
| Master spike-in Total Lysate | 114 | 20 |
| 40S/60S Meiosis Repl. 01 | 115 | 10 |
| 40S/60S Vegetative Repl. 01 | 116 | 10 |
| Master spike-in Polysomes | 117 | 10 |
| Mix 2 | ||
|---|---|---|
| Sample | iTRAQ label | Peptides labeled (μg) |
| Master spike-in Total Lysate | 114 | 20 |
| 40S/60S Vegetative Repl. 02 | 115 | 10 |
| 40S/60S Meiosis Repl. 02 | 116 | 10 |
| Master spike-in Polysomes | 117 | 10 |
All mass spectra were analyzed with the Spectrum Mill software package v4.0 beta (Agilent Technologies) according to (Mertins et al., 2013) using the yeast Uniprot database (UniProt.Yeast.completeIsoforms.UP000002311; strain ATCC 204508 / S288c). For identification, we applied a maximum FDR of 1% separately on the protein and peptide level and proteins were grouped in subgroup specific manner. We calculated intensity ratios relative to iTRAQ channel 117 (“Master spike-in Polysomes”) and subsequently median normalized these ratios for each sample.
Western blotting
Strains were grown in YEPD or SPO, with 3.5 ODs of cells harvested at indicated time points. Cells were fixed in 5% TCA for at least 10 minutes, then washed once with acetone and dried overnight. Samples were resuspended in 50 mM Tris-HCl, 1 mM EDTA, 3 mM DTT, 1.1 mM PMSF (Sigma) and 1× cOmplete mini EDTA-free protease inhibitor cocktail (Roche), then lysed with glass-bead-based agitation for 5 minutes, then boiled in SDS loading buffer for 5 minutes at 95°C. Samples were spun down for 5 min at 20,000 rcf prior to running on a 4–12% Bis-Tris gel at 175 V for 30 minutes. Transfer to nitrocellulose membrane was performed using a Turbo Transfer semi-dry standard 30 minute transfer. Membrane was blocked with 5% milk in PBST for 1 hour, and incubated in primary antibody overnight at 4°C. Primary antibodies were diluted 1:2,000 for mouse anti-GFP (Clontech), 1:10,000 for rabbit anti-hexokinase (Rockland), and 1:10,000 for rat anti-tubulin (Serotec) in PBS blocking buffer (LI-COR). Membrane was washed with PBST 5 times for 5 minutes each time, then incubated in secondary antibody (1:15,000 anti-mouse 800, anti-rabbit 680, or anti-rat 680 (LI-COR) in PBS blocking buffer) for 2 hours, then washed with PBST 5 times for 5 minutes each time. Images were acquired using the LI-COR Odyssey imager, and analysis and quantification was performed in ImageStudio Lite Software (LI-COR).
qPCR
Samples were flash frozen in liquid nitrogen, then resuspended in TES buffer (10 mM Tris 7.5, 10 mM EDTA, 0.5% SDS), with acid-washed glass beads (Sigma) and acid phenol:chloroform:isoamyl alcohol (125:24:1; pH 4.7). Samples were centrifuged for 10 minutes at 21000 rcf at 4°C, then the aqueous phase was removed and added to chloroform. Samples were centrifuged again for 5 minutes at 21000 rcf at RT, then the aqueous phase was removed and added to isopropanol and 0.33 M NaOAc. Samples were precipitated at 4°C overnight, then centrifuged for 20 minutes at 21000 rcf at 4°C. Pellets were washed with 80% ethanol, air-dried, and resuspended in water. The TURBO DNA-free kit (Thermo) was used to treat 2.5 μg RNA with DNAse, then samples were incubated with random hexamers for 5 min at 65°C. DTT, and dNTPs were added, then Superscript III was added and samples were incubated at 25°C for 10 minutes, 42°C for 50 min, and 70°C for 10 minutes. cDNA was quantified by 7500 FAST Real-Time PCR machine with SYBR green mix (Thermo) and the following qPCR primers listed in the Key Resources Table: GFP (oGAB-2736/oGAB-2737), PFY1 (oGAB-3301/oGAB-3302), and HYP2 (oGAB-7864/oGAB-7865).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GFP | Clontech | Cat#632381, RRID:AB_2313808 |
| Rabbit anti-hexokinase antibody | Rockland | Cat#100–4159, RRID:AB_21991 |
| Anti-mouse 800 secondary | LI-COR | Cat#926–32210, RRID:AB_621842 |
| Anti-rabbit 680 secondary | LI-COR | Cat#926–68071, RRID:AB_10956166 |
| Rat anti-tubulin | Serotec | Cat#MCA78G, RRID:AB_325005 |
| Anti-rat 680 secondary | LI-COR | Cat#926–68076, RRID:AB_10956590 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lactimidomycin (LTM) | Millipore | Cat#506291 |
| RNaseI | Ambion | Cat#AM2294 |
| Sequencing grade modified trypsin | Promega | Cat#V5111 |
| PMSF | Sigma | Cat#78830 |
| cOmplete mini EDTA-free protease inhibitor cocktail | Roche | Cat#29384100 |
| Acid-washed glass beads | Sigma | Cat#G8772 |
| PBS Odyssey Blocking Buffer | LI-COR | Cat#927–40100 |
| Superscript III | Thermo | Cat#18080044 |
| SYBR green mix | Thermo | Cat#4309155 |
| Critical Commercial Assays | ||
| Q5 Site Directed Mutagenesis kit | NEB | Cat#E0554S |
| TURBO DNA-free kit | Ambion | Cat#AM1907 |
| Deposited Data | ||
| Raw and analyzed sequencing data | This study | GEO: GSE150375 |
| Experimental Models: Organisms/Strains | ||
| All yeast strains are described in Table S5. | Brar-Ünal Lab and this study | N/A |
| Oligonucleotides | ||
| oGAB-2736 GFP-qPCR_f ctccggtgaaggtgaaggtg | IDT | N/A |
| oGAB-2737 GFP-qPCR_r aggttggccatggaactgg | IDT | N/A |
| oGAB-3301 PFY1-qPCR_f acggtagacatgatgctgagg | IDT | N/A |
| oGAB-3302 PFY1-qPCR_r acggttggtggataatgagc | IDT | N/A |
| oGAB-7864 HYP2-qPCR_f TGTCAAGGCTCCAGAAGGTGA | IDT | N/A |
| oGAB-7865 HYP2-qPCR_r CCCATAGCGGAGATGATGGT | IDT | N/A |
| Recombinant DNA | ||
| pÜB1/pFA6A-KanMX | Addgene | Cat#39296 |
| pÜB189/pFA6A-KanMX-pCUP1 (derived from pFA6A) | Brar-Ünal Lab | N/A |
| pÜB731 (derived from pNH604) | Brar-Ünal Lab | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MochiView | Homann and Johnson, 2010 | http://www.johnsonlab.ucsf.edu/mochi/ |
| ImageStudio Lite Software | LI-COR | https://www.licor.com/bio/products/software/image_studio_lite/index.html |
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of TIS-profiling data
Sequencing data were aligned using bowtie2 (Langmead and Salzberg, 2012), and ORF-RATER was applied to TIS-profiling data and standard profiling data. Genome browser analysis and visualization was done using MochiView (Homann and Johnson, 2010). The distribution of read lengths by this approach was approximately 2 nucleotides longer than seen for standard ribosome profiling (peaking at 30 nt, rather than 28 nt), and we found that the a-site offset typically used for standard ribosome profiling data visualization required shifting of 2 nt upstream, as well. To calculate expression values, footprint values from standard ribosome profiling for annotated genes were averaged, and an expression cutoff greater than or equal to 5 RPKM was used for analysis shown in Figure S2A–B, Table S2.
Footprint quantification and correlation analysis
Standard RPKM calculations were used for cycloheximide profiling. For TIS-profiling, we counted reads mapping to the region spanning 3 nt up- and downstream of the start codon and normalized by total reads at all initiation sites. The spearman correlation between TIS-profiling and standard profiling was calculated for each gene. The distribution of correlation scores was compared to a null distribution generated by shuffling gene names and performing the same correlation analysis. Statistical significance was determined using a K.S. test. For 5’ UTR quantification, read counts were determined for the region from the canonical start to 99 nt upstream. Counts were normalized by total reads at initiation sites to account for library size differences.
Start codon analysis
The region 30–99 nt upstream of canonical starts was used as a proxy for 5’UTRs. The upper cutoff was based on average transcript lengths in yeast and the lower cutoff was matched to the minimum length cutoff used for extensions. Within this region, we counted the number of AUG and near-cognate in-frame start codons that did not also have an in-frame stop codon before the canonical TIS. These counts gave the “expected” distribution of codon usage given no start codon selection bias. The expected counts were compared to the counts that were observed among called extensions. Statistical significance was determined using Fisher’s Exact Test for each individual codon. As a control, we also analyzed the regions within 30 nt upstream of canonical start codons, which would encode short (<10 amino acid) extensions. This class does not show the same start codon bias as is seen for the longer set (Figure S4B, S4C).
Context analysis
Maximum motif score analysis was performed using Mochiview (Homann and Johnson, 2010) for the regions 10 nt up- and down-stream of all annotated genes, recapitulating the known Kozak sequence. The enrichment for this motif in regions 10 basepairs up- and downstream of other start codon classes and control regions were plotted using the maximum motif score enrichment tool in Mochiview.
Conservation analysis
PhyloCSF scores for the extensions were computed using the 7yeast parameter set and the default mle and AsIs options, applied to the extension, starting at the upstream start codon and continuing up to but not including the annotated start codon. Alignments used as input to PhyloCSF and shown in CodAlignView were extracted from the MULTIZ whole genome alignment of seven Saccharomyces species based on the sacCer3 S. cerevisiae S288C reference assembly, obtained from the UCSC Genome Browser (Haeussler et al., 2019). Extensions were first mapped from the SK1 strain assembly to the the S288C strain sacCer3 assembly using an ad hoc alignment created with LASTZ (Harris, 2007). We did not compute PhyloCSF scores for the two extensions of YBR012C because of difficulty mapping to the S288C strain. In some cases, we also computed PhyloCSF scores of 10-codon windows 5’ of the detected TIS to determine if the ancestral extension was longer than the one detected.
Deep proteome identification of peptides and proteins
First, we generated a concatenated search database including all canonical proteins in the yeast UniProt database (release 2014_09, strain ATCC 204508 / S288c), and the newly predicted alternative proteoforms (e.g. N-terminal extensions) and proteins identified by ORF-RATER (a set including scores >0.1). Raw data generated previously to investigate proteome changes during yeast meiosis at deep coverage (Cheng et al., 2018) were analyzed with the MaxQuant software version 1.6.0.16 (Cox and Mann, 2008) against the above mentioned concatenated search database, and MS/MS searches were performed with the following parameters: TMT-11plex labeling on the MS2 level, oxidation of methionine and protein N-terminal acetylation as variable modifications; carbamidomethylation as fixed modification; Trypsin/P as the digestion enzyme; precursor ion mass tolerances of 20 p.p.m. for the first search (used for nonlinear mass re-calibration) and 4.5 p.p.m. for the main search, and a fragment ion mass tolerance of 20 p.p.m. For identification, we applied a maximum FDR of 1% separately on protein and peptide level.
Supplementary Material
Table S2: Annotated ORFs with expression analysis, Related to Figure 2 All annotated ORFs with ORF-RATER scores, if called, and mean expression values.
Table S3: Non-canonical ORF scores, Related to Figure 2 ORF-RATER scores for non-canonical ORFs. This table includes non-AUG extensions, alternative forms with 2 AUGs, and AUG-initiated uORFs.
Table S4: 5’ Extension ORFs, Related to Figures 2, 4, 5, and 6. 5’ Extension ORFs defined by a score cutoff of >0.1, a length of >10 amino acids, and a non-AUG start codon. A total of 2,735 5’ extended ORFs were called by ORF-RATER (for all calls see dataset deposited at NCBI GEO accession number GSE150375). This number was ultimately adjusted to 231 extensions representing 160 unique genes based on the criteria above. Of these, 11 are likely misannotations, resulting in a final class of 149 true extension-containing genes. Shown in column K: ORF-RATER score, N: length of extension, H: start codon, O: PhyloCSF conservation score and R-S: categorization/notes on specific genes, including misannotation information. Columns Q and R indicate whether the associated annotated ORF was also called by ORF-RATER, and if there is any evidence for translation at the annotated AUG, respectively.
Table S5: List of yeast strains, Related to STAR Methods Strain numbers and genotypes for all yeast strains used in this study.
Table S1: Summary of prior studies investigating near-cognate-initiated extensions List of previous studies that have identified and validated near-cognate-initiated extensions, including individually identified examples and genome-wide studies.
Highlights.
TIS-profiling reveals widespread translation of non-canonical ORFs in budding yeast
Production of non-AUG-initiated extended isoforms is prevalent and inefficient
A small subset of possible near-cognate sites are used for translation initiation
eIF5A-based regulation allows conditional unmasking of non-AUG initiation in meiosis
ACKNOWLEDGEMENTS
We thank Elçin Ünal, George Otto, Helen Vander Wende, and Tina Sing for critical reading of this manuscript and members of the Brar and Ünal labs for helpful suggestions. G.A.B., A.R.E., and A.L.H. are supported by the National Institutes of Health [DP2-GM-119138 and 1R35GM134886], investigator awards from the Alfred P. Sloan Foundation [FG-2016-6229] and Pew Charitable Trusts [00029624], and UC-Berkeley start-up funding, including from the Bowes Foundation, to G.A.B. A.L.H. was also funded in part by NSF pre-doctoral fellowship DGE 1752814, and A.R.E. and A.L.H. were funded in part by NIH training grants to the MCB department (T32 GM 0007232 and T32 HG 00047). M.J. is funded by the NIH (R35-GM-128802) and Columbia start-up funding. I.J. is supported by the National Human Genome Research Institute of the National Institutes of Health [U41HG007234 and HG004037].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aitken CE, Lorsch JR, 2012. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol 19, 568–576. doi: 10.1038/nsmb.2303 [DOI] [PubMed] [Google Scholar]
- Almagro Armenteros JJ, Salvatore M, Emanuelsson O, Winther O, Heijne, von G, Elofsson A, Nielsen H, 2019. Detecting sequence signals in targeting peptides using deep learning. Life Sci Alliance 2. doi: 10.26508/lsa.201900429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benne R, Hershey JW, 1978. The mechanism of action of protein synthesis initiation factors from rabbit reticulocytes. Journal of Biological Chemistry 253, 3078–3087. [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS, 2012. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–557. doi: 10.1126/science.1215110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent MR, 2005. Genome annotation past, present, and future: how to define an ORF at each locus 15, 1777–1786. doi: 10.1101/gr.3866105 [DOI] [PubMed] [Google Scholar]
- Celik A, Baker R, He F, Jacobson A, 2017. High-resolution profiling of NMD targets in yeast reveals translational fidelity as a basis for substrate selection. RNA 23 (5), 735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-P, Chen S-J, Lin C-H, Wang T-L, Wang C-C, 2010. A single sequence context cannot satisfy all non-AUG initiator codons in yeast. BMC Microbiol 10, 188. doi: 10.1186/1471-2180-10-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K-J, Wang C-C, 2004. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. Journal of Biological Chemistry 279, 13778–13785. doi: 10.1074/jbc.M311269200 [DOI] [PubMed] [Google Scholar]
- Chen S-J, Lin G, Chang K-J, Yeh L-S, Wang C-C, 2008. Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. Journal of Biological Chemistry 283, 3173–3180. doi: 10.1074/jbc.M706968200 [DOI] [PubMed] [Google Scholar]
- Cheng Z, Otto GM, Powers EN, Keskin A, Mertins P, Carr SA, Jovanovic M, Brar GA, 2018. Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172, 910–923.e16. doi: 10.1016/j.cell.2018.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JM, Laz TM, Sherman F, 1988. Efficiency of translation initiation by non-AUG codons in Saccharomyces cerevisiae. Mol. Cell. Biol 8, 4533–4536. doi: 10.1128/mcb.8.10.4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M, 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26 (12), 1367–1372. [DOI] [PubMed] [Google Scholar]
- David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM, 2006. A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci USA 103, 5320–5325. doi: 10.1073/pnas.0601091103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Grate L, Spingola M, Ares M, 2000. Test of intron predictions reveals novel splice sites, alternatively spliced mRNAs and new introns in meiotically regulated genes of yeast. Nucleic Acids Res 28, 1700–1706. doi: 10.1093/nar/28.8.1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz de Arce AJ, Noderer WL, Wang CL, 2018. Complete motif analysis of sequence requirements for translation initiation at non-AUG start codons. Nucleic Acids Res 46, 985–994. doi: 10.1093/nar/gkx1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields AP, Rodriguez EH, Jovanovic M, Stern-Ginossar N, Haas BJ, Mertins P, Raychowdhury R, Hacohen N, Carr SA, Ingolia NT, Regev A, Weissman JS, 2015. A Regression-Based Analysis of Ribosome-Profiling Data Reveals a Conserved Complexity to Mammalian Translation. Molecular Cell 60, 816–827. doi: 10.1016/j.molcel.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno M, Jiménez A, Vázquez D, 1977. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem 72, 323–330. doi: 10.1111/j.1432-1033.1977.tb11256.x [DOI] [PubMed] [Google Scholar]
- Fritsch C, Herrmann A, Nothnagel M, Szafranski K, Huse K, Schumann F, Schreiber S, Platzer M, Krawczak M, Hampe J, Brosch M, 2012. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting 22, 2208–2218. doi: 10.1101/gr.139568.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa Y, Tsuji J, Fu S-C, Tomii K, Horton P, Imai K, 2015. MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol. Cell Proteomics 14, 1113–1126. doi: 10.1074/mcp.M114.043083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregio APB, Cano VPS, Avaca JS, Valentini SR, Zanelli CF, 2009. eIF5A has a function in the elongation step of translation in yeast. Biochem. Biophys. Res. Commun 380, 785–790. doi: 10.1016/j.bbrc.2009.01.148 [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Shin B-S, Woolstenhulme CJ, Kim J-R, Saini P, Buskirk AR, Dever TE, 2013. eIF5A promotes translation of polyproline motifs. Molecular Cell 51, 35–45. doi: 10.1016/j.molcel.2013.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, Gibson D, Diekhans M, Clawson H, Casper J, Barber GP, Haussler D, Kuhn RM, Kent WJ, 2019. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res 47, D853–D858. doi: 10.1093/nar/gky1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, 2007. Improved Pairwise Alignment of Genomic DNA [Google Scholar]
- Henderson A, Hershey JW, 2011. Eukaryotic translation initiation factor (eIF) 5A stimulates protein synthesis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A 108, 6415–6419. doi: 10.1073/pnas.1008150108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey JW, Smit-McBride Z, Schnier J, 1990. The role of mammalian initiation factor eIF-4D and its hypusine modification in translation. Biochim. Biophys. Acta 1050, 160–162. doi: 10.1016/0167-4781(90)90159-y [DOI] [PubMed] [Google Scholar]
- Heublein M, Ndi M, Vazquez-Calvo C, Vögtle FN, Ott M, 2019. Alternative Translation Initiation at a UUG Codon Gives Rise to Two Functional Variants of the Mitochondrial Protein Kgd4. Journal of Molecular Biology 431, 1460–1467. doi: 10.1016/j.jmb.2019.02.023 [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, 2011. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev 75, 434–67- first page of table of contents. doi: 10.1128/MMBR.00008-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann OR, Johnson AD, 2010. MochiView: versatile software for genome browsing and DNA motif analysis. BMC Biol 8, 49–8. doi: 10.1186/1741-7007-8-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood HM, Neafsey DE, Galagan J, Sachs MS, 2009. Evolutionary roles of upstream open reading frames in mediating gene regulation in fungi. Annu. Rev. Microbiol 63, 385–409. doi: 10.1146/annurev.micro.62.081307.162835 [DOI] [PubMed] [Google Scholar]
- Hossain MA, Rodriguez CM, Johnson TL, 2011. Key features of the two-intron Saccharomyces cerevisiae gene SUS1 contribute to its alternative splicing. Nucleic Acids Res 39, 8612–8627. doi: 10.1093/nar/gkr497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug N, Longman D, Cáceres JF, 2016. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res 44, 1483–1495. doi: 10.1093/nar/gkw010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS, 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. doi: 10.1126/science.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS, 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802. doi: 10.1016/j.cell.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov IP, Shin B-S, Loughran G, Tzani I, Young-Baird SK, Cao C, Atkins JF, Dever TE, 2018. Polyamine Control of Translation Elongation Regulates Start Site Selection on Antizyme Inhibitor mRNA via Ribosome Queuing. Molecular Cell 70, 1–18. doi: 10.1016/j.molcel.2018.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneau K, Nislow C, Davis RW, 2009. Alternative splicing of PTC7 in Saccharomyces cerevisiae determines protein localization. Genetics 183, 185–194. doi: 10.1534/genetics.109.105155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser CA, Botstein D, 1990. Efficiency and diversity of protein localization by random signal sequences. Mol. Cell. Biol 10, 3163–3173. doi: 10.1128/mcb.10.6.3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, Wilusz JE, 2017. Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes & Development 31, 1717–1731. doi: 10.1101/gad.305250.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper WM, Berry KW, Merrick WC, 1976. Purification and properties of rabbit reticulocyte protein synthesis initiation factors M2Balpha and M2Bbeta. Journal of Biological Chemistry 251, 5551–5557. [PubMed] [Google Scholar]
- Kim Guisbert KS, Zhang Y, Flatow J, Hurtado S, Staley JP, Lin S, Sontheimer EJ, 2012. Meiosis-induced alterations in transcript architecture and noncoding RNA expression in S. cerevisiae. RNA 18, 1142–1153. doi: 10.1261/rna.030510.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolitz SE, Takacs JE, Lorsch JR, 2009. Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation. RNA 15, 138–152. doi: 10.1261/rna.1318509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M, 2002. Pushing the limits of the scanning mechanism for initiation of translation. Gene 299, 1–34. doi: 10.1016/j.cell.2011.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M, 1999. Initiation of translation in prokaryotes and eukaryotes. Gene 234, 187–208. doi: 10.1016/s0378-1119(99)00210-3 [DOI] [PubMed] [Google Scholar]
- Kozak M, 1990. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc Natl Acad Sci USA 87, 8301–8305. doi: 10.1073/pnas.87.21.8301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M, 1984. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res 12, 857–872. doi: 10.1093/nar/12.2.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M, 1978. How do eucaryotic ribosomes select initiation regions in messenger RNA? Cell 15, 1109–1123. doi: 10.1016/0092-8674(78)90039-9 [DOI] [PubMed] [Google Scholar]
- Kritsiligkou P, Chatzi A, Charalampous G, Mironov A, Grant CM, Tokatlidis K, 2017. Unconventional Targeting of a Thiol Peroxidase to the Mitochondrial Intermembrane Space Facilitates Oxidative Protein Folding. CellReports 18, 2729–2741. doi: 10.1016/j.celrep.2017.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL, 2012. Fast gapped-read alignment with Bowtie 2. Nature Chemical Biology 9, 357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu B, Lee S, Huang S-X, Shen B, Qian S-B, 2012. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A 109, E2424–32. doi: 10.1073/pnas.1207846109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, Jungreis I, Kellis M, 2011. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275–82. doi: 10.1093/bioinformatics/btr209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR, 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. doi: [DOI] [PubMed] [Google Scholar]
- Lopo AC, Lashbrook CC, Infante D, Infante AA, Hershey JW, 1986. Translational initiation factors from sea urchin eggs and embryos: functional properties are highly conserved. Arch. Biochem. Biophys 250, 162–170. doi: 10.1016/0003-9861(86)90713-7 [DOI] [PubMed] [Google Scholar]
- Machkovech HM, Bloom JD, Subramaniam AR, 2019. Comprehensive profiling of translation initiation in influenza virus infected cells. PLoS Pathog 15, e1007518. doi: 10.1371/journal.ppat.1007518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunath H, Zhang H, Rehfeld F, Han J, Chang T-C, Mendell JT, 2019. Suppression of Ribosomal Pausing by eIF5A Is Necessary to Maintain the Fidelity of Start Codon Selection. CellReports 29, 3134–3146.e6. doi: 10.1016/j.celrep.2019.10.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NC, Hopper AK, 1994. How single genes provide tRNA processing enzymes to mitochondria, nuclei and the cytosol. Biochimie 76, 1161–1167. doi: 10.1016/0300-9084(94)90045-0 [DOI] [PubMed] [Google Scholar]
- Melnikov S, Mailliot J, Shin B-S, Rigger L, Yusupova G, Micura R, Dever TE, Yusupov M, 2016. Crystal Structure of Hypusine-Containing Translation Factor eIF5A Bound to a Rotated Eukaryotic Ribosome. Journal of Molecular Biology 428, 3570–3576. doi: 10.1016/j.jmb.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA, 2013. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nature Chemical Biology 10, 634–637. doi: 10.1038/nmeth.2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteuuis G, Miścicka A, Świrski M, Zenad L, Niemitalo O, Wrobel L, Alam J, Chacinska A, Kastaniotis AJ, Kufel J, 2019. Non-canonical translation initiation in yeast generates a cryptic pool of mitochondrial proteins. Nucleic Acids Res 47, 5777–5791. doi: 10.1093/nar/gkz301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DR, Geballe AP, 2000. Upstream open reading frames as regulators of mRNA translation. Mol. Cell. Biol 20, 8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M, 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. doi: 10.1126/science.1158441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol C, Maréchal-Drouard L, Duchêne A-M, 2007. How can organellar protein N-terminal sequences be dual targeting signals? In silico analysis and mutagenesis approach. Journal of Molecular Biology 369, 356–367. doi: 10.1016/j.jmb.2007.03.015 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y, 2007. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Publishing Group 2, 1896–1906. doi: 10.1038/nprot.2007.261 [DOI] [PubMed] [Google Scholar]
- Saini P, Eyler DE, Green R, Dever TE, 2009. Hypusine-containing protein eIF5A promotes translation elongation. Nature 459, 118–121. doi: 10.1038/nature08034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota D, Lake AM, Yang W, Yang C, Wesseling H, Guise A, Uncu C, Dalal JS, Kraft AW, Lee J-M, Sands MS, Steen JA, Dougherty JD, 2019. Cell-Type-Specific Profiling of Alternative Translation Identifies Regulated Protein Isoform Variation in the Mouse Brain. CellReports 26, 594–607.e7. doi: 10.1016/j.celrep.2018.12.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO, 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nature Chemical Biology 6, 209–217. doi: 10.1038/nchembio.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreier MH, Erni B, Staehelin T, 1977. Initiation of mammalian protein synthesis. I. Purification and characterization of seven initiation factors. Journal of Molecular Biology 116, 727–753. doi: 10.1016/0022-2836(77)90268-6 [DOI] [PubMed] [Google Scholar]
- Schuller AP, Wu CC-C, Dever TE, Buskirk AR, Green R, 2017. eIF5A Functions Globally in Translation Elongation and Termination. Molecular Cell 66, 194–205.e5. doi: 10.1016/j.molcel.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N, Weisburd B, Michalski A, Le VTK, Hein MY, Huang S-X, Ma M, Shen B, Qian S-B, Hengel H, Mann M, Ingolia NT, Weissman JS, 2012. Decoding human cytomegalovirus. Science 338, 1088–1093. doi: 10.1126/science.1227919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara K, Nishiyama Y, Toda S, Komiyama N, Hatori M, Moriyama T, Sawada Y, Kamei H, Konishi M, Oki T, 1992. Lactimidomycin, a new glutarimide group antibiotic. Production, isolation, structure and biological activity. J. Antibiot 45, 1433–1441. doi: 10.7164/antibiotics.45.1433 [DOI] [PubMed] [Google Scholar]
- Suomi F, Suomi F, Menger KE, Menger KE, Monteuuis G, Monteuuis G, Naumann U, Naumann U, Kursu VAS, Kursu VAS, Shvetsova A, Shvetsova A, Kastaniotis AJ, Kastaniotis AJ, 2014. Expression and evolution of the non-canonically translated yeast mitochondrial acetyl-CoA carboxylase Hfa1p. PLoS ONE 9, e114738. doi: 10.1371/journal.pone.0114738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, St Onge RP, Mani R, King OD, Heilbut A, Labunskyy VM, Chen W, Pham L, Zhang LV, Tong AHY, Nislow C, Giaever G, Gladyshev VN, Vidal M, Schow P, Lehár J, Roth FP, 2011. Knocking out multigene redundancies via cycles of sexual assortment and fluorescence selection. Nat Meth 8, 159–164. doi: 10.1038/nmeth.1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H-L, Yeh L-S, Chen N-K, Ripmaster T, Schimmel P, Wang C-C, 2004. Translation of a yeast mitochondrial tRNA synthetase initiated at redundant non-AUG codons. Journal of Biological Chemistry 279, 49656–49663. doi: 10.1074/jbc.M408081200 [DOI] [PubMed] [Google Scholar]
- Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats A-C, Vagner S, 2003. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol. Cell 95, 169–178. doi: 10.1016/s0248-4900(03)00033-9 [DOI] [PubMed] [Google Scholar]
- Yassour M, Kaplan T, Fraser HB, Levin JZ, Pfiffner J, Adiconis X, Schroth G, Luo S, Khrebtukova I, Gnirke A, Nusbaum C, Thompson D-A, Friedman N, Regev A, 2009. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc. Natl. Acad. Sci. U.S.A 106, 3264–3269. doi: 10.1073/pnas.0812841106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogev O, Pines O, 2011. Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochim. Biophys. Acta 1808, 1012–1020. doi: 10.1016/j.bbamem.2010.07.004 [DOI] [PubMed] [Google Scholar]
- Zalatan JG, Coyle SM, Rajan S, Sidhu SS, Lim WA, 2012. Conformational control of the Ste5 scaffold protein insulates against MAP kinase misactivation. Science 337, 1218–1222. doi: 10.1126/science.1220683 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: Annotated ORFs with expression analysis, Related to Figure 2 All annotated ORFs with ORF-RATER scores, if called, and mean expression values.
Table S3: Non-canonical ORF scores, Related to Figure 2 ORF-RATER scores for non-canonical ORFs. This table includes non-AUG extensions, alternative forms with 2 AUGs, and AUG-initiated uORFs.
Table S4: 5’ Extension ORFs, Related to Figures 2, 4, 5, and 6. 5’ Extension ORFs defined by a score cutoff of >0.1, a length of >10 amino acids, and a non-AUG start codon. A total of 2,735 5’ extended ORFs were called by ORF-RATER (for all calls see dataset deposited at NCBI GEO accession number GSE150375). This number was ultimately adjusted to 231 extensions representing 160 unique genes based on the criteria above. Of these, 11 are likely misannotations, resulting in a final class of 149 true extension-containing genes. Shown in column K: ORF-RATER score, N: length of extension, H: start codon, O: PhyloCSF conservation score and R-S: categorization/notes on specific genes, including misannotation information. Columns Q and R indicate whether the associated annotated ORF was also called by ORF-RATER, and if there is any evidence for translation at the annotated AUG, respectively.
Table S5: List of yeast strains, Related to STAR Methods Strain numbers and genotypes for all yeast strains used in this study.
Table S1: Summary of prior studies investigating near-cognate-initiated extensions List of previous studies that have identified and validated near-cognate-initiated extensions, including individually identified examples and genome-wide studies.
Data Availability Statement
The accession number for the sequencing data reported in this paper is GEO: GSE150375.