

Supplemental Digital Content is available in the text.
Keywords: claudin-5, endothelium, junctional adhesion molecule A, tight junctions, vascular permeability
Abstract
Rationale:
Intercellular tight junctions are crucial for correct regulation of the endothelial barrier. Their composition and integrity are affected in pathological contexts, such as inflammation and tumor growth. JAM-A (junctional adhesion molecule A) is a transmembrane component of tight junctions with a role in maintenance of endothelial barrier function, although how this is accomplished remains elusive.
Objective:
We aimed to understand the molecular mechanisms through which JAM-A expression regulates tight junction organization to control endothelial permeability, with potential implications under pathological conditions.
Methods and Results:
Genetic deletion of JAM-A in mice significantly increased vascular permeability. This was associated with significantly decreased expression of claudin-5 in the vasculature of various tissues, including brain and lung. We observed that C/EBP-α (CCAAT/enhancer-binding protein-α) can act as a transcription factor to trigger the expression of claudin-5 downstream of JAM-A, to thus enhance vascular barrier function. Accordingly, gain-of-function for C/EBP-α increased claudin-5 expression and decreased endothelial permeability, as measured by the passage of fluorescein isothiocyanate (FITC)-dextran through endothelial monolayers. Conversely, C/EBP-α loss-of-function showed the opposite effects of decreased claudin-5 levels and increased endothelial permeability. Mechanistically, JAM-A promoted C/EBP-α expression through suppression of β-catenin transcriptional activity, and also through activation of EPAC (exchange protein directly activated by cAMP). C/EBP-α then directly binds the promoter of claudin-5 to thereby promote its transcription. Finally, JAM-A–C/EBP-α–mediated regulation of claudin-5 was lost in blood vessels from tissue biopsies from patients with glioblastoma and ovarian cancer.
Conclusions:
We describe here a novel role for the transcription factor C/EBP-α that is positively modulated by JAM-A, a component of tight junctions that acts through EPAC to up-regulate the expression of claudin-5, to thus decrease endothelial permeability. Overall, these data unravel a regulatory molecular pathway through which tight junctions limit vascular permeability. This will help in the identification of further therapeutic targets for diseases associated with endothelial barrier dysfunction.
Meet the First Author, see p 951
The vascular barrier that separates the blood from the tissues is mainly composed of the endothelium, which is essential for maintenance of vascular hemostasis. Defective barrier function through increased endothelial permeability is a common feature of many pathological processes, such as inflammation and cancers.1 The functional integrity of the endothelial barrier is determined by the fine-tuned organization and activity of the adherens and tight junction complexes localized at the intercellular contacts.2–4
Tight junctions are formed by different types of transmembrane adhesion proteins (eg, occludin, claudins, junctional adhesion molecules) and intracellular proteins (eg, cingulin, afadin, ZO-1–3 [zona occludens 1–3]). Collectively, these constitute a selective barrier to water, solutes, and larger molecules.5 Among the transmembrane proteins, the immunoglobulin superfamily protein JAM-A (junctional adhesion molecule A) has a key role in maintenance of endothelial barrier function.6–10 JAM-A is ubiquitously expressed and regulates several processes, including cell migration, angiogenesis, and stem cell and leukocyte transmigration.11–14 Recent studies have shown that ZO-1 and JAM-A can form a cooperative unit that activates junctional actomyosin, and thus induces endothelial barrier formation.15 Moreover, we have shown previously that JAM-A promotes activation and junctional localization of the small GTPase Rap-1 (Ras-related protein-1) through sustained expression of EPAC (exchange protein directly activated by cAMP)-1 and EPAC-2.13 These proteins are potent activators of Rap-1, which, in turn, enhances endothelial barrier function,16–18 to maintain the correct organization of cell-cell junctions.
The main structural determinant of the paracellular endothelial barrier is claudin-5,19 which belongs to the claudin family. Claudin-5 expression promotes the sealing of tight junctions, and as a consequence, decreased vessel permeability, and thus enhanced endothelial barrier function.20–22 A previous report suggested a link between the expression of JAM-A and claudins. Indeed, in in vitro and in vivo epithelial models, the absence of JAM-A increased the levels of the permeability-enhancing claudin-10 and claudin-15, with no changes in claudin-2 or occludin, which suggested that JAM-A regulates the claudin composition of tight junctions.23 However, the molecular basis of this regulation has received little attention and remains to be better defined.
Claudin-5 expression in endothelial cells is increased by cAMP, a known regulator of endothelial barrier function, through PKA (protein kinase A)-dependent and PKA-independent pathways.24,25 However, the pathway that links JAM-A expression and cAMP-induced claudin-5 expression remains to be clarified.
In the present study, we define a novel signaling circuit that regulates endothelial permeability in vivo and in vitro. We demonstrate that JAM-A induces claudin-5 expression through an EPAC-dependent mechanism that is activated by cAMP. We identify C/EBP-α (CCAAT/enhancer-binding protein-α) as the transcription factor that triggers claudin-5 expression downstream of JAM-A, which decreases endothelial permeability. We reveal that the cAMP-inducible C/EBP-α activity depends on EPAC, whereas C/EBP-α expression is suppressed by active β-catenin.26,27 Indeed, we show that JAM-A deficiency promotes β-catenin signaling, which in turn inhibits C/EBP-α transcription and reduces claudin-5 expression.
It has been reported that the levels of JAM-A, C/EBP-α, and claudin-5 are decreased in different types of cancers.28–31 Here, we observed that the expression of JAM-A, C/EBP-α, and claudin-5 are concomitantly downregulated in the fragile vasculature of glioblastoma multiforme (GBM) and ovarian cancer, which indicates the potential relevance of this signaling pathway for maintenance of vascular integrity in humans.
In summary, our data demonstrate a novel regulatory molecular pathway through which a component of the tight junctions, JAM-A, restricts vascular permeability by enhancing the expression of claudin-5, which is a crucial component of tight junctions. This will help to identify further therapeutic targets for diseases associated with endothelial barrier dysfunction.
Methods
Detailed Methods are available in the Data Supplement.
The data that support the findings of this study are available from the corresponding authors on reasonable request.
Results
JAM-A deficiency reduces the expression of claudin-5, which controls vascular permeability along with Rap-1.
Multiple studies have identified JAM-A as an important component of endothelial and epithelial tight junctions.9,32,33 JAM-A was shown to be required for control of endothelial permeability to plasma solutes and for transmigration of inflammatory and immune cells.23,34 However, the molecular mechanism through which JAM-A limits permeability and maintains the integrity of tight junctions has not been fully elucidated.
To identify transcripts regulated by JAM-A expression, we carried out Affymetrix analysis of gene expression through a comparison of immortalized lung endothelial cells (iLECs) of wild-type (WT) mice and JAM-A–null mice. We examined the endothelial components described in the literature as key molecules for the organization of both tight and adherens junctions. Among the prominently affected genes of the tight junctions, claudin-5 showed greater significant downregulation in the absence of JAM-A (P<0.05, local-pooled error test; fold change >2), whereas adherens junctions molecules were not significantly affected (Figure IA in the Data Supplement).
The differences in the expression levels of claudin-5 were confirmed by real-time quantitative polymerase chain reaction analysis and also at the protein level by immunoblotting and immunofluorescence staining (Figure 1A–1C). Downregulation of claudin-5 expression in iLECs was also observed upon acute depletion of JAM-A with small-interfering RNAs (Figure IB through ID in the Data Supplement).
Figure 1.
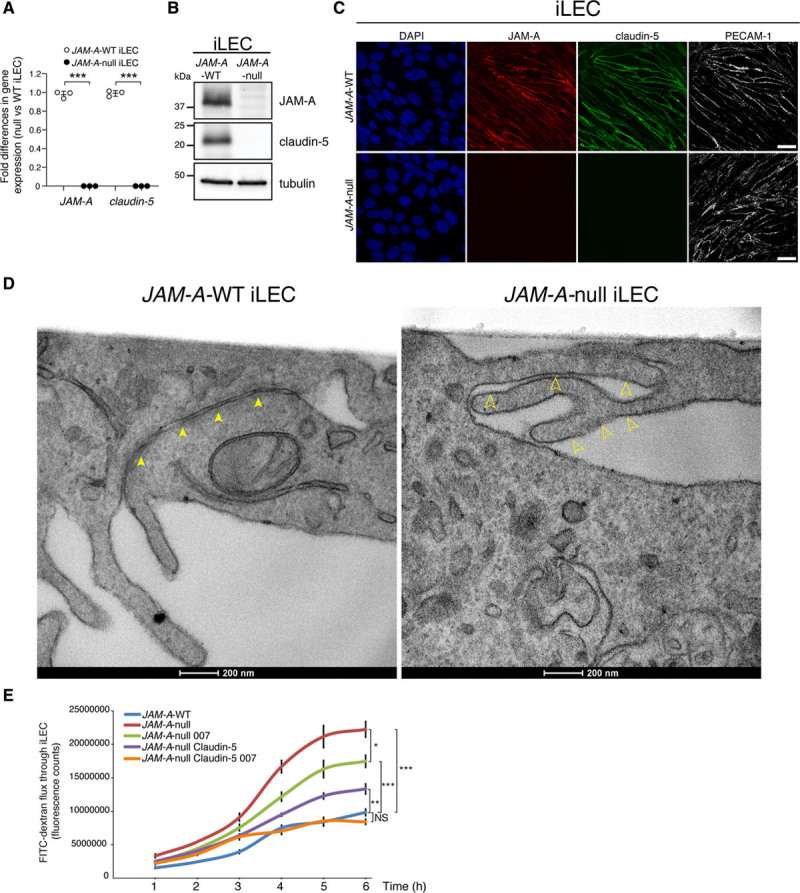
JAM-A (junctional adhesion molecule A) controls endothelial permeability through regulation of claudin-5 expression and Rap-1 (Ras-related protein-1) activation. A, Quantification of fold-differences in JAM-A and claudin-5 gene expression in immortalized lung endothelial cells (iLECs) from JAM-A–wild-type (WT) and JAM-A–null mice following real-time quantitative polymerase chain reaction analysis. Data are means±SD from 3 independent experiments. P value determined by 2-sided unpaired Welch t test. JAM-A–WT vs JAM-A–null: JAM-A ***P=9.54×10–7; claudin-5 ***P=4.17×10–7. B, Representative immunoblotting for JAM-A and claudin-5 JAM-A–WT and JAM-A–null iLECs. Tubulin is shown as the loading control. Data are representative of 3 independent experiments. C, Representative confocal microscopy of JAM-A (red) and claudin-5 (green) expression in JAM-A–WT and JAM-A–null iLECs. PECAM-1 (platelet/endothelial cell adhesion molecule-1; white) is an endothelial junctional marker; DAPI (4′,6-diamidino-2-phenylindole; blue) stains nuclei. Data are representative of 3 independent experiments. Scale bars: 20 µm. D, Representative transmission electron microscopy of JAM-A–WT and JAM-A–null iLECs. Arrows indicate the tight junction strands of the endothelial cells. Scale bars: 200 nm. E, Tracer flux assay. Permeability to fluorescein isothiocyanate (FITC)–dextran of JAM-A–WT and JAM-A–null iLECs and JAM-A–null stably expressing GFP (green fluorescent protein; JAM-A–WT and JAM-A–null) or claudin-5 (JAM-A–null claudin-5) with treatment with vehicle or 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-pCPT-2′-O-Me-cAMP; 007; 100 µmol/L), to activate Rap-1. Data are means±SEM of triplicates from a single experiment, as representative of 3 independent experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. Comparison at 6 h: (overall ****P=5.3×10–7), JAM-A–WT vs JAM-A–null ***P=3×10–4; JAM-A–WT vs JAM-A–null 007 ***P=5×10–4; JAM-A–WT vs JAM-A–null claudin-5 **P=4.3×10–3; JAM-A–WT vs JAM-A–null claudin-5 007, P=0.25, not statistically significant [NS]; JAM-A–null vs JAM-A–null 007 *P=0.0117.
Overall, these data demonstrate a positive role for JAM-A in triggering claudin-5 expression.
To accurately examine tight junction morphology, we used transmission electron microscopy to compare JAM-A–WT and JAM-A–null iLECs. As shown in Figure 1D, in comparison with the control JAM-A–WT, the absence of JAM-A led to discontinuous tight junction strands, with widening of the paracellular spaces, which suggested that these junctions were not formed correctly.
As claudin-5 is the major determinant in the selectivity of paracellular barriers,20–22 the overall data prompted us to postulate that its downregulation in the absence of JAM-A is responsible for increased vascular permeability. To address this point, in vitro permeability was determined through measurement of the passage of fluorescein isothiocyanate (FITC)–labeled dextran across monolayers of JAM-A–WT and JAM-A–null iLECs and after restoring claudin-5 expression in JAM-A–null iLECs. The expression and junctional localization of claudin-5 were evaluated using immunofluorescence and Western blotting, respectively (Figure IE and IF in the Data Supplement). As shown in Figure 1E, endothelial permeability was increased in the absence of JAM-A compared with WT cells. Moreover, reestablishment of claudin-5 expression in these JAM-A–null iLECs showed 72% recovery of the permeability phenotype, which indicated that claudin-5 controls endothelial barrier function downstream of JAM-A (Figure 1E; Figure IE through IG in the Data Supplement).
Our previous study showed that JAM-A promotes activation and junction localization of the small GTPase Rap-1 through sustained expression of EPAC-1 and EPAC-2.13 These last 2 proteins are potent activators of Rap-1, which maintains the correct organization of cell-cell junctions and enhances endothelial barrier function.16–18 Thus, we hypothesized that JAM-A regulates both claudin-5–dependent and Rap-1–dependent control of endothelial permeability. Indeed, complete rescue of endothelial function was obtained by restoring both claudin-5 expression and activating Rap-1 using the EPAC-specific agonist 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-pCPT-2′-O-Me-cAMP; known as 007) in JAM-A–null iLECs (Figure 1E).
Taken together, these data show that JAM-A is an important regulator of vascular permeability that acts through the control of both claudin-5 expression and Rap-1 activation.
Cyclic AMP Increases Claudin-5 Expression Through EPAC Signaling, Which Is Impaired in the Absence of JAM-A
To unravel the molecular mechanisms that underlie reduced expression of claudin-5 in the absence of JAM-A, we analyzed the different factors that might be involved in the regulation of claudin-5 expression. cAMP has been reported to enhance barrier function of tight junctions in brain endothelial cells through both PKA-dependent and PKA-independent induction of claudin-5 expression.24,25,35 Moreover, the main cAMP targets are PKA and EPAC-1/2, which act in concert to tighten the endothelial barrier. Based on this evidence, we hypothesized that PKA- and EPAC-dependent pathways sustain claudin-5 expression in endothelial cells.
Therefore, we investigated different membrane-permeable cAMP analogs for their impact on claudin-5 expression. Treatment of JAM-A–WT iLECs with 8-chlorophenylthio-cAMP, which activates PKA and EPAC-1/2, increased claudin-5 levels by 4-fold, whereas in the absence of JAM-A, this was lower (1.5-fold; Figure 2A). Although pretreatment with the PKA inhibitor H89 did not affect the basal levels of claudin-5, this reduced 8-chlorophenylthio-cAMP–mediated claudin-5 induction by 50%, which confirmed that cAMP increases claudin-5 expression via PKA-dependent and PKA-independent (potentially EPAC-mediated) pathways in iLECs (Figure 2A). Moreover, in the absence of JAM-A, claudin-5 expression induced by 8-chlorophenylthio-cAMP was completely abrogated by H89, which suggests that the claudin-5 expression is controlled by PKA alone. Thus, we hypothesized that in JAM-A–null iLECs, EPAC-dependent claudin-5 expression is affected by the low expression of EPAC-1/2.13 Indeed, treatment with 007 increased the claudin-5 mRNA levels only in the presence of JAM-A (by 2-fold), whereas it failed to do so in the absence of JAM-A, which confirmed that JAM-A regulates claudin-5 expression through EPAC (Figure 2B). Finally, to rule out a role for JAM-A in PKA-dependent regulation of claudin-5, we analyzed the activation of CREB (cAMP response-element binding protein), a downstream effector of PKA.36 Here, comparison of JAM-A–WT and JAM-A–null iLECs showed no differences in CREB phosphorylation at serine residue 13336 (Figure IIA and IIB in the Data Supplement).
Figure 2.
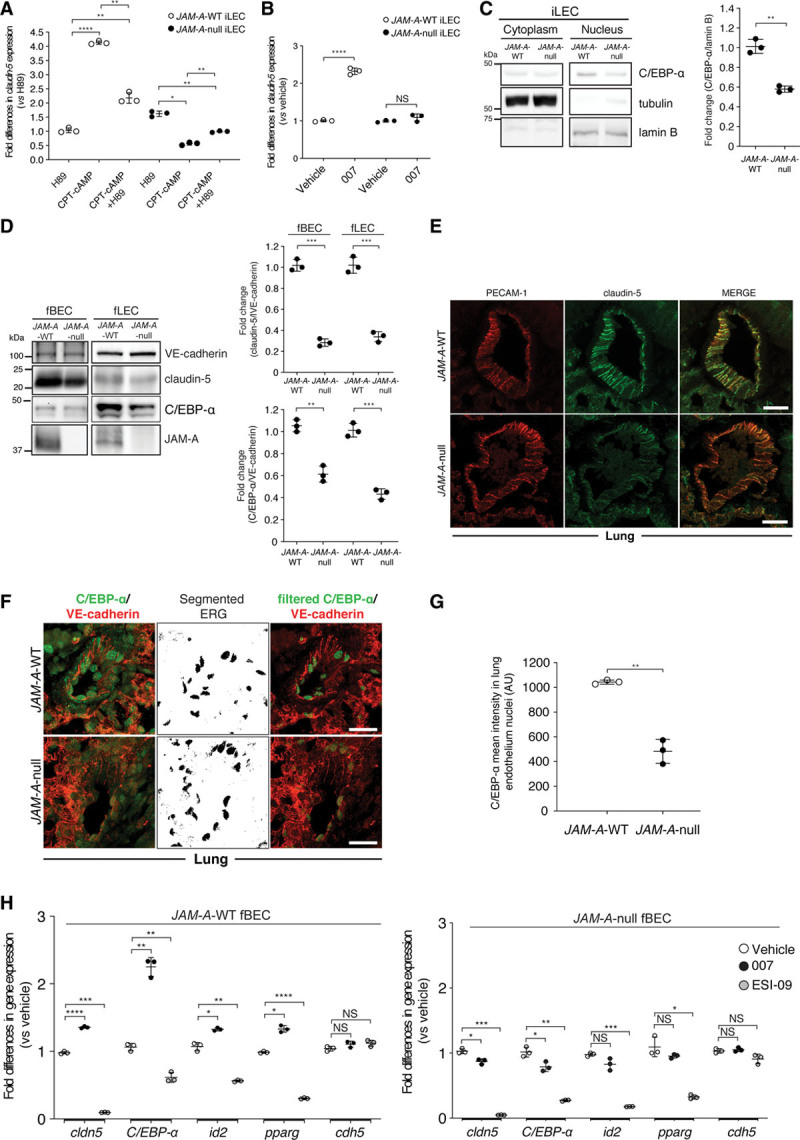
JAM-A (junctional adhesion molecule A) sustains claudin-5 expression through an EPAC (exchange protein directly activated by cAMP)-dependent mechanism and C/EBP-α (CCAAT/enhancer-binding protein-α) regulation. A and B, Quantification of fold-differences in claudin-5 gene expression in JAM-A–wild-type (WT) and JAM-A–null immortalized lung endothelial cells (iLECs) following real-time quantitative polymerase chain reaction (RT-qPCR) analysis. Cells were treated with vehicle (A and B; fold-difference, 1.0) and the specific PKA (protein kinase A) inhibitor H89 (10 µmol/L), and the PKA and EPAC activator CPT-cAMP (8-(4-chlorophenylthio)adenosine 3′,5′-cyclic monophosphate sodium salt; 250 µmol/L) alone and in combination with H89 (A), or selective EPAC activator 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-pCPT-2′-O-Me-cAMP; 007; 100 µmol/L; B). Data are means±SD from 3 independent experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. JAM-A–WT: (overall ***P=10–4), H89 vs CPT-cAMP ****P=2×10–5; H89 vs CPT-cAMP +H89 **P=5.6×10–3; CPT-cAMP vs CPT-cAMP+H89 **P=7×10–3; JAM-A–null: (overall ***P=2.2×10–4), H89 vs CPT-cAMP *P=1.67×10–2; H89 vs CPT-cAMP+H89 **P=2.4×10–3; CPT-cAMP vs CPT-cAMP+H89 **P=10–3. B, P values determined by 2-sided unpaired Welch t test. JAM-A–WT: vehicle vs 007 ***P=6×10–4; JAM-A–null: vehicle vs 007 P=0.23, not statistically significant (NS). C, left, Representative immunoblotting for nuclear/cytoplasm distribution of C/EBP-α in JAM-A–WT and JAM-A–null iLECs. Tubulin and lamin B are shown as cytoplasmic and nuclear markers, respectively. Right, C/EBP-α/lamin B ratio quantified by densitometry scan and expressed as fold changes. Data are means±SD from 3 independent experiments. P value determined by 2-sided unpaired Welch t test. C/EBP-α: JAM-A–WT vs JAM-A–null **P=3.2×10–3. D, left, Representative immunoblotting for JAM-A, claudin-5, and C/EBP-α in freshly isolated endothelial cells from brains (fBECs) and lungs (fLECs) of JAM-A–WT and JAM-A–null mice. VE-cadherin (vascular endothelial cadherin) is shown as loading control for endothelial cell numbers. Right, Claudin-5/VE-cadherin and C/EBP-α/VE-cadherin ratios quantified by densitometry scan and expressed as fold changes. Data are means±SD from 3 independent experiments. P values determined by 2-sided unpaired Welch t test. Claudin-5: fBEC, JAM-A–WT vs JAM-A–null ***P=1.2×10-4; fLEC, JAM-A–WT vs JAM-A–null ***P=5.4×10-4; C/EBP-α: fBEC, JAM-A–WT vs JAM-A–null **P=1.5×10-3; fLEC, JAM-A–WT vs JAM-A–null ***P=2.9×10-4. E, Representative confocal microscopy for PECAM-1 (platelet/endothelial cell adhesion molecule-1; red) and claudin-5 (green) in paraffin-embedded lung sections from JAM-A–WT and JAM-A–null mice (n=3, JAM-A–WT; n=3, JAM-A–null). Sections were 4 μm thick. Scale bars: 20 µm. F, Representative confocal microscopy for C/EBP-α (green) and VE-cadherin (red) in paraffin-embedded lung sections from JAM-A–WT and JAM-A–null mice. As C/EBP-α was detected in the cytoplasm of the perivascular cells in the lung, ERG (ETS-related gene) staining (nuclear marker) was segmented with threshold 350-4.096 to isolate nuclear C/EBP-α (middle), for merged images of filtered C/EBP-α and VE-cadherin (right). Sections were 4 μm thick. Scale bars: 20 μm (n=3, JAM-A–WT; n=3, JAM-A–null). G, Quantification of nuclear C/EBP-α signal in lung vessels sections from JAM-A–WT and JAM-A–null mice as nuclear C/EBP-α mean fluorescence intensity in 4 sections containing at least 10 nuclei, expressed as arbitrary units (AU); n=3, JAM-A–WT; n=3, JAM-A–null, means±SD. P value determined by 2-sided unpaired Welch t test. JAM-A–WT vs JAM-A–null **P=8.3×10-3. H, Quantification of fold-differences for cldn-5, C/EBP-α, id2, pparg, cdh5 expression in JAM-A–WT (left) and JAM-A–null (right) fBECs following RT-qPCR analysis. Cells were treated with vehicle (fold-difference, 1.0), the selective EPAC activator 007 (100 µmol/L), and the specific EPAC inhibitor ESI-09 (10 µmol/L). Data are means±SD from 3 independent experiments (n=12, JAM-A–WT; n=12, JAM-A–null mice; for each experiment). P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. JAM-A–WT fBECs: cldn-5: (overall ****P=6.3×10-8), vehicle vs 007 ****P=2.4×10–5; vehicle vs ESI-09 ***P=2.6×10–4; C/EBP-α: (overall ***P=1.1×10-4), vehicle vs 007 **P=1.4×10–3; vehicle vs ESI-09 **P=1.9×10–3; id2: (overall ***P=6.4×10-4), vehicle vs 007 *P=1.1×10–2; vehicle vs ESI-09 **P=8.4×10–3; pparg: (overall ***P=3.4×10-4), vehicle vs 007 *P=1.1×10–2; vehicle vs ESI-09 ****P=2.9×10–7; cdh5: (overall P=0.12, NS), vehicle vs 007 P=0.21, NS; vehicle vs ESI-09 P=0.13, NS. JAM-A–null fBECs: cldn-5: (overall ****P=5.7×10-6), vehicle vs 007 *P=1.4×10–2; vehicle vs ESI-09 ***P=8.4×10–4; C/EBP-α: (overall ***P=2.5×10-4), vehicle vs 007 *P=3.7×10–2; vehicle vs ESI-09 **P=5.8×10–3; id2: (overall **P=2.6×10-3), vehicle vs 007 P=0.17, NS; vehicle vs ESI-09 ***P=6×10–4; pparg: (overall **P=9.2×10-3), vehicle vs 007 P=0.39, NS; vehicle vs ESI-09 *P=2.1×10–2; cdh5: (overall P=0.058, NS), vehicle vs 007 P=0.74, NS; vehicle vs ESI-09 P=0.14, NS.
Overall, these data demonstrate that JAM-A regulates claudin-5 expression through an EPAC-dependent mechanism.
JAM-A Deficiency Reduces Expression of C/EBP-α
Evidence suggested that cAMP can also regulate gene expression through nonclassical routes that are independent of CREB and PKA.37 In this context, a group of transcription factors known as C/EBPs has been shown to have cAMP-inducible activities that are dependent on EPAC.38,39 To date, 6 C/EBP genes have been identified (α, β, γ, δ, ε, ζ). These all function as master regulators of cellular processes, including cell proliferation and differentiation, and inflammatory responses.40 As we showed that EPAC is involved in the regulation of claudin-5 expression, we investigated whether the expression of specific C/EBP isoforms is affected by JAM-A. First, we compared mRNA and protein expression levels of the different C/EBP isoforms in JAM-A–WT and JAM-A–null iLECs. Real-time polymerase chain reaction and immunoblotting revealed that C/EBP-γ and C/EBP-δ levels remained unchanged, whereas the C/EBP-β isoforms, known as LIP and LAP, were upregulated in JAM-A–null iLECs (Figure IIC and IID in the Data Supplement). Conversely, C/EBP-α was significantly downregulated in the absence of JAM-A, which suggests that it is involved in the regulation of claudin-5 downstream of EPAC (Figure IIC and IID in the Data Supplement).
To further strengthen this evidence, we compared JAM-A–WT and JAM-A–null iLECs in terms of C/EBP-α nuclear localization and transcriptional activity. Nuclear C/EBP-α levels in the JAM-A–null cells were significantly reduced compared with the JAM-A–WT counterpart (Figure 2C; Figure IIE and IIF in the Data Supplement). Moreover, significant reduction in C/EBP-α expression was detected upon small-interfering RNA–mediated acute depletion of JAM-A in iLECs (Figure IIG in the Data Supplement). Consistent with this, when C/EBP-α transcriptional activity was analyzed by monitoring mRNA levels of its known target genes (ie, csf1r, csfr3, id2, pparg),41 these were significantly lower in JAM-A–null iLECs compared with the WT (Figure IIH in the Data Supplement).
To determine the physiological relevance of these in vitro findings, we analyzed claudin-5 and C/EBP-α protein levels in freshly isolated endothelial cells from different organs (ie, freshly isolated lung endothelial cells [fLECs] and freshly isolated brain endothelial cells) obtained from the JAM-A–WT and JAM-A–null mice. We first tested the preparation purity using real-time polymerase chain reaction for cell-specific markers. The endothelial cell enrichment in comparison to other cell types from the brain and lungs was measured. As reported in Figure IIIA in the Data Supplement, both the fLEC and freshly isolated brain endothelial cells preparations showed strong enrichment of endothelial cell–specific markers, with barely detectable contamination of other cell types, such as pericytes, smooth muscle cells, epithelial cells, fibroblasts, and astrocytes. Consistent with the previous in vitro data, the absence of JAM-A led to significant decreases in claudin-5 and C/EBP-α protein levels compared with the WT controls (Figure 2D). Densitometric analysis showed 30% downregulation of claudin-5 expression in both lungs and brain (Figure 2D), whereas C/EBP-α protein levels were reduced by 40% in fLECs and 60% in freshly isolated brain endothelial cells (Figure 2D). In vivo immunofluorescence analysis of the lungs confirmed significant downregulation of claudin-5 and C/EBP-α expression in blood vessels that lacked JAM-A, with no changes in the levels of PECAM-1 (platelet/endothelial cell adhesion molecule-1) and VE-cadherin (vascular endothelial cadherin; Figure 2E through 2G). Furthermore, in both fLECs and iLECs, JAM-A expression did not affect the levels of additional tight junction and adherens junction proteins, which suggests that the iLECs are a suitable cellular model for further studies (Figure IIIB in the Data Supplement).
Finally, to confirm EPAC activity in the brain vessels, we examined the impact of its activation and inhibition in cultured endothelial cells from brain isolated from JAM-A–WT and JAM-A–null mice. Treatment with the EPAC-specific agonist 007 led to a significant increase in the mRNA and protein levels of claudin-5 and C/EBP-α and its targets only in the presence of JAM-A, whereas it failed to do so in the absence of JAM-A (Figure 2H; Figure IIIC in the Data Supplement). Conversely, the EPAC inhibitor ESI-09 reduced mRNA and protein levels of EPAC downstream effectors in both JAM-A–WT and JAM-A–null endothelial cells from brain. As expected, neither of the 007 and ESI-09 treatments affected the mRNA levels of cdh5 (Figure 2H; Figure IIIC in the Data Supplement).
Thus, these data demonstrate that JAM-A sustains C/EBP-α expression, which suggests a role for C/EBP-α in EPAC-mediated claudin-5 induction in endothelial cells.
JAM-A Triggers Claudin-5 Expression Through C/EBP-α and Regulates Endothelial Permeability
To investigate the specific role of C/EBP-α in the regulation of claudin-5 expression, we used several approaches to modulate C/EBP-α expression and its activity as a transcription factor. First, we induced acute C/EBP-α downregulation using small-interfering RNAs. This markedly reduced the C/EBP-α mRNA and protein expression, by ≈75% to 80% compared with the control (Figure 3A and 3B). This reduction of C/EBP-α led to significant decreases in claudin-5 expression at both the mRNA and protein levels, compared with control cells (Figure 3A and 3B). Second, we stably overexpressed full-length C/EBP-α and a 30-kDa C/EBP-α dominant-negative (C/EBP-α-DN)42 in both JAM-A–WT and JAM-A–null iLECs. As shown in Figure 3C and 3D, C/EBP-α-DN decreased claudin-5 mRNA and protein expression, whereas these were upregulated by full-length-C/EBP-α (C/EBP-α-WT). The fold-differences in claudin-5 expression induced by C/EBP-α and C/EBP-α-DN were similar in JAM-A–WT and JAM-A–null cells (Figure 3C). When C/EBP-α-WT was expressed, the absolute level of claudin-5 in JAM-A–null iLECs was ≈4% (ie, 0.0417-fold) that of the WT counterpart, although the levels of C/EBP-α-WT were comparable in both cell types (Figure 3C, p42 isoform, highlighted in blue). This suggests an inhibitory mechanism in JAM-A–null cells, where claudin-5 expression is maintained low even in the presence of C/EBP-α. This inhibitory mechanism is explored further below. Consistent with this, the mRNA levels of the selected C/EBP-α target genes id2 and pparg showed similar trends to that of claudin-5 (Figure 3D).
Figure 3.
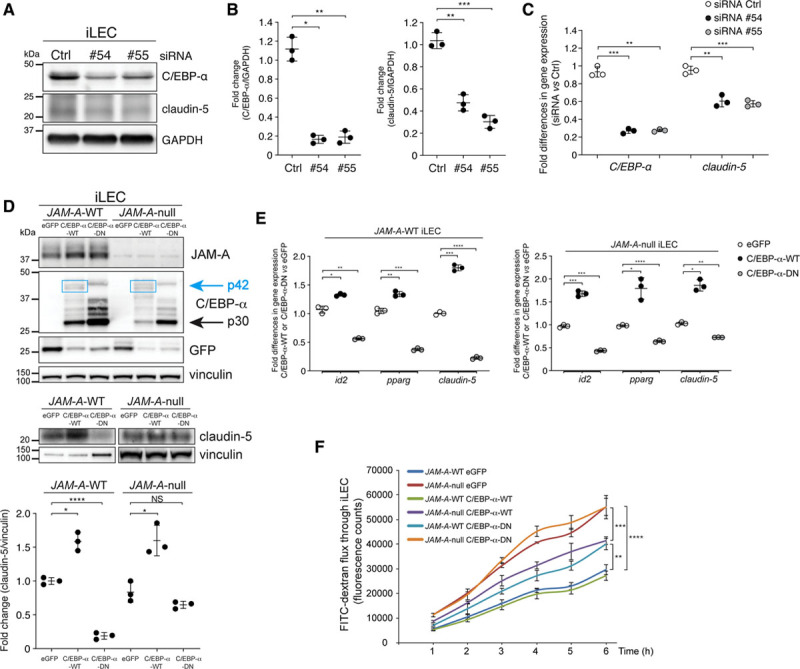
C/EBP-α (CCAAT/enhancer-binding protein-α) controls the endothelial barrier through claudin-5 expression. A, left, Representative immunoblotting for C/EBP-α and claudin-5 in immortalized lung endothelial cells (iLECs) transfected with nontargeting small-interfering RNA (siRNA; Ctrl [control]) or with siRNAs against C/EBP-α (no. 54, no. 55). GAPDH is shown as loading control. Right, C/EBP-α/GAPDH and claudin-5/GAPDH ratios quantified by densitometry scanning and expressed as fold changes. Data are means±SD from 3 independent experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. C/EBP-α: (overall ***P=5.8×10-4), siRNA Ctrl vs siRNA no. 54 *P=1×10-2, siRNA Ctrl vs siRNA no. 55 **P=2.5×10-3. Claudin-5: (overall ****P=3.9×10-5), siRNA Ctrl vs siRNA no. 54 **P=1.3×10-3; siRNA Ctrl vs siRNA no. 55 ***P=2.9×10-4. B, Quantification of fold-differences for C/EBP-α and claudin-5 gene expression in iLECs following real-time quantitative polymerase chain reaction analysis, as shown in A. Cells were treated with nontargeting siRNA (siRNA Ctrl; fold-difference, 1.0) and siRNAs against C/EBP-α (no. 54, no. 55). Data are means±SD from 3 independent experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. C/EBP-α: (overall ***P=2.7×10-4), siRNA Ctrl vs siRNA no. 54 ***P=6.5×10-4; siRNA Ctrl vs siRNA no. 55 **P=4.4×10-3. Claudin-5: (overall ***P=5.1×10-4), siRNA Ctrl vs siRNA no. 54 **P=2.9×10-3; siRNA Ctrl vs siRNA no. 55 ***P=6.3×10-4. C, Representative immunoblotting for JAM-A (junctional adhesion molecule A), C/EBP-α (upper) and claudin-5 (lower) in JAM-A–wild-type (WT) and JAM-A–null iLECs stably expressing full-length C/EBP-α-WT (p42), C/EBP-α-dominant-negative (DN; p30), and eGFP (enhanced green fluorescent protein) as control. Vinculin is shown as loading control. Upper, Blue boxes highlight bands corresponding to full-length C/EBP-α (p42 isoform). Bottom, 10 μg JAM-A–WT and 80 μg JAM-A–null were loaded. Claudin-5/vinculin ratios quantified by densitometry scanning and expressed as fold changes. Data are means±SD from 3 independent experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. JAM-A–WT: (overall ***P=5.5×10-4), eGFP vs C/EBP-α-WT *P=10-2; eGFP vs C/EBP-α-DN ****P=6.7×10-5; JAM-A–null: (overall **P=5.2×10-3), eGFP vs C/EBP-α-WT *P=2.8×10-2; eGFP vs C/EBP-α-DN P=0.27, not statistically significant (NS). D, Quantification of fold-differences for id2, pparg, and claudin-5 gene expression in JAM-A–WT (left) and JAM-A–null (right) iLECs, under stable expression of eGFP (control; fold-difference 1.0) or C/EBP-α-WT or C/EBP-α-DN. Data are means±SD from 3 independent experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. JAM-A–WT iLECs: id2: (overall ***P=6.4×10-4), eGFP vs C/EBP-α-WT *P=1.1×10–2; eGFP vs C/EBP-α-DN **P=8.4×10–3; pparg: (overall ****P=4.1×10-6), eGFP vs C/EBP-α-WT **P=2.6×10–3; eGFP vs C/EBP-α-DN ***P=3×10–4; cldn-5: (overall ****P=2.8×10-6), eGFP vs C/EBP-α-WT ***P=3×10–4; eGFP vs C/EBP-α-DN ****P=4.6×10–6. JAM-A–null iLECs: id2: (overall ****P=3.9×10-5), eGFP vs C/EBP-α-WT ***P=6.6×10–4; eGFP vs C/EBP-α-DN ***P=2.1×10–4; pparg: (overall *P=1.5×10-2), eGFP vs C/EBP-α-WT *P=4.3×10–2; eGFP vs C/EBP-α-DN ****P=5.9×10–5; cldn-5: (overall **P=2.9×10-3), eGFP vs C/EBP-α-WT *P=1.2×10–2; eGFP vs C/EBP-α-DN **P=5.9×10–3. E, Quantification of permeability to fluorescein isothiocyanate (FITC)–dextran (40 kDa) in JAM-A–WT and JAM-A–null iLECs stably expressing full-length C/EBP-α-WT (p42), C/EBP-α-DN (p30), and eGFP as control. Data are means±SEM of triplicates from a single experiment, representative of 3 independent experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. Comparison at 6 h: (overall ****P=7.8×10–7), JAM-A–WT eGFP vs JAM-A–null eGFP ****P=1.3×10–5; JAM-A–WT eGFP vs JAM-A–WT C/EBP-α-DN **P=1.7×10–3; JAM-A–null eGFP vs JAM-A–null C/EBP-α-DN null ***P=6.9×10–4.
Similar data on claudin-5 protein expression and regulation of C/EBP-α target genes at the mRNA level were obtained using transient overexpression of C/EBP-α-WT and C/EBP-α-DN (Figure IIID and IIIE in the Data Supplement). Taken together these data demonstrate that C/EBP-α is a novel regulator of claudin-5 expression.
Finally, we sought to determine whether the increase in claudin-5 expression induced by C/EBP-α can reverse the higher endothelial permeability caused by the absence of JAM-A. For this purpose, we performed in vitro permeability assays to compare JAM-A–WT and JAM-A–null iLECs infected with vectors to stably express full-length C/EBP-α-WT or C/EBP-α-DN, or eGFP (enhanced green fluorescent protein) as the control. As shown in Figure 3E, the expression of C/EBP-α-WT in JAM-A–null cells (JAM-A–null C/EBP-α-WT) decreased the permeability compared with the control cells (JAM-A–null eGFP). This suggests that the increase in claudin-5 expression that is mediated by the transcription factor C/EBP-α induces the tightening of the junctions. As expected, in JAM-A–null C/EBP-α-WT–expressing cells, the permeability defect was not completely rescued, as C/EBP-α failed to re-establish claudin-5 expression to the level of the JAM-A–WT control cells, as previously shown (Figure 3C; Figure IIIF in the Data Supplement).
Conversely, the expression of C/EBP-α-DN in JAM-A–WT iLECs increased the permeability compared with the control cells (JAM-A–WT eGFP), due to the downregulation of claudin-5 (see Figure 3C; Figure IIIF in the Data Supplement). This set of data confirms the novel role of the transcriptional activity of C/EBP-α in the control of endothelial barrier permeability (Figure 3E).
JAM-A Promotes C/EBP-α Binding to the Claudin-5 Promoter and Induces Gene Transcription
We then wanted to determine how C/EBP-α acts on the claudin-5 gene to induce its expression. As 3 putative C/EBP-α binding sites were identified within the claudin-5 promoter region within −6.0 kb of the transcription start site, we hypothesized that the claudin-5 gene is a direct transcriptional target of C/EBP-α (Figure IVA in the Data Supplement).
In chromatin immunoprecipitation assays, C/EBP-α binding was enriched at the selected sites on the claudin-5 promoter in the JAM-A–WT cells, compared with the JAM-A–null iLECs (Figure 4A). This suggested that C/EBP-α sustains claudin-5 transcription by directly binding to its promoter and that this mechanism is compromised in the absence of JAM-A, where C/EBP-α is downregulated (Figure 4A) and less active.
Figure 4.
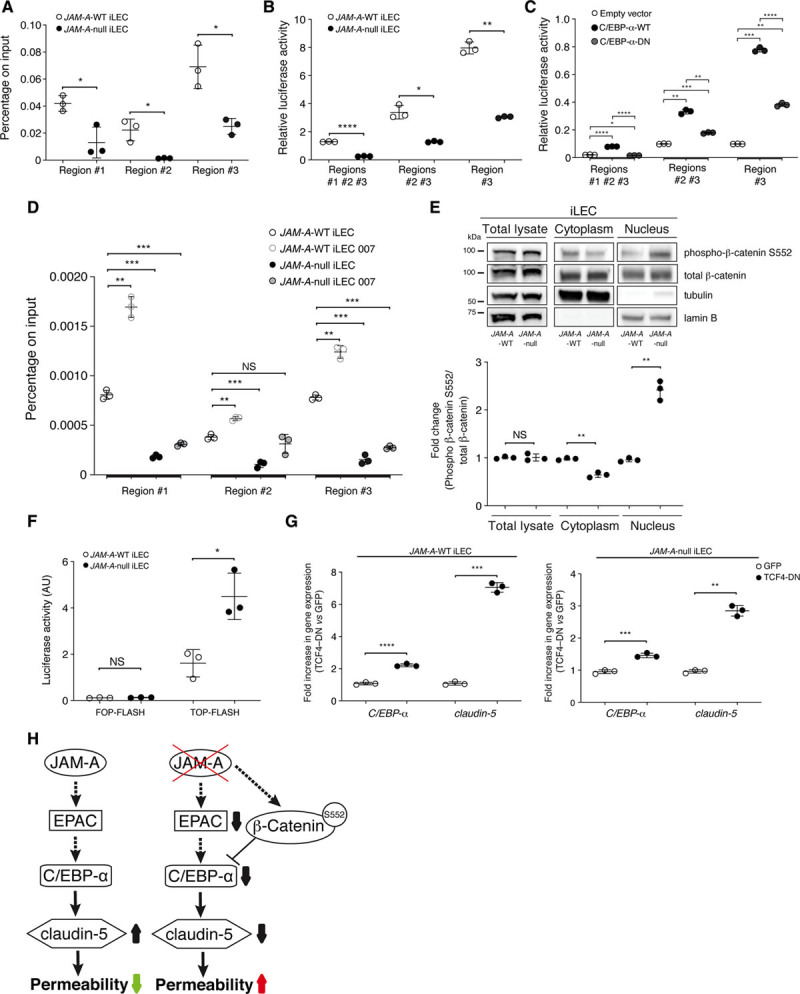
JAM-A (junctional adhesion molecule A) promotes upregulation of claudin-5 expression via C/EBP-α (CCAAT/enhancer-binding protein-α), which is inhibited by active β-catenin. A, Quantification of chromatin immunoprecipitation analysis of C/EBP-α binding to the claudin-5 promoter in JAM-A–wild-type (WT) and JAM-A–null immortalized lung endothelial cells (iLECs; for details see Figure IVA in the Data Supplement). DNA levels were normalized to the input. Data are means±SD of triplicates from a single experiment, as representative of 3 experiments. P values determined by 2-sided unpaired Welch t test. JAM-A–WT vs JAM-A–null: region no. 1 *P=3×10-2; region no. 2 *P=4.5×10-2; region no. 3 *P=2.9×10-2. B, Quantification of transcriptional reporter assays in JAM-A–WT and JAM-A–null iLECs under overexpression of luciferase reporters for all 3 C/EBP-α binding site regions of the claudin-5 promoter (region no. 1, no. 2, no. 3), or both region no. 1 and no. 2 (region no. 1 and no. 2), or only region no. 3 (region no. 3; for details see Figure IVB in the Data Supplement). Data are means±SD of triplicates from a single experiment, representative of 3 experiments. P values determined by 2-sided unpaired Welch t test. JAM-A–WT vs JAM-A–null: region no. 1, no. 2, no. 3 ****P=4.9×10-7; region no. 2 and no. 3 *P=1.6×10-2; region no. 3 **P=1.9×10-3. C, Quantification of transcriptional reporter assays in HEK-293T cells overexpressing claudin-5 promoter-reporter plasmids (as in B) together with empty vector as control, and full-length C/EBP-α-WT, or C/EBP-α–dominant-negative (DN), as indicated. Data are means±SD of triplicates from a single experiment, as representative of 3 experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. Region no. 1, no. 2, and no. 3: (overall ****P=1.8×10-7), empty vector vs C/EBP-α-WT ****P=3.2×10-6; empty vector vs C/EBP-α-DN *P=3.2×10-2; C/EBP-α-WT vs C/EBP-α-DN ****P=2.2×10-5; region no. 2 and no. 3: (overall ***P=7×10-4), empty vector vs C/EBP-α-WT **P=3.3×10-3; empty vector vs C/EBP-α-DN ***P=3×10-4; C/EBP-α-WT vs C/EBP-α-DN **P=8.1×10-3; region no. 3: (overall ****P=9.2×10-7), empty vector vs C/EBP-α-WT ***P=3.7×10-4; empty vector vs C/EBP-α-DN **P=1.3×10-3; C/EBP-α-WT vs C/EBP-α-DN ****P=10-5. D, Quantification of chromatin immunoprecipitation analysis of C/EBP-α binding to the claudin-5 promoter in JAM-A–WT and JAM-A–null iLECs treated with vehicle as control, or with 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-pCPT-2′-O-Me-cAMP; 007). DNA levels were normalized to the input. Data are means±SD of triplicates from a single experiment, representative of 3 experiments. P values determined by Brown-Forsythe ANOVA followed by Dunnett T3 test for multiple comparisons. Region no. 1: (overall ***P=2.1×10-4), JAM-A–WT vs JAM-A–WT 007 **P=2.1×10-3; JAM-A–WT vs JAM-A–null ***P=4.8×10-4; JAM-A–WT vs JAM-A–null 007 ***P=9.3×10-4; region no. 2: (overall **P=7.4×10-3), JAM-A–WT vs JAM-A–WT 007 **P=1.5×10-3; JAM-A–WT vs JAM-A–null ***P=8.7×10-3; JAM-A–WT vs JAM-A–null 007 P=0.64, not statistically significant (NS); region no. 3: (overall ****P=4×10-6), JAM-A–WT vs JAM-A–WT 007 **P=3.2×10-3; JAM-A–WT vs JAM-A–null ***P=5.8×10-4; JAM-A–WT vs JAM-A–null 007 ***P=2.1×10-4. E, Representative immunoblotting for total lysate, cytoplasm and nuclear distribution of total and active β-catenin S552 (phosphor-β-catenin) in JAM-A–WT and JAM-A–null iLECs. Tubulin and lamin B are shown as cytoplasmic and nuclear markers, respectively. Bottom, phospho-β-catenin S552/total- β-catenin ratios quantified by densitometry scan and expressed as fold changes. Data are means±SD from 3 independent experiments. P values determined by 2-sided unpaired Welch t test. Total lysate: JAM-A–WT and JAM-A–null, P=0.94, NS; cytoplasm: JAM-A–WT and JAM-A–null **P=3×10-3; nucleus: JAM-A–WT and JAM-A–null **P=4.9×10-3. F, Quantification of transcriptional activity of TCF (T-cell-factor)–β-catenin in JAM-A–WT and JAM-A–null iLECs underexpression of TOP-FLASH or FOP-FLASH constructs. Data are means±SD from 3 independent experiments. P values determined by 2-sided unpaired Welch t test. JAM-A–WT and JAM-A–null: FOP-FLASH P=0.38, NS; TOP-FLASH, *P=1.9×10-2. G, Quantification of fold-differences of C/EBP-α and claudin-5 gene expression in JAM-A–WT (left) and JAM-A–null (right) iLECs, underexpression of GFP (green fluorescent protein) or TCF4-DN. Data are means±SD from 3 independent experiments. P values determined by 2-sided unpaired Welch t test. C/EBP-α: JAM-A–WT: GFP vs TCF4-DN ****P=8.6×10–6; claudin-5: JAM-A–WT: GFP vs TCF4-DN ***P=2.3×10–4; JAM-A–null: GFP vs TCF4-DN ***P=7.1×10–4; claudin-5: JAM-A–null: GFP vs TCF4-DN **P=10–3. H, Schematic model of JAM-A signaling in the regulation of vascular permeability. Full arrows, direct effects on downstream targets; dashed arrows, regulation of downstream targets (eg, mRNA, protein expression or activity). The molecular mechanisms of this regulatory pathway remain to be defined. AU indicates arbitrary units; and EPAC, exchange protein directly activated by cAMP.
To confirm these data, the sequences of the claudin-5 promoter were cloned upstream of the firefly luciferase gene; these comprised all 3 of the C/EBP-α binding site regions (ie, no. 1, no. 2, no. 3) or regions no. 1 and no. 2, or only region no. 3 (Figure IVB in the Data Supplement). As expected, the transfection of these reporters into iLECs resulted in higher luciferase activities in the presence of JAM-A (Figure 4B).
To demonstrate that the binding of C/EBP-α to the claudin-5 promoter is functionally active, a transcriptional reporter assay was performed in HEK-293T cells, where claudin-5 reporter constructs were cotransfected with plasmids that drive the overexpression of C/EBP-α-WT or C/EBP-α-DN, respectively (Figure IVC in the Data Supplement). Here, C/EBP-α-WT induced claudin-5 promoter activity, and this effect was reduced when the C/EBP-α transcriptional activity was compromised (C/EBP-α-DN; Figure 4C).
Finally, to determine whether EPAC activation regulates the binding of C/EBP-α to the claudin-5 promoter, chromatin immunoprecipitation assays were performed in the presence of 007. Indeed, this treatment strengthened C/EBP-α binding to the claudin-5 promoter only in the presence of JAM-A, with no effects seen in the absence of JAM-A. These last data support the concept that EPAC is required for C/EBP-α binding to the claudin-5 promoter through JAM-A (Figure 4D).
Overall, our data demonstrate that C/EBP-α increases claudin-5 expression in response to EPAC activation. This signaling pathway is positively regulated by JAM-A.
JAM-A Deficiency Increases β-Catenin Signaling, Which In Turn Reduces Claudin-5 Expression Through C/EBP-α Inhibition
Previous studies have demonstrated that activation of Wnt/β-catenin signaling suppresses the expression of C/EBP-α mRNA.26,27 JAM-A negatively regulates β-catenin signaling in epithelial cells.11 Based on this evidence, we hypothesized that JAM-A depletion enhances β-catenin signaling, to thus inhibit C/EBP-α and claudin-5 expression. Therefore, we compared JAM-A–WT and JAM-A–null cells in terms of the nuclear localization and transcriptional signaling of β-catenin. Here, the total amount of active β-catenin that was phosphorylated at Ser552 was similar in the JAM-A–WT and JAM-A–null iLECs and fLECs (Figure 4E; Figure IVD through IVF in the Data Supplement). Conversely, in the absence of JAM-A, there was marked increase in the levels of active nuclear β-catenin, compared with the WT counterpart (Figure 4E; Figure IVE and IVF in the Data Supplement). Consistent with this, β-catenin transcriptional activity was detected by measurement of the expression of its target genes axin2 and ccnd1 and using a TCF (T-cell-factor) luciferase reporter, which showed that it was significantly higher in the absence of JAM-A (Figure 4F; Figure IVG in the Data Supplement). Thus, we can conclude here that JAM–A regulates β–catenin/TCF-mediated gene transcription.
To determine whether β-catenin contributes to C/EBP-α and claudin-5 regulation, JAM-A–WT and JAM-A–null iLECs were treated with Wnt-3a (wingless-type MMTV integration site family, member 3a), which stabilizes β-catenin,43 to trigger the expression of its target genes. Indeed, Wnt-3a upregulated axin2, while it inhibited C/EBP-α and claudin-5 at the mRNA level, compared with vehicle-treated cells (Figure IVH in the Data Supplement). In contrast, inhibition of endogenous β-catenin signaling using a dominant-negative TCF4 construct in JAM-A–WT and JAM-A–null iLECs increased the mRNA expression of C/EBP-α and claudin-5 compared with the control cells (Figure 4G). Taken together, these data demonstrate that JAM-A positively regulates C/EBP-α expression by suppressing β–catenin transcriptional activity. C/EBP-α, in turn, sustains claudin-5 expression, which controls the vascular permeability (Figure 4H).
JAM-A Deficiency Increases Endothelial Permeability in the Brain Microvasculature
Multiple studies have demonstrated that JAM-A is involved in positive regulation of epithelial barrier function.10,23,44 Moreover, claudin-5 is a key component of the tight junction strands, and it is highly expressed in brain endothelial cells, where it decreases permeability to small molecules (<800 Da).19 Given that claudin-5 regulates the size-dependent paracellular pathway in the blood-brain barrier, we investigated whether loss of JAM-A leads to increased endothelial permeability to the low molecular weight (443.43 Da) lysine-reactive biotinylation reagent sulpho–N-hydroxysuccinimide–biotin, from the intravascular space into the brain parenchyma. Compared with JAM-A–WT littermates, JAM-A–null adult mice showed significantly increased leakage of sulpho–N-hydroxysuccinimide–biotin in the cerebral cortex, striatum, hypothalamus, and midbrain (Figure 5A and 5B). Interestingly, the brain areas with widespread leakage also showed downregulation of claudin-5 protein expression, which is consistent with loss of barrier function (Figure 5C; Figure VA in the Data Supplement). To determine the size selectivity of the endothelial barrier dysfunction, we examined extravasation of larger molecular weight tracers in the brain, using cadaverine (950 Da) and dextran (10 kDa). Interestingly, while no leakage of the high molecular weight dextran was detected, in the absence of JAM-A the permeability to cadaverine was slightly, but significantly, increased in the cortex and the hypothalamus (Figure 5D; Figure VB and VC in the Data Supplement). This suggested that JAM-A signaling can regulate the paracellular passage of molecules into the brain in a claudin-5–like size-dependent manner.
Figure 5.
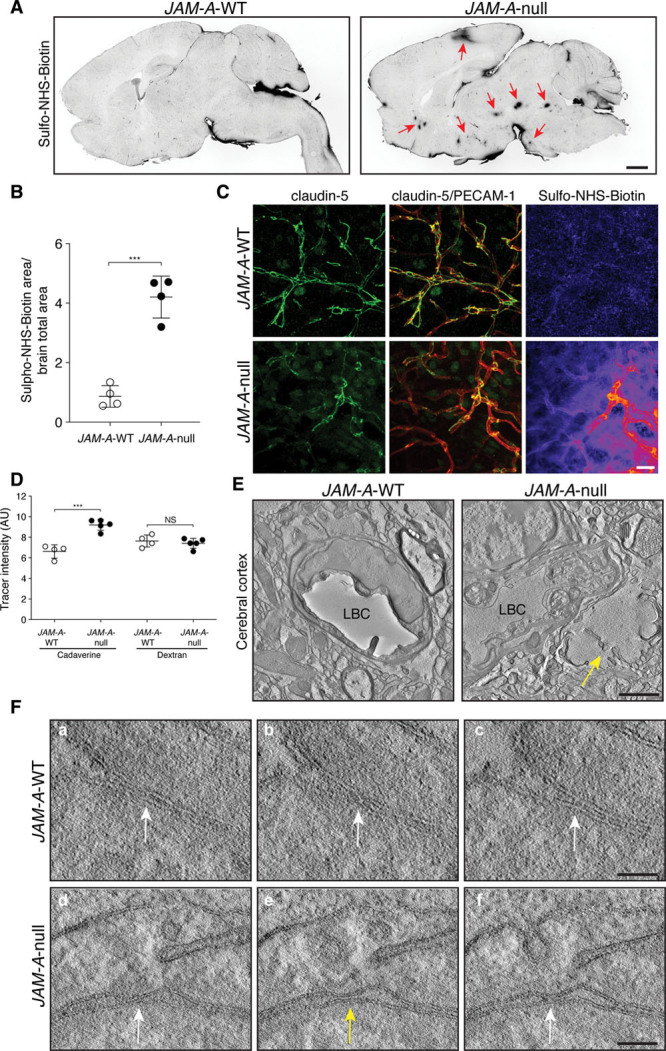
Endothelial permeability is increased in vivo by JAM-A (junctional adhesion molecule A) deficiency in areas of decreased claudin-5. A, Representative confocal microscopy for streptavidin to detect sulpho–N-hydroxysuccinimide–biotin (sulpho-NHS-biotin) leakage in vibratome brain sections (100 μm) from JAM-A–wild-type (WT) and JAM-A–null mice. Red arrows (right) indicate foci of leakage in different brain regions. Scale bar: 1 mm (n=3, JAM-A–WT; n=3, JAM-A–null). B, Quantification of sulpho-NHS-biotin leakage in JAM-A–WT and JAM-A–null mice brain sections, as shown in A. Data are means±SD, as ratio of sulpho-NHS-biotin area/brain total area (n=4, JAM-A–WT; n=4, JAM-A–null mice). P value determined by 2-sided unpaired Welch t test. JAM-A–WT vs JAM-A–null ***P=6.4×10-4. C, Representative confocal microscopy for claudin-5 (green), PECAM-1 (platelet/endothelial cell adhesion molecule-1; red), and streptavidin, for sulpho-NHS-biotin leakage (pseudocolor) in brain sections from JAM-A–WT and JAM-A–null mice. Scale bar: 20 µm (n=3, JAM-A–WT; n=3, JAM-A–null). D, Quantification of cadaverine and dextran leakage in JAM-A–WT and JAM-A–null mice brain sections. Data are means±SD, measured as relative intensities (arbitrary units; AU; n=4, JAM-A–WT; n=5, JAM-A–null mice). P values determined by 2-sided unpaired Welch t test. JAM-A–WT vs JAM-A–null: cadaverine ***P=7.9×10-4; dextran P=0.56, not statistically significant (NS). E, Representative transmission electron microscopy of cerebral cortex from JAM-A–WT and JAM-A–null mice. Arrow denotes locus of edema. Scale bar: 1200 nm (n=3, JAM-A–WT; n=3, JAM-A–null). F, Representative 2-step transmission electron microscopy tomography of cerebral cortex from JAM-A–WT (a–c) and JAM-A–null mice (d–f). Parts (a–c) and (d–f) are 3 consecutive serial tomography sections showing a tight junction strand. White arrows, tight junction that appears as the translucent area (osmiophobic channel) between 2 plasma membranes; yellow arrow; interruption along the pale strand channel. Scale bars (a–c): 80 nm; (d–f) 90 nm (n=3, JAM-A–WT; n=3, JAM-A–null). LBC indicates lumen of brain capillary.
Furthermore, in the cortex of JAM-A–null mice, fluid leakage from blood capillaries and edema were confirmed using 2-step transmission electron microscopy tomography. We sectioned the samples, using serial sections with interleaved thicknesses. Initially, the first section (60 nm) was examined under electron microscopy, and an area where edema was evident in the absence of JAM-A was selected (Figure 5E). Then the first 200-nm section was examined for the cross-section of the blood capillary located near the edematous area (Figure VIA in the Data Supplement). Next, 2 serial tomography boxes were obtained at higher magnification. In the control littermate, the capillaries on consecutive cross-sections of the electron microscopy tomography box were examined. Here, a tight junction appeared as a translucent area (osmiophobic channel) between 2 plasma membranes, where they were attached to each other (external side, relative to the cells), in the dark layers of the plasma membrane (Figure 5F, a through c; Figure VIA in the Data Supplement). This pale channel was observed in all of the consecutive serial tomography sections without any break or interruption, and the strands of the tight junctions were observed in close vicinity to each other (Figure 5F, a through c; Figure VIA in the Data Supplement). In JAM-A–null capillaries located near an edematous locus, the strands were significantly more distant from each other (Figure VIA in the Data Supplement), and in the consecutive sections, there was an interruption along the pale strand channel (Figure 5E, d through f; Figure VIA in the Data Supplement). This pattern typically indicated that the barrier function of these tight junctions was impaired and that this break was responsible for the leakage observed. Thus, the absence of JAM-A can impair the organization of the tight junctions in correspondence with areas of edema, which supports the central role of JAM-A in the control of endothelial barrier function in vivo.
JAM-A Positively Regulates Claudin-5 and C/EBP-α Expression in Cancer Vasculature
The data presented above describe a novel molecular mechanism that links the expression of JAM-A and claudin-5 and that is important for the maintenance of the integrity of the endothelial barrier (Figure 4H).
One of the characteristics of brain tumors is significant peritumoral edema, which can be explained as the result of alterations to the tight junction organization and claudin-5.45 Given the reduction in claudin-5 expression in the GBM vasculature,46,47 we hypothesized that this is related to JAM-A expression. Indeed, downregulation of JAM-A has been described for several solid tumors, such as endometrial, pancreatic, and kidney tumors, and has been shown to correlate with tumor size and progression, as well as with poor prognostic outcome.48–50 Of note, as well as mediating JAM-A–positive regulation of claudin-5 transcription, C/EBP-α has a tumor suppressor role in different solid tumors.28 Hence, we evaluated the expression of JAM-A, claudin-5, and C/EBP-α mRNAs in blood vessels from tissue biopsies of 11 patients with grade IV GBM. As illustrated in Figure 6A, JAM-A, claudin-5, and C/EBP-α mRNA levels were significantly lower in GBM, compared with patient-matched healthy tissues. Moreover, in the healthy tissue, JAM-A and claudin-5 showed intense staining patterns at the endothelial cell-to-cell contacts, whereas in the GBM vessels, neither JAM-A nor claudin-5 were detectable (Figure 6B). In parallel, nuclear C/EBP-α staining in the tumor vasculature was lower compared with the vasculature of the healthy tissue (Figure 6C).
Figure 6.
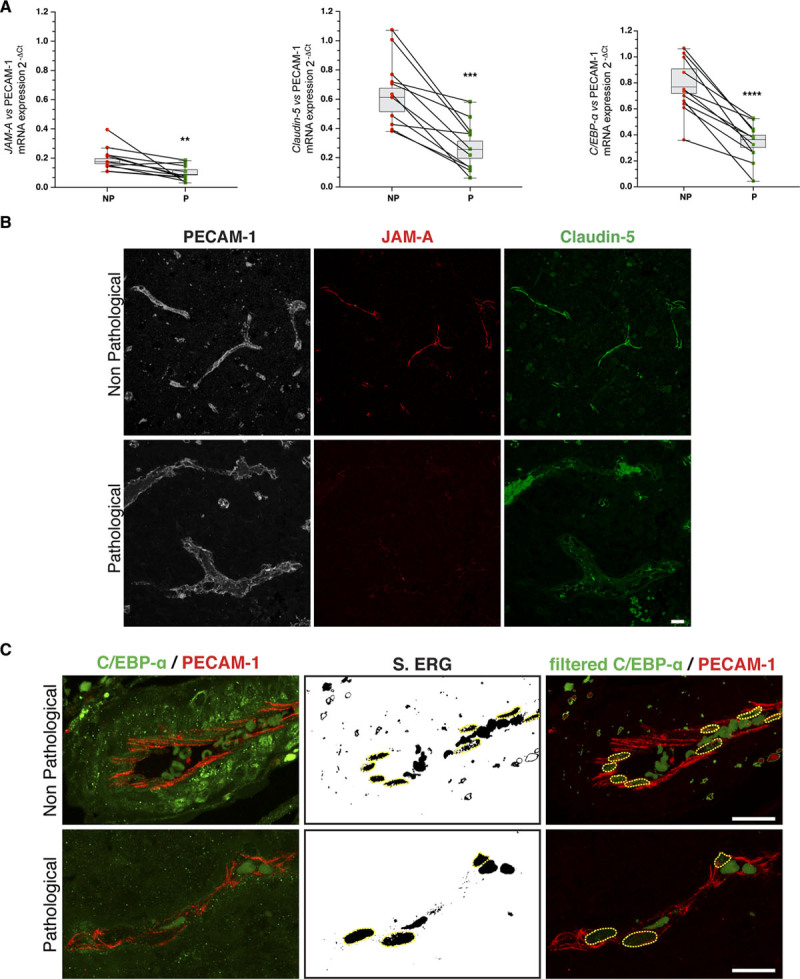
JAM-A (junctional adhesion molecule A) positively regulates claudin-5 and C/EBP-α (CCAAT/enhancer-binding protein-α) expression in glioblastoma multiforme. A, Quantification of JAM-A, claudin-5, and C/EBP-α expression in tissue biopsies from 11 patients with grade IV glioblastoma multiforme (GBM) following real-time quantitative polymerase chain reaction analysis. mRNA expression for each gene was normalized to PECAM-1 (platelet/endothelial cell adhesion molecule-1). The lines link values between GBM and patient-matched healthy tissues. P values determined by 2-sided paired t test. Non-pathological (NP) vs pathological (P): JAM-A **P=6.8×10-3; claudin-5 ***P=1.3×10-4; C/EBP-α ****P=2.1×10-5. B, Representative confocal microscopy for PECAM-1 (white), JAM-A (red), and claudin-5 (green) in paraffin-embedded tissue sections (4 µm) from GBM and patient-matched healthy tissue. Images are representative of 11 patients. Scale bars: 20 µm. C, Representative confocal microscopy for C/EBP-α (green) and PECAM-1 (red) in paraffin-embedded tissue sections (4 μm) from GBM and patient-matched healthy tissue. ERG (ETS-related gene) staining was segmented with threshold 350-4.096 (S. ERG) for merged images of filtered C/EBP-α and PECAM-1 (filtered C/EBP-α PECAM-1). Yellow dashed circles outline ERG-positive nuclei to exclude signals belonging to circulating cells. Scale bars: 20 µm.
Finally, we extended this approach to high-grade serous ovarian cancer, where the expression of JAM-A, claudin-5, and C/EBP-α in blood vessels was evaluated in patient-derived tissue biopsies by immunofluorescence. The endothelial levels of JAM-A, claudin-5, and C/EBP-α were significantly lower in the ovarian cancer tissues, compared with the healthy ovarian controls (Figure VIIA through VIIE in the Data Supplement).
Taken together, these data suggest that the dysregulation of these molecular mechanisms that involve the tight junction proteins JAM-A and claudin-5 has pathological implications.
Discussion
One of the hallmarks of deteriorating vascular integrity is increased vessel permeability, which occurs in pathological contexts such as inflammation and tumor growth. The vascular barrier is mainly maintained through the stability of the endothelial cell-to-cell junctions.51 Claudin-5 is the major constituent of endothelial cell-to-cell tight junctions, and it has a critical role in the maintenance of the vascular barrier.52 We describe here a novel role for the transcription factor C/EBP-α that acts downstream of JAM-A (a component of cell-to-cell tight junctions) and EPAC to promote the transcription of claudin-5, thus strengthening the endothelial barrier in vivo and in vitro. Therefore, as a component of tight junctions, JAM-A contributes to the organization and function of tight junctions through enhanced expression of claudin-5.
Claudin-5 transcription can be controlled by various signaling pathways, and its activity can be regulated post-translationally.53 Here, we have shown that C/EBP-α binds directly to the claudin-5 promoter to induce transcription of the claudin-5 gene, thus enhancing claudin-5 expression.
The mechanism by which JAM-A activates C/EBP-α requires EPAC, which in turn activates C/EBP-α. In this context, in contrast, β-catenin acts as a negative regulator of claudin-5 expression. Indeed, JAM-A deficiency increases β-catenin transcriptional activity, which, in turn, inhibits the expression of C/EBP-α, thus reducing claudin-5 transcription. A role for active β-catenin in suppression of the expression of C/EBP-α is in agreement with previous reports in other cell types.26,27,38,39 Indeed, our data indicate that JAM-A deficiency increases β-catenin nuclear localization and signaling, which in turn reduces claudin-5 expression through inhibition of C/EBP-α. Therefore, our data and previous findings20 indicate that β-catenin can repress the transcription of claudin-5 through different parallel mechanisms: indirectly through inhibition of C/EBP-α expression; and directly through interactions with FoxO1 (forkhead box O1) and TCF.20
We have shown here that EPAC activates C/EBP-α downstream of JAM-A in endothelial cells. However, the molecular mechanisms of this regulation remain to be explored further. Covalent modification of C/EBP-α by the intermediate EPAC-activated protein kinases might occur. In this respect, it has been demonstrated that certain C/EBP isoforms are substrates for ERK (extracellular signal-regulated kinase), ribosomal S6 kinase, and protein kinase C protein kinases. The resulting phosphorylation enhances the activity of C/EBPs, although the different C/EBP proteins are regulated by specific mechanisms without functional redundancy.39,40
We have previously demonstrated that the absence of JAM-A impairs EPAC-mediated activation of Rap-1, which is required to restrict transmigration of leukocytes and mesoangioblasts.13 Here, we now shown that JAM-A regulates vascular permeability via the control of the strength of tight junctions through claudin-5 expression. This highlights a dual role for JAM-A, as an important regulator of the vascular barrier properties to both solutes and circulating cells.
Interestingly, although increasing evidence has suggested that the mechanisms underlying vascular leakage might vary in different organ-specialized vasculatures,54 the signaling pathway of JAM-A–EPAC–C/EBP-α that we have identified here is conserved in diverse tissues, such as brain and lung. This underlines the possible broad-spectrum relevance of this signaling in the maintenance of vascular integrity in physiology and its possible alterations in pathologies. Indeed, it has been reported that the levels of JAM-A, C/EBP-α, and claudin-5 are decreased in several diseases55,56 and different cancers,28–30 which also implies a role for these molecules as tumor suppressors. Our findings show that reduced JAM-A in the human GBM and ovarian carcinoma vasculature parallels the downregulation of claudin-5 and C/EBP-α, which suggests a correlation between these players in pathological situations in humans.
In conclusion, our data demonstrate a novel regulatory loop through which a component of the tight junctions, JAM-A, restricts vascular permeability by enhancing the expression of claudin-5, a crucial functional component of the tight junctions themselves. This pathway should help to identify novel therapeutic targets for diseases associated with vascular barrier dysfunction.
Acknowledgments
We thank Amanda Oldani for image processing support and Fabrizio Orsenigo for sharing luciferase claudin-5 promoter constructs. We thank Christopher P. Berrie for editorial assistance, Lorenzo Bello for collecting patient informed consent according to the clinical procedure (IRB1299) and Giorgio Binelli for statistical support. We are indebted to Ruud D. Fontijn for providing the lentiviral human claudin-5 constructs, and with Daniel Tenen for providing the retroviral C/EBP-α constructs. M. Giannotta and E. Dejana conceived and designed the study; L. Ferrari, M. Giannotta, and A.A. Scalise performed the in vitro experiments and analyzed the results with C. Giampietro; C. Maderna, M. Ravà and N. Kakogiannos performed the in vivo experiments and analyzed the results with M. Giannotta; F. Pisati performed paraffin-embedded tissue sectioning; E. Martini and I. Costa performed imaging analysis. M.G. Lampugnani, C. Giampietro, N. Kakogiannos, M. Malinverno, and A. Taddei contributed to scientific discussions. M. Lupia and U. Cavallaro provided tissue biopsies from healthy and ovarian cancer patients and analyzed the data with M. Giannotta; B. Fernandes and N. Rudini provided tissue biopsies from healthy patients and patients with glioblastoma multiforme and analyzed the data with M. Giannotta; G.V. Beznoussenko, and A.A. Mironov performed the electron microscopy experiments; M. Giannotta and M.G. Lampugnani wrote the article, which was revised by E. Dejana, C. Giampietro, and A. Taddei. The final form of the article has been approved by all of the authors.
Sources of Funding
This study was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC; Investigator Grants 18683 and 21320), the AIRC 5x1000 call Metastatic disease: the key unmet need in oncology of the MYNERVA project, no. 21267 (Myeloid Neoplasms Research Venture AIRC), the European Research Council (project EC-ERC-V-EPC, contract 742922), Initial Training Networks BtRAIN grant 675619, the CARIPLO (Cassa di Risparmio delle Provincie Lombarde) Foundation (2014-1038, 2016-0461), the Italian Ministry of Health (Ricerca Finalizzata PE-2016-02362551), the Telethon Foundation (GGP19202), the IEO-CCM (Istituto Europeo di Oncologia-Centro Cardiologico Monzino) Foundation, the Umberto Veronesi Foundation, the Swedish Science Council, and the Knut and Alice Wallenberg Foundation.
Disclosures
None.
Supplemental Materials
Major Resources Table
Detailed Methods
Online Tables I and II
Online Figures I–VII
Full Unedited Gels
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- 007
- 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-pCPT-2′-O-Me-cAMP)
- C/EBP-α
- CCAAT/enhancer-binding protein-α
- CREB
- cAMP response-element binding protein
- DN
- dominant negative
- eGFP
- enhanced green fluorescent protein
- EPAC
- exchange protein directly activated by cAMP
- ERK
- extracellular signal-regulated kinase
- fLECs
- freshly isolated lung endothelial cells
- FoxO
- forkhead box protein O
- GBM
- glioblastoma multiforme
- iLECs
- immortalized lung endothelial cells
- JAM-A
- junctional adhesion molecule A
- PECAM-1
- platelet/endothelial cell adhesion molecule-1
- PKA
- protein kinase A
- TCF
- T-cell factor
- VE-cadherin
- vascular endothelial cadherin
- WT
- wild-type
- ZO
- zona occludens
N.K. and L.F. contributed equally to this article.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.316742.
For Sources of Funding and Disclosures, see page 1071.
Novelty and Significance
What Is Known?
Intercellular tight junctions are crucial for the regulation of endothelial barrier function and the control of vascular permeability. Their composition and integrity are affected in several pathological contexts, which include inflammation and cancers.
JAM-A (junctional adhesion molecule A) is a transmembrane component of tight junctions with a role in maintenance of endothelial barrier function.
Besides JAM-A, proteins of the claudin family are key components of tight junctions. Claudin-5 is an endothelial-specific protein that contributes to the selectivity of paracellular barriers.
What New Information Does This Article Contribute?
It defines the mechanism of action of JAM-A in the control of claudin-5 expression and, hence, in the regulation of the vascular barrier.
It identifies the transcription factor C/EBP-α (CCAAT/enhancer-binding protein-α) as a key player in controlling claudin-5 expression and endothelial permeability.
It describes the dysregulation of the novel JAM-A–C/EBP-α–claudin-5 pathway in pathological conditions, such as cancer.
A detailed understanding of the molecular and cellular pathways that regulate vascular permeability is critical to the design of efficient and safe therapies for the targeted treatment of a wide range of human diseases. We describe a signaling circuit that involves JAM-A and claudin-5 and regulates endothelial permeability in vivo and in vitro. We show that the transcription factor C/EBP-α induces the expression of claudin-5 downstream of JAM-A, thus restricting endothelial permeability. This signaling cascade is negatively regulated by active β-catenin, which represses C/EBP-α expression in the absence of JAM-A. Finally, JAM-A–C/EBP-α–mediated regulation of claudin-5 is dysregulated in the ovarian cancer and glioblastoma vasculature, implicating this circuit in aberrant tumor vessels. The discovery of this pathway can help to identify new therapeutic targets for diseases associated with vascular barrier dysfunction.
References
- 1.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005 [DOI] [PubMed] [Google Scholar]
- 2.Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36:149–155. doi: 10.1042/BST0360149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357 [DOI] [PubMed] [Google Scholar]
- 4.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003 [DOI] [PubMed] [Google Scholar]
- 5.Lampugnani MG. Endothelial cell-to-cell junctions: adhesion and signaling in physiology and pathology. Cold Spring Harb Perspect Med. 2012;2:a006528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113pt 132363–2374 [DOI] [PubMed] [Google Scholar]
- 7.Mandell KJ, Babbin BA, Nusrat A, Parkos CA. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J Biol Chem. 2005;280:11665–11674. doi: 10.1074/jbc.M412650200 [DOI] [PubMed] [Google Scholar]
- 8.Mandell KJ, Holley GP, Parkos CA, Edelhauser HF. Antibody blockade of junctional adhesion molecule-A in rabbit corneal endothelial tight junctions produces corneal swelling. Invest Ophthalmol Vis Sci. 2006;47:2408–2416. doi: 10.1167/iovs.05-0745 [DOI] [PubMed] [Google Scholar]
- 9.Mandell KJ, Berglin L, Severson EA, Edelhauser HF, Parkos CA. Expression of JAM-A in the human corneal endothelium and retinal pigment epithelium: localization and evidence for role in barrier function. Invest Ophthalmol Vis Sci. 2007;48:3928–3936. doi: 10.1167/iovs.06-1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monteiro AC, Parkos CA. Intracellular mediators of JAM-A-dependent epithelial barrier function. Ann N Y Acad Sci. 2012;1257:115–124. doi: 10.1111/j.1749-6632.2012.06521.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nava P, Capaldo CT, Koch S, Kolegraff K, Rankin CR, Farkas AE, Feasel ME, Li L, Addis C, Parkos CA, et al. JAM-A regulates epithelial proliferation through Akt/β-catenin signalling. EMBO Rep. 2011;12:314–320. doi: 10.1038/embor.2011.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA. Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and enhance cell migration. Mol Biol Cell. 2009;20:1916–1925. doi: 10.1091/mbc.e08-10-1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giannotta M, Benedetti S, Tedesco FS, Corada M, Trani M, D’Antuono R, Millet Q, Orsenigo F, Gálvez BG, Cossu G, et al. Targeting endothelial junctional adhesion molecule-A/EPAC/Rap-1 axis as a novel strategy to increase stem cell engraftment in dystrophic muscles. EMBO Mol Med. 2014;6:239–258. doi: 10.1002/emmm.201302520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nourshargh S, Krombach F, Dejana E. The role of JAM-A and PECAM-1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J Leukoc Biol. 2006;80:714–718. doi: 10.1189/jlb.1105645 [DOI] [PubMed] [Google Scholar]
- 15.Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, Schwartz MA, Matter K, Balda MS. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. 2015;208:821–838. doi: 10.1083/jcb.201404140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–1955. doi: 10.1182/blood-2004-05-1987 [DOI] [PubMed] [Google Scholar]
- 17.Kooistra MR, Dubé N, Bos JL. Rap1: a key regulator in cell-cell junction formation. J Cell Sci. 2007;120:17–22. doi: 10.1242/jcs.03306 [DOI] [PubMed] [Google Scholar]
- 18.Dubé N, Kooistra MR, Pannekoek WJ, Vliem MJ, Oorschot V, Klumperman J, Rehmann H, Bos JL. The RapGEF PDZ-GEF2 is required for maturation of cell-cell junctions. Cell Signal. 2008;20:1608–1615. doi: 10.1016/j.cellsig.2008.05.006 [DOI] [PubMed] [Google Scholar]
- 19.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752 [DOI] [PubMed] [Google Scholar]
- 21.Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol. 2008;6:179–192. doi: 10.2174/157015908785777210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9:23 doi: 10.1186/2045-8118-9-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishizaki T, Chiba H, Kojima T, Fujibe M, Soma T, Miyajima H, Nagasawa K, Wada I, Sawada N. Cyclic AMP induces phosphorylation of claudin-5 immunoprecipitates and expression of claudin-5 gene in blood-brain-barrier endothelial cells via protein kinase A-dependent and -independent pathways. Exp Cell Res. 2003;290:275–288. doi: 10.1016/s0014-4827(03)00354-9 [DOI] [PubMed] [Google Scholar]
- 25.Beese M, Wyss K, Haubitz M, Kirsch T. Effect of cAMP derivates on assembly and maintenance of tight junctions in human umbilical vein endothelial cells. BMC Cell Biol. 2010;11:68 doi: 10.1186/1471-2121-11-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang S, Bennett CN, Gerin I, Rapp LA, Hankenson KD, Macdougald OA. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007;282:14515–14524. doi: 10.1074/jbc.M700030200 [DOI] [PubMed] [Google Scholar]
- 27.Kawai M, Mushiake S, Bessho K, Murakami M, Namba N, Kokubu C, Michigami T, Ozono K. Wnt/Lrp/beta-catenin signaling suppresses adipogenesis by inhibiting mutual activation of PPARgamma and C/EBPalpha. Biochem Biophys Res Commun. 2007;363:276–282. doi: 10.1016/j.bbrc.2007.08.088 [DOI] [PubMed] [Google Scholar]
- 28.Lourenço AR, Coffer PJ. A tumor suppressor role for C/EBPα in solid tumors: more than fat and blood. Oncogene. 2017;36:5221–5230. doi: 10.1038/onc.2017.151 [DOI] [PubMed] [Google Scholar]
- 29.Zhao C, Lu F, Chen H, Zhao X, Sun J, Chen H. Dysregulation of JAM-A plays an important role in human tumor progression. Int J Clin Exp Pathol. 2014;7:7242–7248 [PMC free article] [PubMed] [Google Scholar]
- 30.Ma SC, Li Q, Peng JY, Zhouwen JL, Diao JF, Niu JX, Wang X, Guan XD, Jia W, Jiang WG. Claudin-5 regulates blood-brain barrier permeability by modifying brain microvascular endothelial cell proliferation, migration, and adhesion to prevent lung cancer metastasis. CNS Neurosci Ther. 2017;23:947–960. doi: 10.1111/cns.12764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morini MF, Giampietro C, Corada M, Pisati F, Lavarone E, Cunha SI, Conze LL, O’Reilly N, Joshi D, Kjaer S, et al. VE-cadherin-mediated epigenetic regulation of endothelial gene expression. Circ Res. 2018;122:231–245. doi: 10.1161/CIRCRESAHA.117.312392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7:467–477. doi: 10.1038/nri2096 [DOI] [PubMed] [Google Scholar]
- 33.Martìn-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, Haskard DO, Dejana E, Krombach F, Nourshargh S. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood. 2007;110:1848–1856. doi: 10.1182/blood-2006-09-047431 [DOI] [PubMed] [Google Scholar]
- 35.Soma T, Chiba H, Kato-Mori Y, Wada T, Yamashita T, Kojima T, Sawada N. Thr(207) of claudin-5 is involved in size-selective loosening of the endothelial barrier by cyclic AMP. Exp Cell Res. 2004;300:202–212. doi: 10.1016/j.yexcr.2004.07.012 [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5 [DOI] [PubMed] [Google Scholar]
- 37.Wilson HL, Roesler WJ. CCAAT/enhancer binding proteins: do they possess intrinsic cAMP-inducible activity? Mol Cell Endocrinol. 2002;188:15–20. doi: 10.1016/s0303-7207(01)00754-7 [DOI] [PubMed] [Google Scholar]
- 38.Yarwood SJ, Borland G, Sands WA, Palmer TM. Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J Biol Chem. 2008;283:6843–6853. doi: 10.1074/jbc.M710342200 [DOI] [PubMed] [Google Scholar]
- 39.Borland G, Bird RJ, Palmer TM, Yarwood SJ. Activation of protein kinase Calpha by EPAC1 is required for the ERK- and CCAAT/enhancer-binding protein beta-dependent induction of the SOCS-3 gene by cyclic AMP in COS1 cells. J Biol Chem. 2009;284:17391–17403. doi: 10.1074/jbc.M109.015370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramji DP, Foka P. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J. 2002;365:561–575. doi: 10.1042/BJ20020508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thiel AT, Feng Z, Pant DK, Chodosh LA, Hua X. The trithorax protein partner menin acts in tandem with EZH2 to suppress C/EBPα and differentiation in MLL-AF9 leukemia. Haematologica. 2013;98:918–927. doi: 10.3324/haematol.2012.074195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–270. doi: 10.1038/85820 [DOI] [PubMed] [Google Scholar]
- 43.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandell KJ, McCall IC, Parkos CA. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem. 2004;279:16254–16262. doi: 10.1074/jbc.M309483200 [DOI] [PubMed] [Google Scholar]
- 45.Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018;135:311–336. doi: 10.1007/s00401-018-1815-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liebner S, Fischmann A, Rascher G, Duffner F, Grote EH, Kalbacher H, Wolburg H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000;100:323–331. doi: 10.1007/s004010000180 [DOI] [PubMed] [Google Scholar]
- 47.Karnati HK, Panigrahi M, Shaik NA, Greig NH, Bagadi SA, Kamal MA, Kapalavayi N. Down regulated expression of Claudin-1 and Claudin-5 and up regulation of β-catenin: association with human glioma progression. CNS Neurol Disord Drug Targets. 2014;13:1413–1426. doi: 10.2174/1871527313666141023121550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koshiba H, Hosokawa K, Kubo A, Tokumitsu N, Watanabe A, Honjo H. Junctional adhesion molecule A [corrected] expression in human endometrial carcinoma. Int J Gynecol Cancer. 2009;19:208–213. doi: 10.1111/IGC.0b013e31819bc6e9 [DOI] [PubMed] [Google Scholar]
- 49.Fong D, Spizzo G, Mitterer M, Seeber A, Steurer M, Gastl G, Brosch I, Moser P. Low expression of junctional adhesion molecule A is associated with metastasis and poor survival in pancreatic cancer. Ann Surg Oncol. 2012;19:4330–4336. doi: 10.1245/s10434-012-2381-8 [DOI] [PubMed] [Google Scholar]
- 50.Gutwein P, Schramme A, Voss B, Abdel-Bakky MS, Doberstein K, Ludwig A, Altevogt P, Hansmann ML, Moch H, Kristiansen G, et al. Downregulation of junctional adhesion molecule-A is involved in the progression of clear cell renal cell carcinoma. Biochem Biophys Res Commun. 2009;380:387–391. doi: 10.1016/j.bbrc.2009.01.100 [DOI] [PubMed] [Google Scholar]
- 51.Murakami M, Simons M. Regulation of vascular integrity. J Mol Med (Berl). 2009;87:571–582. doi: 10.1007/s00109-009-0463-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jia W, Lu R, Martin TA, Jiang WG. The role of claudin-5 in blood-brain barrier (BBB) and brain metastases (review). Mol Med Rep. 2014;9:779–785. doi: 10.3892/mmr.2013.1875 [DOI] [PubMed] [Google Scholar]
- 53.Lv J, Hu W, Yang Z, Li T, Jiang S, Ma Z, Chen F, Yang Y. Focusing on claudin-5: A promising candidate in the regulation of BBB to treat ischemic stroke. Prog Neurobiol. 2018;161:79–96. doi: 10.1016/j.pneurobio.2017.12.001 [DOI] [PubMed] [Google Scholar]
- 54.Claesson-Welsh L. Vascular permeability–the essentials. Ups J Med Sci. 2015;120:135–143. doi: 10.3109/03009734.2015.1064501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kealy J, Greene C, Campbell M. Blood-brain barrier regulation in psychiatric disorders. Neurosci Lett. 2020;726:133664 doi: 10.1016/j.neulet.2018.06.033 [DOI] [PubMed] [Google Scholar]
- 56.Forrest JC, Campbell JA, Schelling P, Stehle T, Dermody TS. Structure-function analysis of reovirus binding to junctional adhesion molecule 1. Implications for the mechanism of reovirus attachment. J Biol Chem. 2003;278:48434–48444. doi: 10.1074/jbc.M305649200 [DOI] [PubMed] [Google Scholar]
- 57.Cera MR, Del Prete A, Vecchi A, Corada M, Martin-Padura I, Motoike T, Tonetti P, Bazzoni G, Vermi W, Gentili F, et al. Increased DC trafficking to lymph nodes and contact hypersensitivity in junctional adhesion molecule-A-deficient mice. J Clin Invest. 2004;114:729–738. doi: 10.1172/JCI21231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F, De Angelis N, Villa A, Bossi M, Staszewsky LI, et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2005;102:10634–10639. doi: 10.1073/pnas.0500147102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bussolino F, De Rossi M, Sica A, Colotta F, Wang JM, Bocchietto E, Padura IM, Bosia A, DeJana E, Mantovani A. Murine endothelioma cell lines transformed by polyoma middle T oncogene as target for and producers of cytokines. J Immunol. 1991;147:2122–2129 [PubMed] [Google Scholar]
- 60.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79:707–717. doi: 10.1078/0171-9335-00101 [DOI] [PubMed] [Google Scholar]
- 62.Calabria AR, Weidenfeller C, Jones AR, de Vries HE, Shusta EV. Puromycin-purified rat brain microvascular endothelial cell cultures exhibit improved barrier properties in response to glucocorticoid induction. J Neurochem. 2006;97:922–933. doi: 10.1111/j.1471-4159.2006.03793.x [DOI] [PubMed] [Google Scholar]
- 63.Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol. 2013;83:122–128. doi: 10.1124/mol.112.080689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lluis F, Pedone E, Pepe S, Cosma MP. Periodic activation of Wnt/beta-catenin signaling enhances somatic cell reprogramming mediated by cell fusion. Cell Stem Cell. 2008;3:493–507. doi: 10.1016/j.stem.2008.08.017 [DOI] [PubMed] [Google Scholar]
- 65.Fontijn RD, Rohlena J, van Marle J, Pannekoek H, Horrevoets AJ. Limited contribution of claudin-5-dependent tight junction strands to endothelial barrier function. Eur J Cell Biol. 2006;85:1131–1144. doi: 10.1016/j.ejcb.2006.07.005 [DOI] [PubMed] [Google Scholar]
- 66.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwieger M, Löhler J, Fischer M, Herwig U, Tenen DG, Stocking C. A dominant-negative mutant of C/EBPalpha, associated with acute myeloid leukemias, inhibits differentiation of myeloid and erythroid progenitors of man but not mouse. Blood. 2004;103:2744–2752. doi: 10.1182/blood-2003-07-2280 [DOI] [PubMed] [Google Scholar]
- 68.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spagnuolo R, Corada M, Orsenigo F, Zanetta L, Deuschle U, Sandy P, Schneider C, Drake CJ, Breviario F, Dejana E. Gas1 is induced by VE-cadherin and vascular endothelial growth factor and inhibits endothelial cell apoptosis. Blood. 2004;103:3005–3012. doi: 10.1182/blood-2003-07-2459 [DOI] [PubMed] [Google Scholar]
- 70.Lampugnani MG, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110 (pt 17):2065–2077 [DOI] [PubMed] [Google Scholar]
- 71.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yen JC, Chang FJ, Chang S. A new criterion for automatic multilevel thresholding. IEEE Trans Image Process. 1995;4:370–378. doi: 10.1109/83.366472 [DOI] [PubMed] [Google Scholar]
- 73.Li CH, Tam PKS. An iterative algorithm for minimum cross entropy thresholding. Pattern Recogn Lett. 1998;19:771–776 [Google Scholar]
- 74.Steger C. An unbiased detector of curvilinear structures. IEEE Trans Pattern Anal. 1998;20:113–125 [Google Scholar]
- 75.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beznoussenko GV, Mironov AA. Correlative video-light-electron microscopy of mobile organelles. Methods Mol Biol. 2015;1270:321–346. doi: 10.1007/978-1-4939-2309-0_23 [DOI] [PubMed] [Google Scholar]
- 77.Corada M, Orsenigo F, Bhat GP, Conze LL, Breviario F, Cunha SI, Claesson-Welsh L, Beznoussenko GV, Mironov AA, Bacigaluppi M, et al. Fine-tuning of Sox17 and Canonical Wnt coordinates the permeability properties of the blood-brain barrier. Circ Res. 2019;124:511–525. doi: 10.1161/CIRCRESAHA.118.313316 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.