

Supplemental Digital Content is available in the text.
Keywords: blood platelets, brain ischemia, infarction, stroke, T lymphocytes
Abstract
Rationale:
Ischemic stroke is a leading cause of morbidity and mortality worldwide. Recanalization of the occluded vessel is essential but not sufficient to guarantee brain salvage. Experimental and clinical data suggest that infarcts often develop further due to a thromboinflammatory process critically involving platelets and T cells, but the underlying mechanisms are unknown.
Objective:
We aimed to determine the role of CD (cluster of differentiation)-84 in acute ischemic stroke after recanalization and to dissect the underlying molecular thromboinflammatory mechanisms.
Methods and Results:
Here, we show that mice lacking CD84—a homophilic immunoreceptor of the SLAM (signaling lymphocyte activation molecule) family—on either platelets or T cells displayed reduced cerebral CD4+ T-cell infiltration and thrombotic activity following experimental stroke resulting in reduced neurological damage. In vitro, platelet-derived soluble CD84 enhanced motility of wild-type but not of Cd84−/− CD4+ T cells suggesting homophilic CD84 interactions to drive this process. Clinically, human arterial blood directly sampled from the ischemic cerebral circulation indicated local shedding of platelet CD84. Moreover, high platelet CD84 expression levels were associated with poor outcome in patients with stroke.
Conclusions:
These results establish CD84 as a critical pathogenic effector and thus a potential pharmacological target in ischemic stroke.
In This Issue, see p 949
Meet the First Author, see p 950
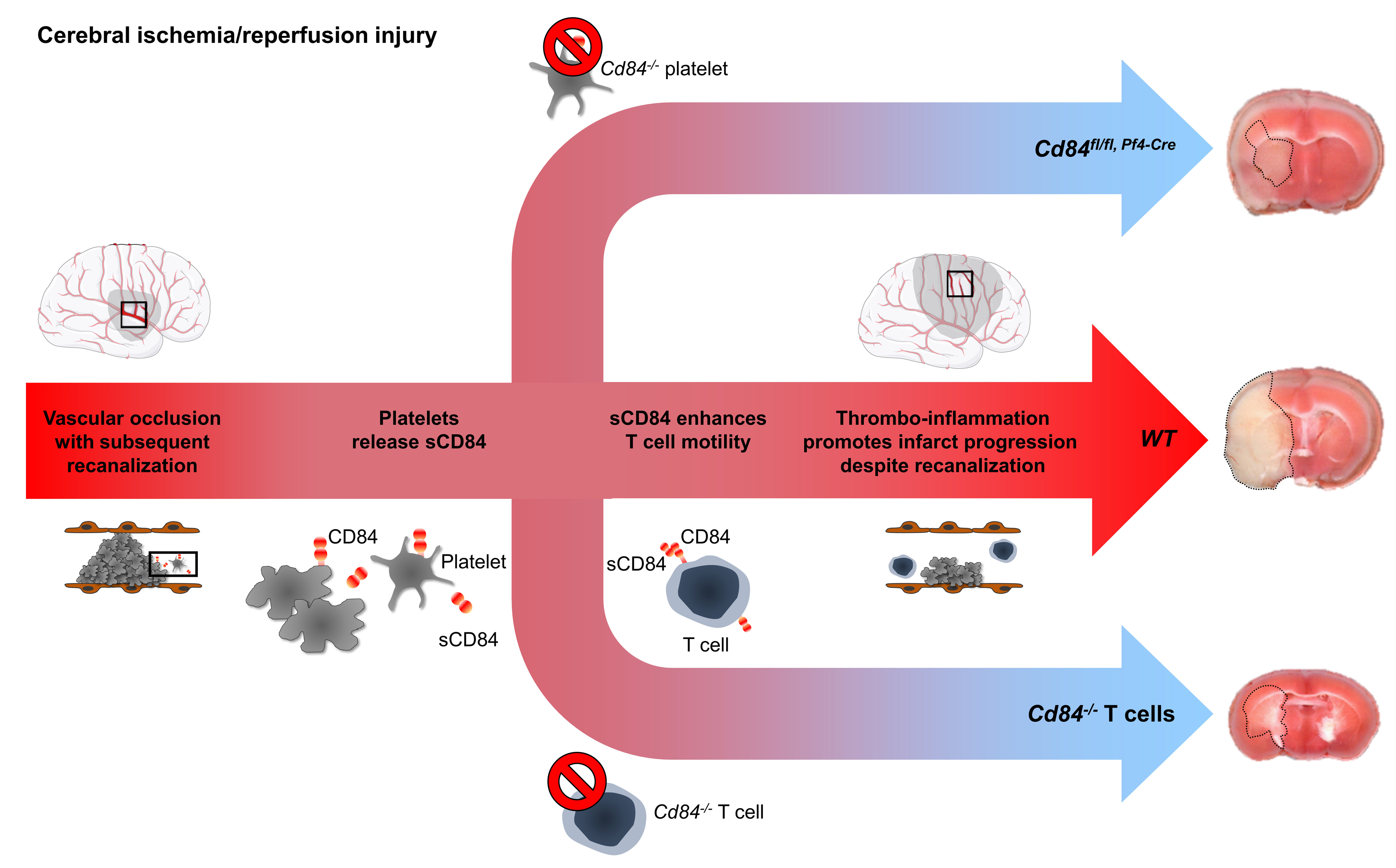
Stroke is one of the major causes of death and disability worldwide with limited treatment options. In acute ischemic stroke (IS), the primary therapeutic goal is the rapid reconstitution of cerebral blood flow, achievable either by treatment with recombinant tPA (tissue-type plasminogen activator) or mechanical thrombectomy (MTE). MTE, which is experimentally mimicked by the most widely used animal stroke model, the transient middle cerebral artery occlusion (tMCAO) model—is particularly effective for severe clinical syndromes caused by occlusion of large cerebral arteries as the internal carotid artery (ICA) or the middle cerebral artery.1,2 However, despite recent advances in MTE with high recanalization rates of up to ≈80%, the therapeutic efficacy of vessel reopening remains limited. Some studies indicate that infarction may progress significantly during reperfusion,3–5 and for many patients, infarction has already progressed unfavorably before hospital arrival preventing any recanalization treatment.4–6 The progression of cerebral infarction is frequently attributed to ischemia/reperfusion (I/R) injury, which has long been recognized as progressive tissue injury during blood flow return after transient organ ischemia. I/R is not a peculiarity of the ischemic brain but firmly established also, for example, in the heart, kidney, and liver.3,7,8 In the brain, T cells significantly contribute to I/R injury following tMCAO in an antigen-independent manner, which is demonstrated by protection of the brain from I/R injury in immunodeficient Rag1−/− mice, while adoptive transfer of CD (cluster of differentiation) 4+ T cells fully reconstituted stroke susceptibility.9,10 Besides T cells, platelets have been identified as key orchestrators of experimental cerebral I/R injury that promote inflammation and subsequent thrombotic activity in the microcirculation. Microvascular thromboinflammation can be efficiently prevented by blocking early steps of platelet adhesion and activation via GP (glycoprotein) Ib and GPVI receptors, respectively.11 Importantly, the pathogenic effect of platelets is linked to that of T cells as depletion of platelets in Rag1−/− mice abolished the detrimental effect of adoptively transferred CD4+ T cells.12 These findings led to the recent concept of thromboinflammation as the driving force underlying I/R injury in the ischemic brain.13–15 Thereby platelets act in concert with T cells to cause further brain injury, but the underlying mechanisms and the molecular link between the two cell types have remained elusive.
CD84 is a member of the SLAM (signaling lymphocyte activation molecule) family and acts as a homophilic cell adhesion molecule highly expressed on different immune cell populations and platelets.16–18 The N-terminal ectodomain of CD84 mediates the homophilic interaction between CD84 proteins,17,18 whereas the C-terminal intracellular portion of CD84 bears 2 immunoreceptor tyrosine-based switch motifs, which can bind the intracellular adapters SAP (SLAM-associated protein; also termed SH2D1A) and EAT-2 (Ewing sarcoma activated transcript 2).16,19 CD84 ligation triggers immunoreceptor tyrosine-based switch motif phosphorylation and SAP recruitment, resulting in enhanced IFN (interferon)-γ production and proliferation in T cells stimulated with low doses of anti-CD3 antibodies.17,19 CD84 has been established as a functional coreceptor in lymphocytes that facilitates prolonged B:T-cell interactions required for germinal center formation.20 Thus, while CD84 appears to have a role in immune cell activation, its function in platelets is not well understood. Upon platelet activation, CD84 is shed from the platelet surface,21 but the (patho)physiological significance of soluble CD84 (sCD84) is unknown. Notably, CD84 deficiency had no effect on the hemostatic or thrombotic function of platelets in mice in vitro and in vivo.22
Here, we show that platelet-derived sCD84 acts on CD4+ T-cell CD84 leading to enhanced CD4+ T-cell motility in vitro and aggravated infarct growth following cerebral I/R. Finally, we corroborated the clinical relevance of these findings by directly demonstrating reduced platelet CD84 levels in the ischemic circulation of stroke patients indicating CD84 shedding during the hyperacute stage of stroke. Furthermore, human platelet CD84 expression levels were associated with poor outcome in acute IS.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. All animal experiments and the analysis of the corresponding data were performed blinded.
Experimental Animals
Mice used in this study were matched for age (7–14 weeks), sex, and genetic background. Experiments were conducted in accordance with the regulations of the local authorities (Regierung von Unterfranken) and performed in accordance with the current Animal Research: Reporting of In Vivo Experiments guidelines (https://www.nc3rs.org.uk/arrive-guidelines). Exclusion criteria and dropout rates are provided in Table I in the Data Supplement.
Sample Size Calculation for tMCAO Experiments
Assuming a reduction of infarct volume of 30% as functionally relevant and an SD of 20% to the respective mean values, a group size of 8 to 10 was necessary to show this effect with a power of 0.8 and a probability of a type I error of <0.05 (calculated with GraphPad StatMate 2.00).
Animal Treatment
Heart rate and blood pressure were determined in isoflurane anesthetized mice using a Pressure Meter LE5001 (Harvard Apparatus, Boston, MA) according to the manufacturer’s instructions. For T-cell transfer experiments into Rag1−/− and Cd84−/− mice, splenic CD4+ T cells were isolated by negative selection (Miltenyi Biotech). Cells were injected intravenously (750 000 cells/mouse) 1 day before tMCAO, and successful T-cell transfer was controlled 1 hour after iv injection by flow cytometry of blood immune cells.10
Focal Ischemia Model
Focal cerebral ischemia was induced in 10- to 14-week-old C57BL/6, Cd84−/−,21 Rag1−/−,23 Cd84fl/fl,PF4-Cre-neg, or Cd84fl/fl,PF4-Cre mice by tMCAO as described previously.24 Inhalation anesthesia was induced by 2% isoflurane. The duration of the surgical procedure per animal was kept below 10 minutes. A silicon rubber-coated 6.0 nylon monofilament (6021PK10; Doccol, Redlands, CA) was advanced through the carotid artery up to the origin of the middle cerebral artery, causing an middle cerebral artery infarction. After an occlusion time of 60 minutes, the filament was removed allowing reperfusion. Animals were sacrificed 23 hours after reperfusion, and brains were checked for intracerebral hemorrhages. Neurological function was analyzed calculating a neuroscore (score, 0–10) based on the direct sum of the Grip test (score, 0–5) and the inverted Bederson score (score, 0–5).25–27 For assessment of the long-term neurological outcome, the occlusion time was reduced to 30 minutes, and Bederson score, modified neurological severity score, corner test, body swing test, grip test, latency to move, and adhesive tape removal test were performed daily as described previously.28
Infarction Size Measurement
The extent of infarction was quantitatively assessed 24 hours after occlusion. Animals were sacrificed, and brains were cut in three 2-mm-thick coronal sections. The slices were stained for 20 minutes at 37°C with 2% 2,3,5 triphenyltetrazolium chloride (Sigma-Aldrich; 2% [w/v] solution) to visualize the infarctions.29 Edema-corrected infarct volumes were calculated by planimetry (ImageJ Software; National Institutes of Health) according to the following equation: Vindirect (mm3)=Vinfarct×(1−[VI−VC]/VC). VI−VC represents the volume difference between the ischemic hemisphere (VI) and the control hemisphere (VC), and (VI−VC)/VC expresses this difference as a percentage of the control hemisphere.
Histology
Histology and immunohistochemistry were performed according to standard procedures, and all antibodies were validated using isotype controls (not shown).24 Cryo-embedded coronal brain sections (2 mm) were cut into 10-µm-thick slices. Every tenth slice was used for evaluation. The following antibodies were used: monoclonal antibodies: anti-CD11b (MCA711; Serotec), anti-Ly6B (MCA771G; Bio-Rad), and anti-CD31 (MCA2388; Bio-Rad) and polyclonal antibodies: anti-CD4 (100506; BioLegend) and anti-albumin (ab207327; Abcam). Apoptotic neurons were visualized by double immunolabeling of mouse anti-NeuN (neuronal nuclei) antibody (MAB377; Merck, Darmstadt, Germany) and TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) in situ cell death detection kit TMR red (12156792910; Merck). For quantification of occluded microvessels, brain slices were stained with hematoxylin and eosin. Afterward, the numbers of occluded and opened vessels per hemisphere were counted to determine the percentage of occlusions as described previously.24 All immunohistologic stainings were analyzed and acquired using a Nikon Eclipse 50i microscope or a Leica SP8 confocal microscope. Representative images were chosen among the images of one genotype to best reflect the mean values of this genotype.
Cell Separation and Flow Cytometry
For platelet analysis, peripheral blood was collected in heparin (20 U/mL in TBS [Tris-buffered saline]) and diluted in Tyrode-HEPES buffer and incubated with the indicated antibodies. CD84 expression (in-house generated JER1-FITC21 [fluorescein isothiocyanate]) of GPllb/IIIa–positive platelets (in-house generated JON6-PE30) and expression of major platelet receptors was analyzed using FACS Celesta or FACS Calibur (Becton Dickinson, Heidelberg, Germany). In the remaining blood, RBCs (red blood cells) were lysed using ACK (ammonium-chloride-potassium) buffer and immune cells were stained with the indicated antibodies for 30 minutes on ice in FACS buffer. For the isolation of brain-infiltrating mononuclear cells, a Percoll (GE Healthcare) density gradient (50%/30%) was used as described.12 Cells were washed and subsequently incubated with the dead cell marker (LIFE/DEAD Fixable Aqua Dead Cell Stain Kit; Thermo Fisher) for 30 minutes on ice. CD84 expression was analyzed using FACS Celesta. The following antibodies were used: anti-CD45-A700 (clone 30-F11), anti-Ly6G-A647 (clone 1A8) anti-CD3-BV786 (clone 145-2C11), anti-CD4-BV605 (clone RM4-5), anti-CD8a-PerCP/Cy5 (clone 53-6.7), anti-CD11b-APC/Cy7: clone M1/70), anti-CD19-BV650 (clone 6D5—all from BioLegend), and clone HB197 (produced in-house) was used to block Fc receptors. Gating strategy is indicated in Figure IVA in the Data Supplement.
Protein Extraction and Western Blot Analysis
Western blot analysis was performed according to standard procedures using antibodies against albumin (ab106582; Abcam), CD84 (in-house generated JER121), GAPDH (G9545; Sigma), and anti-β-actin (A5441; Sigma-Aldrich).31
Real-Time Polymerase Chain Reaction
Tissue homogenization, RNA isolation, and real-time polymerase chain reaction were performed as described.31 Relative gene expression levels of TNFα (tumor necrosis factor-alpha; assay ID: Mm 00443258_m1; Applied Biosystems), IL (interleukin)-1β (assay ID: Mm 00434228_m1; Applied Biosystems), MMP (matrix metalloproteinase)-2 (assay ID: Mm 00439498_m1; Applied Biosystems), and MMP-9 (assay ID: Mm 00442991_m1; Applied Biosystems) were analyzed with a fluorescent TaqMan technology. As an endogenous control Gapdh (TaqMan Predeveloped Assay Reagent for gene expression, part number 4352339E; Applied Biosystems) was used. Polymerase chain reaction was performed using the StepOnePlus Real-Time PCR System (Applied Biosystems).
T-Cell Migration Assays
The migratory capacity of CD4+ T cells was measured using ibidi µ-Slides VI0.4 coated with poly-D-lysine (10 µg/mL; Merck) and laminin (20 µg/mL; Merck) or ibidi Glass Bottom µ-Slides 8 well cultured with primary primary murine brain microvascular endothelial cells as described previously.32 The migration assays were either performed in DMEM high glucose (31053-028; Thermo Fisher) with penicillin/streptomycin (1%, P4333; Merck) and B27 supplement (2%, 17504044; Thermo Fisher) or Roswell Park Memorial Institute (RPMI) (12633020; Sigma) with glutamine (1%, 59202C; Merck) and penicillin/streptomycin (1%, P4333; Merck). Each condition (phorbol-12-myristate-13-acetate [8 µg/µL; Merck] and CCL20 [C-C chemokine cysteine motif chemokine ligand 20; 24.5 µg/µL; PeproTech]), Fc-proteins (0.2 µg/mL), washed WT or Cd84−/− platelets (diluted 50:1 with CD4+ T cells), platelet-releasate (PLT-R; 1:1 diluted with migration media) was performed in duplicates or triplicates. PLT-R was obtained by stimulating platelets (500 000/µL) with 10 µg/mL of the GPVI agonist CRP (collagen-related peptide) for 15 minutes; subsequently, the platelet supernatant was harvested (5 minutes with 800 g, followed by 5 minutes with 22000 g, 4°C). The GPVI-Fc fusion protein (control-Fc)33 that did not affect CD4+ T-cell migration compared with vehicle (not shown) served as control for CD84-Fc (recombinant sCD84 fused to the Fc part of human IgG1); both Fc fusion proteins were purified in-house from transfected HEK 293 (human embryonic kidney) cells using standard techniques. For time-lapse video microscopy, MACS (magnetic-activated cell sorting) isolated mouse CD4+ T cells (130-104-454; Miltenyi; 100 cells/µL) were added to the chamber. Images were collected every 30 seconds for 30 minutes on a Leica DMI8 inverted microscope with 20× objective. Cells were tracked with LAS X Imaging software and analyzed with ImageJ 1.51 software.
Analysis of Human Blood Samples
Both the local ischemic and systemic blood samples were drawn into citrate phosphate dextrose adenine monovettes (S-Monovette; Sarstedt, Nümbrecht, Germany). Samples were diluted after blood collection in PBS, and platelets were stained using anti-CD42b-FITC (SZ2; Immunotech SAS, Marseille, France) and anti-CD84-PE antibodies (CD84.1.21; BioLegend, San Diego). After an incubation period of 30 minutes, the samples were analyzed by flow cytometry instrument FACS Calibur (Becton Dickinson, Franklin Lakes). CD84 expression was defined as mean fluorescence intensity of PE (phycoerythrin) in platelets gated by CD42b positivity.
Clinical Observation of Local Human Arterial Blood Samples
The protocol of the prospective study of local ischemic and systemic arterial blood samples during hyperacute stroke was approved by the local ethics committee (approval number 135/17) and performed as described previously.34 During first-ever IS, it was determined by CT angiography that the embolic occlusion location matched the ICA-T/M1 segment. MTE was then indicated based on clinical and radiological criteria recommended by current guidelines.35 All procedures were performed by board-certified neurointerventional experts or by supervised neurointerventional fellows. A detailed description of the MTE technical procedural steps used in this study is given elsewhere.36 Briefly, microcatheter navigation was performed into the occluded vascular field via a 0.0014 microinch microwire over a coaxial guide catheter system positioned in the cervical common carotid arteries and ICAs. Having reached this position, one local arterial blood sample was drawn by microcatheter aspiration. Two further samples were drawn after recanalization at the systemic levels of the proximal internal carotid and common femoral arteries.
Clinical Observation of the SICFAIL Cohort
To assess the transferability of the findings from mouse model into humans, the association of CD84 expression on platelets and stroke severity at day 3 after hospital admission was analyzed in a substudy within the SICFAIL study (Stroke-Induced Cardiac Failure in Mice and Men). SICFAIL is an ongoing, prospective cohort study, aiming to describe the natural course of cardiac function in 750 unselected acute IS patients (DRKS00011615). The subsample of the SICFAIL study population, analyzed in this study, included 98 patients with the diagnosis of acute IS according to World Health Organization definition37 aged ≥18 years, who were hospitalized at the stroke unit of the University Hospital Würzburg between June 2016 and January 2017. Written informed consent was obtained prior study enrollment. The Ethics Committee of the Medical Faculty of the University of Würzburg (vote 176/13) approved the study. All patients or their legal representatives provided written informed consent to participate.
All patients underwent standard treatment and stroke unit monitoring. Information on stroke severity and acute treatment (eg, intravenous thrombolysis) was retrieved by review of patient records. Stroke severity was assessed by a neurologist according to the National Institutes of Health Stroke Scale (NIHSS).38 Additionally, all study participants underwent a personal standardized interview to collect information on demographic characteristics and comorbidities. Based on previously published data,39 poor outcome was defined before analyses as NIHSS ≥5 on day 3 of hospitalization, representing moderate or severe stroke.
For CD84 analysis, blood was drawn the morning after study enrollment in tubes containing citrate phosphate dextrose adenine and analyzed as described above.
Statistical Analyses
All data from animal experiments are given as box plots including median with the 25th percentile, the 75th percentile, minimum deviation, and maximum deviation except for the migration assays, which are depicted as scatter plots including the mean and SEM. Human data are presented as median and interquartile range (IQR; 25th and 75th percentile) or, in case of categorical data, frequencies and percentages, respectively. Cell migration data were analyzed by 1-way ANOVA with Bonferroni post hoc tests. All other data were analyzed by nonparametric tests (specified in the respective figure and table legends) due to small sample sizes. Corrections for multiple tests within one experimental assay in animal studies were done by appropriate post hoc tests (eg, Dunn test for Kruskal-Wallis tests) or the Holm-Sidak method. No corrections for multiple testing were made across assays. For nonparametric repeated measures of human data, we used Friedman rank test followed by Dunn multiple comparison test. In case of frequencies, χ2 test or Fisher exact test was used to compare groups. Multivariable logistic regression analysis, adjusted for age and NIHSS at baseline, was used to identify the association of CD84 expression and poor outcome on day 3 of hospitalization (NIHSS score, ≥5). For sensitivity analyses, the model was recalculated by further adjusting for comorbidities including previous stroke or transient ischemic attack, coronary heart disease, hypertension, type 2 diabetes mellitus, and atrial fibrillation. The α-level was set to 0.05 (2 sided). P values displayed in figures are corrected for multiple testing, as described above; P>0.05 is not displayed in the figures (albeit all data were analyzed with the statistical tests provided in the figure or table legends). For statistical analysis, the GraphPad Prism 5.0 and 8.2.1 software package (GraphPad Software) and SAS 9.4 (SAS Institute, Inc, Cary, NC) were used.
Results
Cd84−/− Mice Have Less CD4+ T-Cell Infiltration Into the Ischemic Hemisphere and Smaller Infarcts Following IS
We have previously shown that Cd84−/− mice display no overt phenotype and exhibit no defects in platelet function in hemostasis or thrombosis.21,22 Of note, brain morphology was unaltered in Cd84−/− mice as assessed by ink staining of the vasculature and Nissl staining of brain slices (Figure I in the Data Supplement). To test for a possible role of CD84 in thromboinflammation, Cd84−/− mice were subjected to 1 hour of tMCAO and 23 hours of reperfusion. Strikingly, median infarct volumes in the mutant mice were significantly reduced compared with WT littermates at 24 hours after tMCAO as measured by triphenyltetrazolium chloride staining (97.8 [IQR, 75.7–124.2]) versus 83.8 [IQR, 45.1–92.2] mm3; P=0.0198; Figure 1A and 1B), while heart rate (median, 532.8 bpm [IQR, 520.2–544.4] versus 533.5 bpm [IQR, 512.6–588.2]) and blood pressure in naive mice were indistinguishable (not shown) and local cerebral blood flow during and after middle cerebral artery occlusion was comparable (Figure IIA in the Data Supplement). The reduction in infarct volumes corresponded with improved neurological outcome in Cd84−/− mice as reflected by a higher mean neuroscore (4.3±1.0 versus 5.2±1.5, respectively; P=0.0269; Figure 1C). Sex can have a significant impact on stroke outcome.40 Therefore, we subjected male and female mice to 60 minutes of tMCAO. Gender-specific evaluation revealed significantly smaller infarctions in both male and female Cd84−/− mice (Figure IIB and IIC in the Data Supplement). To investigate the role of CD84 in long-term functional outcomes following stroke, we analyzed sensorimotor deficits of male Cd84−/− mice and control littermates over 7 days after 30 minutes of tMCAO (Figure 2). In line with the results following 60-minute tMCAO, Cd84−/− mice displayed less severe functional defects on day 1 following 30-minute tMCAO as compared with WT mice (modified neurological severity score, 3 [IQR, 2.5–4.0] versus 6 [IQR, 4.8–8.3]; P=0.0111; Figure 2A). This difference between the 2 genotypes persisted for the 7-day observation period as confirmed by the modified neurological severity score or different additional tests (Figure 2B and 2C; Figure III in the Data Supplement). Three WT but no Cd84−/− mice had to be euthanized due to severe impairments (Figure IIIA in the Data Supplement).
Figure 1.
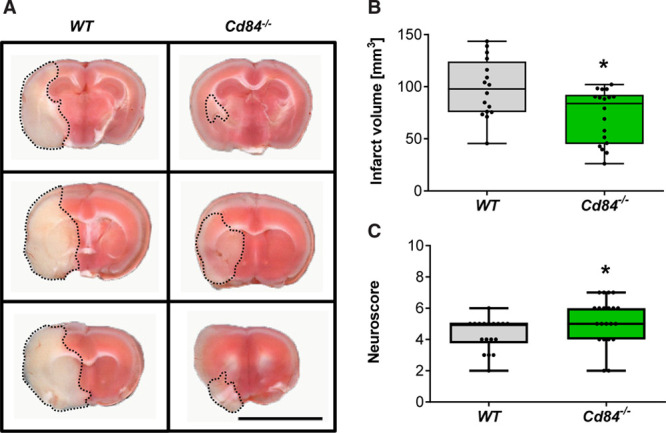
Cd84−/− mice are protected after transient middle cerebral artery occlusion (tMCAO).
A, Representative triphenyltetrazolium chloride–stained 2-mm brain slices from 1 WT and 1 Cd84−/− mouse 24 h after tMCAO. Bar=1 cm. B, Infarct volumes 24 h after tMCAO of WT (n=16) and Cd84−/− mice (n=18; P=0.0198). C, WT (n=18) and Cd84−/− (n=21) neuroscore at 24 h after tMCAO (P=0.0269). Statistical significances analyzed by Mann-Whitney U test. *P<0.05.
Figure 2.
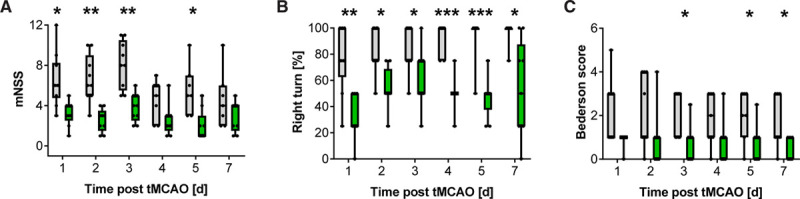
Role of CD (cluster of differentiation) 84 in long-term functional outcome following stroke.
Neurological outcome over time in WT (gray boxes) and Cd84−/− mice (green boxes) as assessed by modified neurological severity score (mNSS; A), Corner test (B), and Bederson score (C) after 30-min transient middle cerebral artery occlusion (tMCAO; n=7–10 mice per group). Each symbol represents one individual. Statistical significances between WT and Cd84−/− mice were determined using the Holm-Sidak correction on P obtained by Mann-Whitney U tests. *P<0.05, **P<0.01, and P<0.001; exact n-numbers and P are listed in Table V in the Data Supplement.
Next, we assessed accumulation of lymphocytes and monocytes in the brain after tMCAO. Cd84−/− mice showed significantly reduced numbers of CD4+ T cells in the infarcted brain (reduction by ≈50%) compared with WT mice (Figure 3A), whereas the amount of CD11b+ cells (Figure 3B) and the accumulation of neutrophils (Ly6B.2+ cells; Figure 3C) was comparable between the 2 groups. These data were confirmed by flow cytometry (Figure IV in the Data Supplement). Furthermore, the percentages of occluded vessels in the ipsilateral hemisphere was reduced by ≈35% in Cd84−/− mice 24 hours after stroke induction (Figure 3D), and less platelet deposition in the ipsilateral hemisphere of Cd84−/− mice was observed (not shown). These data demonstrated that CD84 deficiency is protective after tMCAO and that CD84 contributes to CD4+ T-cell recruitment into the infarcted brain territory. We analyzed neuronal apoptosis and blood-brain barrier damage in WT and Cd84−/− mice 24 hours after stroke induction (Figure VA through VC in the Data Supplement). In line with smaller infarct volumes, neuronal survival and blood–brain barrier (BBB) function in the mutant mice were significantly improved, whereas the expression levels of TNFα, IL-1β, MMP-2, and MMP-9 did not differ significantly between WT and Cd84−/− mice (Figure VD through VG in the Data Supplement).
Figure 3.
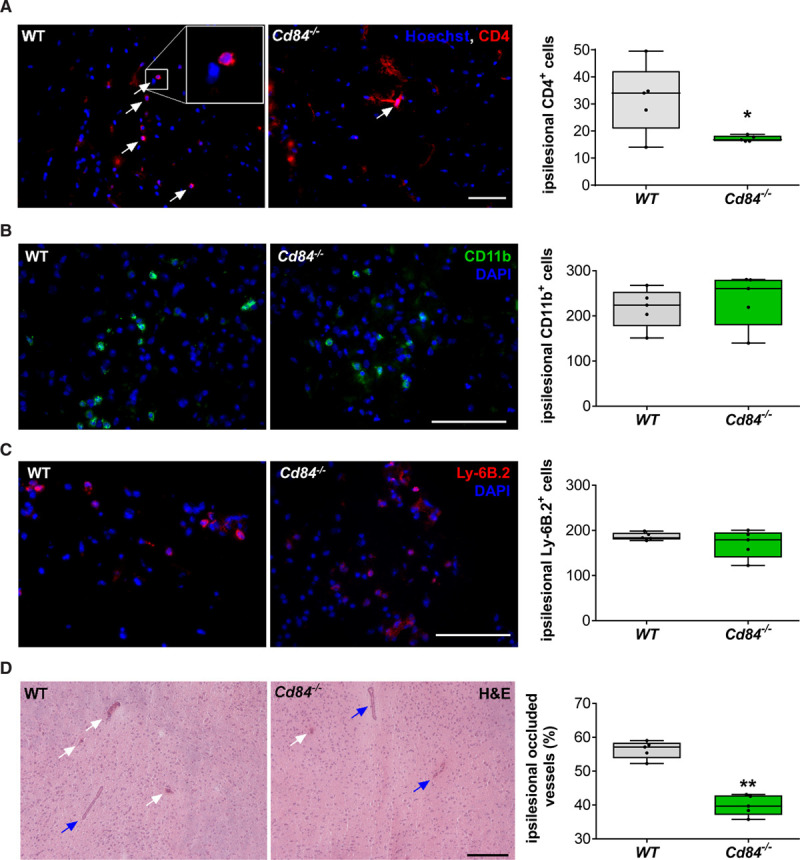
CD (cluster of differentiation) 4+ T-cell recruitment to the postischemic brain is diminished in CD84-deficient mice.
A, Representative immunocytologic stainings and quantification of brain-infiltrating CD4+ T lymphocytes in the ipsilateral hemisphere on day 1 after transient middle cerebral artery occlusion (tMCAO) of WT (wild type) and Cd84−/− mice (n=5 mice per group; P=0.0328). Bar=100 µm. B, Representative CD11b immunoreactivity and quantification of CD11b+ cells in the ipsilateral hemisphere 24 h after tMCAO of WT and Cd84−/− mice (n=5 mice per group; P=0.5476). Bar=100 µm. C, Representative images and quantification of Ly6B.2+ cells in the ipsilateral hemisphere 24 h after tMCAO of WT and Cd84−/− mice (n=5 mice per group; P=0.5794). Bar=100 µm. D, Representative hematoxylin and eosin (H&E) staining and quantification of the percentage of occluded vessels in the infarcted hemispheres of WT and Cd84−/− mice (n=5 mice per group; P=0.0079). White arrows indicate occluded vessels; blue arrows indicate patent vessels. Bar=100 µm. Statistical significances analyzed by Mann-Whitney U test. *P<0.05 and **P<0.01. DAPI indicates 4′,6-diamidino-2-phenylindole.
CD84 Expression on CD4+ T Cells and Platelets Is Required to Promote Infarct Growth After tMCAO
Then, we assessed the relevance of CD4+ T cell–expressed CD84 for infarct progression by performing adoptive T-cell transfer experiments. Immunodeficient Rag1−/− mice, which lack B and T cells, or Cd84−/− mice received CD4+ T cells from WT or Cd84−/− mice 24 hours before subjecting them to tMCAO. While Rag1−/− mice displayed small infarcts 24 hours after tMCAO,9,10,41 fully evolved infarctions were found in Rag1−/− mice after adoptive transfer of WT CD4+ T cells (Figure 4A and 4B). In sharp contrast, Rag1−/− mice reconstituted with Cd84−/− CD4+ T cells had significantly smaller infarcts (median, 100.9 [IQR, 74.8–126.0] versus 28.6 [IQR, 22.2–111.4] mm3; P=0.0050; Figure 4A and 4B) and better neurological outcome (Figure VI in the Data Supplement) at day 1 after stroke than those transplanted with WT CD4+ T cells. Of note, lymphocyte and thymocyte subsets were indistinguishable between WT and Cd84−/− mice (not shown), nor did the secretion of typical cytokines from stimulated CD4+ T cells differ (Table II in the Data Supplement).
Figure 4.
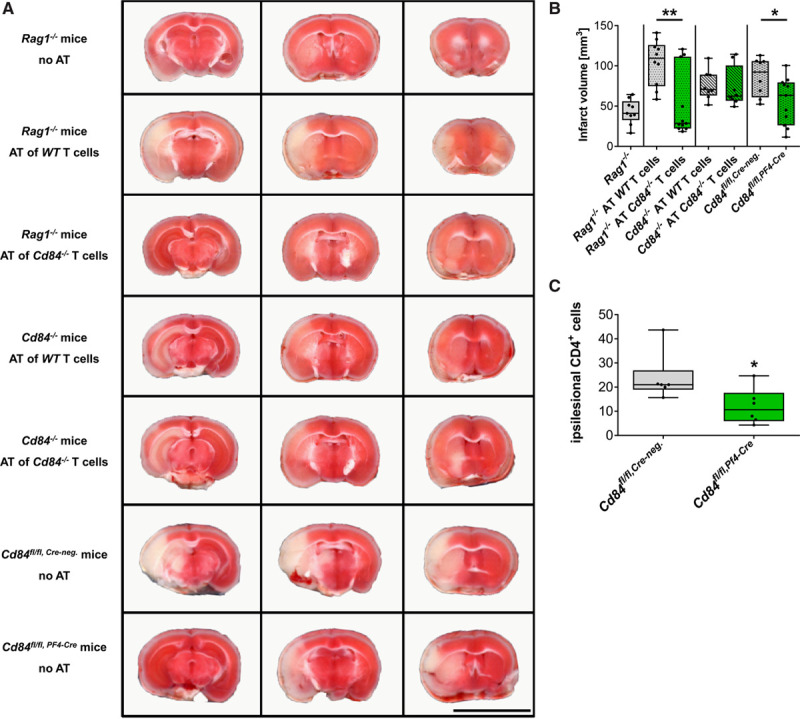
CD (cluster of differentiation) 84 expression on CD4+ T cells and platelets is required to promote infarct growth after transient middle cerebral artery occlusion (tMCAO).
Representative triphenyltetrazolium chloride–stained 2-mm brain slices of (A, top to bottom) and infarct volumes 24 h after tMCAO (B) of Rag1−/− mice (n=9), Rag1−/− mice with adoptive transfer of WT (n=10), or Cd84−/− CD4+ T cells (n=11; P=0.0050) at day 1 before surgery, Cd84−/− mice with adoptive transfer of WT (n=8) or Cd84−/− CD4+ T cells (n=8) at day 1 before surgery, and mice specifically lacking the expression of CD84 on platelets (n=11) compared with control mice (n=8; P=0.0409), at day 1 after tMCAO. Bar=1 cm. C, Quantification of brain-infiltrating CD4+ T lymphocytes in the ipsilateral hemisphere on day 1 after tMCAO of Cd84fl/fl, Cre-neg and Cd84fl/fl, Pf4-Cre mice (n=6 mice per group; P=0.0368). Statistical significances analyzed by Mann-Whitney U test. *P<0.05. AT indicates adoptive transfer.
In contrast to adoptive CD4+ T-cell transfer in Rag1−/−, infarct volumes and functional outcome did not differ when adoptively transferring WT or Cd84−/− CD4+ T cells into Cd84−/− mice (Figure 4A and 4B) indicating that an interaction of CD84+ T cells with another CD84-expressing cell type is required to induce I/R injury. To address a possible role of platelets in this setting, we generated Cd84fl/fl,PF4-Cre mice. Knockout efficacy was confirmed using flow cytometry and Western blot (Table III in the Data Supplement; Figure VIIA through VIIC in the Data Supplement). As reported previously,22 CD84 deficiency did not affect classical platelet activation (Figure VIID and VIIE in the Data Supplement). Cd84fl/fl,PF4-Cre mice developed significantly (≈30%) smaller infarcts at day 1 after tMCAO when compared with littermate control mice (Cd84fl/fl, Cre-neg; median, 92.3 [IQR, 61.0–105.9] versus 63.4 [IQR, 26.2–79.4] mm3; P=0.041; Figure 4A and 4B) suggesting that platelet CD84 is required for the development of CD4+ T cell–dependent cerebral I/R injury. This is in agreement with the previous observation that platelet depletion abolished detrimental CD4+ T-cell effects during I/R in stroke after adoptive transfer into immune-deficient Rag1−/− mice.12 Of note, also in mice lacking CD84 only on megakaryocytes and platelets, less CD4+ T cells were detected in the ischemic hemisphere 24 hours after tMCAO (Figure 4C), while the expression of Cre recombinase under the Pf4 promoter did not affect infarct sizes (Figure VIII in the Data Supplement).
sCD84 Promotes CD4+ T-Cell Migration
To assess whether CD84 is required for T-cell migration, we measured the motility of WT and Cd84−/− CD4+ T cells in a laminin/PDL (poly-D-lysine)-coated 2-dimensional in vitro system and did not observe differences between the 2 genotypes in response to phorbol-12-myristate-13-acetate or the CCL20 (Figure 5A; Figure IXA in the Data Supplement). As we have previously shown that the activatory platelet receptor GPVI plays a critical role in cerebral ischemia,11,42 we tested the effect of the releasate from GPVI–stimulated WT platelets on CD4+ T-cell motility in this assay. Treatment of WT CD4+ T cells with PLT-R induced an increase of ≈25% in distance and speed of migrating CD4+ T cells, whereas this effect was absent in Cd84−/− T cells (Figure 5B; Figure IXB in the Data Supplement). Given the established homophilic binding of CD84 and the efficient ectodomain shedding of CD84 in activated platelets,21 we suspected platelet-derived sCD84 to mediate this effect. Indeed, the releasate of Cd84−/− platelets failed to increase the migratory capacity of WT T cells, strongly suggesting that platelet-derived sCD84 acts as a costimulatory factor on CD4+ T cells (Figure 5C; Figure IXC in the Data Supplement). To test this directly, we treated WT and Cd84−/− T cells with CD84-Fc or a control-Fc protein and compared their migratory behavior to that of untreated CD4+ T cells (control). Notably, WT but not Cd84−/− T cells responded to the presence of CD84-Fc with an increase in velocity and migrated distance compared with the control groups, indicating that the homophilic CD84 interactions are critical for the T-cell modulating effects of sCD84 (Figure 5D; Figure IXD in the Data Supplement). Of note, no significant difference in T-cell motility was observed in the presence of WT or Cd84−/− platelets (Figure IXE and IXF in the Data Supplement). In contrast, Cd84−/− PLT-R supplemented with CD84-Fc restored CD4+ T-cell motility to a level comparable to that induced with WT PLT-R (Figure 5E; Figure IXG in the Data Supplement) demonstrating that platelet-derived sCD84 enhances CD4+ T-cell migration. Notably, the migration-promoting effect of sCD84 on WT T cells was also observed in a migration assay in the presence of primary brain-derived microvascular endothelial cells, and results were indistinguishable on WT or Cd84−/− endothelial cells (Figure 5F; Figure IXH in the Data Supplement). Together, these data demonstrated that platelet CD84 exerts a (co)stimulatory effect on CD4+ T cells and is a critical determinant of thromboinflammatory infarct progression in a murine model of IS.
Figure 5.
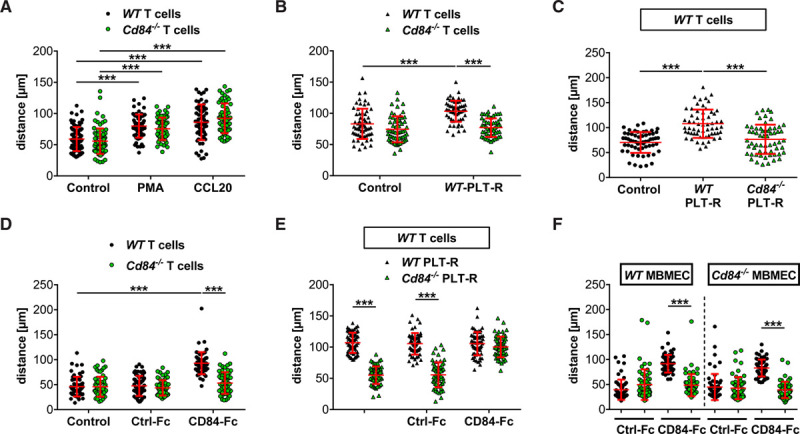
Soluble CD (cluster of differentiation) 84 promotes CD4+ T-cell migration.
A, Migrated distance of CD4+ WT and Cd84−/− T cells treated with vehicle, phorbol-12-myristate-13-acetate (PMA), or CCL20 (C-C motif chemokine ligand 20). B, WT and Cd84−/− CD4+ T-cell migration in response to stimulation with WT platelet-releasate (PLT-R) compared with vehicle (control). C, WT CD4+ T-cell migration in response to stimulation with WT or Cd84−/− PLT-R compared with control. D, Migrated distance of CD4+ WT and Cd84−/− T cells treated with vehicle, recombinant control-Fc, or CD84-Fc (recombinant soluble CD84 fused to the Fc part of human IgG1) protein. E, WT CD4+ T-cell migration in response to stimulation with WT or Cd84−/− PLT-R in the presence of control-Fc or recombinant CD84-Fc protein. F, Migrated distance of CD4+ WT and Cd84−/− T cells treated with control-Fc or CD84-Fc on primary murine brain microvascular endothelial cells (MBMECs) of WT or Cd84−/− mice. Each dot represents the migrated distance over 30 min of one CD4+ T cell (n=59–80 cells per group of 3–4 independent experiments). Horizontal red lines correspond to the mean and SD. Statistical significances analyzed by 1-way ANOVA with Bonferroni post hoc test or Kruskal-Wallis test with Dunn multiple comparison test, the exact test n-numbers and P are listed in Table VI in the Data Supplement. ***P<0.001.
Downregulation of Platelet CD84 Surface Levels in the Ischemic Arterial Compartment
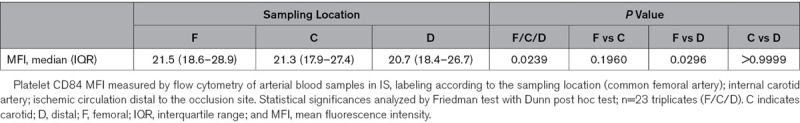
Finally, we applied 2 strategies of clinical observation to analyze the potential role of platelet CD84 in acute stroke patients. First, we established an endovascular clinical method of local human in vivo sampling of ischemic arterial blood already during the (hyper)acute stage of stroke with the samples taken directly within the occluded arterial vascular field. In this manner, local aspirates of 1.0 mL ischemic arterial blood were obtained via transfemoral arterial access immediately after endovascular microcatheter navigation into the center of the ischemic cerebral vascular field and before vessel reopening.34 This step was immediately ensued by therapeutic recanalization (ie, MTE). It was considered a particularly relevant a priori criterion for eligibility that the occlusion location had to precisely match the experimental situation (ie, ICA-T/M1 segment embolic occlusions).34 In a prospective cohort of large vessel occlusion stroke patients (n=23), we could then show that platelet surface abundance of CD84 (Table 1) was reduced locally within the ischemic circulation as compared with nonischemic intraindividual systemic platelet CD84 surface abundance indicating local shedding of CD84 from the platelet surface.
Table 1.
Downregulation of Platelet CD84 Surface Levels in the Ischemic Arterial Compartment
High Platelet CD84 Surface Levels Correlate With Poor Outcome in Stroke Patients
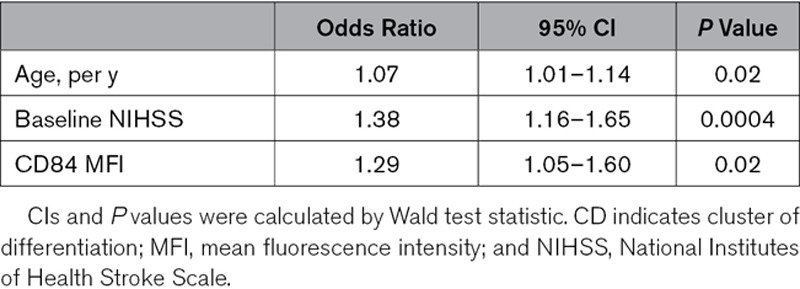
Second, local findings during the hyperacute stage were further extended by observation of a large prospective cohort in which we analyzed platelet CD84 expression in 98 participants of the SICFAIL study, to assess a possible link between platelet CD84 expression and disease course in acute IS patients. Median age was 67.0 years (IQR, 55.0–76.0 years), 66% of the patients were men, and the median NIHSS score on admission was 3 (IQR, 2–6; Table IV in the Data Supplement). Nineteen patients had poor outcome at day 3 of hospitalization defined as NIHSS ≥5. Patients with poor outcome tended to be older, had a history of hypertension, and had higher NIHSS scores on admission. Although not statistically significant in univariate analysis (P=0.18), platelet CD84 surface abundance was identified to be independently associated with poor outcome after adjustment of age and baseline NIHSS score (odds ratio, 1.29 [95% CI, 1.05–1.60]; Table 2; Table IV in the Data Supplement) in an exploratory multivariable logistic regression approach. After further adjusting the model for comorbidities, CD84 remained independently associated with NIHSS score ≥5 at day 3 (odds ratio, 1.27 [95% CI, 1.02–1.59]) in sensitivity analyses (data not shown).
Table 2.
Multivariable Logistic Regression to Assess the Association of Platelet CD84 Expression and Poor Outcome (NIHSS Score, ≥5) at Day 3 of Hospitalization, Adjusted for Age and NIHSS at Admission
Discussion
Our data strongly suggest that the homophilic interaction of platelet-derived sCD84 and CD4+ T cell–expressed CD84 represents a key pathogenic mechanism in cerebral I/R injury. In T cells, CD84 has been shown to support interactions with B cells and TCR-induced IFNγ secretion.17,20 To date, the function of CD84 in platelets is not well understood since Cd84−/− mice display normal platelet function in hemostasis and thrombosis.22 We and others have reported that platelet activation results in rapid ectodomain shedding of CD84,21,43 and here we show that sCD84 directly acts on CD4+ T cells and modulates their migratory behavior indicative of a (co)stimulatory effect. Studies in mice have shown that platelets are recruited very early to the postischemic cerebral microvasculature, and this appears to be mediated by GPlb-VWF (von Willebrand factor) interaction followed by GPVI–mediated cellular activation,11,42,44 the latter efficiently inducing release of sCD84.21 These findings support the assumption that sCD84 accumulates in the ischemic cerebral microvasculature during I/R and locally exerts its proinflammatory effects by acting on CD4+ T cells to promote their recruitment and migratory capacity, resulting in aggravated cerebral I/R injury. Thus, our data establish CD84 as a critical mechanistic link between T cells and platelets in acute IS that may represent a new therapeutic target to reduce neuronal damage in acute IS. It appears as a paradox that platelet CD84 is not critical for arterial thrombosis22 but contributes to tissue damage during I/R in the brain. However, the pathophysiology of I/R injury does not require rethrombosis14 as substantiated by the fact that blockade of GPllb/IIIa is ineffective.11 To investigate the relevance of this novel mechanism for acute stroke in humans, considerable efforts of clinical observation were undertaken. First, very early during the hyperacute phase of IS, and at the exact same occlusion location as in the experimental setting (ie, ICA-T/M1 segment occlusions),34 we observed a reduction of local platelet CD84 surface abundance. This reduction of CD84 is in line with ectodomain shedding of CD84 in activated platelets, locally within the center of the human ischemic arterial brain circulation.21
Furthermore, platelet CD84 surface abundance in peripheral venous blood of patients with acute IS was identified to be independently associated with poor outcome after adjustment of age and baseline NIHSS in 98 participants of the prospective SICFAIL cohort study in an exploratory approach. Despite the limitations of this analysis, including the small sample size and relatively mild affected IS patients, these results clearly point to a pathogenic role of CD84 in the development of IS in human patients.
Collectively, our data indicate that targeting CD84 might be effective in reducing antithromboinflammatory neuronal damage in the acute ischemic brain with the potential to improve stroke outcomes. In this regard, it is noteworthy that attempts to further improve clinical outcome after thrombolysis by conventional antiplatelet drugs, for example, acetylsalicylic acid, clearly failed due to an increase in intracranial hemorrhages.45 In our study, both, the data from complementary in vivo and in vitro experimental mouse models and the observations in human stroke patients point toward a role of CD84 in cerebral ischemia. Nevertheless, our results should be validated using further preclinical studies applying additional transient and permanent stroke models. In addition, it is important to note that, although the overall structure of the immune system in mice is similar to that in humans, existing differences in innate and adaptive immunity between mice and humans could interfere with clinical translation when targeting the interactions of T cells and platelets in acute stroke. Despite these potential differences, the concept of thromboinflammation as driving force in cerebral I/R injury has opened new therapeutic avenues based on the targeting of immunomodulatory functions of platelets while leaving their hemostatic response largely unaffected. Recently, several randomized controlled pilot trials could be completed testing the immunosuppressant drug FTY720, which rapidly reduces the number of circulating lymphocytes and is approved for the treatment of relapsing-remitting multiple sclerosis.46 Stroke patients treated immediately with FTY720 alone or in conjunction with r-tPA thrombolysis or thrombectomy showed decreased infarction.47–49 Our study now shows that targeting T-cell/platelet CD84 may become an alternative therapeutic strategy, which similarly does not affect hemostasis in the ischemic brain that is highly susceptible to bleeding complications. Our findings are likely to have broader implications also beyond the brain, since T cells have been identified as key players in I/R injury in many organs system such as the heart, kidney, and lung.
Acknowledgments
We thank Gabriele Köllner for excellent technical support. We are indebted to the Interventional Stroke Team of the Department of Neuroradiology (Thomas Günthner-Lengsfeld, Alexander März, Brigitte Bison, and Yanyan Xiong) for their cooperation in obtaining local arterial samples during hyperacute stroke treatment. The SICFAIL study (Stroke-Induced Cardiac Failure in Mice and Men) group consists of Christoph Kleinschnitz, Daniel Mackenrodt, Viktoria Rücker, Kathrin Ungethüm, Stefan Frantz, Karl Georg Haeusler, and Silke Wiedmann whom the authors wish to thank for designing and coordinating the human SICFAIL cohort study. We are grateful to Kathrin Ungethüm and Cornelia Fiessler for the statistical review of the SICFAIL study, as well as Tobias Geisler and Dominik Rath for flow cytometry (FACS) training of M. Seyhan. Finally, the authors thank the patients and their families for participating in the SICFAIL study. Parts of the results of the analysis of the human SICFAIL cohort study were presented at the European Stroke Organisation Conference 2018 in Gothenburg. M. Seyhan was supported by a scholarship of the Medical Faculty and the Graduate School of Life Sciences, University of Würzburg. M.K. Schuhmann, G. Stoll, B. Nieswandt, and D. Stegner designed the animal studies. M.K. Schuhmann, M. Bieber, T. Vögtle, S. Hofmann, V. Klaus, P. Kraft, L. Papp, and D. Stegner performed animal experiments. M.K. Schuhmann, G. Stoll, B. Nieswandt, and D. Stegner analyzed data from animal experiments, discussed results, and provided scientific input throughout the study. A.M. Kollikowski and M.K. Schuhmann analyzed the blood samples from stroke patients undergoing thrombectomy and M. Pham designed that part of the study. M. Seyhan performed FACS analyses in SICFAIL samples. M. Seyhan and P.U. Heuschmann analyzed SICFAIL data and wrote the corresponding part of the manuscript. M.K. Schuhmann, G. Stoll, B. Nieswandt, and D. Stegner wrote the paper with input and approval from all authors.
Sources of Funding
This work was supported by the Deutsche Forschungsgemeinschaft (project number 374031971–CRC/TR 240 project B06 to M.K. Schuhmann and D. Stegner, B02 to G. Stoll and M. Pham, B07 to B. Nieswandt, and Z03 to P.U. Heuschmann) and by the European Union (Thrombo-Inflame, Europäischer Fonds für regionale Entwicklung, Bavaria). The authors have no competing financial interests. The human SICFAIL study (Stroke-Induced Cardiac Failure in Mice and Men) was supported by the German Ministry of Research and Education within the Comprehensive Heart Failure Centre Würzburg (grant numbers BMBF 01EO1004 and 01EO1504).
Disclosures
M.K. Schuhmann, G. Stoll, B. Nieswandt, and D. Stegner have filled a patent (EP 19183678.2) related to this study.
Supplemental Material
Online Figures I–IX
Online Tables I–VI
Full Unedited Gels
Major Resources Table
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- CCL20
- C-C chemokine cysteine motif chemokine ligand 20
- CD
- cluster of differentiation
- CD84-Fc
- recombinant soluble CD84 fused to the Fc part of human IgG1
- CRP
- collagen-related peptide
- EAT-2
- Ewing sarcoma activated transcript 2
- GP
- glycoprotein
- I/R
- ischemia/reperfusion
- ICA
- internal carotid artery
- IFN
- interferon
- IL
- interleukin
- IQR
- interquartile range
- IS
- ischemic stroke
- MMP
- matrix metalloproteinase
- MTE
- mechanical thrombectomy
- NIHSS
- National Institutes of Health Stroke Scale
- PE
- phycoerythrin
- PLT-R
- platelet-releasate
- SAP
- SLAM-associated protein
- sCD84
- soluble CD84
- SICFAIL
- Stroke-Induced Cardiac Failure in Mice and Men
- SLAM
- signaling lymphocyte activation molecule
- tMCAO
- transient middle cerebral artery occlusion
- TNFα
- tumor necrosis factor-alpha
- tPA
- tissue-type plasminogen activator
- VWF
- von Willebrand factor
On behalf of the SICFAIL study group.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.316655.
For Sources of Funding and Disclosures, see page 1034.
Novelty and Significance
What Is Known?
Ischemia/reperfusion (I/R) injury in cerebral ischemia depends on the concerted action of platelets and T cells, a process referred to as thromboinflammation, but the molecular basis of the interplay of these 2 cell types is only partially understood.
CD (cluster of differentiation) 84 is highly expressed on different immune cell populations and platelets. It acts as a homophilic cell adhesion molecule and serves as a functional coreceptor in lymphocytes.
In activated platelets, CD84 has been shown to undergo ectodomain shedding, resulting in the release of soluble CD84. However, the (patho)physiological significance of soluble CD84 remains elusive.
What New Information Does This Article Contribute?
Mouse models of experimental stroke identified CD84 as critical pathogenic effector in ischemic stroke.
Platelet surface abundance of CD84 is reduced locally and directly within the ischemic circulation during hyperacute total vascular occlusion in stroke patients.
High platelet CD84 expression levels are associated with poor outcome in stroke patients.
CD84 links platelet and T-cell activities during cerebral I/R injury and aggravates neuronal damage.
In acute ischemic stroke, the primary therapeutic goal is reconstitution of cerebral blood flow. However, in the majority of patients, infarcts worsen despite successful recanalization due to I/R injury. Here, we demonstrate that CD84 promotes infarct progression following cerebral I/R injury by linking T-cell and platelet activities in the acutely ischemic brain, as mice lacking CD84 in platelets or T cells displayed smaller infarct sizes. In vitro studies revealed that platelet-derived soluble CD84 enhances motility of wild-type but not of Cd84-deficient T cells, suggesting that homophilic soluble CD84:CD84 interactions trigger this response. A critical role for CD84 in stroke progression was further supported by direct human observation during the hyperacute stroke phase of total vascular occlusion. Local probing of occlusive ischemic blood showed that platelet expression of CD84 decreases in the ischemic arterial compartment during persisting vessel occlusion, indicating shedding of platelet CD84. In addition, high systemic platelet CD84 expression levels were associated with poor outcome in stroke patients. Collectively, our data establish CD84 as key molecule linking detrimental T-cell and platelet activities in acute stroke and indicating that this receptor might be a promising pharmacological target to limit I/R injury.
References
- 1.Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, Schonewille WJ, Vos JA, Nederkoorn PJ, Wermer MJ, et al. ; MR CLEAN Investigators A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372:11–20. doi: 10.1056/NEJMoa1411587 [DOI] [PubMed] [Google Scholar]
- 2.BD 2015 Neurological Disorders Collaborator Group Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the global burden of disease study 2015. Lancet Neurol. 2017;16:877–897.. doi: 10.1016/S1474-4422(17)30299-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizuma A, You JS, Yenari MA. Targeting reperfusion injury in the age of mechanical thrombectomy. Stroke. 2018;49:1796–1802. doi: 10.1161/STROKEAHA.117.017286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, Dávalos A, Majoie CB, van der Lugt A, de Miquel MA, et al. ; HERMES collaborators Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet. 2016;387:1723–1731. doi: 10.1016/S0140-6736(16)00163-X [DOI] [PubMed] [Google Scholar]
- 5.Church EW, Gundersen A, Glantz MJ, Simon SD. Number needed to treat for stroke thrombectomy based on a systematic review and meta-analysis. Clin Neurol Neurosurg. 2017;156:83–88. doi: 10.1016/j.clineuro.2017.03.005 [DOI] [PubMed] [Google Scholar]
- 6.Froehler MT, Saver JL, Zaidat OO, Jahan R, Aziz-Sultan MA, Klucznik RP, Haussen DC, Hellinger FR, Jr, Yavagal DR, Yao TL, et al. ; STRATIS Investigators Interhospital transfer before thrombectomy is associated with delayed treatment and worse outcome in the STRATIS registry (Systematic Evaluation of Patients Treated With Neurothrombectomy Devices for Acute Ischemic Stroke). Circulation. 2017;136:2311–2321. doi: 10.1161/CIRCULATIONAHA.117.028920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hallenbeck JM, Dutka AJ. Background review and current concepts of reperfusion injury. Arch Neurol. 1990;47:1245–1254. doi: 10.1001/archneur.1990.00530110107027 [DOI] [PubMed] [Google Scholar]
- 8.Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046 [DOI] [PubMed] [Google Scholar]
- 10.Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, Austinat M, Nieswandt B, Wiendl H, Stoll G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115:3835–3842. doi: 10.1182/blood-2009-10-249078 [DOI] [PubMed] [Google Scholar]
- 11.Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115:2323–2330. doi: 10.1161/CIRCULATIONAHA.107.691279 [DOI] [PubMed] [Google Scholar]
- 12.Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Göbel K, Schuhmann MK, Langhauser F, Helluy X, Schwarz T, Bittner S, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121:679–691. doi: 10.1182/blood-2012-04-426734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stoll G, Kleinschnitz C, Nieswandt B. Combating innate inflammation: a new paradigm for acute treatment of stroke? Ann NY Acad Sci. 2010;1207:149–154. doi: 10.1111/j.1749-6632.2010.05730.x [DOI] [PubMed] [Google Scholar]
- 14.Stoll G, Nieswandt B. Thrombo-inflammation in acute ischaemic stroke - implications for treatment. Nat Rev Neurol. 2019;15:473–481. doi: 10.1038/s41582-019-0221-1 [DOI] [PubMed] [Google Scholar]
- 15.Nieswandt B, Kleinschnitz C, Stoll G. Ischaemic stroke: a thrombo-inflammatory disease? J Physiol. 2011;589:4115–4123. doi: 10.1113/jphysiol.2011.212886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nanda N, Andre P, Bao M, Clauser K, Deguzman F, Howie D, Conley PB, Terhorst C, Phillips DR. Platelet aggregation induces platelet aggregate stability via SLAM family receptor signaling. Blood. 2005;106:3028–3034. doi: 10.1182/blood-2005-01-0333 [DOI] [PubMed] [Google Scholar]
- 17.Martin M, Romero X, de la Fuente MA, Tovar V, Zapater N, Esplugues E, Pizcueta P, Bosch J, Engel P. CD84 functions as a homophilic adhesion molecule and enhances IFN-gamma secretion: adhesion is mediated by Ig-like domain 1. J Immunol. 2001;167:3668–3676. doi: 10.4049/jimmunol.167.7.3668 [DOI] [PubMed] [Google Scholar]
- 18.Romero X, Benítez D, March S, Vilella R, Miralpeix M, Engel P. Differential expression of SAP and EAT-2-binding leukocyte cell-surface molecules CD84, CD150 (SLAM), CD229 (Ly9) and CD244 (2B4). Tissue Antigens. 2004;64:132–144. doi: 10.1111/j.1399-0039.2004.00247.x [DOI] [PubMed] [Google Scholar]
- 19.Tangye SG, Nichols KE, Hare NJ, van de Weerdt BC. Functional requirements for interactions between CD84 and Src homology 2 domain-containing proteins and their contribution to human T cell activation. J Immunol. 2003;171:2485–2495. doi: 10.4049/jimmunol.171.5.2485 [DOI] [PubMed] [Google Scholar]
- 20.Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, Wakeland EK, Germain RN, Schwartzberg PL. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity. 2010;32:253–265. doi: 10.1016/j.immuni.2010.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hofmann S, Vögtle T, Bender M, Rose-John S, Nieswandt B. The SLAM family member CD84 is regulated by ADAM10 and calpain in platelets. J Thromb Haemost. 2012;10:2581–2592. doi: 10.1111/jth.12013 [DOI] [PubMed] [Google Scholar]
- 22.Hofmann S, Braun A, Pozgaj R, Morowski M, Vögtle T, Nieswandt B. Mice lacking the SLAM family member CD84 display unaltered platelet function in hemostasis and thrombosis. PLoS One. 2014;9:e115306 doi: 10.1371/journal.pone.0115306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g [DOI] [PubMed] [Google Scholar]
- 24.Schuhmann MK, Kraft P, Stoll G, Lorenz K, Meuth SG, Wiendl H, Nieswandt B, Sparwasser T, Beyersdorf N, Kerkau T, et al. CD28 superagonist-mediated boost of regulatory T cells increases thrombo-inflammation and ischemic neurodegeneration during the acute phase of experimental stroke. J Cereb Blood Flow Metab. 2015;35:6–10. doi: 10.1038/jcbfm.2014.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuhmann MK, Kraft P, Bieber M, Haarmann A, Homola GA, Pham M, Nieswandt B, Stoll G. Influence of thrombolysis on the safety and efficacy of blocking platelet adhesion or secretory activity in acute ischemic stroke in mice. Transl Stroke Res. 2018;9:493–498. doi: 10.1007/s12975-017-0606-7 [DOI] [PubMed] [Google Scholar]
- 26.Moran PM, Higgins LS, Cordell B, Moser PC. Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human beta-amyloid precursor protein. Proc Natl Acad Sci USA. 1995;92:5341–5345. doi: 10.1073/pnas.92.12.5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472 [DOI] [PubMed] [Google Scholar]
- 28.Bieber M, Gronewold J, Scharf AC, Schuhmann MK, Langhauser F, Hopp S, Mencl S, Geuss E, Leinweber J, Guthmann J, et al. Validity and reliability of neurological scores in mice exposed to middle cerebral artery occlusion. Stroke. 2019;50:2875–2882. doi: 10.1161/STROKEAHA.119.026652 [DOI] [PubMed] [Google Scholar]
- 29.Junge CE, Sugawara T, Mannaioni G, Alagarsamy S, Conn PJ, Brat DJ, Chan PH, Traynelis SF. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc Natl Acad Sci USA. 2003;100:13019–13024. doi: 10.1073/pnas.2235594100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nieswandt B, Bergmeier W, Rackebrandt K, Gessner JE, Zirngibl H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood. 2000;96:2520–2527 [PubMed] [Google Scholar]
- 31.Langhauser F, Göb E, Kraft P, Geis C, Schmitt J, Brede M, Göbel K, Helluy X, Pham M, Bendszus M, et al. Kininogen deficiency protects from ischemic neurodegeneration in mice by reducing thrombosis, blood-brain barrier damage, and inflammation. Blood. 2012;120:4082–4092. doi: 10.1182/blood-2012-06-440057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bittner S, Ruck T, Schuhmann MK, Herrmann AM, Moha ou Maati H, Bobak N, Göbel K, Langhauser F, Stegner D, Ehling P, et al. Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat Med. 2013;19:1161–1165. doi: 10.1038/nm.3303 [DOI] [PubMed] [Google Scholar]
- 33.Grüner S, Prostredna M, Koch M, Miura Y, Schulte V, Jung SM, Moroi M, Nieswandt B. Relative antithrombotic effect of soluble GPVI dimer compared with anti-GPVI antibodies in mice. Blood. 2005;105:1492–1499. doi: 10.1182/blood-2004-06-2391 [DOI] [PubMed] [Google Scholar]
- 34.Kollikowski AM, Schuhmann MK, Nieswandt B, Müllges W, Stoll G, Pham M. Local leukocyte invasion during hyperacute human ischemic stroke. Ann Neurol. 2020;87:466–479. doi: 10.1002/ana.25665 [DOI] [PubMed] [Google Scholar]
- 35.Turc G, Bhogal P, Fischer U, Khatri P, Lobotesis K, Mazighi M, Schellinger PD, Toni D, de Vries J, White P, et al. European stroke organisation (eso) - european society for minimally invasive neurological therapy (esmint) guidelines on mechanical thrombectomy in acute ischemic stroke. J NeuroInterv Surg. 2019neurintsurg-2018-014569. doi: 10.1136/neurintsurg-2018-014569 [DOI] [PubMed] [Google Scholar]
- 36.Humphries W, Hoit D, Doss VT, Elijovich L, Frei D, Loy D, Dooley G, Turk AS, Chaudry I, Turner R, et al. Distal aspiration with retrievable stent assisted thrombectomy for the treatment of acute ischemic stroke. J Neurointerv Surg. 2015;7:90–94. doi: 10.1136/neurintsurg-2013-010986 [DOI] [PubMed] [Google Scholar]
- 37.Hatano S. Experience from a multicentre stroke register: a preliminary report. Bull World Health Organ. 1976;54:541–553 [PMC free article] [PubMed] [Google Scholar]
- 38.Berger K, Weltermann B, Kolominsky-Rabas P, Meves S, Heuschmann P, Böhner J, Neundörfer B, Hense HW, Büttner T. [The reliability of stroke scales. The german version of NIHSS, ESS and Rankin scales]. Fortschr Neurol Psychiatr. 1999;67:81–93. doi: 10.1055/s-2007-993985 [DOI] [PubMed] [Google Scholar]
- 39.Adams HP, Jr, Davis PH, Leira EC, Chang KC, Bendixen BH, Clarke WR, Woolson RF, Hansen MD. Baseline NIH stroke scale score strongly predicts outcome after stroke: a report of the trial of Org 10172 in acute stroke treatment (TOAST). Neurology. 1999;53:126–131. doi: 10.1212/wnl.53.1.126 [DOI] [PubMed] [Google Scholar]
- 40.Roy-O’Reilly M, McCullough LD. Sex differences in stroke: the contribution of coagulation. Exp Neurol. 2014;259:16–27. doi: 10.1016/j.expneurol.2014.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuhmann MK, Langhauser F, Kraft P, Kleinschnitz C. B cells do not have a major pathophysiologic role in acute ischemic stroke in mice. J Neuroinflammation. 2017;14:112 doi: 10.1186/s12974-017-0890-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherpokova D, Bender M, Morowski M, Kraft P, Schuhmann MK, Akbar SM, Sultan CS, Hughes CE, Kleinschnitz C, Stoll G, et al. SLAP/SLAP2 prevent excessive platelet (hem)ITAM signaling in thrombosis and ischemic stroke in mice. Blood. 2015;125:185–194. doi: 10.1182/blood-2014-06-580597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fong KP, Barry C, Tran AN, Traxler EA, Wannemacher KM, Tang HY, Speicher KD, Blair IA, Speicher DW, Grosser T, et al. Deciphering the human platelet sheddome. Blood. 2011;117:e15–e26. doi: 10.1182/blood-2010-05-283838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goebel S, Li Z, Vogelmann J, Holthoff HP, Degen H, Hermann DM, Gawaz M, Ungerer M, Münch G. The GPVI-Fc fusion protein Revacept improves cerebral infarct volume and functional outcome in stroke. PLoS One. 2013;8:e66960 doi: 10.1371/journal.pone.0066960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zinkstok SM, Beenen LF, Majoie CB, Marquering HA, de Haan RJ, Roos YB. Early deterioration after thrombolysis plus aspirin in acute stroke: a post hoc analysis of the antiplatelet therapy in combination with recombinant t-PA thrombolysis in ischemic stroke trial. Stroke. 2014;45:3080–3082. doi: 10.1161/STROKEAHA.114.006268 [DOI] [PubMed] [Google Scholar]
- 46.Kappos L, Antel J, Comi G, Montalban X, O’Connor P, Polman CH, Haas T, Korn AA, Karlsson G, Radue EW; FTY720 D2201 Study Group Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–1140. doi: 10.1056/NEJMoa052643 [DOI] [PubMed] [Google Scholar]
- 47.Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, Han W, Xue R, Liu Q, Hao J, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci USA. 2014;111:18315–18320. doi: 10.1073/pnas.1416166111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian DC, Shi K, Zhu Z, Yao J, Yang X, Su L, Zhang S, Zhang M, Gonzales RJ, Liu Q, et al. Fingolimod enhances the efficacy of delayed alteplase administration in acute ischemic stroke by promoting anterograde reperfusion and retrograde collateral flow. Ann Neurol. 2018;84:717–728. doi: 10.1002/ana.25352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, Dong Y, Xu X, Liu Q, Huang D, et al. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: a pilot trial. Circulation. 2015;132:1104–1112. doi: 10.1161/CIRCULATIONAHA.115.016371 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.