

Abstract
Once focused exclusively on the creation of tissues to repair or replace diseased or damaged organs, the field of tissue engineering has undergone an important evolution in recent years. Namely, tissue engineering techniques are increasingly being applied to intentionally generate pathological conditions. Motivated in part by the wide gap between 2D cultures and animal models in the current disease modeling continuum, disease-inspired tissue-engineered platforms have numerous potential applications, and may serve to advance our understanding and clinical treatment of various diseases. This review will focus on recent progress toward generating tissue-engineered models of cardiovascular diseases, including cardiac hypertrophy, fibrosis, and ischemia reperfusion injury, atherosclerosis, and calcific aortic valve disease, with an emphasis on how these disease-inspired platforms can be used to decipher disease etiology. Each pathology is discussed in the context of generating both disease-specific cells as well as disease-specific extracellular environments, with an eye toward future opportunities to integrate different tools to yield more complex and physiologically relevant culture platforms. Ultimately, the development of effective disease treatments relies upon our ability to develop appropriate experimental models; as cardiovascular diseases are the leading cause of death worldwide, the insights yielded by improved in vitro disease modeling could have substantial ramifications for public health and clinical care.
Keywords: myocardial infarction, atherosclerosis, aortic valve disease, in vitro disease modeling
Graphical Abstract
Introduction
From its inception, tissue engineering has promised to yield a source of healthy neo-tissues targeted at replacing, repairing, or regenerating diseased or damaged tissues1. Myriad tools have been developed to advance this pursuit, including generation/identification of new cell sources2–3, controlled delivery of biophysical and biochemical stimuli4–5, and the synthesis of increasingly complex 3D scaffolds to mimic the extracellular environment6–8. Successful products have been developed1, with the most recent market evaluation identifying 21 U.S. companies with commercialized tissue-engineered products, yielding $9 billion in sales in 20179. Despite these successes and advancements, the development of plentiful replacement organs has been a much slower and winding road than was predicted for the field10.
Thus, the field of tissue engineering has been experiencing a subtle, but important, evolution. Although it remains a common assumption that one’s goal in performing tissue engineering is to create healthy tissues - even in 2019, encyclopedias, journal articles, and the NIH still define “tissue engineering” as creating materials for “restoration or replacement of a damaged or diseased body part”9, 11–12 - an increasing share of tissue engineering researchers have turned their attention toward the intentional creation of diseased tissues. The purpose of these tissue-engineered disease models varies with the specific application and tissue, and may range from interrogating specific pathological pathways to examining patient-specific responses to a drug. In some cases, the shift to disease tissue engineering creates a somewhat ironic scenario where one is “doing tissue engineering to avoid doing tissue engineering” – i.e., if a disease treatment can be elucidated using a tissue-engineered disease model, it may obviate the need to ultimately replace the tissue (theoretically, with an engineered substitute).
While tissue engineering’s original goal of creating healthy tissue replacements remains a necessary and important pursuit, the potential impact of tissue-engineered disease models is significant, and has the ability to yield advancements in the clinic on a shorter time scale than ‘traditional’ tissue engineering. These platforms are poised to occupy an important and unique position on the disease model continuum, which typically spans from 2D studies to animals and then humans. The limitations of 2D cultures have been reviewed at length13, and researchers continue to discover how much of our cellular knowledge is specific to the 2D, tissue culture polystyrene context, as different responses have been achieved in more physiological environments14–15. By allowing independent tuning of variables, control over conditions, and good throughput, the in vitro 3D culture environment enables prospective studies of specific biological questions that would be challenging to achieve in animals. Additionally, some studies have indicated that even simplistic 2D cultures with human cells produce more predictive results than studies performed in rodents16, and there are diseases for which appropriate animal models have yet to be identified. These limitations in both 2D and animal platforms outline a distinct gap which 3D engineered tissues seem ideally suited to fill.
Our ability to understand disease mechanisms and develop therapies relies heavily upon our ability to develop appropriate experimental models. In combination with the delivery of physiological risk factors, 3D tissue-engineered platforms can help us identify conditions that support both the onset of pathology and its progression. These environments can be manipulated and probed to gain mechanistic insight on the specific signaling pathways involved in pathogenesis, thereby revealing potential treatment targets. Tissue-engineered disease platforms can be employed to test potential disease treatments, acting as an extra measure of in vitro drug screening that may significantly reduce the use of animals in subsequent in vivo studies. Finally, disease tissue engineering is not completely divorced from traditional healthy tissue engineering, as investigating the physical and biochemical factors that promote disease pathogenesis also serves to identify factors that promote maintenance of a healthy tissue environment, and this information may be applied to inform the design of healthy engineered tissues.
This review article will focus on progress made toward the development of tissue-engineered models of cardiovascular diseases, addressing pathologies found in vessels, cardiac muscle, and the aortic heart valve. This is a burgeoning area of investigation and distinct from the pursuit of generating healthy engineered tissues to perform cardiotoxicity screening of drugs17. In theory, any healthy engineered tissue may become a disease model if combined with the appropriate stimuli, but this review will concentrate solely on instances where mimicking the disease environment was the specified goal.
Cardiac
Cardiovascular diseases affect nearly half of all American adults and are responsible for 18 million deaths per year worldwide, or 1 out of every 3 deaths in the U.S.18. These diseases span both congenital and acquired conditions, but almost half of the deaths from cardiovascular disease are attributed to complications resulting from a myocardial infarction (MI)18. While the immediate post-MI survival rate has been increasing in recent years19, patients that survive the initial acute MI injury remain at elevated risk of eventual heart failure due to the tissue death and remodeling that occur in response to ischemic injury19–20. Meanwhile, it is the heart’s own healing and compensatory mechanisms that cause further dysfunction, as attempts to normalize the mechanical stresses to account for the loss of functional contractile tissue can ultimately lead to cardiac fibrosis and pathological hypertrophy21. Post-MI treatment regimens generally consist of blood pressure-lowering and anti-platelet drugs22, which do not directly address the mechanistic cascades causing hypertrophy or fibrosis. Ischemic damage to the heart can also occur in a chronic fashion, not associated with an acute cardiac event; such is the case of chronic ischemic heart disease due to atherosclerosis23. Similarly, pathological cardiac hypertrophy can result from non-acute events, such as systemic hypertension24. In both of these conditions, pharmacological treatment tends to focus upon modifying risk factors, such as cholesterol levels and blood pressure, as well as lifestyle modification (e.g., smoking cessation, exercise)22, 25.
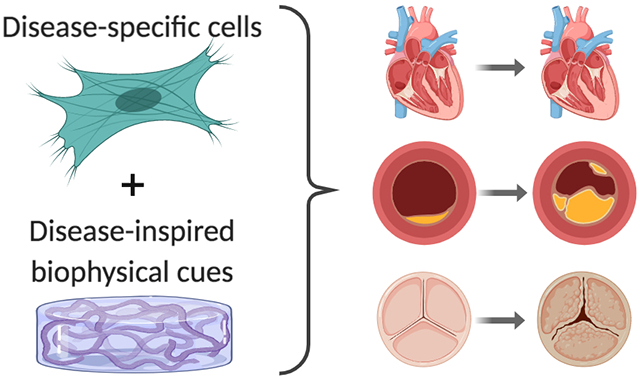
The prevalence and severity of these various types of myocardial dysfunction has motivated a wealth of studies aimed at creating healthy tissue-engineered myocardium to regenerate or repair this lost function. While we leave a thorough discussion of healthy cardiac tissue engineering to other reviews26–29, it is worth noting the foundational work in this area that ultimately set the stage for the pursuit of disease-inspired cardiac models. In particular, the engineered heart tissue (EHT) approach described by Eschenhagen et al. in 199730 has become a widely used platform for 3D myocardial culture. EHTs have evolved to take many forms31 and have been an enabling technology with respect to the creation of cardiac disease models. While the vast majority of cardiac tissue engineering research remains focused on creating healthy tissues, the application of these approaches to specifically study the pathology of cardiac dysfunction or treatment of diseased tissues is an area of steady growth. Such disease-mimetic platforms are poised to address ongoing concerns about the predictive ability of current in vitro and animal models17 across all of the pathologies mentioned above. Additionally, our mechanistic knowledge of these pathologies remains extremely limited. Advancements in the area of cell source have dramatically altered the landscape of this research field32 and made it easier for researchers to apply 3D tissue engineering techniques, which often require large numbers of cells. In the sections below, we highlight recent work aimed at creating tissue-engineered disease-mimetic platforms for studying both disease pathophysiology and potential treatments (Figure 1).
Figure 1.

Overview of disease-inspired tissue engineering approaches to mimic cardiac pathologies
Disease-specific cells
The majority of work in cardiac disease modeling has been in the form of creating disease-specific cardiomyocytes33–36. For decades, the logistics of culturing cardiomyocytes in vitro were challenging and often prohibitive in terms of throughput. Due to their limited proliferation capacity, cardiomyocytes had to be obtained as primary cultures, without the ability to sub-culture. Neonatal rodents still serve as the most common source of in vitro cardiomyocytes, but their applicability to modeling human cells is questionable37. The ability to generate cardiomyocytes from human pluripotent stem cells (hPSCs)38, followed by dramatic improvements in differentiation efficiency39, have been transformative events in cardiomyocyte culture, and the culture of hPSC-CMs in 3D hydrogels (e.g., EHTs) is increasingly used to make healthy myocardial cultures in vitro31. The maturity of hPSC-CMs does remain a concern and can significantly impact their use as predictive models40. But, given the current trajectory, the use of hPSC-derived cardiomyocytes (hPSC-CMs) may soon surpass the use of animal-derived primary cells for in vitro studies.
The advent of induced pluripotent stem cell (iPSC) techniques has also enabled the generation of plentiful diseased CM cultures from patients with congenital cardiac conditions (e.g., arrhythmias, cardiomyopathies)41. Further revolutionizing the in vitro usage of diseased cardiomyocytes was the development of genome editing techniques, which have expanded the ability of researchers to create disease-specific cardiomyocytes from any cell type, modeling both congenital and acquired conditions42. Diseased hPSC-CMs produced via both patient-specific and gene-edited approaches are even sold commercially (Cellular Dynamics, Inc., Madison, WI). In most cases, these diseased hPSC-CMs have been used in a traditional (2D) context, although an increasing number of studies have embedded these cells in collagen I and/or fibrin, an approach that is commonly used to enable quantification of contractile function, but is not necessarily modeled upon mimicking the native extracellular environment. Because much of the work with disease-specific hPSC-CMs has not yet involved the creation of 3D disease-inspired environments, we leave more thorough coverage of these cells to other reviews35, 42. However, they are positioned to become a critical element in the generation of future 3D tissue-engineered disease models, so we will highlight some of the key advances in this area (Table 1).
Table 1:
Summary of approaches used to mimic cardiac diseases
Congenital arrhythmias, including Long QT Syndromes (LQTS), comprise the bulk of current diseased hPSC-CM research35, 43–44. LQTS hPSC-CMs in standard 2D culture exhibit prolonged action potential duration and responsiveness to current and potential anti-arrhythmic drugs, indicating that these cells are capable of recapitulating the disease phenotype and predicting response to pharmacological treatments45–46. In the case of cardiomyopathies, gene-edited hPSC-CMs have primarily been used to characterize the genetic and molecular contributors to disease pathogenesis47 in traditional in vitro culture environments, with a focus on genes related to contraction. EHTs have been formed using hPSC-CMs to yield 3D constructs intended to improve iPSC maturation and the physiological relevance of disease models48–49. For example, both patient-specific and gene-edited iPSC-CMs were cultured in EHTs to examine the pathogenicity of different titin gene mutations in dilated cardiomyopathy (DCM), as well as elucidate the downstream pathways by which these mutations ultimately caused decreased force generation49. In this case, 2D iPSC-CM cultures failed to recapitulate the disease phenotype observed in patients, while the EHT constructs revealed mechanistic information about the pathogenesis of DCM. While the aforementioned studies have generally been confined to monogenic cardiac diseases, recent work employed the novel Biowire platform50 to investigate the differences between hypertensive individuals who develop left ventricular hypertrophy (LVH) and those who do not, an issue which is likely polygenic in nature. iPSC-CMs were derived from hypertensive individuals with or without LVH, cultured in combination with cardiac fibroblasts in collagen/matrigel EHTs, and subjected to regimes of electrical stimulation for up to 8 months51. This platform enabled monitoring of contractile function and yielded distinct gene expression profiles across the patient groups, which may help in understanding the pathogenesis of this dysfunction. Finally, the construction of EHTs using patient-derived iPSC-CMs has also fueled commercial developments in cardiac disease modeling. InvivoSciences (Madison, WI) generates EHTs using patient-specific iPSC-CMs52 in order to define personalized approaches to treating various cardiac diseases as well as to perform rare disease drug discovery.
The generation of a diseased cardiac phenotype does not necessarily need to come from the use of cells with genetic mutations. Manipulating the composition of the cell population in a co-culture model can also provide a cell-driven approach to yield a disease-mimetic culture platform53–54. For example, the Biowire platform50 was recently used as the basis to generate not only healthy and fibrotic engineered myocardium, but also an integrated model of adjacent healthy and fibrotic tissue, to mimic the post-infarct border zone and scar53. This 3D culture employed healthy iPSC-CMs and cardiac fibroblasts embedded in a mixture of fibrin and matrigel, with fibrosis mimicked by increasing the ratio of fibroblasts to cardiomyocytes. These tissues exhibited a physiologically relevant fibrotic phenotype and may be used to test both the efficacy and timing of anti-fibrotic therapies53.
Disease-inspired physical cues
Several types of biomaterials have been used in the pursuit of creating healthy tissue-engineered myocardium55–56. In general, these scaffolds have been in the form of hydrogels, which are attractive for myocardial applications for several reasons, including mechanical properties with broad similarities to native cardiac tissue, allowing functional contraction of encapsulated cardiomyocytes. However, scaffolds used in the context of disease-inspired cardiac tissue engineering have not branched out to the same extent, and the majority of 3D disease platforms use collagen I, fibrin, Matrigel, or a mixture of these materials55. These materials have been well-established for encapsulation of cardiomyocytes and supporting cell types, but are not necessarily aimed at recapitulating native healthy or disease-specific cardiac ECM composition. Rather, the disease-mimicking aspect of these systems has generally come in the form of using disease-specific cells (discussed above), or application of external stressors: exogenous biomolecules associated with disease pathogenesis (e.g., transforming growth factor beta 1, TGF-β1), hypoxic conditions, or pathophysiological mechanical stresses.
During a myocardial infarction, vessel occlusion limits blood flow to the heart, leading to hypoxia, metabolic waste accumulation, and nutrient deficiency57. While reperfusion of the occluded vessel is necessary to reduce acute ischemic injury and minimize infarct size, it can also cause additional injury, termed ischemia reperfusion injury (IRI)58. Current preclinical models of IRI have not successfully translated to the clinic59, leading to the recent emergence of engineered cardiac tissues to test potential therapeutic strategies to protect the heart from IRI damage60. In one of the first examples of this application, collagen I-based EHTs were constructed using neonatal rat cardiomyocytes and transiently cultured in hypoxic conditions in the presence or absence of potential cytoprotective agents cyclosporine A and acetylcholine61. The application of hypoxic conditions initiated a physiologically relevant ischemic cellular response, and treatment of these constructs with cytoprotective agents was able to successfully inhibit hypoxia-induced damage, thereby indicating potential for use of EHTs for studying the cardiac response to stressors, as well as for screening pharmacological agents that may help treat myocardial injuries. However, the cytoprotective agents in this study were administered prior to the onset of hypoxia, which would not be relevant to the clinical setting. Subsequent studies using neonatal rat cardiomyocytes in EHTs further refined the IRI-mimicking conditions and found moderate cardioprotective effects offered by a nitric oxide donor or B-type natriuretic peptide62. A recent study was the first to use human cells in a tissue-engineered environment to study IRI63. This work encapsulated hPSC-derived CMs in collagen I/fibrin scaffolds and further improved upon the physiological relevance of the IRI-mimicking conditions by subjecting the engineered constructs to both hypoxia and a small volume of ischemia-mimicking buffer solution to reproduce the metabolic waste accumulation, nutrient deprivation, hyperkalemia, low pH, and high lactate concentration found during ischemia. Using this novel system, the authors investigated both IRI pathophysiology (e,g., distinguishing between ischemic and reperfusion injury) and possible treatments, with potential cardioprotective agents added at the time of reperfusion63. Importantly, this work also addressed how hPSC-CM maturity affected the ischemic response and employed a strategy to further hPSC-CM maturation. Together, these studies have established a strong platform for evaluation of potential strategies to inhibit IRI.
Pathological hypertrophy occurs in response to volume or pressure overload in the heart64. Although it can be intended to compensate for altered wall stresses post-injury, it is always a maladaptive process in the adult myocardium, leading to extensive cardiac remodeling and heart dysfunction or failure64–65. Despite many years of considerable effort to elucidate the mechanisms of pathological hypertrophy, there is still much to be learned about its pathogenesis, which is critical to develop effective treatments for this condition. Limitations of traditional in vitro cell cultures and animal models are often named as major factors that have impeded progress on this front66. In combination with primary or hPSC-derived cardiomyocytes, fibrin-based EHTs have been used as a platform for applying both biochemical and mechanical hypertrophic stimuli67–68. Humoral factors such as endothelin-1 and phenylephrine are commonly used to stimulate hypertrophic behaviors, but do not recapitulate the chronically increased hemodynamic load that triggers hypertrophy. To examine the latter, novel methods have been applied to mimic increased afterload in these 3D cultures. EHTs are typically strips of tissue anchored to a flexible post on either end, where contraction of the EHT results in post deflection69. Afterload in EHTs containing rat cardiomyocytes was increased by 12-fold by fitting metal braces to these posts, without alteration to the beating rate. Using this approach, the authors found that a hypertrophic gene program was enacted by this application of pathological afterload, with significant increases in cardiomyocyte size, glycolysis, and fibrotic ECM remodeling67. This platform of enhanced afterload laid the foundation for multiple subsequent studies that have uncovered mechanistic features of pathological hypertrophy, such as differential regulation of multiple microRNAs70 and characterization of DNA methylation signatures71. In the EHT platform, miR-21–5p was strongly upregulated by hypertrophic afterload, consistent with its elevation in human heart failure72; additionally, antagonism of this microRNA attenuated the afterload-induced hypertrophic response in EHTs70. In another demonstration of the potential for the hypertrophic EHT platform to discover new treatment targets, the DNA methyltransferase inhibitor RG108 was found to attenuate some hypertrophic features in the EHTs71, which also held true in a follow-up study in rats with pressure overload-induced hypertrophy73. These studies have been performed using animal CM sources, but others have employed a similar system to apply increased afterload to fibrin EHTs containing hPSC-CMs68. Although performed for the purpose of using afterload to enhance the maturity of hPSC-CMs, the authors observed that application of high levels of afterload (~9 μN/μm) significantly upregulated markers of pathological hypertrophy and fibrosis.
Efforts made by the heart to ameliorate the biological and mechanical insufficiency of the post-MI environment include not only hypertrophy of resident cardiomyocytes but also extensive remodeling of the extracellular matrix (ECM) by cardiac fibroblasts74–75. Cardiac fibroblasts differentiate into a myofibroblast phenotype and produce excessive amounts of ECM proteins such as collagen I and fibronectin76. While these changes can stabilize heart tissue integrity in the short-term, the increase in tissue stiffness ultimately leads to impaired ability to contract and increased risk of arrhythmia77. A few recent efforts have employed tissue engineering techniques to intentionally create fibrotic cardiac tissues78–81. A proof of concept for this approach was provided by encapsulating neonatal rat cardiac fibroblasts and cardiomyocytes within scaffolds of methacrylated gelatin (GelMA) and then treating with a known pro-fibrotic agent, TGF-β178. The use of GelMA enabled mechanical tuning of the 3D environment to best match the in vivo setting and promote normal, quiescent cardiac fibroblast behavior as a healthy baseline. Addition of TGF-β1 stimulated a fibrotic response, as measured by both phenotypic indicators as well as altered functional activity (e.g., asynchronous beating), demonstrating the ability of this approach to serve as a platform for modeling cardiac fibrosis. Mechanical cues have been introduced into these tissue-engineered systems by constructing microdevices that applied cyclic compressive strain to GelMA79 or fibrin82 hydrogels containing cardiac fibroblasts, resulting in increasing fibrotic activity in a strain-dependent manner. The value of taking a tissue engineering-based approach to study cardiac fibrosis was also illustrated by work in which collagen density and fibroblast number were independently varied in order to decipher their relative influences on contraction, revealing that fibroblast number affected contractile tissue function more than collagen density54. Recent studies have begun to construct cardiac fibrosis platforms using entirely human-derived cells80–81 and demonstrated similar successes in mimicking fibrotic outcomes upon treatment with pro-fibrotic stimuli. Together, these advancements have provided an important foundation for constructing tissue-engineered models of cardiac fibrosis that will permit elucidation of its pathogenesis and testing of potential therapeutics.
Future Opportunities
As described in the preceding sections, many types of disease-specific cardiomyocytes have been generated, as have several approaches to provide disease-specific physical environments. However, relatively little research has been done at the intersection of these areas (i.e., placing disease-specific cells in disease-specific extracellular environments). Furthermore, the diseased ECM has been highly underappreciated in the context of mimicking cardiac diseases83, and most of the aforementioned approaches have not attempted to replicate disease on the ECM level. Increased ECM deposition is a hallmark of hypertrophy, cardiomyopathy, and heart failure84–86, and merging diseased hPSC-CMs with disease-mimicking 3D scaffolds has significant applicability to the study of these conditions. For example, ECM alterations are thought to be a driving force in the pathogenesis of cardiomyopathies87, but these potentially influential features are omitted when cardiomyopathies are studied using a model that is based solely on disease-specific cells. Recreating a dysfunctional ECM environment is even important in pathologies that are typically only cell-focused (e.g., arrhythmias). Although arrhythmias are not known to cause changes to the ECM environment, the occurrence of ECM changes – due to other cardiac events or simply the fibrosis that happens with natural aging83 – can strongly influence the arrhythmia and its drug responsiveness88. The design and implementation of pathological scaffold environments, combined with innovations in delivering pathological stimuli such as hypoxia89, could greatly advance the ability of these environments to faithfully mimic native disease conditions.
Vascular
Myocardial infarctions are generally caused by coronary artery disease (CAD), or atherosclerosis of the vessels supplying the heart. CAD is the most common type of cardiovascular disease and is the leading cause of death worldwide18. Yet, the onset and progression of ischemic vascular diseases tend to go undetected until symptoms indicate an advanced stage of disease90. The pursuit of tissue-engineered vascular grafts is one of the earliest examples of tissue engineering, when Weinberg and Bell described seeding endothelial cells, smooth muscle cells, and fibroblasts within a collagen I matrix in 198691. As reviewed elsewhere92, numerous innovative advancements in vascular tissue engineering have followed, ultimately yielding multiple different approaches that have reached clinical tests in humans93–95.
While promising strides have been made to construct tissue engineered blood vessels for the purposes of vascular grafting96–98, engineered models of vascular diseases could also prove useful for earlier detection of disease as well as the development of preventative drugs and therapies. Significant challenges face the development of these models, given the complexity of native vessel composition, architecture, and pathogenesis of disease. The native arterial wall is concentrically composed of endothelial cells (ECs), smooth muscle cells, and fibroblasts, each residing within a different ECM environment. Endothelial cells in the healthy blood vessel intima provide a selectively permeable layer that does not allow infiltration of lipoproteins into the subendothelial space. However, hypercholesterolemia or other pathological stimuli can disrupt the integrity of this barrier, allowing the infiltration and accumulation of low density lipoproteins (LDL)99. Following LDL entrapment, monocytes infiltrate the vessel wall through the compromised endothelial layer, uptake LDL, and become foam cells100. The inflammatory state that is then assumed ultimately culminates in the characteristic lesion and plaque formation seen in atherosclerosis. As described in subsequent sections, progress toward fabricating disease-inspired models of atherosclerosis has focused on cellular-based methods to produce diseased EC phenotypes as well as the construction of complex scaffold environments to replicate key risk factors and features of disease onset (Figure 2).
Figure 2.

Overview of disease-inspired tissue engineering approaches to mimic atherosclerosis
Disease-specific cells
Endothelial cells, smooth muscle cells, and fibroblasts that comprise the vessel structure are each uniquely impacted throughout the progression of atherosclerosis and other vascular pathologies. EC dysfunction has long been considered an initiating event in atherosclerosis, in which there is a reduction in EC-produced nitric oxide and increase in inflammatory cytokine production by ECs101. EC dysfunction also affects processes such as angiogenesis, which is crucial to restoring myocardial function in response to ischemic conditions. Sourcing and culturing ECs has not been limited by the same challenges described above for cardiomyocytes, and in vitro experiments have often utilized human umbilical vein endothelial cells (HUVECs) as the cell source. However, efforts are increasingly moving towards the use of iPSC-derived ECs or direct reprogramming of somatic cells (e.g., fibroblasts) into ECs as an alternative102–104. Similar to the case of cardiomyocytes, these derivation approaches have opened up the possibility of generating disease-specific EC cultures, either by starting with cells from diseased individuals, or by the application of gene editing techniques to introduce targeted mutations. For example, using CRISPR/Cas9 gene editing, iPSC-ECs deficient in hypoxia inducible factor-1 alpha (HIF-1α) were recently generated105; HIF-1α plays a crucial role in regulating the response to ischemia but is impaired with advanced age. HIF1A knockout iPSC-ECs expressed key EC characteristics and, in normoxic conditions, exhibited few differences compared to wild type ECs. However, their response to ischemic conditions was significantly altered, as these diseased iPSC-ECs demonstrated decreased viability and angiogenic potential, both in traditional 2D conditions and in a GelMA-based 3D environment105. This study provides an important foundation for generating tissue-engineered models of EC dysfunction.
Overall, the creation of disease-specific ECs is in a relatively nascent stage in comparison to the extensive work done thus far with cardiomyocytes, but iPSC-ECs have been derived to represent other conditions associated with high risk of cardiovascular disease, such as familial pulmonary arterial hypertension106, Fabry disease107, or diet-induced obesity108. Smooth muscle cells (SMCs) are in a similar position109, with a few cardiovascular disease-specific phenotypes being generated thus far (e.g., elastin mutation in supravalvular aortic stenosis110 or fibrillin-1 mutation to mimic Marfan syndrome111), but the integration of these cells within the context of 3D disease modeling has not been explored.
Disease-inspired physical cues
The effect of altered shear stresses and atherogenic stimuli on endothelial dysfunction has been investigated for several decades using traditional 2D environments, providing many insights on atherosclerosis and thrombosis mechanisms112–114. The recent development and refinement of microfluidic cell culture techniques has enabled further advancements in disease-inspired vascular platforms115. Many of these investigations remain focused on what is functionally a 2D culture setup (i.e., ECs lining a channel)116–117. These systems may be used to introduce tunable channel constrictions that mimic atherosclerotic plaques of varying severity, thereby allowing an examination of EC dysfunction and inflammatory cell adhesion in response to disease-mimicking hemodynamic alterations. However, few of these current vascular platforms represent the vessel as a 3D, multi-layered structure, which is important for studying inflammatory cell infiltration and plaque formation. Additionally, in the human body, the vessels that are most affected by atherosclerosis are not micro in size, raising concerns about whether a microfluidic approach is best suited to investigate atherosclerosis-mimicking flow profiles.
Thus, more traditional methods of fabricating tissue-engineered blood vessels (TEBVs) have been employed to create platforms that mimic elements of atherosclerosis. Early work in this area described the production of fibrin-based scaffolds containing smooth muscle cells and an EC lining to study LDL accumulation in normal and hypercholesterolemic conditions118. LDL deposition was dose-dependent and associated with increased monocyte infiltration and foam cell formation, illustrating the ability of a tissue-engineered vessel to replicate key features of atherosclerosis. However, these studies were not done in the presence of fluid flow, which has been viewed as a significant limitation. Thus, more recent iterations have focused upon TEBVs cultured in the presence of flow. For example, TEBVs made from smooth muscle cells embedded in tubular polymeric scaffolds119 or collagen I hydrogels120 and an EC-populated lumen were used to study lipoprotein accumulation119 as well as inflammatory cell adhesion and transendothelial migration119–120 under physiological fluid flow. These platforms were able to mimic early inflammatory events in atherogenesis. Additionally, the EC dysfunction induced by the inflammatory factor tumor necrosis factor-alpha was reduced by administration of anti-inflammatory drugs, suggesting the potential for this platform to be used to evaluate potential therapeutics120.
Future Opportunities
Advances in both microfluidics and TEBV creation have yielded 3D culture platforms with great promise for studying vascular disease onset, progression, and treatment. Work in these areas has already progressed to mimicking multiple complex features of the in vivo environment, including delivery of shear stresses, presence of systemic risk factors (e.g., hypercholesterolemia), and presence of inflammatory cell types. The tunability of these existing platforms can be further exploited in the future to mimic key features of aging or common comorbidities. Advanced age is the strongest risk factor for atherosclerosis121 and is well known to be associated with significant arterial stiffening and ECM remodeling122. Current in vitro models of atherosclerosis do not account for these changes, but evidence that monocyte behavior is stiffness-dependent123–124 suggests that age-related stiffening may impact disease onset and progression. There is also room to expand these models to mimic different stages of atherosclerosis, as existing studies have focused upon only the earliest events in disease onset. These efforts would require tailoring scaffold features to mimic the ECM and biophysical alterations that occur with progression of atherosclerosis and plaque development125. Until recently, no models had been described to mimic the advanced atherosclerotic plaque itself. However, new work describes an innovative approach to mimic late-stage atherosclerotic lesions in a spheroid culture model126, creating a future opportunity to combine tissue-engineered atherosclerosis models with advances in organoid technology to produce plaque-containing vessels.
Aortic Heart Valve
Non-rheumatic calcific aortic valve disease (CAVD) leads to the fibrotic thickening and calcification of the aortic heart valve, and is the most common heart valve disorder in developed countries127–128. CAVD affects more than 25% of individuals over the age of 65, and the number of individuals with CAVD is expected to triple by the year 2050129. However, much of our knowledge of this disease is derived from end-point analyses of diseased valves, and the only treatment option remains surgical valve replacement130–131. Because total valve replacement is the sole treatment for CAVD, numerous advances have been made toward creating healthy tissue-engineered valves. As reviewed elsewhere132, pioneering work in this area has been performed by multiple individuals, with Shinoka and Mayer describing large animal implantation of a tissue-engineered valve in 1995133, followed by further work in sheep by Hoerstrup134 and in humans by Dohmen et al.135 in the early 2000s. Preclinical studies of tissue engineered heart valves have been promising136, and these foundational studies have laid the groundwork for the ongoing development of novel valve scaffolds and cell sources. However, there is also a strong desire to develop strategies that target CAVD progression prior to the disease advancing to the stage that requires valve replacement surgery.
Despite sharing numerous risk factors and pathological similarities with atherosclerosis, therapies and lifestyle changes that are efficacious in slowing atherosclerosis have not been met with the same success in CAVD137–139, illustrating that CAVD has a unique pathogenesis. Our incomplete understanding of CAVD etiology ultimately acts as a major limiting factor in developing CAVD treatments. Thus, the development of 3-D in vitro models of valvular calcification is being investigated in order to both characterize the cellular and molecular events that lead to the initiation and progression of CAVD. Such in vitro engineered diseased models could aid in the development of therapeutics to slow or reverse CAVD progression, but would need to account for changes in valve cell phenotypes and leaflet architecture at different stages of disease, as we discuss below (Figure 3).
Figure 3.

Overview of disease-inspired tissue engineering approaches to mimic calcific aortic valve disease
Disease-specific cells
Aortic valve leaflets are lined with valvular endothelial cells while the most abundant cell type within the leaflets are valvular interstitial cells (VICs). In healthy valves, VICs exhibit a quiescent, fibroblastic phenotype. Following injury, quiescent VICs become activated to a myofibroblastic phenotype and begin to proliferate and remodel their surrounding ECM140. While VIC activation is necessary to maintain valve homeostasis, when myofibroblastic VICs fail to apoptose, the valve progressively transitions to a diseased state characterized by an aberrant ECM140 and formation of calcified nodules via dystrophic mechanisms141. To a lesser extent, heterotopic ossification also contributes to calcification as VICs may transdifferentiate into an osteoblast like phenotype141. In contrast to the other cardiovascular tissues discussed in this review, VICs have not yet been directly derived from hPSCs. Because VICs obtained from porcine valves are viewed by many as an adequate mimic for human VICs, the need to identify alternative cell sources has not been as pressing as for other cell types. Additionally, human valves that are explanted at the time of valve replacement surgery have been able to act as a relatively accessible source of diseased VICs142–143. However, iPSC sources offer greater potential for manipulation and improved consistency, and recent efforts have achieved the generation of VICs from mesenchymal stem cells that had been derived from iPSCs144. It is also postulated that new efforts describing the derivation of second heart field progenitors from iPSCs may be used to generate VICs145.
Rather, efforts to mimic disease-specific cell phenotypes have generally relied on exogenous administration of biochemical stimuli, such as TGF-β1 for myofibroblastic VIC activation146–147 and mineralization media for generating osteoblastic VICs (obVICs)148–149. Interestingly, it is actually the generation of healthy VICs (qVICs) that has been more challenging. Even when creating disease-inspired environments, the ability to support a healthy phenotype is essential for generation of appropriate baseline or control conditions. But, when VICs are cultured on standard tissue culture polystyrene, they rapidly assume a myofibroblastic phenotype150–151, which is problematic when expanding a cell population for use in an experiment. Only recently have researchers been able to prevent myofibroblastic differentiation or reverse this phenotype to a quiescent state in human152 and porcine151 VICs with the use of a fibroblastic media formulation in 2D cultures. Others have found that qVICs can also be generated by adjusting the elastic modulus of the culture substrate153–154, which provides an alternative method for qVIC expansion in addition to informing the construction of 3D scaffolds.
Disease-inspired physical cues
The ECM of the native aortic valve leaflet exhibits a trilayered arrangement of collagen, glycosaminoglycans (GAGs), and elastin. This ECM composition and arrangement impacts both the mechanical and biochemical behavior of the valve155. In particular, the fibrosa layer on the aortic outflow side of the valve mainly consists of collagen and a small amount of elastin, allowing it to withstand much of the mechanical stress as the leaflets repeatedly coapt. In contrast, the ventricularis layer on the inflow side is mainly composed of elastin and a small amount of collagen, accommodating for shear stress due to blood flow. The spongiosa layer, between the fibrosa and ventricularis, is composed of GAGs and proteoglycans (PGs) that act to dampen pressure differences between the fibrosa and ventricularis. During CAVD pathogenesis, the valvular ECM undergoes progressive alterations156. In early-stage CAVD, the valve becomes highly enriched in GAGs and PGs157, leading to early leaflet thickening and stiffening. As CAVD progresses, there is elastin fragmentation and increased deposition of disorganized collagen I, particularly in the spongiosa158. Several 2D studies have illustrated that ECM composition and substrate stiffness can modulate VIC differentiation to a myofibroblastic phenotype159–162, thereby setting the stage to create 3D tissue-engineered models of CAVD.
One of the first detectable hallmarks of CAVD is thickening of valve leaflets due to increased deposition of PGs and GAGs157. Motivated by the field’s poor understanding of CAVD pathogenesis, particularly during early stages of the disease, our group recently created tissue-engineered models of early CAVD163. Scaffolds were constructed using GelMA as the base material, to mimic the collagen foundation of the normal valve structure, and copolymerized with either healthy or diseased amounts of methacrylated hyaluronic acid (HA) or chondroitin sulfate (CS) to mimic disease-associated GAG enrichment. Using this early CAVD platform, we demonstrated that increased GAGs decreased inflammatory cytokine production by VICs, indicating that this ECM alteration was not directly responsible for the development of an inflammatory environment that is seen in early CAVD. Rather, the function of the GAG enrichment varied by GAG type, with CS indirectly stimulating inflammatory activity. Specifically, CS enrichment enabled binding and retention of LDL and oxidized LDL (oxLDL) to the matrix; oxLDL, in turn, stimulated the production of numerous inflammatory cytokines by VICs163. Meanwhile, HA enrichment was not linked to inflammatory activity, but did significantly increase the production of angiogenic factors, another hallmark of CAVD. Moreover, the oxLDL retained by the CS stimulated further production of CS and HA, thereby creating a positive feedback loop. This in vitro disease-inspired culture platform allowed us to recreate and decipher a complex, multi-step mechanism in CAVD pathogenesis163. Additionally, it illustrated the benefits of using tissue-engineered systems to investigate disease etiology, such as being able to separately study individual ECM components to discover differential functions of each one in promoting disease progression.
The creation of diseased ECM conditions has also been pursued using a top-down tissue engineering approach, wherein the ECM composition of native valve leaflets was altered to produce pathological changes164–165. Motivated by observations that collagen I and HA promote quiescent (i.e., ‘healthy’) behavior by VICs, these studies examined the effects of collagen I or HA degradation on VIC behavior in ex vivo leaflet organ cultures. Mild enzyme treatments yielded alterations in collagen I or HA without affecting other ECM components and showed that disruption of either of these ECM components was sufficient to promote VIC differentiation to a myofibroblastic phenotype and significant tissue mineralization164–165. This top-down tissue engineering approach offers a unique complement to the traditional bottom-up engineered tissues; although it is not as precisely controlled as bottom-up engineering, it retains the complexity of the intact, native tissue and can thus be used to validate the physiological relevance of findings from more reductionist systems.
Others have exploited this ECM-based control of diseased VIC phenotype to construct systems that investigate disease pathogenesis. Consistent with the aforementioned findings that VIC interactions with HA are necessary to maintain a healthy phenotype, recent work found that VICs in GelMA-HA hybrid scaffolds did not undergo the spontaneous myofibroblastic differentiation seen in GelMA-only scaffolds166. These scaffolds provided a platform for examining how soluble factors (e.g., TGF-β1 or osteogenic medium) enacted development of disease features and mineralization and postulated a sequence by which VICs can become osteoblastic VICs167. A different approach to gaining mechanistic insight is the use of peptide-modified polyethylene glycol (PEG)-based hydrogels, which allow the investigation of how individual ECM-derived peptides influence VIC behavior. Modification of PEG hydrogels with peptides derived from different valve ECM proteins revealed that key markers of VIC fibrosis (e.g., myofibroblastic differentiation, ECM production) could be differentially modulated by individual peptides168, thus providing a platform for studying how specific cell-ligand interactions may be regulating disease pathogenesis. A similar PEG-based platform has also been implemented to study how exogenously added agents may be used to attenuate or halt fibrotic events in VIC-populated scaffolds169.
Finally, the mechanical stiffening that occurs in CAVD has also been replicated in 3D tissue-engineered platforms to create disease-inspired conditions. Interestingly, these studies have not yielded the results that were anticipated. Despite numerous 2D studies demonstrating increased myofibroblastic differentiation on stiffer substrates154, 161–162, 170 and stiffening being associated with native disease progression, VICs encapsulated in modified HA scaffolds171 as well as dynamically-stiffened PEG-based hydrogels172 exhibited decreased myofibroblastic differentiation in stiffer scaffolds. The source of this discordance remains unclear, but provides an interesting mechanobiological question to probe using these tailored microenvironments.
Future Opportunities
The pursuit to decipher CAVD pathogenesis has yielded great advancements in the design of ECM-inspired scaffold environments. It is believed that CAVD will be most treatable in its earliest stages, which is why initial development of disease-inspired environments have focused on early events163. However, there is still a need to elucidate subsequent steps in CAVD pathogenesis, necessitating the construction of tissue-engineered platforms that mimic the biochemical and biophysical stimuli that are characteristic of more advanced stages of the disease. Additionally, the majority of tissue-engineered CAVD platforms have focused upon examining how the disease-mimicking microenvironment impacts VIC differentiation to a myofibroblastic phenotype. However, VIC differentiation is just one part of CAVD, and CAVD is not solely driven by VICs. Infiltration of inflammatory cells such as macrophages and T lymphocytes also occurs fairly early in CAVD157, 173, and these cells are thought to significantly contribute to CAVD progression174–175. The pathological ECM environment can indirectly163 and directly176 influence inflammatory activity, motivating future work to merge disease-inspired CAVD platforms with inflammatory cell co-culture; many of the biomaterials approaches described by researchers developing tissue-engineered tumor microenvironments may prove useful in this endeavor176. Additionally, evaluating immune cell behavior in these systems may aid in understanding ECM remodeling and angiogenesis throughout CAVD progression, as macrophages also contribute to these processes177–178.
Conclusions and Recommendations
Let’s Redefine “Tissue Engineering”
Other recent reviews have covered advances made toward generating tissue-engineered disease models for many other tissue types, ranging from bone to tumors179–180. Herein, we present highlights from the cardiovascular disease modeling, an emerging field that is poised to have dramatic impact, with over 90 million people in the U.S. alone affected by the cardiovascular diseases noted above. Yet, as noted at the start of this review, the textbook definition of “tissue engineering” still focuses on the creation of healthy constructs; it is time to revisit this definition and expand to include disease-inspired tissue engineering. The current definition fails to accurately capture the diverse ways in which the application of tissue engineering techniques can advance both our mechanistic knowledge and discovery of therapeutic treatments for a disease.
Sex as a Biological Variable
Most researchers are now familiar with sex as a biological variable within the NIH-mandated framework of justifying the sex of vertebrate animals used in experiments. However, cellular-scale sex is often overlooked as a potentially influential factor, despite calls to account for cellular-scale sex in in vitro cultures181–182. Many cardiovascular diseases exhibit strong sexual dimorphism182–185, and sex-separated cultures of several types of cardiovascular cells have illustrated that cellular sex can have a powerful influence on disease pathogenesis and responsiveness to extracellular stimuli186–189. Ideally, our tissue-engineered disease models will take into account these cellular-scale sex differences, thereby offering a potentially powerful platform for investigating sources of sexual dimorphism in cardiovascular pathologies and perhaps even the development of sex-specific treatments.
Integration Across the Cardiovascular System
Although the myocardium, aortic valves, and vessels all fall under the umbrella of cardiovascular tissues, these areas tend to be quite siloed from each other. There is surprisingly little overlap across the researchers creating tissue-engineered disease models of the myocardium, aortic valves, and vessels, with individual labs generally specializing in one of the three. However, while each of these tissues experiences a unique suite of pathologies, there are also numerous commonalities across these tissues that could be better shared and exploited to advance disease model creation. For example, heart valve researchers have been investigating myofibroblastic differentiation in ECM-inspired 3D environments for over a decade – experience that could be harnessed to inform the relatively nascent field of tissue engineering cardiac fibrosis. In a similar vein, atherosclerosis shares many common elements with aortic valve stenosis190, but there is relatively little crosstalk across these fields to examine how the more developed mechanistic knowledge of atherosclerosis may be applied to model CAVD. Greater advancements and innovation in tissue-engineered cardiovascular disease modeling will grow from increased communication across researchers specializing in the different cardiovascular tissues.
Cited References
- (1).Shafiee A; Atala A, Tissue Engineering: Toward a New Era of Medicine. Annu Rev Med 2017, 68, 29–40. 10.1146/annurev-med-102715-092331: 10.1146/annurev-med-102715-092331 [DOI] [PubMed] [Google Scholar]
- (2).Wang Y; Yin P; Bian GL; Huang HY; Shen H; Yang JJ; Yang ZY; Shen ZY, The combination of stem cells and tissue engineering: an advanced strategy for blood vessels regeneration and vascular disease treatment. Stem Cell Res Ther 2017, 8 (1), 194 10.1186/s13287-017-0642-y: 10.1186/s13287-017-0642-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hirschi KK; Li S; Roy K, Induced pluripotent stem cells for regenerative medicine. Annu Rev Biomed Eng 2014, 16, 277–94. 10.1146/annurev-bioeng-071813-105108: 10.1146/annurev-bioeng-071813-105108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rambhia KJ; Ma PX, Controlled drug release for tissue engineering. J Control Release 2015, 219, 119–128. 10.1016/j.jconrel.2015.08.049: 10.1016/j.jconrel.2015.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vats K; Benoit DS, Dynamic manipulation of hydrogels to control cell behavior: a review. Tissue Eng Part B Rev 2013, 19 (6), 455–69. 10.1089/ten.TEB.2012.0716: 10.1089/ten.TEB.2012.0716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Naderi H; Matin MM; Bahrami AR, Review paper: critical issues in tissue engineering: biomaterials, cell sources, angiogenesis, and drug delivery systems. J Biomater Appl 2011, 26 (4), 383–417. 10.1177/0885328211408946: 10.1177/0885328211408946 [DOI] [PubMed] [Google Scholar]
- (7).Chen FM; Liu X, Advancing biomaterials of human origin for tissue engineering. Prog Polym Sci 2016, 53, 86–168. 10.1016/j.progpolymsci.2015.02.004: 10.1016/j.progpolymsci.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).O’brien FJ, Biomaterials & scaffolds for tissue engineering. Materials today 2011, 14 (3), 88–95. [Google Scholar]
- (9).Kim YS; Smoak MM; Melchiorri AJ; Mikos AG, An Overview of the Tissue Engineering Market in the United States from 2011 to 2018. Tissue Eng Part A 2019, 25 (1–2), 1–8. 10.1089/ten.TEA.2018.0138: 10.1089/ten.TEA.2018.0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ikada Y, Challenges in tissue engineering. J R Soc Interface 2006, 3 (10), 589–601. 10.1098/rsif.2006.0124: 10.1098/rsif.2006.0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Katari R; Peloso A; Orlando G, Tissue engineering and regenerative medicine: semantic considerations for an evolving paradigm. Front Bioeng Biotechnol 2014, 2, 57 10.3389/fbioe.2014.00057: 10.3389/fbioe.2014.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).National Institute of Biomedical Imaging and Bioengineering (NIBIB), Tissue Engineering and Regenerative Medicine. https://www.nibib.nih.gov/science-education/science-topics/tissue-engineering-and-regenerative-medicine (accessed 7/8/19).
- (13).Duval K; Grover H; Han LH; Mou Y; Pegoraro AF; Fredberg J; Chen Z, Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology (Bethesda) 2017, 32 (4), 266–277. 10.1152/physiol.00036.2016: 10.1152/physiol.00036.2016 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- (14).Baker BM; Chen CS, Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci 2012, 125 (Pt 13), 3015–24. 10.1242/jcs.079509: 10.1242/jcs.079509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mabry KM; Payne SZ; Anseth KS, Microarray analyses to quantify advantages of 2D and 3D hydrogel culture systems in maintaining the native valvular interstitial cell phenotype. Biomaterials 2016, 74, 31–41. 10.1016/j.biomaterials.2015.09.035: 10.1016/j.biomaterials.2015.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Voskoglou-Nomikos T; Pater JL; Seymour L, Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res 2003, 9 (11), 4227–39. [PubMed] [Google Scholar]
- (17).Savoji H; Mohammadi MH; Rafatian N; Toroghi MK; Wang EY; Zhao Y; Korolj A; Ahadian S; Radisic M, Cardiovascular disease models: A game changing paradigm in drug discovery and screening. Biomaterials 2019, 198, 3–26. 10.1016/j.biomaterials.2018.09.036: 10.1016/j.biomaterials.2018.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Benjamin EJ; Virani SS; Callaway CW; Chamberlain AM; Chang AR; Cheng S; Chiuve SE; Cushman M; Delling FN; Deo R; de Ferranti SD; Ferguson JF; Fornage M; Gillespie C; Isasi CR; Jimenez MC; Jordan LC; Judd SE; Lackland D; Lichtman JH; Lisabeth L; Liu S; Longenecker CT; Lutsey PL; Mackey JS; Matchar DB; Matsushita K; Mussolino ME; Nasir K; O’Flaherty M; Palaniappan LP; Pandey A; Pandey DK; Reeves MJ; Ritchey MD; Rodriguez CJ; Roth GA; Rosamond WD; Sampson UKA; Satou GM; Shah SH; Spartano NL; Tirschwell DL; Tsao CW; Voeks JH; Willey JZ; Wilkins JT; Wu JH; Alger HM; Wong SS; Muntner P; American Heart Association Council on, E.; Prevention Statistics, C.; Stroke Statistics, S., Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137 (12), e67–e492. 10.1161/CIR.0000000000000558: 10.1161/CIR.0000000000000558 [DOI] [PubMed] [Google Scholar]
- (19).Johansson S; Rosengren A; Young K; Jennings E, Mortality and morbidity trends after the first year in survivors of acute myocardial infarction: a systematic review. BMC Cardiovasc Disord 2017, 17 (1), 53 10.1186/s12872-017-0482-9: 10.1186/s12872-017-0482-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Rapsomaniki E; Thuresson M; Yang E; Blin P; Hunt P; Chung SC; Stogiannis D; Pujades-Rodriguez M; Timmis A; Denaxas SC; Danchin N; Stokes M; Thomas-Delecourt F; Emmas C; Hasvold P; Jennings E; Johansson S; Cohen DJ; Jernberg T; Moore N; Janzon M; Hemingway H, Using big data from health records from four countries to evaluate chronic disease outcomes: a study in 114 364 survivors of myocardial infarction. Eur Heart J Qual Care Clin Outcomes 2016, 2 (3), 172–183. 10.1093/ehjqcco/qcw004: 10.1093/ehjqcco/qcw004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kong P; Christia P; Frangogiannis NG, The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014, 71 (4), 549–74. 10.1007/s00018-013-1349-6: 10.1007/s00018-013-1349-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jones K; Saxon L; Cunningham W; Adams P; guideline Development, G., Secondary prevention for patients after a myocardial infarction: summary of updated NICE guidance. BMJ 2013, 347, f6544 10.1136/bmj.f6544: 10.1136/bmj.f6544 [DOI] [PubMed] [Google Scholar]
- (23).Pursnani S; Korley F; Gopaul R; Kanade P; Chandra N; Shaw RE; Bangalore S, Percutaneous coronary intervention versus optimal medical therapy in stable coronary artery disease: a systematic review and meta-analysis of randomized clinical trials. Circ Cardiovasc Interv 2012, 5 (4), 476–90. 10.1161/CIRCINTERVENTIONS.112.970954: 10.1161/CIRCINTERVENTIONS.112.970954 [DOI] [PubMed] [Google Scholar]
- (24).Cuspidi C; Sala C; Negri F; Mancia G; Morganti A; Italian Society of, H., Prevalence of left-ventricular hypertrophy in hypertension: an updated review of echocardiographic studies. J Hum Hypertens 2012, 26 (6), 343–9. 10.1038/jhh.2011.104: 10.1038/jhh.2011.104 [DOI] [PubMed] [Google Scholar]
- (25).Taylor RS; Brown A; Ebrahim S; Jolliffe J; Noorani H; Rees K; Skidmore B; Stone JA; Thompson DR; Oldridge N, Exercise-based rehabilitation for patients with coronary heart disease: systematic review and meta-analysis of randomized controlled trials. The American journal of medicine 2004, 116 (10), 682–692. [DOI] [PubMed] [Google Scholar]
- (26).Jawad H; Ali NN; Lyon AR; Chen QZ; Harding SE; Boccaccini AR, Myocardial tissue engineering: a review. J Tissue Eng Regen Med 2007, 1 (5), 327–42. 10.1002/term.46: 10.1002/term.46 [DOI] [PubMed] [Google Scholar]
- (27).Domenech M; Polo-Corrales L; Ramirez-Vick JE; Freytes DO, Tissue Engineering Strategies for Myocardial Regeneration: Acellular Versus Cellular Scaffolds? Tissue Eng Part B Rev 2016, 22 (6), 438–458. 10.1089/ten.TEB.2015.0523: 10.1089/ten.TEB.2015.0523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dai Y; Foley A, Tissue engineering approaches to heart repair. Crit Rev Biomed Eng 2014, 42 (3–4), 213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Coulombe KL; Bajpai VK; Andreadis ST; Murry CE, Heart regeneration with engineered myocardial tissue. Annu Rev Biomed Eng 2014, 16, 1–28. 10.1146/annurev-bioeng-071812-152344: 10.1146/annurev-bioeng-071812-152344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Eschenhagen T; Fink C; Remmers U; Scholz H; Wattchow J; Weil J; Zimmermann W; Dohmen HH; Schafer H; Bishopric N; Wakatsuki T; Elson EL, Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: a new heart muscle model system. FASEB J 1997, 11 (8), 683–94. 10.1096/fasebj.11.8.9240969: 10.1096/fasebj.11.8.9240969 [DOI] [PubMed] [Google Scholar]
- (31).Zuppinger C, 3D Cardiac Cell Culture: A Critical Review of Current Technologies and Applications. Front Cardiovasc Med 2019, 6, 87 10.3389/fcvm.2019.00087: 10.3389/fcvm.2019.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zwi-Dantsis L; Gepstein L, Induced pluripotent stem cells for cardiac repair. Cell Mol Life Sci 2012, 69 (19), 3285–99. 10.1007/s00018-012-1078-2: 10.1007/s00018-012-1078-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liang P; Lan F; Lee AS; Gong T; Sanchez-Freire V; Wang Y; Diecke S; Sallam K; Knowles JW; Wang PJ; Nguyen PK; Bers DM; Robbins RC; Wu JC, Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127 (16), 1677–91. 10.1161/CIRCULATIONAHA.113.001883: 10.1161/CIRCULATIONAHA.113.001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Callaghan NI; Hadipour-Lakmehsari S; Lee SH; Gramolini AO; Simmons CA, Modeling cardiac complexity: Advancements in myocardial models and analytical techniques for physiological investigation and therapeutic development in vitro. APL Bioeng 2019, 3 (1), 011501 10.1063/1.5055873: 10.1063/1.5055873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Brandao KO; Tabel VA; Atsma DE; Mummery CL; Davis RP, Human pluripotent stem cell models of cardiac disease: from mechanisms to therapies. Dis Model Mech 2017, 10 (9), 1039–1059. 10.1242/dmm.030320: 10.1242/dmm.030320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Musunuru K; Sheikh F; Gupta RM; Houser SR; Maher KO; Milan DJ; Terzic A; Wu JC; American Heart Association Council on Functional, G.; Translational B; Council on Cardiovascular Disease in the, Y.; Council on, C.; Stroke N, Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ Genom Precis Med 2018, 11 (1), e000043 10.1161/HCG.0000000000000043: 10.1161/HCG.0000000000000043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Peter AK; Bjerke MA; Leinwand LA, Biology of the cardiac myocyte in heart disease. Mol Biol Cell 2016, 27 (14), 2149–60. 10.1091/mbc.E16-01-0038: 10.1091/mbc.E16-01-0038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhang J; Wilson GF; Soerens AG; Koonce CH; Yu J; Palecek SP; Thomson JA; Kamp TJ, Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res 2009, 104 (4), e30–41. 10.1161/CIRCRESAHA.108.192237: 10.1161/CIRCRESAHA.108.192237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lian X; Hsiao C; Wilson G; Zhu K; Hazeltine LB; Azarin SM; Raval KK; Zhang J; Kamp TJ; Palecek SP, Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A 2012, 109 (27), E1848–57. 10.1073/pnas.1200250109: 10.1073/pnas.1200250109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tzatzalos E; Abilez OJ; Shukla P; Wu JC, Engineered heart tissues and induced pluripotent stem cells: Macro- and microstructures for disease modeling, drug screening, and translational studies. Adv Drug Deliv Rev 2016, 96, 234–244. 10.1016/j.addr.2015.09.010: 10.1016/j.addr.2015.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Savla JJ; Nelson BC; Perry CN; Adler ED, Induced pluripotent stem cells for the study of cardiovascular disease. J Am Coll Cardiol 2014, 64 (5), 512–9. 10.1016/j.jacc.2014.05.038: 10.1016/j.jacc.2014.05.038 [DOI] [PubMed] [Google Scholar]
- (42).Christidi E; Huang HM; Brunham LR, CRISPR/Cas9-mediated genome editing in human stem cell-derived cardiomyocytes: Applications for cardiovascular disease modelling and cardiotoxicity screening. Drug Discov Today Technol 2018, 28, 13–21. 10.1016/j.ddtec.2018.06.002: 10.1016/j.ddtec.2018.06.002 [DOI] [PubMed] [Google Scholar]
- (43).Itzhaki I; Maizels L; Huber I; Zwi-Dantsis L; Caspi O; Winterstern A; Feldman O; Gepstein A; Arbel G; Hammerman H; Boulos M; Gepstein L, Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471 (7337), 225–9. 10.1038/nature09747: 10.1038/nature09747 [DOI] [PubMed] [Google Scholar]
- (44).Sala L; Gnecchi M; Schwartz PJ, Long QT Syndrome Modelling with Cardiomyocytes Derived from Human-induced Pluripotent Stem Cells. Arrhythm Electrophysiol Rev 2019, 8 (2), 105–110. 10.15420/aer.2019.1.1: 10.15420/aer.2019.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Duncan G; Firth K; George V; Hoang MD; Staniforth A; Smith G; Denning C, Drug-Mediated Shortening of Action Potentials in LQTS2 Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Dev 2017, 26 (23), 1695–1705. 10.1089/scd.2017.0172: 10.1089/scd.2017.0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lahti AL; Kujala VJ; Chapman H; Koivisto AP; Pekkanen-Mattila M; Kerkela E; Hyttinen J; Kontula K; Swan H; Conklin BR; Yamanaka S; Silvennoinen O; Aalto-Setala K, Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech 2012, 5 (2), 220–30. 10.1242/dmm.008409: 10.1242/dmm.008409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mosqueira D; Mannhardt I; Bhagwan JR; Lis-Slimak K; Katili P; Scott E; Hassan M; Prondzynski M; Harmer SC; Tinker A; Smith JGW; Carrier L; Williams PM; Gaffney D; Eschenhagen T; Hansen A; Denning C, CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J 2018, 39 (43), 3879–3892. 10.1093/eurheartj/ehy249: 10.1093/eurheartj/ehy249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Goldfracht I; Efraim Y; Shinnawi R; Kovalev E; Huber I; Gepstein A; Arbel G; Shaheen N; Tiburcy M; Zimmermann WH; Machluf M; Gepstein L, Engineered heart tissue models from hiPSC-derived cardiomyocytes and cardiac ECM for disease modeling and drug testing applications. Acta Biomater 2019, 92, 145–159. 10.1016/j.actbio.2019.05.016: 10.1016/j.actbio.2019.05.016 [DOI] [PubMed] [Google Scholar]
- (49).Hinson JT; Chopra A; Nafissi N; Polacheck WJ; Benson CC; Swist S; Gorham J; Yang L; Schafer S; Sheng CC; Haghighi A; Homsy J; Hubner N; Church G; Cook SA; Linke WA; Chen CS; Seidman JG; Seidman CE, HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349 (6251), 982–6. 10.1126/science.aaa5458: 10.1126/science.aaa5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Nunes SS; Miklas JW; Liu J; Aschar-Sobbi R; Xiao Y; Zhang B; Jiang J; Masse S; Gagliardi M; Hsieh A; Thavandiran N; Laflamme MA; Nanthakumar K; Gross GJ; Backx PH; Keller G; Radisic M, Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods 2013, 10 (8), 781–7. 10.1038/nmeth.2524: 10.1038/nmeth.2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhao Y; Rafatian N; Feric NT; Cox BJ; Aschar-Sobbi R; Wang EY; Aggarwal P; Zhang B; Conant G; Ronaldson-Bouchard K; Pahnke A; Protze S; Lee JH; Davenport Huyer L; Jekic D; Wickeler A; Naguib HE; Keller GM; Vunjak-Novakovic G; Broeckel U; Backx PH; Radisic M, A Platform for Generation of Chamber-Specific Cardiac Tissues and Disease Modeling. Cell 2019, 176 (4), 913–927 e18. 10.1016/j.cell.2018.11.042: 10.1016/j.cell.2018.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wakatsuki T Engineered cardiac tissues and methods of using them. Patent #10,113,150, 2018.
- (53).Wang EY; Rafatian N; Zhao Y; Lee A; Lai BFL; Lu RX; Jekic D; Davenport Huyer L; Knee-Walden EJ; Bhattacharya S; Backx PH; Radisic M, Biowire Model of Interstitial and Focal Cardiac Fibrosis. ACS Cent Sci 2019, 5 (7), 1146–1158. 10.1021/acscentsci.9b00052: 10.1021/acscentsci.9b00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).van Spreeuwel ACC; Bax NAM; van Nierop BJ; Aartsma-Rus A; Goumans MTH; Bouten CVC, Mimicking Cardiac Fibrosis in a Dish: Fibroblast Density Rather than Collagen Density Weakens Cardiomyocyte Function. J Cardiovasc Transl Res 2017, 10 (2), 116–127. 10.1007/s12265-017-9737-1: 10.1007/s12265-017-9737-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Weinberger F; Mannhardt I; Eschenhagen T, Engineering Cardiac Muscle Tissue: A Maturating Field of Research. Circ Res 2017, 120 (9), 1487–1500. 10.1161/CIRCRESAHA.117.310738: 10.1161/CIRCRESAHA.117.310738 [DOI] [PubMed] [Google Scholar]
- (56).Jawad H; Lyon AR; Harding SE; Ali NN; Boccaccini AR, Myocardial tissue engineering. Br Med Bull 2008, 87, 31–47. 10.1093/bmb/ldn026: 10.1093/bmb/ldn026 [DOI] [PubMed] [Google Scholar]
- (57).Yellon DM; Hausenloy DJ, Myocardial reperfusion injury. N Engl J Med 2007, 357 (11), 1121–35. 10.1056/NEJMra071667: 10.1056/NEJMra071667 [DOI] [PubMed] [Google Scholar]
- (58).Hausenloy DJ; Yellon DM, Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 2013, 123 (1), 92–100. 10.1172/JCI62874: 10.1172/JCI62874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Lindsey ML; Bolli R; Canty JM Jr.; Du XJ; Frangogiannis NG; Frantz S; Gourdie RG; Holmes JW; Jones SP; Kloner RA; Lefer DJ; Liao R; Murphy E; Ping P; Przyklenk K; Recchia FA; Schwartz Longacre L; Ripplinger CM; Van Eyk JE; Heusch G, Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol 2018, 314 (4), H812–H838. 10.1152/ajpheart.00335.2017: 10.1152/ajpheart.00335.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Chen T; Vunjak-Novakovic G, In vitro Models of Ischemia-Reperfusion Injury. Regen Eng Transl Med 2018, 4 (3), 142–153. 10.1007/s40883-018-0056-0: 10.1007/s40883-018-005-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Katare RG; Ando M; Kakinuma Y; Sato T, Engineered heart tissue: a novel tool to study the ischemic changes of the heart in vitro. PLoS One 2010, 5 (2), e9275 10.1371/journal.pone.0009275: 10.1371/journal.pone.0009275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Gorbe A; Eder A; Varga ZV; Paloczi J; Hansen A; Ferdinandy P; Eschenhagen T, Protection by the NO-Donor SNAP and BNP against Hypoxia/Reoxygenation in Rat Engineered Heart Tissue. PLoS One 2015, 10 (7), e0132186 10.1371/journal.pone.0132186: 10.1371/journal.pone.0132186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Chen T; Vunjak-Novakovic G, Human Tissue-Engineered Model of Myocardial Ischemia-Reperfusion Injury. Tissue Eng Part A 2019, 25 (9–10), 711–724. 10.1089/ten.TEA.2018.0212: 10.1089/ten.TEA.2018.0212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Frey N; Olson EN, Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 2003, 65, 45–79. 10.1146/annurev.physiol.65.092101.142243: 10.1146/annurev.physiol.65.092101.142243 [DOI] [PubMed] [Google Scholar]
- (65).Ho KK; Pinsky JL; Kannel WB; Levy D, The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol 1993, 22 (4 Suppl A), 6A–13A. [DOI] [PubMed] [Google Scholar]
- (66).Ovchinnikova E; Hoes M; Ustyantsev K; Bomer N; de Jong TV; van der Mei H; Berezikov E; van der Meer P, Modeling Human Cardiac Hypertrophy in Stem Cell-Derived Cardiomyocytes. Stem Cell Reports 2018, 10 (3), 794–807. 10.1016/j.stemcr.2018.01.016: 10.1016/j.stemcr.2018.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Hirt MN; Sorensen NA; Bartholdt LM; Boeddinghaus J; Schaaf S; Eder A; Vollert I; Stohr A; Schulze T; Witten A; Stoll M; Hansen A; Eschenhagen T, Increased afterload induces pathological cardiac hypertrophy: a new in vitro model. Basic Res Cardiol 2012, 107 (6), 307 10.1007/s00395-012-0307-z: 10.1007/s00395-012-0307-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Leonard A; Bertero A; Powers JD; Beussman KM; Bhandari S; Regnier M; Murry CE; Sniadecki NJ, Afterload promotes maturation of human induced pluripotent stem cell derived cardiomyocytes in engineered heart tissues. J Mol Cell Cardiol 2018, 118, 147–158. 10.1016/j.yjmcc.2018.03.016: 10.1016/j.yjmcc.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Schaaf S; Eder A; Vollert I; Stohr A; Hansen A; Eschenhagen T, Generation of strip-format fibrin-based engineered heart tissue (EHT). Methods Mol Biol 2014, 1181, 121–9. 10.1007/978-1-4939-1047-2_11: 10.1007/978-1-4939-1047-2_11 [DOI] [PubMed] [Google Scholar]
- (70).Hirt MN; Werner T; Indenbirken D; Alawi M; Demin P; Kunze AC; Stenzig J; Starbatty J; Hansen A; Fiedler J; Thum T; Eschenhagen T, Deciphering the microRNA signature of pathological cardiac hypertrophy by engineered heart tissue- and sequencing-technology. J Mol Cell Cardiol 2015, 81, 1–9. 10.1016/j.yjmcc.2015.01.008: 10.1016/j.yjmcc.2015.01.008 [DOI] [PubMed] [Google Scholar]
- (71).Stenzig J; Hirt MN; Loser A; Bartholdt LM; Hensel JT; Werner TR; Riemenschneider M; Indenbirken D; Guenther T; Muller C; Hubner N; Stoll M; Eschenhagen T, DNA methylation in an engineered heart tissue model of cardiac hypertrophy: common signatures and effects of DNA methylation inhibitors. Basic Res Cardiol 2016, 111 (1), 9 10.1007/s00395-015-0528-z: 10.1007/s00395-015-0528-z [DOI] [PubMed] [Google Scholar]
- (72).Yan M; Chen C; Gong W; Yin Z; Zhou L; Chaugai S; Wang DW, miR-21–3p regulates cardiac hypertrophic response by targeting histone deacetylase-8. Cardiovasc Res 2015, 105 (3), 340–52. 10.1093/cvr/cvu254: 10.1093/cvr/cvu254 [DOI] [PubMed] [Google Scholar]
- (73).Stenzig J; Schneeberger Y; Loser A; Peters BS; Schaefer A; Zhao RR; Ng SL; Hoppner G; Geertz B; Hirt MN; Tan W; Wong E; Reichenspurner H; Foo RS; Eschenhagen T, Pharmacological inhibition of DNA methylation attenuates pressure overload-induced cardiac hypertrophy in rats. J Mol Cell Cardiol 2018, 120, 53–63. 10.1016/j.yjmcc.2018.05.012: 10.1016/j.yjmcc.2018.05.012 [DOI] [PubMed] [Google Scholar]
- (74).Deddens JC; Sadeghi AH; Hjortnaes J; van Laake LW; Buijsrogge M; Doevendans PA; Khademhosseini A; Sluijter JP, Modeling the Human Scarred Heart In Vitro: Toward New Tissue Engineered Models. Adv Healthc Mater 2017, 6 (3). 10.1002/adhm.201600571: 10.1002/adhm.201600571 [DOI] [PubMed] [Google Scholar]
- (75).Hinderer S; Schenke-Layland K, Cardiac fibrosis - A short review of causes and therapeutic strategies. Adv Drug Deliv Rev 2019. 10.1016/j.addr.2019.05.011: 10.1016/j.addr.2019.05.011 [DOI] [PubMed] [Google Scholar]
- (76).Travers JG; Kamal FA; Robbins J; Yutzey KE; Blaxall BC, Cardiac Fibrosis: The Fibroblast Awakens. Circ Res 2016, 118 (6), 1021–40. 10.1161/CIRCRESAHA.115.306565: 10.1161/CIRCRESAHA.115.306565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Disertori M; Mase M; Ravelli F, Myocardial fibrosis predicts ventricular tachyarrhythmias. Trends Cardiovasc Med 2017, 27 (5), 363–372. 10.1016/j.tcm.2017.01.011: 10.1016/j.tcm.2017.01.011 [DOI] [PubMed] [Google Scholar]
- (78).Sadeghi AH; Shin SR; Deddens JC; Fratta G; Mandla S; Yazdi IK; Prakash G; Antona S; Demarchi D; Buijsrogge MP; Sluijter JPG; Hjortnaes J; Khademhosseini A, Engineered 3D Cardiac Fibrotic Tissue to Study Fibrotic Remodeling. Adv Healthc Mater 2017, 6 (11). 10.1002/adhm.201601434: 10.1002/adhm.201601434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Kong M; Lee J; Yazdi IK; Miri AK; Lin YD; Seo J; Zhang YS; Khademhosseini A; Shin SR, Cardiac Fibrotic Remodeling on a Chip with Dynamic Mechanical Stimulation. Adv Healthc Mater 2019, 8 (3), e1801146 10.1002/adhm.201801146: 10.1002/adhm.201801146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Mastikhina O; Moon B-U; Williams K; Hatkar R; Gustafson D; Sun X; Koo M; Lam AYL; Sun Y; Fish JE; Young EWK; Nunes SS, Human cardiac-fibrosis-on-a-chip model recapitulates disease hallmarks and can serve as a platform for drug screening. bioRxiv 2019, 632406 10.1101/632406: 10.1101/632406 [DOI] [PubMed] [Google Scholar]
- (81).Lee MO; Jung KB; Jo SJ; Hyun SA; Moon KS; Seo JW; Kim SH; Son MY, Modelling cardiac fibrosis using three-dimensional cardiac microtissues derived from human embryonic stem cells. J Biol Eng 2019, 13, 15 10.1186/s13036-019-0139-6: 10.1186/s13036-019-0139-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Occhetta P; Isu G; Lemme M; Conficconi C; Oertle P; Raz C; Visone R; Cerino G; Plodinec M; Rasponi M; Marsano A, A three-dimensional in vitro dynamic micro-tissue model of cardiac scar formation. Integr Biol (Camb) 2018, 10 (3), 174–183. 10.1039/c7ib00199a: 10.1039/c7ib00199a [DOI] [PubMed] [Google Scholar]
- (83).Spadaccio C; Rainer A; Mozetic P; Trombetta M; Dion RA; Barbato R; Nappi F; Chello M, The role of extracellular matrix in age-related conduction disorders: a forgotten player? J Geriatr Cardiol 2015, 12 (1), 76–82. 10.11909/j.issn.1671-5411.2015.01.009: 10.11909/j.issn.1671-5411.2015.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Berk BC; Fujiwara K; Lehoux S, ECM remodeling in hypertensive heart disease. J Clin Invest 2007, 117 (3), 568–75. 10.1172/JCI31044: 10.1172/JCI31044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Leonard BL; Smaill BH; LeGrice IJ, Structural remodeling and mechanical function in heart failure. Microsc Microanal 2012, 18 (1), 50–67. 10.1017/S1431927611012438: 10.1017/S1431927611012438 [DOI] [PubMed] [Google Scholar]
- (86).Rienks M; Papageorgiou AP; Frangogiannis NG; Heymans S, Myocardial extracellular matrix: an ever-changing and diverse entity. Circ Res 2014, 114 (5), 872–88. 10.1161/CIRCRESAHA.114.302533: 10.1161/CIRCRESAHA.114.302533 [DOI] [PubMed] [Google Scholar]
- (87).Kapelko VI, Extracellular matrix alterations in cardiomyopathy: The possible crucial role in the dilative form. Exp Clin Cardiol 2001, 6 (1), 41–9. [PMC free article] [PubMed] [Google Scholar]
- (88).Jeevaratnam K; Chadda KR; Salvage SC; Valli H; Ahmad S; Grace AA; Huang CL, Ion channels, long QT syndrome and arrhythmogenesis in ageing. Clin Exp Pharmacol Physiol 2017, 44 Suppl 1, 38–45. 10.1111/1440-1681.12721: 10.1111/1440-1681.12721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Li C; Chaung W; Mozayan C; Chabra R; Wang P; Narayan RK, A New Approach for On-Demand Generation of Various Oxygen Tensions for In Vitro Hypoxia Models. PLoS One 2016, 11 (5), e0155921 10.1371/journal.pone.0155921: 10.1371/journal.pone.0155921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Cohn PF, Silent ischemia: a timely aspect in coronary artery disease. Herz 1987, 12 (5), 314–7. [PubMed] [Google Scholar]
- (91).Weinberg CB; Bell E, A blood vessel model constructed from collagen and cultured vascular cells. Science 1986, 231 (4736), 397–400. 10.1126/science.2934816: 10.1126/science.2934816 [DOI] [PubMed] [Google Scholar]
- (92).Pashneh-Tala S; MacNeil S; Claeyssens F, The Tissue-Engineered Vascular Graft-Past, Present, and Future. Tissue Eng Part B Rev 2016, 22 (1), 68–100. 10.1089/ten.teb.2015.0100: 10.1089/ten.teb.2015.0100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Lawson JH; Glickman MH; Ilzecki M; Jakimowicz T; Jaroszynski A; Peden EK; Pilgrim AJ; Prichard HL; Guziewicz M; Przywara S; Szmidt J; Turek J; Witkiewicz W; Zapotoczny N; Zubilewicz T; Niklason LE, Bioengineered human acellular vessels for dialysis access in patients with end-stage renal disease: two phase 2 single-arm trials. Lancet 2016, 387 (10032), 2026–34. 10.1016/S0140-6736(16)00557-2: 10.1016/S0140-6736(16)00557-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Wystrychowski W; McAllister TN; Zagalski K; Dusserre N; Cierpka L; L’Heureux N, First human use of an allogeneic tissue-engineered vascular graft for hemodialysis access. J Vasc Surg 2014, 60 (5), 1353–1357. 10.1016/j.jvs.2013.08.018: 10.1016/j.jvs.2013.08.018 [DOI] [PubMed] [Google Scholar]
- (95).Olausson M; Patil PB; Kuna VK; Chougule P; Hernandez N; Methe K; Kullberg-Lindh C; Borg H; Ejnell H; Sumitran-Holgersson S, Transplantation of an allogeneic vein bioengineered with autologous stem cells: a proof-of-concept study. Lancet 2012, 380 (9838), 230–7. 10.1016/S0140-6736(12)60633-3: 10.1016/S0140–6736(12)60633-3 [DOI] [PubMed] [Google Scholar]
- (96).L’Heureux N; Paquet S; Labbe R; Germain L; Auger FA, A completely biological tissue-engineered human blood vessel. FASEB J 1998, 12 (1), 47–56. 10.1096/fasebj.12.1.47: 10.1096/fasebj.12.1.47 [DOI] [PubMed] [Google Scholar]
- (97).Gui L; Dash BC; Luo J; Qin L; Zhao L; Yamamoto K; Hashimoto T; Wu H; Dardik A; Tellides G; Niklason LE; Qyang Y, Implantable tissue-engineered blood vessels from human induced pluripotent stem cells. Biomaterials 2016, 102, 120–9. 10.1016/j.biomaterials.2016.06.010: 10.1016/j.biomaterials.2016.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Jung Y; Ji H; Chen Z; Fai Chan H; Atchison L; Klitzman B; Truskey G; Leong KW, Scaffold-free, Human Mesenchymal Stem Cell-Based Tissue Engineered Blood Vessels. Sci Rep 2015, 5, 15116 10.1038/srep15116: 10.1038/srep15116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Linton MRF; Yancey PG; Davies SS; Jerome WG; Linton EF; Song WL; Doran AC; Vickers KC, The Role of Lipids and Lipoproteins in Atherosclerosis In Endotext [Internet], Feingold KR; Anawalt B; Boyce A, Eds. MDtext Inc.: Dartmouth, MA, 2019; p Available from: https://www.ncbi.nlm.nih.gov/books/NBK343489/. [Google Scholar]
- (100).Bergheanu SC; Bodde MC; Jukema JW, Pathophysiology and treatment of atherosclerosis: Current view and future perspective on lipoprotein modification treatment. Neth Heart J 2017, 25 (4), 231–242. 10.1007/s12471-017-0959-2: 10.1007/s12471-017-0959-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Deanfield JE; Halcox JP; Rabelink TJ, Endothelial function and dysfunction: testing and clinical relevance. Circulation 2007, 115 (10), 1285–95. 10.1161/CIRCULATIONAHA.106.652859: 10.1161/CIRCULATIONAHA.106.652859 [DOI] [PubMed] [Google Scholar]
- (102).Lee S; Lee SJ; Yoon YS, Vascular Regeneration With New Sources of Endothelial Cells. Circ Res 2019, 124 (1), 29–31. 10.1161/CIRCRESAHA.118.314195: 10.1161/CIRCRESAHA.118.314195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Klein D, iPSCs-based generation of vascular cells: reprogramming approaches and applications. Cell Mol Life Sci 2018, 75 (8), 1411–1433. 10.1007/s00018-017-2730-7: 10.1007/s00018-017-2730-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Jang S; Collin de l’Hortet A; Soto-Gutierrez A, Induced Pluripotent Stem Cell-Derived Endothelial Cells: Overview, Current Advances, Applications, and Future Directions. Am J Pathol 2019, 189 (3), 502–512. 10.1016/j.ajpath.2018.12.004: 10.1016/j.ajpath.2018.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Acun A; Zorlutuna P, CRISPR/Cas9 Edited Induced Pluripotent Stem Cell-Based Vascular Tissues to Model Aging and Disease-Dependent Impairment. Tissue Eng Part A 2019, 25 (9–10), 759–772. 10.1089/ten.TEA.2018.0271: 10.1089/ten.TEA.2018.0271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Gu M; Shao NY; Sa S; Li D; Termglinchan V; Ameen M; Karakikes I; Sosa G; Grubert F; Lee J; Cao A; Taylor S; Ma Y; Zhao Z; Chappell J; Hamid R; Austin ED; Gold JD; Wu JC; Snyder MP; Rabinovitch M, Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20 (4), 490–504 e5. 10.1016/j.stem.2016.08.019: 10.1016/j.stem.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Tseng WL; Chou SJ; Chiang HC; Wang ML; Chien CS; Chen KH; Leu HB; Wang CY; Chang YL; Liu YY; Jong YJ; Lin SZ; Chiou SH; Lin SJ; Yu WC, Imbalanced Production of Reactive Oxygen Species and Mitochondrial Antioxidant SOD2 in Fabry Disease-Specific Human Induced Pluripotent Stem Cell-Differentiated Vascular Endothelial Cells. Cell Transplant 2017, 26 (3), 513–527. 10.3727/096368916X694265: 10.3727/096368916X694265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Gu M; Mordwinkin NM; Kooreman NG; Lee J; Wu H; Hu S; Churko JM; Diecke S; Burridge PW; He C; Barron FE; Ong SG; Gold JD; Wu JC, Pravastatin reverses obesity-induced dysfunction of induced pluripotent stem cell-derived endothelial cells via a nitric oxide-dependent mechanism. Eur Heart J 2015, 36 (13), 806–16. 10.1093/eurheartj/ehu411: 10.1093/eurheartj/ehu411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Ayoubi S; Sheikh SP; Eskildsen TV, Human induced pluripotent stem cell-derived vascular smooth muscle cells: differentiation and therapeutic potential. Cardiovasc Res 2017, 113 (11), 1282–1293. 10.1093/cvr/cvx125: 10.1093/cvr/cvx125 [DOI] [PubMed] [Google Scholar]
- (110).Ge X; Ren Y; Bartulos O; Lee MY; Yue Z; Kim KY; Li W; Amos PJ; Bozkulak EC; Iyer A; Zheng W; Zhao H; Martin KA; Kotton DN; Tellides G; Park IH; Yue L; Qyang Y, Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation 2012, 126 (14), 1695–704. 10.1161/CIRCULATIONAHA.112.116996: 10.1161/CIRCULATIONAHA.112.116996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Granata A; Serrano F; Bernard WG; McNamara M; Low L; Sastry P; Sinha S, An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet 2017, 49 (1), 97–109. 10.1038/ng.3723: 10.1038/ng.3723 [DOI] [PubMed] [Google Scholar]
- (112).Koslow AR; Stromberg RR; Friedman LI; Lutz RJ; Hilbert SL; Schuster P, A Flow System for the Study of Shear Forces Upon Cultured Endothelial Cells. Journal of Biomechanical Engineering 1986, 108 (4), 338–341. 10.1115/1.3138625: 10.1115/1.3138625 [DOI] [PubMed] [Google Scholar]
- (113).Harding IC; Mitra R; Mensah SA; Herman IM; Ebong EE, Pro-atherosclerotic disturbed flow disrupts caveolin-1 expression, localization, and function via glycocalyx degradation. J Transl Med 2018, 16 (1), 364 10.1186/s12967-018-1721-2: 10.1186/s12967-018-1721-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Vion AC; Kheloufi M; Hammoutene A; Poisson J; Lasselin J; Devue C; Pic I; Dupont N; Busse J; Stark K; Lafaurie-Janvore J; Barakat AI; Loyer X; Souyri M; Viollet B; Julia P; Tedgui A; Codogno P; Boulanger CM; Rautou PE, Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc Natl Acad Sci U S A 2017, 114 (41), E8675–E8684. 10.1073/pnas.1702223114: 10.1073/pnas.1702223114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Young EW; Simmons CA, Macro- and microscale fluid flow systems for endothelial cell biology. Lab Chip 2010, 10 (2), 143–60. 10.1039/b913390a: 10.1039/b913390a [DOI] [PubMed] [Google Scholar]
- (116).Zheng W; Huang R; Jiang B; Zhao Y; Zhang W; Jiang X, An Early-Stage Atherosclerosis Research Model Based on Microfluidics. Small 2016, 12 (15), 2022–34. 10.1002/smll.201503241: 10.1002/smll.201503241 [DOI] [PubMed] [Google Scholar]
- (117).Mannino RG; Myers DR; Ahn B; Wang Y; Margo R; Gole H; Lin AS; Guldberg RE; Giddens DP; Timmins LH; Lam WA, “Do-it-yourself in vitro vasculature that recapitulates in vivo geometries for investigating endothelial-blood cell interactions”. Sci Rep 2015, 5, 12401 10.1038/srep12401: 10.1038/srep12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Dorweiler B; Torzewski M; Dahm M; Ochsenhirt V; Lehr HA; Lackner KJ; Vahl CF, A novel in vitro model for the study of plaque development in atherosclerosis. Thromb Haemost 2006, 95 (1), 182–9. [PubMed] [Google Scholar]
- (119).Robert J; Weber B; Frese L; Emmert MY; Schmidt D; von Eckardstein A; Rohrer L; Hoerstrup SP, A three-dimensional engineered artery model for in vitro atherosclerosis research. PLoS One 2013, 8 (11), e79821 10.1371/journal.pone.0079821: 10.1371/journal.pone.0079821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Chen Z; Tang M; Huang D; Jiang W; Li M; Ji H; Park J; Xu B; Atchison LJ; Truskey GA; Leong KW, Real-time observation of leukocyte-endothelium interactions in tissue-engineered blood vessel. Lab Chip 2018, 18 (14), 2047–2054. 10.1039/c8lc00202a: 10.1039/c8lc00202a [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Head T; Daunert S; Goldschmidt-Clermont PJ, The Aging Risk and Atherosclerosis: A Fresh Look at Arterial Homeostasis. Front Genet 2017, 8, 216 10.3389/fgene.2017.00216: 10.3389/fgene.2017.00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Lee HY; Oh BH, Aging and arterial stiffness. Circ J 2010, 74 (11), 2257–62. [DOI] [PubMed] [Google Scholar]
- (123).Blakney AK; Swartzlander MD; Bryant SJ, The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J Biomed Mater Res A 2012, 100 (6), 1375–86. 10.1002/jbm.a.34104: 10.1002/jbm.a.34104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Adlerz KM; Aranda-Espinoza H; Hayenga HN, Substrate elasticity regulates the behavior of human monocyte-derived macrophages. Eur Biophys J 2016, 45 (4), 301–9. 10.1007/s00249-015-1096-8: 10.1007/s00249-015-1096-8 [DOI] [PubMed] [Google Scholar]
- (125).Strobel HA; Hookway TA; Piola M; Fiore GB; Soncini M; Alsberg E; Rolle MW, Assembly of Tissue-Engineered Blood Vessels with Spatially Controlled Heterogeneities. Tissue Eng Part A 2018, 24 (19–20), 1492–1503. 10.1089/ten.TEA.2017.0492: 10.1089/ten.TEA.2017.0492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Mallone A; Stenger C; Von Eckardstein A; Hoerstrup SP; Weber B, Biofabricating atherosclerotic plaques: In vitro engineering of a three-dimensional human fibroatheroma model. Biomaterials 2018, 150, 49–59. 10.1016/j.biomaterials.2017.09.034: 10.1016/j.biomaterials.2017.09.034 [DOI] [PubMed] [Google Scholar]
- (127).Schoen FJ; Levy RJ, Calcification of tissue heart valve substitutes: progress toward understanding and prevention. Ann Thorac Surg 2005, 79 (3), 1072–80. [DOI] [PubMed] [Google Scholar]
- (128).Mohler ER 3rd, Mechanisms of aortic valve calcification. Am J Cardiol 2004, 94 (11), 1396–402, A6. [DOI] [PubMed] [Google Scholar]
- (129).Takkenberg JJ; Rajamannan NM; Rosenhek R; Kumar AS; Carapetis JR; Yacoub MH; Society for Heart Valve, D., The need for a global perspective on heart valve disease epidemiology. The SHVD working group on epidemiology of heart valve disease founding statement. J Heart Valve Dis 2008, 17 (1), 135–9. [PubMed] [Google Scholar]
- (130).Otto CM; Lind BK; Kitzman DW; Gersh BJ; Siscovick DS, Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med 1999, 341 (3), 142–7. 10.1056/NEJM199907153410302: 10.1056/NEJM199907153410302 [DOI] [PubMed] [Google Scholar]
- (131).Mohler ER 3rd; Wang H; Medenilla E; Scott C, Effect of statin treatment on aortic valve and coronary artery calcification. J Heart Valve Dis 2007, 16 (4), 378–86. [PubMed] [Google Scholar]
- (132).Mendelson K; Schoen FJ, Heart valve tissue engineering: concepts, approaches, progress, and challenges. Ann Biomed Eng 2006, 34 (12), 1799–819. 10.1007/s10439-006-9163-z: 10.1007/s10439-006-9163-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Shinoka T; Breuer CK; Tanel RE; Zund G; Miura T; Ma PX; Langer R; Vacanti JP; Mayer JE Jr., Tissue engineering heart valves: valve leaflet replacement study in a lamb model. Ann Thorac Surg 1995, 60 (6 Suppl), S513–6. 10.1016/0003-4975(95)00733-4: 10.1016/0003-4975(95)00733-4 [DOI] [PubMed] [Google Scholar]
- (134).Hoerstrup SP; Sodian R; Daebritz S; Wang J; Bacha EA; Martin DP; Moran AM; Guleserian KJ; Sperling JS; Kaushal S; Vacanti JP; Schoen FJ; Mayer JE Jr., Functional living trileaflet heart valves grown in vitro. Circulation 2000, 102 (19 Suppl 3), III44–9. 10.1161/01.cir.102.suppl_3.iii-44: 10.1161/01.cir.102.suppl_3.iii-44 [DOI] [PubMed] [Google Scholar]
- (135).Dohmen PM; Lembcke A; Hotz H; Kivelitz D; Konertz WF, Ross operation with a tissue-engineered heart valve. Ann Thorac Surg 2002, 74 (5), 1438–42. 10.1016/s0003-4975(02)03881-x: 10.1016/s0003-4975(02)03881-x [DOI] [PubMed] [Google Scholar]
- (136).Zhang BL; Bianco RW; Schoen FJ, Preclinical Assessment of Cardiac Valve Substitutes: Current Status and Considerations for Engineered Tissue Heart Valves. Front Cardiovasc Med 2019, 6, 72 10.3389/fcvm.2019.00072: 10.3389/fcvm.2019.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Schlotter F; Matsumoto Y; Mangner N; Schuler G; Linke A; Adams V, Regular exercise or changing diet does not influence aortic valve disease progression in LDLR deficient mice. PLoS One 2012, 7 (5), e37298 10.1371/journal.pone.0037298: 10.1371/journal.pone.0037298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Wierzbicki AS; Viljoen A; Chambers JB, Aortic stenosis and lipids: does intervention work? Curr Opin Cardiol 2010, 25 (4), 379–84. 10.1097/HCO.0b013e3283393c9b: 10.1097/HCO.0b013e3283393c9b [DOI] [PubMed] [Google Scholar]
- (139).Zhao Y; Nicoll R; He YH; Henein MY, The effect of statins on valve function and calcification in aortic stenosis: A meta-analysis. Atherosclerosis 2016, 246, 318–24. 10.1016/j.atherosclerosis.2016.01.023: 10.1016/j.atherosclerosis.2016.01.023 [DOI] [PubMed] [Google Scholar]
- (140).Liu AC; Joag VR; Gotlieb AI, The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol 2007, 171 (5), 1407–18. 10.2353/ajpath.2007.070251: 10.2353/ajpath.2007.070251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Rutkovskiy A; Malashicheva A; Sullivan G; Bogdanova M; Kostareva A; Stenslokken KO; Fiane A; Vaage J, Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J Am Heart Assoc 2017, 6 (9). 10.1161/JAHA.117.006339: 10.1161/JAHA.117.006339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Poggio P; Sainger R; Branchetti E; Grau JB; Lai EK; Gorman RC; Sacks MS; Parolari A; Bavaria JE; Ferrari G, Noggin attenuates the osteogenic activation of human valve interstitial cells in aortic valve sclerosis. Cardiovasc Res 2013, 98 (3), 402–10. 10.1093/cvr/cvt055: 10.1093/cvr/cvt055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Branchetti E; Sainger R; Poggio P; Grau JB; Patterson-Fortin J; Bavaria JE; Chorny M; Lai E; Gorman RC; Levy RJ; Ferrari G, Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler Thromb Vasc Biol 2013, 33 (2), e66–74. 10.1161/ATVBAHA.112.300177: 10.1161/ATVBAHA.112.300177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Nachlas ALY; Li S; Jha R; Singh M; Xu C; Davis ME, Human iPSC-derived mesenchymal stem cells encapsulated in PEGDA hydrogels mature into valve interstitial-like cells. Acta Biomater 2018, 71, 235–246. 10.1016/j.actbio.2018.02.025: 10.1016/j.actbio.2018.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Zhang J; Tao R; Campbell KF; Carvalho JL; Ruiz EC; Kim GC; Schmuck EG; Raval AN; da Rocha AM; Herron TJ; Jalife J; Thomson JA; Kamp TJ, Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat Commun 2019, 10 (1), 2238 10.1038/s41467-019-09831-5: 10.1038/s41467-019-09831-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Walker GA; Masters KS; Shah DN; Anseth KS; Leinwand LA, Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res 2004, 95 (3), 253–60. 10.1161/01.RES.0000136520.07995.aa: 10.1161/01.RES.0000136520.07995.aa [DOI] [PubMed] [Google Scholar]
- (147).Liu AC; Gotlieb AI, Transforming growth factor-beta regulates in vitro heart valve repair by activated valve interstitial cells. Am J Pathol 2008, 173 (5), 1275–85. 10.2353/ajpath.2008.080365: 10.2353/ajpath.2008.080365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Monzack EL; Masters KS, Can valvular interstitial cells become true osteoblasts? A side-by-side comparison. J Heart Valve Dis 2011, 20 (4), 449–63. [PMC free article] [PubMed] [Google Scholar]
- (149).Goto S; Rogers MA; Blaser MC; Higashi H; Lee LH; Schlotter F; Body SC; Aikawa M; Singh SA; Aikawa E, Standardization of Human Calcific Aortic Valve Disease in vitro Modeling Reveals Passage-Dependent Calcification. Front Cardiovasc Med 2019, 6, 49 10.3389/fcvm.2019.00049: 10.3389/fcvm.2019.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Taylor PM; Allen SP; Yacoub MH, Phenotypic and functional characterization of interstitial cells from human heart valves, pericardium and skin. J Heart Valve Dis 2000, 9 (1), 150–8. [PubMed] [Google Scholar]
- (151).Porras AM; Engeland NC; Marchbanks E; McCormack A; Yacoub MH; Bouten CV; Latif N; Masters KS, Robust Generation of Quiescent Porcine Valvular Interstitial Cell Cultures. J Am Heart Assoc 2017, 6, e005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Latif N; Quillon A; Sarathchandra P; McCormack A; Lozanoski A; Yacoub MH; Chester AH, Modulation of human valve interstitial cell phenotype and function using a fibroblast growth factor 2 formulation. PLoS One 2015, 10 (6), e0127844 10.1371/journal.pone.0127844: 10.1371/journal.pone.0127844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Wang H; Haeger SM; Kloxin AM; Leinwand LA; Anseth KS, Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS One 2012, 7 (7), e39969 10.1371/journal.pone.0039969: 10.1371/journal.pone.0039969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Ma H; Killaars AR; DelRio FW; Yang C; Anseth KS, Myofibroblastic activation of valvular interstitial cells is modulated by spatial variations in matrix elasticity and its organization. Biomaterials 2017, 131, 131–144. 10.1016/j.biomaterials.2017.03.040: 10.1016/j.biomaterials.2017.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Wang H; Leinwand LA; Anseth KS, Cardiac valve cells and their microenvironment--insights from in vitro studies. Nat Rev Cardiol 2014, 11 (12), 715–27. 10.1038/nrcardio.2014.162: 10.1038/nrcardio.2014.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Fondard O; Detaint D; Iung B; Choqueux C; Adle-Biassette H; Jarraya M; Hvass U; Couetil JP; Henin D; Michel JB; Vahanian A; Jacob MP, Extracellular matrix remodelling in human aortic valve disease: the role of matrix metalloproteinases and their tissue inhibitors. Eur Heart J 2005, 26 (13), 1333–41. 10.1093/eurheartj/ehi248: 10.1093/eurheartj/ehi248 [DOI] [PubMed] [Google Scholar]
- (157).Porras AM; Shanmuganayagam D; Meudt JJ; Krueger CG; Hacker TA; Rahko PS; Reed JD; Masters KS, Development of Aortic Valve Disease in Familial Hypercholesterolemic Swine: Implications for Elucidating Disease Etiology. J Am Heart Assoc 2015, 4 (10), e002254 10.1161/JAHA.115.002254: 10.1161/JAHA.115.002254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Hutson HN; Marohl T; Anderson M; Eliceiri K; Campagnola P; Masters KS, Calcific Aortic Valve Disease Is Associated with Layer-Specific Alterations in Collagen Architecture. PLoS One 2016, 11 (9), e0163858 10.1371/journal.pone.0163858: 10.1371/journal.pone.0163858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Rodriguez KJ; Masters KS, Regulation of valvular interstitial cell calcification by components of the extracellular matrix. J Biomed Mater Res A 2009, 90 (4), 1043–53. 10.1002/jbm.a.32187: 10.1002/jbm.a.32187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Benton JA; Kern HB; Anseth KS, Substrate properties influence calcification in valvular interstitial cell culture. J Heart Valve Dis 2008, 17 (6), 689–99. [PMC free article] [PubMed] [Google Scholar]
- (161).Yip CY; Chen JH; Zhao R; Simmons CA, Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol 2009, 29 (6), 936–42. 10.1161/ATVBAHA.108.182394: 10.1161/ATVBAHA.108.182394 [DOI] [PubMed] [Google Scholar]
- (162).Kirschner CM; Alge DL; Gould ST; Anseth KS, Clickable, photodegradable hydrogels to dynamically modulate valvular interstitial cell phenotype. Adv Healthc Mater 2014, 3 (5), 649–57. 10.1002/adhm.201300288: 10.1002/adhm.201300288 [DOI] [PubMed] [Google Scholar]
- (163).Porras AM; Westlund JA; Evans AD; Masters KS, Creation of disease-inspired biomaterial environments to mimic pathological events in early calcific aortic valve disease. Proc Natl Acad Sci U S A 2018, 115 (3), E363–E371. 10.1073/pnas.1704637115: 10.1073/pnas.1704637115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Rodriguez KJ; Piechura LM; Masters KS, Regulation of valvular interstitial cell phenotype and function by hyaluronic acid in 2-D and 3-D culture environments. Matrix Biol 2011, 30 (1), 70–82. 10.1016/j.matbio.2010.09.001: 10.1016/j.matbio.2010.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Rodriguez KJ; Piechura LM; Porras AM; Masters KS, Manipulation of valve composition to elucidate the role of collagen in aortic valve calcification. BMC Cardiovasc Disord 2014, 14, 29 10.1186/1471-2261-14-29: 10.1186/1471-2261-14-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Hjortnaes J; Camci-Unal G; Hutcheson JD; Jung SM; Schoen FJ; Kluin J; Aikawa E; Khademhosseini A, Directing valvular interstitial cell myofibroblast-like differentiation in a hybrid hydrogel platform. Adv Healthc Mater 2015, 4 (1), 121–30. 10.1002/adhm.201400029: 10.1002/adhm.201400029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Hjortnaes J; Goettsch C; Hutcheson JD; Camci-Unal G; Lax L; Scherer K; Body S; Schoen FJ; Kluin J; Khademhosseini A; Aikawa E, Simulation of early calcific aortic valve disease in a 3D platform: A role for myofibroblast differentiation. J Mol Cell Cardiol 2016, 94, 13–20. 10.1016/j.yjmcc.2016.03.004: 10.1016/j.yjmcc.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Gould ST; Anseth KS, Role of cell-matrix interactions on VIC phenotype and tissue deposition in 3D PEG hydrogels. J Tissue Eng Regen Med 2016, 10 (10), E443–E453. 10.1002/term.1836: 10.1002/term.1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Gonzalez Rodriguez A; Schroeder ME; Walker CJ; Anseth KS, FGF-2 inhibits contractile properties of valvular interstitial cell myofibroblasts encapsulated in 3D MMP-degradable hydrogels. APL Bioeng 2018, 2 (4), 046104 10.1063/1.5042430: 10.1063/1.5042430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Wang H; Tibbitt MW; Langer SJ; Leinwand LA; Anseth KS, Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway. Proc Natl Acad Sci U S A 2013, 110 (48), 19336–41. 10.1073/pnas.1306369110: 10.1073/pnas.1306369110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Duan B; Hockaday LA; Kapetanovic E; Kang KH; Butcher JT, Stiffness and adhesivity control aortic valve interstitial cell behavior within hyaluronic acid based hydrogels. Acta Biomater 2013, 9 (8), 7640–50. 10.1016/j.actbio.2013.04.050: 10.1016/j.actbio.2013.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Mabry KM; Lawrence RL; Anseth KS, Dynamic stiffening of poly(ethylene glycol)-based hydrogels to direct valvular interstitial cell phenotype in a three-dimensional environment. Biomaterials 2015, 49, 47–56. 10.1016/j.biomaterials.2015.01.047: 10.1016/j.biomaterials.2015.01.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Otto CM; Kuusisto J; Reichenbach DD; Gown AM; O’Brien KD, Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994, 90 (2), 844–53. 10.1161/01.cir.90.2.844: 10.1161/01.cir.90.2.844 [DOI] [PubMed] [Google Scholar]
- (174).Garcia-Rodriguez C; Parra-Izquierdo I; Castanos-Mollor I; Lopez J; San Roman JA; Sanchez Crespo M, Toll-Like Receptors, Inflammation, and Calcific Aortic Valve Disease. Front Physiol 2018, 9, 201 10.3389/fphys.2018.00201: 10.3389/fphys.2018.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Mahler GJ; Butcher JT, Inflammatory regulation of valvular remodeling: the good(?), the bad, and the ugly. Int J Inflam 2011, 2011, 721419 10.4061/2011/721419: 10.4061/2011/721419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Springer NL; Fischbach C, Biomaterials approaches to modeling macrophage-extracellular matrix interactions in the tumor microenvironment. Curr Opin Biotechnol 2016, 40, 16–23. 10.1016/j.copbio.2016.02.003: 10.1016/j.copbio.2016.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Madsen DH; Leonard D; Masedunskas A; Moyer A; Jurgensen HJ; Peters DE; Amornphimoltham P; Selvaraj A; Yamada SS; Brenner DA; Burgdorf S; Engelholm LH; Behrendt N; Holmbeck K; Weigert R; Bugge TH, M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol 2013, 202 (6), 951–66. 10.1083/jcb.201301081: 10.1083/jcb.201301081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Sunderkotter C; Steinbrink K; Goebeler M; Bhardwaj R; Sorg C, Macrophages and angiogenesis. J Leukoc Biol 1994, 55 (3), 410–22. 10.1002/jlb.55.3.410: 10.1002/jlb.55.3.410 [DOI] [PubMed] [Google Scholar]
- (179).Villasante A; Vunjak-Novakovic G, Tissue-engineered models of human tumors for cancer research. Expert Opin Drug Discov 2015, 10 (3), 257–68. 10.1517/17460441.2015.1009442: 10.1517/17460441.2015.1009442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Benam KH; Dauth S; Hassell B; Herland A; Jain A; Jang KJ; Karalis K; Kim HJ; MacQueen L; Mahmoodian R; Musah S; Torisawa YS; van der Meer AD; Villenave R; Yadid M; Parker KK; Ingber DE, Engineered in vitro disease models. Annu Rev Pathol 2015, 10, 195–262. 10.1146/annurev-pathol-012414-040418: 10.1146/annurev-pathol-012414-040418 [DOI] [PubMed] [Google Scholar]
- (181).Pollitzer E, Biology: Cell sex matters. Nature 2013, 500 (7460), 23–4. 10.1038/500023a: 10.1038/500023a [DOI] [PubMed] [Google Scholar]
- (182).Ventura-Clapier R; Dworatzek E; Seeland U; Kararigas G; Arnal JF; Brunelleschi S; Carpenter TC; Erdmann J; Franconi F; Giannetta E; Glezerman M; Hofmann SM; Junien C; Katai M; Kublickiene K; Konig IR; Majdic G; Malorni W; Mieth C; Miller VM; Reynolds RM; Shimokawa H; Tannenbaum C; D’Ursi AM; Regitz-Zagrosek V, Sex in basic research: concepts in the cardiovascular field. Cardiovasc Res 2017, 113 (7), 711–724. 10.1093/cvr/cvx066: 10.1093/cvr/cvx066 [DOI] [PubMed] [Google Scholar]
- (183).Simard L; Cote N; Dagenais F; Mathieu P; Couture C; Trahan S; Bosse Y; Mohammadi S; Page S; Joubert P; Clavel MA, Sex-Related Discordance Between Aortic Valve Calcification and Hemodynamic Severity of Aortic Stenosis: Is Valvular Fibrosis the Explanation? Circ Res 2017, 120 (4), 681–691. 10.1161/CIRCRESAHA.116.309306: 10.1161/CIRCRESAHA.116.309306 [DOI] [PubMed] [Google Scholar]
- (184).Vaccarino V; Badimon L; Corti R; de Wit C; Dorobantu M; Manfrini O; Koller A; Pries A; Cenko E; Bugiardini R, Presentation, management, and outcomes of ischaemic heart disease in women. Nat Rev Cardiol 2013, 10 (9), 508–18. 10.1038/nrcardio.2013.93: 10.1038/nrcardio.2013.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Arnold AP; Cassis LA; Eghbali M; Reue K; Sandberg K, Sex Hormones and Sex Chromosomes Cause Sex Differences in the Development of Cardiovascular Diseases. Arterioscler Thromb Vasc Biol 2017, 37 (5), 746–756. 10.1161/ATVBAHA.116.307301: 10.1161/ATVBAHA.116.307301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).McCoy CM; Nicholas DQ; Masters KS, Sex-related differences in gene expression by porcine aortic valvular interstitial cells. PLoS One 2012, 7 (7), e39980 10.1371/journal.pone.0039980: 10.1371/journal.pone.0039980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Cattaneo MG; Vanetti C; Decimo I; Di Chio M; Martano G; Garrone G; Bifari F; Vicentini LM, Sex-specific eNOS activity and function in human endothelial cells. Sci Rep 2017, 7 (1), 9612 10.1038/s41598-017-10139-x: 10.1038/s41598-017-10139-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Malorni W; Straface E; Matarrese P; Ascione B; Coinu R; Canu S; Galluzzo P; Marino M; Franconi F, Redox state and gender differences in vascular smooth muscle cells. FEBS Lett 2008, 582 (5), 635–42. 10.1016/j.febslet.2008.01.034: 10.1016/j.febslet.2008.01.034 [DOI] [PubMed] [Google Scholar]
- (189).Parks RJ; Howlett SE, Sex differences in mechanisms of cardiac excitation-contraction coupling. Pflugers Arch 2013, 465 (5), 747–63. 10.1007/s00424-013-1233-0: 10.1007/s00424-013-1233-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Yutzey KE; Demer LL; Body SC; Huggins GS; Towler DA; Giachelli CM; Hofmann-Bowman MA; Mortlock DP; Rogers MB; Sadeghi MM; Aikawa E, Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol 2014, 34 (11), 2387–93. 10.1161/ATVBAHA.114.302523: 10.1161/ATVBAHA.114.302523 [DOI] [PMC free article] [PubMed] [Google Scholar]