

Abstract
Prions are unorthodox pathogens that cause fatal neurodegenerative diseases in humans and other mammals. Prion propagation occurs through the self-templating of the pathogenic conformer PrPSc, onto the cell-expressed conformer, PrPC. Here we study the conversion of PrPC to PrPSc using a recombinant mouse PrPSc conformer (mouse protein-only recPrPSc) as a unique tool that can convert bank vole but not mouse PrPC substrates in vitro. Thus, its templating ability is not dependent on sequence homology with the substrate. In the present study, we used chimeric bank vole/mouse PrPC substrates to systematically determine the domain that allows for conversion by Mo protein-only recPrPSc. Our results show that that either the presence of the bank vole amino acid residues E227 and S230 or the absence of the second N-linked glycan are sufficient to allow PrPC substrates to be converted by Mo protein-only recPrPSc and several native infectious prion strains. We propose that residues 227 and 230 and the second glycan are part of a C-terminal domain that acts as a linchpin for bank vole and mouse prion conversion.
Author summary
Prions are unconventional infectious agents that lack nucleic acids such as DNA and RNA, and the mechanism by which prions replicate is not fully understood. It has been established that a central feature of the replication mechanism involves the misfolding of a host protein (PrPC) into an infectious shape termed PrPSc, but it is unclear how this misfolding occurs. Interestingly, it has been observed that a particular animal species, the European bank vole, is unusually susceptible to prion infection and that this near-universal susceptibility is caused by the specific PrPC sequence of this protein. Here we use a powerful and unique biochemical system to determine the specific region of bank vole PrPC that is primarily responsible for its propensity to misfold into PrPSc. This critical region, which is located at the extreme C-terminal end of the protein, appears to act as a linchpin domain that normally stabilizes the shape of PrPC and thereby regulates its misfolding into PrPSc.
Introduction
Prions are unorthodox infectious agents of fatal neurological diseases that affect many mammalian species, including humans. The central pathogenic event underlying prion infection is the self-propagating conformational change of a host-encoded glycoprotein (PrPC) into a misfolded conformer (PrPSc)[1].
The protein-only hypothesis states that infectious prions are composed solely of PrPSc[2, 3]. However, much evidence indicates that cofactor molecules are required for the formation of infectious prions[4–7]. In direct support of this concept, our laboratory generated two self-propagating bacterially-expressed recombinant (rec) PrPSc conformers (cofactor recPrPSc and protein-only recPrPSc) by propagating the same original seed in the presence and absence of purified phospholipids, respectively[7, 8]. Whereas cofactor recPrPSc effectively seeds mouse (Mo) brain homogenate (BH) sPMCA reactions in vitro and potently infects wild-type mice in vivo, protein-only recPrPSc cannot seed Mo BH sPMCA reactions and fails to infect wild-type mice[7].
Most animal species have transmission barriers that render them resistant to the majority of prion strains from other species. Some species, such as rabbits, dogs, and horses are resistant to naturally occurring prion strains[9]. In contrast, the European bank vole (Myodes glareolus) is a highly susceptible host for a wide variety of prion diseases[10–14]. Experiments with transgenic mice showed that the susceptibility of bank voles to prion infection is ultimately encoded by the amino acid sequence of bank vole (BV) PrPC[13, 15], and BV PrP has been shown to be a highly susceptible substrate for in vitro conversion assays [16]. Several residues and domains in the BV PrPC sequence have previously been identified through in vitro and in vivo approaches to play important roles in the susceptibility by specific prions from other species[17–22]. While the residues and domains identified in these experiments appear to play roles for the specific species barriers studied, it is difficult to distinguish whether they do so because of sequence mismatch between seed and substrate or because they control the structural transition of PrPC to PrPSc.
Recently, we discovered that protein-only recPrPSc is able to potently seed BV but not Mo PrPC substrate in sPMCA reactions, and the PrPSc molecules produced in these reactions are highly infectious[23]. Interestingly, we observed that the formation of infectious PrPSc molecules from non-infectious protein-only recPrPSc seed required several factors: (1) bank vole rather than mouse PrPC substrate, (2) native PrPC rather than recPrP substrate lacking post-translational modifications (PTMs), and (3) cofactor molecules.
In the present study, we exploit this system to identify PrPC domains that might serve as “linchpins” to control PrP conformational change. Uniquely, this system has the critical advantage of not being influenced by species barriers[24] since mouse protein-only recPrPSc can seed BV PrPC but not Mo PrPC in this system. Therefore, we can test the ability of native Mo-BV PrPC chimeric substrates to convert in sPMCA reactions driven by Mo protein-only recPrPSc seed, with confidence that this process depends only upon the enhanced ability of BV PrPC (and chimeric substrates) to convert into PrPSc, rather than the degree of amino acid sequence similarity between seed and substrate. Similarly, this system also provides a unique opportunity to study the effect of PTMs on PrPSc formation since protein-only recPrPSc, which lacks PTMs, requires native PrPC substrate to produce PrPSc.
Results
BV PrP is uniquely able to propagate Mo protein-only recPrPSc
The amino acid sequence of the processed region of BV PrP is a natural chimera of Mo PrP and Ha PrP, with the exception of two unique residues at the extreme C-terminus (Fig 1A). As a preliminary experiment, we first compared the conversion ability of BV BH, Ha BH, and Mo BH to propagate Mo protein-only recPrPSc. The results show that BV BH, but not Ha BH or Mo BH, is capable of propagating Mo protein-only recPrPSc (Fig 1B, third column). BV BH is also capable of propagating both the hamster prion strain 139H and the mouse prion strain RML. In contrast, Mo BH is capable of propagating only RML, and Ha BH is capable of propagating only 139H (Fig 1B). Taken together, these results show that BV PrP is a uniquely susceptible substrate to Mo protein-only recPrPSc and native prion strains from other species.
Fig 1. Susceptibility of various rodent species in BH sPMCA.

(A) Amino acid comparison of the processed regions of Mo PrP, Ha PrP, and BV PrP. The sequence bar highlights the regions of BV PrP homologous to Mo PrP (orange) or Ha PrP (green), or are unique to BV PrP (blue). Black arrowheads denote the location of N-linked glycans. The locations of various structural domains are shown in black. OR = octapeptide repeat; PB = polybasic domain; GPI = glycophosphaditylinositol. Sequence alignments were performed using Multalin[25]. (B) Western blots showing three-round sPMCA reactions using normal brain homogenates (BH) from the species used in (A) as the substrates and initially seeded on day 0 with various seeds, as indicated. Day 0 samples are seeded reactions not subject to sonication. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed.
We next developed a system that allowed us to design and produce a wide variety of PrPC substrates in HEK 293 cells. HEK 293 are human cells that can express secretory and membrane-bound proteins with post-translational modifications, including N-linked glycans and a GPI anchor[26, 27], at high level. All HEK-expressed constructs were partially deglycosylated using PNGase F as previously described (S1 Fig) to improve the conversion efficiency of Mo PrPC[28] (S2 Fig). This effect is likely due to the previously observed inhibitory effects of PrPC glycosylation on the propagation of mouse prion strains[28, 29], which may be due to negatively charged sialic acid groups within the glycan structure[30], and which may be further aggravated by hyper-glycosylation in HEK 293 cells. To ensure our system faithfully reproduces similar seed specificity as brain homogenate substrates in sPMCA, we first tested the susceptibility of cell-expressed BV PrP and Mo PrP in sPMCA (Fig 2). The susceptibility of each construct was tested in three-round reconstituted BH sPMCA reactions containing PrP0/0 brain homogenate and a partially purified PrPc construct. The results show that, like the crude BH substrates, cell-expressed BV PrPC is capable of propagating Mo protein-only recPrPSc, 139H, and RML (Fig 2, top row). Cell-expressed BV PrPC can also propagate Mo cofactor recPrPSc. However, significant PrPSc formation was not observed in either the 139H-seeded or Mo cofactor recPrPSc seeded lanes until round 3 of sPMCA (Fig 2, top row, second and third panels). In contrast, cell-expressed Mo PrPC was capable of propagating RML and Mo cofactor recPrPSc, but not Mo protein-only recPrPSc or 139H (Fig 2, bottom row). Taken together, the results show that the cell-expressed PrPC substrates have similar susceptibilities to crude BH substrates from the same species.
Fig 2. Susceptibility of BV PrPC and Mo PrPC substrates in reconstituted sPMCA.

Western blots showing three-round reconstituted sPMCA reactions using either BV or Mo partially purified PrPC substrates supplemented with PrP0/0 BH and seeded with various seeds, as indicated. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed.
BV PrP C-terminal domain is required for efficient conversion of sPMCA reactions seeded with Mo protein-only recPrPSc
To identify the specific amino acid residue(s) required to enable BV PrPC to propagate Mo protein-only recPrPSc, we designed a series of chimeric constructs based on the BV PrPC backbone sequence that, working from the C- towards the N- terminus, become progressively substituted with Mo PrP residues (Fig 3A), and tested the ability of these chimeric constructs to be seeded by Mo protein-only recPrPSc in sPMCA reactions. Within this systematic paradigm, BV PrP substitution(s) that are specifically required to propagate Mo protein-only recPrPSc are identified when the resulting chimera is no longer capable of propagating Mo protein-only recPrPSc. The results show that substitution of residues 227 and 230 from BV sequence to Mo sequence (i.e. E227D and S230R) inhibited conversion of Mo protein-only recPrPSc seeded reactions by >10-fold (Fig 3A, third row), chimera BV(DR)). We observed some variability in sPMCA patterns obtained with BV(DR) substrate from experiment-to-experiment; we detected no bands at all in round three in 3/5 Mo protein-only recPrPSc-seeded independent sPMCA experiments. In other cases, such as the example shown (Fig 3B, second row right panel), we observed a near-complete absence of the ~24 kDa PK-resistant by round three of the reaction, while the lower molecular weight bands at ~16 and 19 kDa are present in round three, but have reduced signal intensity. In comparison, the positive control reaction containing BV(DR) PrPC and seeded with RML PrPSc showed consistent three-round propagation (Fig 3B, second row, left panel).
Fig 3. Determining the minimum PrP sequence required for propagation of Mo protein-only recPrPSc seed.

(A) Table summarizing the ability of various BV/Mo PrPC chimeras to propagate Mo protein-only recPrPSc. All constructs in this series are based on the BV PrPC backbone and become progressively substituted with Mo residues from the C-terminus towards the N-terminus, except for the last two constructs which contain different combinations of 3 Mo substitutions. (B) Western blots of experiments summarized in (A), showing three-round reconstituted sPMCA reactions using partially purified PrPC substrates supplemented with PrP0/0 BH and seeded with various seeds, as indicated. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed.
BV(DR) chimeras with additional step-wise N-terminal Mo substitutions (i.e. BV(S170DR), BV(YS170DR) and BV(LYS170DR) displayed no significant conversion in reactions seeded with Mo protein-only recPrPSc (Fig 3A, rows 4–6; and Fig 3B, rows 3–5, right panel). In contrast, BV(LDR) and BV(YDR) substrates were able to propagate Mo protein-only recPrPSc at ~1/3 the conversion efficiency of BV PrPC (Fig 3A, rows 7–8; and Fig 3B, rows 6–7, right panel). Taken together, our results indicate that residues E227 and S230 are required for efficient conversion of sPMCA reactions seeded by Mo protein-only recPrPSc, and additional substitutions of other non-homologous residues also influence the degree of conversion efficiency.
Finally, we also designed and tested the ability of a series of chimeric constructs, which substituted single Mo PrP amino acid residues to their cognate BV PrP residue, to propagate Mo protein-only recPrPSc (Fig 4A and Fig 4B). This analysis showed that neither E227D nor S230R substitution alone reduced the conversion efficiency of BV PrPC (Fig 4A, rows 3–4; and Fig 4B, rows 1–2, right panel). On the other hand, BV(S170) and BV(L) displayed lower conversion efficiencies than BV PrPC (Fig 4A, rows 5 and 7; and Fig 4B, rows 3 and 5, right panel) These data indicate that the E227D and S230R substitutions work synergistically to block propagation of Mo protein-only recPrPSc, whereas substitution of other individual residues can reduce the conversion efficiency of chimeric PrPC molecules.
Fig 4. Determining the effect of single Mo amino acid mutations on the propagation of Mo protein-only recPrPSc.

(A) Table summarizing the effect of single Mo amino acid substitutions on the ability of BV/Mo PrPC chimeras to propagate Mo protein-only recPrPSc. All constructs are based on the BV PrPC backbone. (B) Western blots of experiments summarized in (A), showing three-round reconstituted sPMCA reactions using partially purified PrPC substrates supplemented with PrP0/0 BH and seeded with various seeds, as indicated. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed.
The extreme C-terminal domain of BV PrPC is sufficient to propagate Mo protein-only recPrPSc
We next sought to identify bank vole amino acid residue(s) that are sufficient to propagate Mo protein-only recPrPSc. Therefore, we designed a series of chimeric constructs based on the Mo PrPC backbone sequence that, working from the C- to the N-terminus, become progressively substituted with BV PrP residues (Fig 5A). and tested the ability of these chimeric constructs to be seeded by Mo protein-only recPrPSc in sPMCA reactions. The results show that substitution of the two non-homologous residues in extreme C-terminus (227 and 230) from DR to ES is sufficient to propagate Mo protein-only recPrPSc (Fig 5B, first row; Mo(ES), right panel).
Fig 5. Determining the BV amino acids that are sufficient to propagate protein-only recPrPSc.
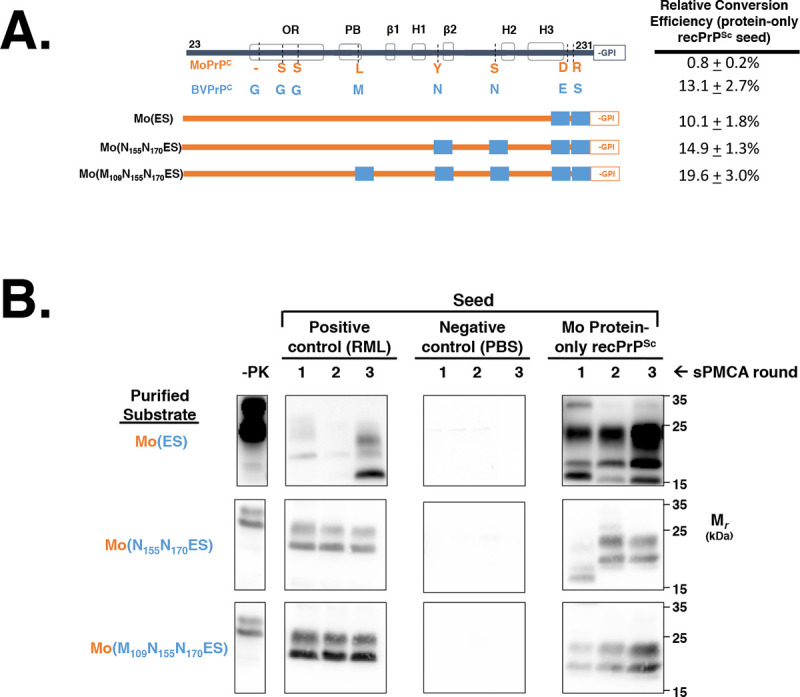
(A) Table summarizing the ability of BV/Mo PrPC chimeras to propagate Mo protein-only recPrPSc. All constructs are based on the Mo PrPC backbone and become progressively substituted with BV residues from the C-terminus towards the N-terminus. (B) Western blots of experiments summarized in (A), showing three-round reconstituted sPMCA reactions using partially purified PrPC substrates supplemented with PrP0/0 BH and initially seeded with various seeds, as indicated. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed.
We also designed and tested the ability of a series of chimeric constructs that substitute single BV PrP amino acid residues to their cognate Mo PrP residue to propagate Mo protein-only recPrPSc (Fig 6A). The results show that this series of chimeric PrPC substrates are unable to propagate Mo protein-only recPrPSc for three rounds in sPMCA (Fig 6B, right panels). In each case, we observed a decrease in PK-resistant PrPSc over each round of sPMCA, resulting in the near absence of PK-resistant PrPSc by round three in each reaction tested (Fig 6B, right panels). In contrast, we found that the positive control RML PrPSc efficiently propagates in reactions this series of chimeric PrPC substrates for three rounds of sPMCA (Fig 6B, left panels). Thus, no single amino acid mutation from Mo to BV PrPC sequence is sufficient to restore the ability to propagate Mo protein-only recPrPSc. Overall, our results show that both the E227D and S230R substitutions are required to enable Mo PrPC chimeras to propagate Mo protein-only recPrPSc.
Fig 6. Determining the effect of single BV amino acid mutations on the propagation of Mo protein-only recPrPSc.

(A) Table summarizing the effect of single BV amino acid substitutions on the ability of BV/Mo PrPC chimeras to propagate Mo protein-only recPrPSc. All constructs are based on the Mo PrPC backbone. (B) Western blots of experiments summarized in (A), showing three-round reconstituted sPMCA reactions using partially purified PrPC substrates supplemented with PrP0/0 BH and initially seeded with various seeds, as indicated. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed.
The extreme C-terminus also controls propagation of native infectious prion strains
To evaluate whether 227 and 230 also controls the ability of PrPC substrate to propagate native infectious prion strains, we tested the ability of hamster Sc237, sheep scrapie, and deer CWD to seed the critical chimeric substrates affecting residues 227 and 230, BV(DR) and Mo(ES). Like protein-only recPrPSc, all three of these native strains can seed BV but not Mo PrPC substrate (Fig 7, top and third rows) (. Unlike BV PrPC, the BV(DR) chimera was unable to propagate Sc237, sheep scrapie, or CWD (Fig 7, second row). Conversely, unlike Mo PrPC, the Mo(ES) chimera was competent substrate for propagation of these three native strains (Fig 7, bottom row). As expected, all of the wild-type and chimeric substrates could successfully propagate mouse RML, a strain that infects both mice and bank voles efficiently (Fig 7, first seed column). Taken together, these results indicate that residues 227 and 230 play a critical role in determining the enhanced susceptibility of BV PrPC to seeding by a variety of native prion strains.
Fig 7. Determining the effect of specific BV and Mo amino acids on the species barrier.

Western blots of experiments showing three-round reconstituted sPMCA reactions using partially purified PrPC substrates supplemented with PrP0/0 BH and initially seeded on day 0 with various seeds, as indicated. Day 0 samples are a seeded reaction not subject to sonication. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed.
In some reactions, we observed changes in the migration pattern of PK-resistant bands between sPMCA rounds (Fig 7, RML- and sheep scrapie-seeded BVPrPC and CWD-seeded Mo(ES)) These shifts may represent strain adaptation, which can occur rapidly and stochastically during sPMCA propagation [31], and which is more likely to occur during cross-species propagation[32].
Specific PrPC glycosylation mutant is also able to propagate Mo protein-only recPrPSc
Finally, we also sought to investigate the influence of PTMs (rather than amino acid sequence) on PrPC susceptibility to Mo protein-only recPrPSc. To do this, we used brain homogenates from transgenic mice expressing various mutant Mo PrPC molecules with specific PTM defects as substrates in sPMCA reactions. We first analyzed a series of previously described glycosylation mutants with known differences in susceptibility to infection by various prion strains in vivo[33]. Within this series, G1 mice express PrPC with a point mutation at the first N-linked glycosylation site, G2 mice express PrPC with a point mutation at the second N-linked glycosylation site, and G3 mice express PrPC containing both mutations[34, 35]. We carried out sPMCA experiments using G1, G2, or G3 brain homogenate substrate seeded with Mo protein-only recPrPSc or RML. Remarkably, the results show that G2 PrPC was able to convert into PrPSc when seeded by Mo protein-only recPrPSc (Fig 8, middle panel). The conversion process appears to be slower for G2 PrPC than for susceptible chimeric PrPC substrates, since G2 PrPSc became detectable only after 4 rounds of sPMCA propagation. In contrast, neither G1 nor G3 PrPC appeared to be susceptible to seeding by protein-only recPrPSc (Fig 8, top and bottom panels).
Fig 8. Propagation of Mo protein-only recPrPSc in sPMCA reactions with glycosylation-deficient PrPC substrates.

Western blots of sPMCA reactions using brain homogenates from glycosylation mutants G1, G2, or G3 as substrates, as indicated. Reactions were seeded with Mo protein-only recPrPSc or RML, as indicated. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed. Mouse amino acid numbering scheme is used to show locations of N-linked glycans on Mo PrP.
Discussion
Here we leveraged a unique in vitro conversion system to identify a specific domain within the prion protein (containing residues 227 and 230) that appears to serve as a linchpin that controls the conformational change of PrPC to PrPSc. The amino acid sequences of mature bank vole PrPC and mouse PrPC differ at only 8 residues[12, 13], and our system allowed us to systematically examine which of these non-homologous amino-acid residues are responsible for the remarkable susceptibility of BV PrPC to convert into PrPSc. For the purposes of this study, our system provided two critical technical advantages. (1) The ability of Mo protein-only recPrPSc to convert BV PrPC does not depend on amino acid complementarity between seed and substrate, allowing us to interpret our results without the potentially confounding factor of sequence mismatch. (2) Our cell-based expression system allowed us to produce multiple BV-Mo chimeric PrPC constructs rapidly and inexpensively, allowing us to perform a thorough analysis of potential permutations in an unbiased manner.
Identification of a C-terminal “linchpin” domain that controls prion protein conformational change
The major finding of this study is that the propensity of BV PrPC to undergo conformational change to PrPSc appears to be primarily controlled by two residues (227 and 230) located within the extreme C-terminus of mature PrP. Mutation of these two residues within the Mo PrPC backbone to their corresponding BV sequence identities was sufficient to allow seeded conversion of the resulting chimera by Mo protein-only recPrPSc. Conversely, mutation of these same two residues within the BV PrPC backbone to their corresponding Mo sequence identities largely inhibited conversion seeded by protein-only recPrPSc. Based on these findings, we propose that the extreme C-terminal domain of PrPC may serve as a key “linchpin” for its conformational change. We specifically infer that the E227 and S230 residues destabilize this domain within BV PrPC to facilitate its conformational change into a variety of PrPSc conformers and prion strains. In support of this concept, we found that these two amino acids also control susceptibility to hamster Sc237, sheep scrapie, and deer CWD prion strains. Notably, Kobayashi et al. reported that ~40% of transgenic mice overexpressing Mo PrPC containing the E227 and S230 mutations develop a spontaneous prion disease [36]. Taken together, the results of our systematic analysis using seeded conversion assays and the spontaneous disease reported by Kobayashi et al. provide complementary and compelling evidence for the hypothesis that the extreme C-terminus is a linchpin domain, which controls the conversion (either seeded or unseeded) of PrPC into PrPSc.
It is worth noting that, among the 8 BV residues that do not have homologous match in the Mo PrP sequence, E227 and S230 are unique to vole species (Fig 1). In contrast, the other 6 mismatched BV residues can be found in other mammalian species; for instance, all 6 are homologous to the corresponding residues within the hamster (Ha) PrP sequence (Fig 1). Together, the observations that (1) residues E227 and S230 are unique to vole species and (2) voles are uniquely susceptible to prion diseases[10–14] are consistent with the hypothesis that the extreme C-terminus plays a key role in prion formation, possibly by stabilizing PrPC. This hypothesis is also consistent with a number of previous results. (1) This region displays decreased solvent accessibility upon transition from PrPC to PrPSc [37], indicating it undergoes conformational change. (2) Residues 225 and 226 of deer PrPC (222 and 223 of BV PrPC) appear to play critical roles in interspecies prion conversion [38]. (3) Specific polymorphisms and mutations in the C-terminus can produce dominant negative PrPC molecules [39]. (4) Y145stop, a PrP mutant lacking the C-terminus, appears to convert spontaneously into amyloid fibers[40] (the absence of the C-terminus in this truncated mutant may destabilize PrP structure and promote misfolding). Finally, it is interesting to note that the GPI anchor is attached to the C-terminus of PrP. It is possible that attachment to this bulky modification may normally stabilize the C-terminal domain to prevent the conformational change of native PrPC into PrPSc. Consistent with this notion, transgenic mice that overexpress Mo PrPC lacking a GPI anchor spontaneously develop prion disease[41].
It is important to acknowledge the experimental limitations of our work. (1) We have not confirmed that the various chimeric PrPSc molecules produced in our sPMCA reactions are infectious. However, we have previously shown that similar reactions containing BV PrPC substrate and seeded with protein-only recPrPSc produces fully infectious prions[23], making it likely that we are also studying a process relevant to infectious prion formation in this study. (2) Since we are using Mo protein-only recPrPSc as a tool to study prion conversion, we can only say that our results are sequence-independent, and may not hold for every prion strain (we have only been able to test the few native strains that show differential ability to seed BV vs. Mo PrPC substrate in reconstituted sPMCA reactions). Additional in vitro and in vivo studies are needed to fully characterize the role played by the C-terminal domain in the propagation of naturally occurring prion strains (3) Finally, our PrPC substrates are partially deglycosylated, and the glycosylation status of PrPC has been shown to modulate its susceptibility to prion infection in vivo[35, 42], raising the possibility that the deglycosylation step might artifactually change the results of our chimeric analysis. However, this appears to be unlikely because we found that our partially deglycosylated WT BV PrPC and Mo PrPC substrates display similar patterns of seeding specificity as their fully glycosylated counterparts in sPMCA reactions.
The influence of other non-homologous residues on seeded conversion by protein-only recPrPSc
In addition to identifying residues 227 and 230 as a linchpin domain, our work also showed that other non-homologous residues could influence the ability of chimeric molecules to propagate Mo protein-only recPrPSc. Specifically, we observed that additional substitution of either M109L or N155Y partially restored the conversion ability of the BV(DR) chimera (Fig 3A, rows 7–8), whereas additional substitution with N170S inhibited conversion of BV(DR) (Fig 3A row 4) as well as BV PrPC (Fig 4A, row 5). The former effects are expected outcomes of increasing homology between substrate and seed in non-linchpin domains, but the latter effect at residue 170 appears paradoxical, since increasing homology to the MoPrP sequence actually reduces the ability of chimeric substrates to propagate Mo protein-only recPrPSc.
It has been previously reported that homology between PrPC and PrPSc at position 170 correlates with interspecies prion conversion in vitro[43] and is important in allowing transmission across the species barrier in vivo[44]. Additionally, residues 155 and 170 have been shown to control the conversion efficiency of BVPrPC in cell-free conversion (CFC) assays in vitro[19] and control susceptibility to prion infection in rodents[17]. Surprisingly, our results paradoxically show that increasing homology at position 170 between PrPC and PrPSc blocks conversion with Mo protein-only recPrPSc. PrP forms a β2- α2 loop in the region 165–171[20], and mutations that modify this structure can alter host susceptibility[45]. Mice, which have serine at position 170, have a disordered loop [46, 47], while bank voles which have asparagine at position 170 have a rigid loop [20]. Our data supports a model whereby structural or biophysical elements dictated by the amino acid sequence of PrPC that correlate with loop rigidity, rather than amino acid sequence homology between PrPC and PrPSc, promote the conversion of PrPC to PrPSc. This model is supported by our finding that a mutation that increases loop homology but decreases rigidity blocks conversion by Mo protein-only recPrPSc together with a series of observations from other investigators. (1) Transgenic mice that 2-3x overexpress PrP containing 2 point mutations (170N,174T) that increase the rigidity of the a β2- α2 loop develop a spontaneous prion disease[48]. (2) The 170N, 174T double mutation was also found to increase the propensity of recPrP to form amyloid fibrils in vitro[49]. (3) Transgenic mice overexpressing 3-5x PrP containing another mutation that increases the rigidity of the β2- α2 loop (D167S) develop a spontaneous neurologic disease [50].
It is worth noting that the impact of 170 substitution on PrP conversion is asymmetrical. In other words, the inverse substitution (S170N) into the Mo PrPC backbone does not produce a chimera that is capable of propagating Mo protein-only recPrPSc (Fig 6A, row 5). We can therefore infer that the extreme C-terminal domain likely plays a larger role than residue 170 in facilitating propagation of Mo protein-only recPrPSc.
PrPC glycosylation adjacent to C-terminus also inhibits prion conversion
In addition to the amino acid sequence of the extreme C-terminus, we found that the propensity of PrPC to undergo prion conversion could also be affected by the adjacent N-linked glycan. Specifically, we found that mutant Mo PrPC lacking the second N-linked glycan (G2) at position 196 (mouse numbering) near the C-terminus can also propagate mouse protein-only recPrPSc in vitro. The effect of G2 deglycosylation does not appear to be as important as the effect of the E227 and S230 mutations since G2 PrPSc formation was not observed until round 4. In contrast, mutant PrPC lacking the other (G1), or both (G3) N-linked glycans remained resistant to conversion. It is surprising that Mo G2 PrPC should be more susceptible than Mo G3 PrPC to seeding by Mo protein-only recPrPSc, since Mo G3 PrPC is more similar to recPrP (in that both proteins are completely devoid of N-linked glycans). However, in line with our results, it has been previously reported that transgenic mice expressing Mo G2 PrPC are more susceptible than G1, G3, and wild-type mice to cross-species infection with human prions[33], suggesting that the first N-linked glycan may serve to facilitate PrPSc formation for several different prion strains. However, the first N-linked glycan of PrPC may not be important for propagating all prion strains (e.g. Kim et al. produced a synthetic prion strain is infectious to mice expressing PrPC lacking both glycans[42]).
Conclusion
We have leveraged a unique in vitro conversion system to show that the extreme C-terminus serves as a linchpin to control the seeded conversion of PrPC to PrPSc. Specifically, our data indicate that BV-specific residues at positions 227 and 230 are sufficient to promote prion conversion independent of seed/substrate sequence homology. We also found that the second N-linked glycan (adjacent to the C-terminal domain) inhibits seeding by Mo protein-only recPrPSc. While PrPC from both bank voles and G2 mutant mice are susceptible to conversion by recombinant protein-only protein seeds and other natural strains, both are relatively resistant to BSE-derived prions [17, 35], which is surprising since BSE prions readily infect wild-type mice[51] and many other animal species. Taken together, these observations suggest that Mo G2 PrPC and BV PrPC may share a common folding mechanism to form PrPSc, and that this mechanism may be able to accommodate a wide variety of PrPSc conformations (except for BSE, which may propagate by a different folding mechanism). It is possible that the G2 glycan, which is attached to residue N196, normally serves to stabilize the C-terminal domain of PrPC, and that specific residues perform a similar function, such as D227 and R230 in mouse PrP. Given that this linchpin controls the conversion of PrPC into PrPSc, we propose that stabilization of this region potentially represents an attractive target for potential prion therapeutics.
Materials and methods
Ethics statement
The Guide for the Care and Use of Laboratory Animals of the National Research Council was strictly followed for all animal experiments. All experiments conducted at Dartmouth College involving voles and mice in this study were conducted in accordance with protocol supa.su.1 as reviewed and approved by Dartmouth College’s Institutional Animal Care and Use Committee, operating under the regulations/guidelines of the NIH Office of Laboratory Animal Welfare (assurance number A3259-01) and the United States Department of Agriculture. All experiments conducted at the Roslin Institute involving mice were approved by The Roslin Institute’s Animal Welfare Ethical Review Board (internal protocol number; A471) and were conducted according to the regulations of the 1986 United Kingdom Home Office Animals (Scientific Procedures) Act.
Native prion strains
The mouse prion strain RML and the hamster prion strains 139H and sc237 were gifts from Stanley Prusiner (University of California, San Francisco, USA). The deer CWD prion strain and sheep scrapie prion strain were gifts from Mark Hall (USDA, Ames, Iowa, USA). The hamster strain Hyper was a gift from Suzette Priola (Rocky Mountain Laboratories, Hamilton, MT).
General sPMCA methods
The general sPMCA experimental method was adapted from Castilla et al.[52]. All PMCA reactions were sonicated in microplate horns at 37°C using a Misonix S-4000 power supply (Qsonica, Newtown, CT) set to power 70 for three rounds. One round of PMCA is equal to 24 hr. The first round of PMCA was seeded with a volume of PrPSc equal to 10% of the total reaction volume. RML prion-infected brain homogenates used as PrPSc seeds were used at a final reaction concentration of 1.0% (v/v), and 139H prion-infected brain homogenates used as PrPSc seeds were used at a final reaction concentration of 0.1% (v/v). To propagate the reaction between PMCA rounds, 10% of the reaction volume was transferred into a new, unseeded, substrate mixture. Due to the sensitivity of sPMCA[53], measures were undertaken to prevent sample contamination. Sample conical tubes were sealed with Parafilm (Bemis Company, Oshkosh, WI) and the sonicator horn was soaked in 100% bleach between experiments to prevent cross-contamination. The experimenter wore two pairs of gloves and changed the outer layer of gloves when handling a new sample. Sample conical tubes were spun at 500 x g for 5 sec to remove liquid off the conical tube lids before propagation and samples were propagated individually using aerosol resistant pipette tips. With each experiment, a sentinel conical tube (a conical tube containing the entire sPMCA reaction mixture but lacking seed) was also placed in the sonicator horn to monitor reactions for contamination.
Preparation of cofactor and protein-only recPrPSc by sPMCA
RecPrP, purified as described by Breydo et al.[54] was used to generate cofactor recPrPSc and protein-only recPrPSc, as described[7, 23]. Briefly, 200 μL reactions containing 6 μg/mL recombinant mouse PrP 23–230 or M109 bank vole PrP 23–231 in conversion buffer [20 mM Tris pH 7.5, 135 mM NaCl, 5 mM EDTA pH 7.5, 0.15% Triton X-100] were supplemented with either brain-derived cofactor[7] for cofactor recPrPSc propagation, or water for protein-only recPrPSc propagation. Reactions were seeded with 20 μL of converted cofactor recPrPSc or protein-only recPrPSc and sonicated with 15 sec pulses every 30 min for 24 hr at 37°C.
sPMCA with brain homogenate
A 10% (w/v) brain homogenate was prepared initially by Potter homogenization in PBS. The crude homogenate was spun at 400 x g for 1 min, and the supernatant was removed and kept. Triton X-100 was added to the supernatant for a final concentration of 1% (v/v), and the supernatant was solubilized on ice for 10 min. Brains were taken from European bank voles with the M109 genotype, Syrian hamsters, C57BL/6J mice, PrP0/0 mice or transgenic mice expressing glycosylation mutants (G1, G2, G3) as previously described[35]. One-hundred microliter reactions were seeded with 10 μL of PrPSc and sonicated with 20 sec pulses every 30 min for 24 hr at 37°C.
Reconstituted sPMCA with HEK 293-expressed PrP
Fifty-five microliters of HEK expressed PrPC substrate was mixed with 10 μL of 10X cell PMCA buffer (1% Triton X-100, 150 mM NaCl, 500 mM imidazole pH 7.5, 50 mM EDTA pH 7.5), 25 μL of 10% PrP0/0 brain homogenate, and seeded with 10 μL of a sPMCA reaction or 10% brain homogenate. One-hundred microliter reactions were seeded with 10 μL of PrPSc and sonicated with 20 sec pulses every 30 min for 24 hr at 37°C. The final concentration of PrPC in each reaction was 4–5 μg/mL.
Detection of PrPSc in sPMCA reactions
Formation of PrPSc was monitored by digestion of PMCA samples with proteinase K (PK) and western blotting. Samples were digested with 64 μg/mL PK (Roche, Basel, Switzerland) at 37°C with shaking at 750 r.p.m. Samples from sPMCA reactions using recPrP as the substrate were treated for 30 min, while samples using HEK 293 expressed PrP or brain homogenate as the substrate were treated for 60 min. Digestions were quenched by adding SDS-PAGE loading buffer and heating to 95°C for 15 min. SDS-PAGE and western blotting were performed as described previously [7] using mAb 27/33. Twenty microliters of a sPMCA reaction was subjected to PK digestion. The minus PK (-PK) lane is used to determine the conversion efficiency of a sPMCA reaction. For reactions using recombinant PrP or crude BH as the substrate, the -PK lane contains the same volume (20 μL) of a sPMCA reaction as a PK-digested sample. For reactions using HEK 293 expressed PrP the -PK lane contains a volume (2 μL) equivalent to one-tenth used in the PK-digested samples. This is because these substrates result in a much lower expected conversion efficiency. Images were quantified using Image Studio Lite (Licor) software to obtain the background-subtracted average signal intensity for the -PK and rounds 1–3 bands. The -PK value was multiplied by 10 to account for loading differences between it and rounds 1–3. The percent-conversion was then calculated for round 3 by dividing the background-subtracted average signal intensity for each round by the background-subtracted average signal intensity for the -PK round. The average percent conversion ±SEM for round 3 was calculated using between 1 and 5 biological replicates and 1 and 3 technical replicates.
We also confirmed that the final round PrPSc molecules produced in sPMCA reactions using HEK-expressed PrPC substrates could subsequently seed bank vole brain homogenate PMCA reactions (S3 Fig).
Mutagenesis and expression of HEK 293 PrP constructs
MoPrP on pcDNA 3.1 was generated by excising MoPrP from pcDNA5/FRT. BV PrP M109 pcDNA 3.1[23] and MoPrP pcDNA 5/FRT were cut using ApaI and HindIII (New England Biolabs, Ipswich, MA, USA). MoPrP was then ligated into the pcDNA 3.1 backbone. Chimeric constructs were generated using site directed mutagenesis using either the GeneTailor Site Directed Mutagenesis System (Invitrogen, Carlsbad, CA, USA) or GeneArt Site Directed Mutagenesis System (Invitrogen)(Table 1). All constructs were confirmed by sequencing. Constructs were then transfected into HEK 293 Freestyle-F cells (Invitrogen, Carlsbad, CA, USA)[55]. After 48 hr, 320 mL of cells were harvested in 80 mL aliquots by spinning at 500 x g for 5 min and stored at -80°C until purification.
Table 1. Primers and template plasmids used to generate chimeric BV/Mo PrP plasmids.
| CONSTRUCT | TEMPLATE PLASMID | PRIMERS |
|---|---|---|
| BV(DR) | BV PrP pcDNA 3.1 |
F: AGTCCCAGGCCTACTACGATGGGAGAAG ATCCCGCGCCGTGCTGCTCTTCTC R: TCGTAGTAGGCCTGGGACTCCTTCT GATACTG |
| BV(S170DR) | BV(DR) |
F: CCGGTGGACCAGTACAGCAACCAGAA CAACT R: TGTACTGGTCCACCGGCCGGTAGT ACACTT |
| BV(YS170DR) | BV(S170DR) |
F: TACTACCGTGAAAACATGTACCGGTAC CCTAA R: CATGTTTTCACGGTAGTAGCGGTC CTCC |
| BV(LYS170DR) | BV(YS170DR) |
F: CAGTAAGCCAAAAACCAACCTGAAGCA TGTGG R: GTTGGTTTTTGGCTTACTGGGCTTG TTCCACT |
| Mo(ES) | Mo PrP pcDNA 5 |
F: TCAGGCCTATTACGAGGGGAGAAGCTC CAGCAGC R: GCTGCTGGAGCTTCTCCCCTCGTAATAG GCCTGA |
| Mo(N155N170ES) | Mo(N170ES) |
F: ACCGTGAAAACATGAACCGATATCCTAA CCAAGTG R: CACTTGGTTAGGATATCGGTTCATGTTTT CACGGT |
| Mo(M109N155N170ES) | Mo(N155N170ES) |
F: CCAAAAACCAACATGAAGCACGTGGCA GG R: CCTGCCACGTGCTTCATGTTGGTTTTTGG |
| BV(R) | BV PrP pcDNA 3.1 |
F: TACTACGAAGGGAGAAGATCCCGCGCC GTGCT R: AGCACGGCGCGGGATCTTCTCCCTTCGTA GTA |
| BV(D) | BV PrP pcDNA 3.1 |
F: CCCAGGCCTACTACGACGGGAGAAGTT CCC R: GGGAACTTCTCCCGTCGTAGTAGGCCT GGG |
| BV(S170) | BV PrP pcDNA 3.1 |
F: GTGGACCAGTACAGCAACCAGAACAAC R: GTTGTTCTGGTTGCTGTACTGGTCCAC |
| BV(Y) | BV PrP pcDNA 3.1 |
F: ACCGTGAAAACATGTACCGGTACCCTAA CC R: GGTTAGGGTACCGGTACATGTTTTCACG GT |
| BV(L) | BV PrP pcDNA 3.1 |
F: GCCAAAAACCAACCTGAAGCATGTGGCA G R: CTGCCACATGCTTCAGGTTGGTTTTTGGC |
| Mo(S) | Mo PrP pcDNA 5 |
F: TTACGACGGGAGAAGCTCCAGCAGCAC R: GTGCTGCTGGAGCTTCTCCCGTCGTAA |
| Mo(E) | Mo PrP pcDNA 5 |
F: CAGGCCTATTACGAGGGGAGAAGATCC AGCA R: TGCTGGATCTTCTCCCCTCGTAATAGGC CTG |
| Mo(N170) | Mo PrP pcDNA 5 |
F: GTGGATCAGTACAATAACCAGAACAACT TCG R: CGAAGTTGTTCTGGTTATTGTACTGATC CAC |
| Mo(N155) | Mo PrP pcDNA 5 |
F: ACCGTGAAAACATGAACCGATATCCTAA CCAAGTG R: CACTTGGTTAGGATATCGGTTCATGTTTT CACGGT |
| Mo(M109) | Mo PrP pcDNA 5 |
F: CCAAAAACCAACATGAAGCACGTGGCAG G R: CCTGCCACGTGCTTCATGTTGGTTTTTGG |
| BV(LDR) | BV(DR) |
F: CAGTAAGCCAAAAACCAACCTGAAGCAT GTGG R: GTTGGTTTTTGGCTTACTGGGCTTG TTCCACT |
| BV(YDR) | BV(DR) |
F: TACTACCGTGAAAACATGTACCGGTACC CTAA R: CATGTTTTCACGGTAGTAGCGGTCC TCC |
Purification of HEK 293 expressed PrP constructs
Pellets thawed on ice were re-suspended in 20 mL of lysis buffer [20 mM MOPS pH 7.0, 0.15 M NaCl, 1% Triton X-100, 1% DOC, CompleteTM mini EDTA-free protease inhibitor (Roche, Basel, Switzerland)]. The mixture was solubilized on ice for 30 min, then centrifuged at 100,000 x g for 35 min. The solubilized supernatant was applied to a 2 mL IMAC copper sulfate column made with chelating Sepharose (GE Healthcare, Chicago, USA) that was pre-equilibrated with equilibration and wash buffer [20 mM MOPS pH 7.0, 0.15 M NaCl, 10 mM imidazole, 1% Triton X-100]. The column was then washed with 20 mL of equilibration and wash buffer. The sample was eluted with 8 mL of elution buffer [20 mM MOPS pH 7.5, 0.15 M NaCl, 0.15 M imidazole pH 7.0, 1% Triton X-100]. The eluate was diluted 1:1 with pre-SP buffer [20 mM MES pH 5.4, 0.15 M imidazole pH 7.0, 0.15 M NaCl, and 1% Triton X-100]. Next, the eluate was applied over a 2 mL SP Sepharose fast flow (Sigma-Aldrich, St. Louis, MO, USA) exchange column pre-equilibrated with SP wash buffer [20 mM MOPS pH 7.0, 250 mM NaCl, and 1% Triton X-100]. The column was washed with 10 mL SP was buffer and eluted with 8 mL SP elution buffer [20 mM MOPS pH 7.5, 500 mM NaCl, and 1% Triton X-100]. The eluate was then partially deglycosylated by adding 80 μL of Glycerol Free PNGase F (New England Biolabs, Ipswich, MA, USA) to 8 mL of eluate. The mixture was incubated for 24 hr at 37°C with shaking at 350 r.p.m. The partially deglycosylated substrate was then concentrated and repurified by applying over a 400 μL copper column pre-equilibrated with equilibration and wash buffer. The column was washed with 20 mL of equilibration and wash buffer and eluted with 2 mL of IMAC-CuSO4-elution buffer [20 mM MES pH 6.4, 0.15 M imidazole pH 7.0, 150 mM NaCl, and 1% Triton X-100]. The eluate was loaded into a 3500 MWCO Slide-A-Lyser (Thermo Fisher, Waltham, MA, USA) and dialyzed overnight into dialysis buffer [44].
To ensure that substitutions near the C-terminus of PrPC do not interfere with addition of the C-terminal GPI-anchor, we treated chimeric PrPC molecules with either single or double C-terminal substitutions with PI-PLC (S4 Fig). It has been previously reported that PI-PLC treatment of GPI-anchored proteins such as PrPC cause the treated proteins to bind less well to PVDF membranes and migrate more slowly than untreated controls [56]. Accordingly, we observed that all of the HEK-expressed PrPC chimeras tested, as well as HEK-expressed BV PrPC native brain PrPC immunopurified from mouse brain, showed displayed slower migration and lower intensity on western blot after PI-PLC treatment (S4 Fig). In contrast, bacterially-expressed recPrP, which lacks a GPI-anchor, is apparently unaffected by PI-PLC treatment (S4 Fig). Thus, all of the chimeric PrPC substrates used in this study appear to have proper post-translational modifications (S1 Fig and S4 Fig) and folding (as judged by solubility in non-denaturing solution and normal behavior during IMAC and ion exchange chromatography. These observations indicate that substitution of homologous residues between mouse and bank vole PrPC does not cause aberrant metabolism or trafficking of chimeric molecules in HEK 293 cells.
Supporting information
Western blots showing PrP partially purified from cell lysates expressing the indicated protein, or no protein, before and after partial enzymatic deglycosylation with PNGase F (+ PF), as indicated. (A) Wild-type constructs. (B) Chimeric constructs.
(TIF)
Western blots showing three-round reconstituted sPMCA reactions using partially purified Mo PrPC substrate supplemented with PrP0/0 BH and seeded with mouse RML. Substrates were either untreated or partially deglycosylated with PNGase F during purification, as indicated. -PK = samples not subjected to proteinase K digestion; all other samples were proteolyzed.
(TIF)
Western blots showing PMCA reactions using crude bank vole brain homogenate substrate seeded with various HEK-expressed PrPSc molecules, as indicated. The chimeric PrPSc molecules are final round products of 3-round sPMCA reactions using HEK-expressed PrPC substrates originally seeded with Mo protein-only recPrPSc or buffer, as indicated.
(TIF)
Western blots of various purified PrP substrates (bacterially-expressed recPrP, immunopurified native brain PrPC, different HEK-expressed PrPC chimeras with substitutions at residues 227 and/or 230 as indicated, or HEK expressed bank vole PrPC) treated either with (+) or without (-) 0.25U/mL Phosphoinositide phospholipase C (PI-PLC) for 14 hr at 37 oC with shaking at 800 r.p.m. as indicated. Proteins were transferred from a 12.5% polyacrylamide gel onto a Millipore Immobilon-P PVDF membrane by semi-dry electroblotting, and probed with mAb 27/33.
(TIF)
Acknowledgments
We thank Emilie Shipman and Jason McLellan for much-appreciated assistance with Freestyle™ cell transfection and culture; Jan Stöhr, Stanley Prusiner, Cathy Chang, and Ta Yuan Chang for providing mouse brains; and Tamutenda Chidawanyika, and Romolo Nonno for valuable advice and discussion.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was supported by NIH grants R01NS102301, R01NS118796, R01NS117276 to S.S., NIH IDeA award to Dartmouth BioMT P20-GM113132, and NIH grant T32AI007519 to C.B. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363–83. 10.1073/pnas.95.23.13363 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffith JS. Self-replication and scrapie. Nature. 1967;215(105):1043–4. 10.1038/2151043a0 . [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–44. 10.1126/science.6801762 . [DOI] [PubMed] [Google Scholar]
- 4.Supattapone S. Synthesis of High Titer Infectious Prions with Cofactor Molecules. J Biol Chem. 2014;289(29):19850–4. Epub 2014/05/27. 10.1074/jbc.R113.511329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104(23):9741–6. 10.1073/pnas.0702662104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang F, Wang X, Yuan CG, Ma J. Generating a Prion with Bacterially Expressed Recombinant Prion Protein. Science. 2010; 327(5969):1132–5. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, et al. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A. 2012;109(28):E1938–E46. Epub 2012/06/20. 1206999109 [pii] 10.1073/pnas.1206999109 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, et al. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A. 2012;109(22):8546–51. Epub 2012/05/16. 1204498109 [pii] 10.1073/pnas.1204498109 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Funez P, Zhang Y, Sanchez-Garcia J, Jensen K, Zou WQ, Rincon-Limas DE. Pulling rabbits to reveal the secrets of the prion protein. Communicative & integrative biology. 2011;4(3):262–6. Epub 2011/10/08. 10.4161/cib.4.3.15054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Bari MA, Chianini F, Vaccari G, Esposito E, Conte M, Eaton SL, et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. The Journal of general virology. 2008;89(Pt 12):2975–85. Epub 2008/11/15. 10.1099/vir.0.2008/005520-0 . [DOI] [PubMed] [Google Scholar]
- 11.Di Bari MA, Nonno R, Castilla J, D'Agostino C, Pirisinu L, Riccardi G, et al. Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog. 2013;9(3):e1003219 Epub 2013/03/19. 10.1371/journal.ppat.1003219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell'Omo G, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006;2(2):e12 Epub 2006/03/07. 10.1371/journal.ppat.0020012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog. 2014;10(4):e1003990 Epub 2014/04/05. 10.1371/journal.ppat.1003990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandler RL, Turfrey BA. Inoculation of voles, Chinese hamsters, gerbils and guinea-pigs with scrapie brain material. Research in veterinary science. 1972;13(3):219–24. Epub 1972/05/01. . [PubMed] [Google Scholar]
- 15.Espinosa JC, Nonno R, Di Bari M, Aguilar-Calvo P, Pirisinu L, Fernandez-Borges N, et al. PrPC governs prion strain susceptibility in bank vole while others host factors modulate strain features. J Virol. 2016. Epub 2016/09/23. 10.1128/JVI.01592-16 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orru CD, Groveman BR, Raymond LD, Hughson AG, Nonno R, Zou W, et al. Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog. 2015;11(6):e1004983 Epub 2015/06/19. 10.1371/journal.ppat.1004983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agrimi U, Nonno R, Dell'Omo G, Di Bari MA, Conte M, Chiappini B, et al. Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog. 2008;4(7):e1000113 Epub 2008/07/26. 10.1371/journal.ppat.1000113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davenport KA, Henderson DM, Mathiason CK, Hoover EA. Assessment of the PrPc amino-terminal domain in prion species barriers. J Virol. 2016. Epub 2016/09/23. 10.1128/JVI.01121-16 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piening N, Nonno R, Di Bari M, Walter S, Windl O, Agrimi U, et al. Conversion efficiency of bank vole prion protein in vitro is determined by residues 155 and 170, but does not correlate with the high susceptibility of bank voles to sheep scrapie in vivo. J Biol Chem. 2006;281(14):9373–84. Epub 2006/02/04. 10.1074/jbc.M512239200 . [DOI] [PubMed] [Google Scholar]
- 20.Christen B, Perez DR, Hornemann S, Wuthrich K. NMR structure of the bank vole prion protein at 20 degrees C contains a structured loop of residues 165–171. J Mol Biol. 2008;383(2):306–12. Epub 2008/09/09. 10.1016/j.jmb.2008.08.045 . [DOI] [PubMed] [Google Scholar]
- 21.Kurt TD, Aguilar-Calvo P, Jiang L, Rodriguez JA, Alderson N, Eisenberg DS, et al. Asparagine and glutamine ladders promote cross-species prion conversion. J Biol Chem. 2017;292(46):19076–86. 10.1074/jbc.M117.794107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otero A, Hedman C, Fernandez-Borges N, Erana H, Marin B, Monzon M, et al. A Single Amino Acid Substitution, Found in Mammals with Low Susceptibility to Prion Diseases, Delays Propagation of Two Prion Strains in Highly Susceptible Transgenic Mouse Models. Mol Neurobiol. 2019;56(9):6501–11. Epub 2019/03/09. 10.1007/s12035-019-1535-0 [pii]. . [DOI] [PubMed] [Google Scholar]
- 23.Burke CM, Walsh DJ, Steele AD, Agrimi U, Di Bari MA, Watts JC, et al. Full restoration of specific infectivity and strain properties from pure mammalian prion protein. PLoS Pathog. 2019;15(3):e1007662 10.1371/journal.ppat.1007662 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Priola SA. Prion protein and species barriers in the transmissible spongiform encephalopathies. Biomed Pharmacother. 1999;53(1):27–33. 10.1016/s0753-3322(99)80057-2 . [DOI] [PubMed] [Google Scholar]
- 25.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic acids research. 1988;16(22):10881–90. 10.1093/nar/16.22.10881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paulick MG, Forstner MB, Groves JT, Bertozzi CR. A chemical approach to unraveling the biological function of the glycosylphosphatidylinositol anchor. Proc Natl Acad Sci U S A. 2007;104(51):20332–7. 10.1073/pnas.0710139104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Croset A, Delafosse L, Gaudry JP, Arod C, Glez L, Losberger C, et al. Differences in the glycosylation of recombinant proteins expressed in HEK and CHO cells. J Biotechnol. 2012;161(3):336–48. 10.1016/j.jbiotec.2012.06.038 . [DOI] [PubMed] [Google Scholar]
- 28.Nishina KA, Deleault NR, Mahal SP, Baskakov I, Luhrs T, Riek R, et al. The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry. 2006;45(47):14129–39. 10.1021/bi061526k . [DOI] [PubMed] [Google Scholar]
- 29.Burke CM, Walsh DJ, Mark KMK, Deleault NR, Nishina KA, Agrimi U, et al. Cofactor and glycosylation preferences for in vitro prion conversion are predominantly determined by strain conformation. PLoS Pathog. 2020;16(4):e1008495 10.1371/journal.ppat.1008495 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katorcha E, Makarava N, Savtchenko R, Baskakov IV. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Scientific reports. 2015;5:16912 10.1038/srep16912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyerett C, Michel B, Pulford B, Spraker TR, Nichols TA, Johnson T, et al. In vitro strain adaptation of CWD prions by serial protein misfolding cyclic amplification. Virology. 2008;382(2):267–76. 10.1016/j.virol.2008.09.023 . [DOI] [PubMed] [Google Scholar]
- 32.Castilla J, Gonzalez-Romero D, Saa P, Morales R, De Castro J, Soto C. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell. 2008;134(5):757–68. 10.1016/j.cell.2008.07.030 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiseman FK, Cancellotti E, Piccardo P, Iremonger K, Boyle A, Brown D, et al. The glycosylation status of PrPC is a key factor in determining transmissible spongiform encephalopathy transmission between species. J Virol. 2015;89(9):4738–47. Epub 2015/02/13. 10.1128/JVI.02296-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancellotti E, Wiseman F, Tuzi NL, Baybutt H, Monaghan P, Aitchison L, et al. Altered glycosylated PrP proteins can have different neuronal trafficking in brain but do not acquire scrapie-like properties. J Biol Chem. 2005;280(52):42909–18. 10.1074/jbc.M509557200 . [DOI] [PubMed] [Google Scholar]
- 35.Tuzi NL, Cancellotti E, Baybutt H, Blackford L, Bradford B, Plinston C, et al. Host PrP glycosylation: a major factor determining the outcome of prion infection. PLoS Biol. 2008;6(4):e100 10.1371/journal.pbio.0060100 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi A, Matsuura Y, Takeuchi A, Yamada M, Miyoshi I, Mohri S, et al. A domain responsible for spontaneous conversion of bank vole prion protein. Brain Pathol. 2019;29(2):155–63. Epub 2018/07/28. 10.1111/bpa.12638 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gong B, Ramos A, Vazquez-Fernandez E, Silva CJ, Alonso J, Liu Z, et al. Probing structural differences between PrP(C) and PrP(Sc) by surface nitration and acetylation: evidence of conformational change in the C-terminus. Biochemistry. 2011;50(22):4963–72. Epub 2011/04/30. 10.1021/bi102073j . [DOI] [PubMed] [Google Scholar]
- 38.Angers R, Christiansen J, Nalls AV, Kang HE, Hunter N, Hoover E, et al. Structural effects of PrP polymorphisms on intra- and interspecies prion transmission. Proc Natl Acad Sci U S A. 2014;111(30):11169–74. Epub 2014/07/19. 1404739111 [pii] 10.1073/pnas.1404739111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noble GP, Walsh DJ, Miller MB, Jackson WS, Supattapone S. Requirements for mutant and wild-type prion protein misfolding in vitro. Biochemistry. 2015;54(5):1180–7. Epub 2015/01/15. 10.1021/bi501495j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kundu B, Maiti NR, Jones EM, Surewicz KA, Vanik DL, Surewicz WK. Nucleation-dependent conformational conversion of the Y145Stop variant of human prion protein: structural clues for prion propagation. Proc Natl Acad Sci U S A. 2003;100(21):12069–74. Epub 2003/10/02. 10.1073/pnas.2033281100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stohr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen HO, et al. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A. 2011;108(52):21223–8. Epub 2011/12/14. 10.1073/pnas.1117827108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim C, Xiao X, Chen S, Haldiman T, Smirnovas V, Kofskey D, et al. Artificial strain of human prions created in vitro. Nature communications. 2018;9(1):2166 10.1038/s41467-018-04584-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurt TD, Telling GC, Zabel MD, Hoover EA. Trans-species amplification of PrP(CWD) and correlation with rigid loop 170N. Virology. 2009;387(1):235–43. Epub 2009/03/10. S0042-6822(09)00137-8 [pii] 10.1016/j.virol.2009.02.025 . [DOI] [PubMed] [Google Scholar]
- 44.Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Fernandez-Borges N, Schwarz P, et al. A molecular switch controls interspecies prion disease transmission in mice. The Journal of clinical investigation. 2010;120(7):2590–9. Epub 2010/06/17. 10.1172/JCI42051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurt TD, Bett C, Fernandez-Borges N, Joshi-Barr S, Hornemann S, Rulicke T, et al. Prion transmission prevented by modifying the beta2-alpha2 loop structure of host PrPC. J Neurosci. 2014;34(3):1022–7. Epub 2014/01/17. 10.1523/JNEUROSCI.4636-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121–321). Nature. 1996;382(6587):180–2. 10.1038/382180a0 . [DOI] [PubMed] [Google Scholar]
- 47.Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci U S A. 2005;102(3):646–50. Epub 2005/01/14. 0409008102 [pii] 10.1073/pnas.0409008102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sigurdson CJ, Nilsson KP, Hornemann S, Heikenwalder M, Manco G, Schwarz P, et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci U S A. 2009;106(1):304–9. 10.1073/pnas.0810680105 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dutta A, Chen S, Surewicz WK. The effect of beta2-alpha2 loop mutation on amyloidogenic properties of the prion protein. FEBS Lett. 2013;587(18):2918–23. Epub 2013/07/31. S0014-5793(13)00552-8 [pii] 10.1016/j.febslet.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sigurdson CJ, Joshi-Barr S, Bett C, Winson O, Manco G, Schwarz P, et al. Spongiform encephalopathy in transgenic mice expressing a point mutation in the beta2-alpha2 loop of the prion protein. J Neurosci. 2011;31(39):13840–7. Epub 2011/10/01. 10.1523/JNEUROSCI.3504-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fraser H, Bruce ME, Chree A, McConnell I, Wells GA. Transmission of bovine spongiform encephalopathy and scrapie to mice. The Journal of general virology. 1992;73(Pt 8):1891–7. 10.1099/0022-1317-73-8-1891 . [DOI] [PubMed] [Google Scholar]
- 52.Castilla J, Saa P, Morales R, Abid K, Maundrell K, Soto C. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Methods in enzymology. 2006;412:3–21. Epub 2006/10/19. S0076-6879(06)12001-7 [pii] 10.1016/S0076-6879(06)12001-7 . [DOI] [PubMed] [Google Scholar]
- 53.Cosseddu GM, Nonno R, Vaccari G, Bucalossi C, Fernandez-Borges N, Di Bari MA, et al. Ultra-Efficient PrP Amplification Highlights Potentialities and Pitfalls of PMCA Technology. PLoS Pathog. 2011;7(11):e1002370 Epub 2011/11/25. 10.1371/journal.ppat.1002370 PPATHOGENS-D-11-01410 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breydo L, Makarava N, Baskakov IV. Methods for conversion of prion protein into amyloid fibrils. Methods Mol Biol. 2008;459:105–15. 10.1007/978-1-59745-234-2_8 . [DOI] [PubMed] [Google Scholar]
- 55.Gilman MS, Moin SM, Mas V, Chen M, Patel NK, Kramer K, et al. Characterization of a Prefusion-Specific Antibody That Recognizes a Quaternary, Cleavage-Dependent Epitope on the RSV Fusion Glycoprotein. PLoS Pathog. 2015;11(7):e1005035 10.1371/journal.ppat.1005035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishina KA, Supattapone S. Immunodetection of glycophosphatidylinositol-anchored proteins following treatment with phospholipase C. Anal Biochem. 2007;363(2):318–20. 10.1016/j.ab.2007.01.032 . [DOI] [PMC free article] [PubMed] [Google Scholar]