

Abstract
Despite the efforts of the global medical and scientific community, tuberculosis remains the leading cause of death from infectious diseases. The expectation of success associated with the development of new anti-TB drugs was not justified, and the attention of researchers was largely drawn to the creation of new mycobacterial strains for vaccination against tuberculosis. The proposed review contains current information on the existing vaccine strains and the development of new, genetically engineered strains for the prevention of tuberculosis and the prevention and treatment of other diseases. The review includes relevant information on the correlation between BCG vaccination and the frequency and severity of COVID-19 infection.
Keywords: tuberculosis, BCG, vaccine, recombinant vaccine
INTRODUCTION
Tuberculosis is an infectious disease caused by Mycobacterium tuberculosis (MTB). It remains the leading cause of death from infectious diseases in many countries [1]. Today, tuberculosis is treated with a combination antibiotic therapy for 6–9 months, which causes significant side effects. The tuberculosis control strategy of the World Health Organization is currently aimed at strengthening disease prevention [2], and active work is underway around the world to develop improved tuberculosis vaccines, some of which have entered the clinical research phase.
Today, Bacillus Calmett–Guerin (BCG) is still the only vaccine against tuberculosis. It successfully protects against tuberculosis (TB) in children, but it provides only partial protection against respiratory forms of TB in adolescents and adults. Albert Kalmet and Camille Guerin obtained BCG from the cattle TB pathogen Mycobacterium bovis via continuous passaging for several years, which led to a weakening of the strain. BCG has been used for vaccination since 1921. It also leads to a decrease in total infant mortality due to the nonspecific immunomodulating effect of mycobacterial vaccination. Although BCG has made a significant contribution to TB prevention [3], the loss of several dominant antigens and key molecular signs of pathogenic mycobacteria may explain its limitations in the prevention of adult TB. The proposed review contains current data on the existing BCG strains, on the development of genetically modified vaccine strains that may be capable of replacing BCG, and the optimal route for vaccine delivery to the body, and it discusses the prospects for the use of BCG to fight other diseases.
BCG strains and their genealogy. The main difference between BCG and the original M. bovis strain is the absence of several segments of the genome, in particular, the so-called region of difference (region of difference 1, RD1) [4]. It encodes immunodominant antigens, such as ESAT6, CFP10, and the unique mycobacterial secretion system ESX-1 type II (T7SS) [5, 6]. The loss of ESX-1 led to the loss of the BCG ability to penetrate the cytosol from the phagosome, which was a powerful factor in strain attenuation.
Quite a large number of BCG strains have been isolated due to the peculiarities of the conditions for the maintenance of lines in different countries [7]. It is interesting to note that the first daughter BCG strain obtained from the original culture was registered in Russia in 1940 [8].
Behr et al. [9] studied the genealogy of BCG distribution and grouped the strains into groups based on information on the number of copies of the IS6110 gene (an insertion element characteristic only for the group of mycobacteria that cause TB) and the presence or absence of the mpt64 gene (conserved MTB antigen used for diagnosis). The study found synonymous strains under different names and were combined to simplify typing [10].
Speaking of the genetic characteristics of BCG strains, it is interesting to consider one of them in more detail. In research on the BCG-Prague strain, a phoP gene mutation was detected in the form of a single nucleotide insert, which led to the destruction of the C-terminal DNA-binding domain [11, 12]. This mutation is specific for BCG-Prague and is not found in other BCG strains [13]. PhoP is the regulator of the response of the two-component PhoP-PhoR system. It positively regulates more than 40 genes in MTB, including two important protective antigens (Ag85A, PPE18), which are used to construct subunit vaccines [14, 15]. Thus, it was suggested that the low immunogenicity of BCG-Prague may result from a phoP mutation. It was shown that the complementation of mutations with an allele M. bovis phoP actually restores the immunogenicity of BCG-Prague. Overexpression of the M. bovis allele phoP-phoR in BCG-Japan with a full copy of phoP-phoR further enhances the immunogenicity and protective efficacy of this vaccine strain. The vaccination of C57BL/6 mice with the recombinant rBCG-Japan/PhoPR strain induced higher levels of the production of interferon-γ (IFN-γ) CD4+ T cells than that with BCG-Japan. RBCG-Japan/PhoPR vaccinated guinea pigs were better protected against MTB infection than those immunized with BCG-Japan, showing significantly longer survival times, reduced bacterial loads, and less severe pathology. These studies identified a genetic modification that can be used to create new recombinant BCG vaccines.
Methods to protect mycobacteria from the immune system of an infected host. After bodily penetration, MTB cells face the first echelon of defense in the form of phagocytic cells. A phagocytized bacterium is exposed to acid stress, the effects of reactive oxygen and nitrogen (ROS and RNS), hydrolytic enzymes, and cationic antimicrobial peptides. The low pH inside the maturing phagosome activates enzymes that break down bacterial lipids and proteins [16, 17]. However, mycobacteria have developed various means to adapt to the bactericidal environment of maturing phagosomes. In particular, they evade the action of bactericidal mechanisms by blocking the proton “pump” that acidifies the contents of phagosomes and inhibit the fusion of phagosomes and lysosomes [18, 19]. With a decrease in pH, MTB can change its metabolism and use the glyoxylate pathway instead of the Krebs cycle, which reduces NADH synthesis and the release of CO2. Thus, additional carbon can then be used to synthesize lipids or other metabolites to further reduce stress management [20]. Thus, according to studies, the synthesis of hyperphosphorylated guanine nucleotides (p) ppGpp is necessary for the survival of mycobacteria in adverse conditions, including redox stress [21]. Another mechanism for the response to acidification is the production of ammonia to increase the pH. MTBs use the ompATb operon (which is necessary for functioning at a low pH); asparagine serves as a source of ammonia [22]. The urease operon also contributes to the alkalization of the medium [23]. It should be added that mycobacterial urease is involved in the prevention of the normal expression of MHC class-II molecules, which are involved in the binding of antigens and their presentation to T cells, on the cell surface [24]. In addition, MTB also uses ergothioneine, thiol, and thioredoxin systems, and the PhoP-PhoR system described above [25] in response to oxidative stress.
By blocking the formation of phagolysosomes, mycobacteria destroys the phagosome membrane with the ESX-1 secretion system, which allows TB antigens and bacteria themselves to enter the cytosol [19, 26]. Conversely, BCG does not have ESX-1 and remains inside the host phagosome; therefore, antigens and bacterial DNA do not enter the cytosol [5, 6, 27]. Studies have shown that the immunoresponse of mycobacterium-infected macrophages is characterized by a reduced expression of MHC class-II genes and other genes induced by IFN-βγ. One of the mechanisms for the suppression of MHC II expression is specific inhibition via histone deacetylation, which leads to a decrease in response of CD4+ T cells [28, 29]. CD4+ T cells differentiate into central memory T cells (TCM), which, in turn, differentiate into cells of the effector memory TEM, effector cells Th1 or Th17; then they migrate and carry out their effector functions in infected tissues. When CD4+ T cells arrive at the infection site, they collide with aggregates of MTB-containing macrophages and other immune cells and together form a dense cell structure called granuloma. CD4+ T cells secrete cytokines that activate infected macrophages to control bacterial growth and attract more immune cells to granuloma [30]. The presence of mycobacterial DNA in the cytosol leads to the activation of NLPR3 and AIM-2 inflammosomes, the release of interferons, and an increase in autophagy and apoptosis [20, 31, 32], which may ultimately lead to improved induction of T-cell responses [33].
During disease development, the amount of MBT-specific CD8+ T-cells increases, but their role in protective immunity is not clear. It was shown that depletion of CD8+ cells reduces the efficacy of BCG vaccination [34]. At the same time, even high levels of vaccination-induced CD8+-cells does not affect MTB proliferation and does not lead to the recognition of MTB-infected macrophages [35].
The role of humoral immunity in MTB infection remains controversial, but it was shown that individuals with latent TB infection and active disease have different MTB-specific humoral responses and have different antibody glycosylation patterns [36].
Methods to increase BCG protectiveness. BCG :: ESX-1. Several candidates for BCG recombinant vaccines were designed to ensure the destruction of the phagosome membrane by cells of the vaccine strain (Fig. 1) [37]. As discussed above, the BCG strain, which was designed for the secretion of proteins that play a key role in host interactions with the pathogen, has lost the genes necessary for type-VII secretion (T7s) ESX-1 [38, 39].
Fig. 1.
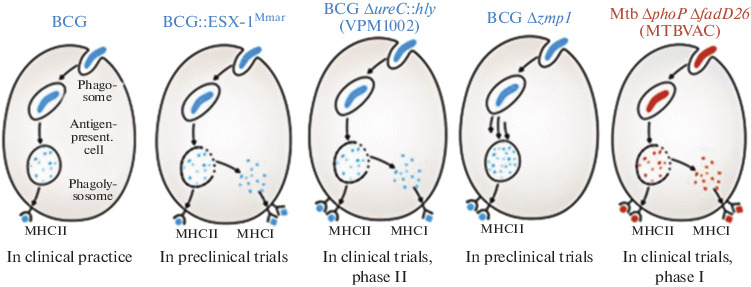
Candidate vaccine strains with a permeable phagosome [37].
The main ESX-1 system consists of EccB-D ATPases, which form a transmembrane structure in the form of a channel. The cytosolic components of the ESX-1 system are ATPase EccA, EspD-H chaperones, and the secreted substrate proteins EsxAB, PE35-PPE68, EspA-C, and EspE. Other important effectors that are secreted by this system include the early secreted antigenic target of 6 kDa ESAT-6) and 10-kDa culture-filtrate protein (culture-filtrate protein of 10 kDa, CFP-10). Proteins are secreted as a heterodimer and bind to the nuclear components of the ESX-1 system [40], which causes a cascade of nonspecific immunoresponses [41–43]: the activation of AIM-2 and NLRP3, increased secretion of interleukin-1b (IL-1b) and/or IL-18 [30, 31, 44], and the activation of cyclic synthase GMP-AMP (cGas), an interferon gene stimulator (STING) and TANK binding kinase 1 (TBK1) The last signaling cascade leads to the formation of type-I interferons (IFN) [17]. In addition to nonspecific immune activation, the secreted effectors of ESX-1 also induce specific Th1-cell responses with strong protective potential [45].
Via the heterologous expression of the esx-1 region of Mycobacterium marinum, recombinant BCG with ESX-1 activity (BCG :: ESX-1Mmar) was created. It induces the cGas/STING/TBK1/IRF-3/interferon I chain and enhances the inflammatory activity of AIM2 and NLRP3, which stimulates the formation of the effector CD8+ and CD4+ Th1 cells (Fig. 2) [46]. It is important to note that the BCG :: ESX-1Mmar strain is characterized by low virulence and provided more effective protection than the original BCG strain when infected with highly virulent MTB.
Fig. 2.
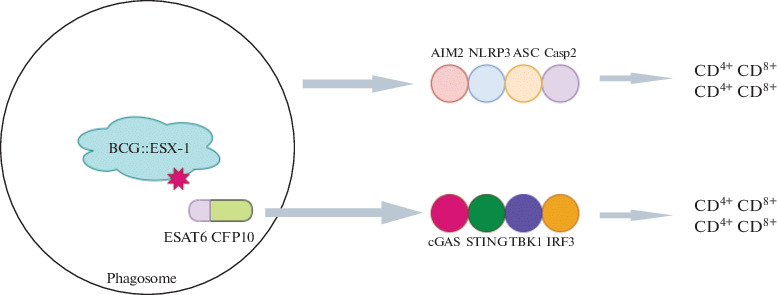
Schematic mechanism of enhanced protection of BCG::ESX-1Mmar [46].
BCG ΔureC :: hly (VPM1002) and derivatives. VPM1002 is a recombinant rBCG strain ΔureC :: hly, in which the urease C gene was replaced by the hly gene, which encodes listeriolysin O (LLO) from Listeria monocytogenes [47]. Urease C prevents the acidification of phagosomes containing mycobacteria via the formation of ammonia, and it thereby inhibits phagolysosome maturation and promotes mycobacterial survival inside macrophages [48, 49]. A decrease in urease C production leads to phagosome acidification, which contributes to phagolysosome formation [50]. LLO is a cholesterol-dependent cytolysin that forms transmembrane pores in the phagolysosome, allowing L. monocytogenes to enter the cytosol [50, 51]. Its expression in VPM1002 led to the release of antigens and bacterial DNA into the cytosol, which resulted in autophagy, the activation of inflammation, and apoptosis. VPM1002 demonstrated a significant increase in immunogenicity, efficacy, and safety as compared with BCG in preclinical studies and successfully passed Phases I and II of clinical trials. Phase II/III clinical trials are ongoing in India. The vaccine passed the first phase of clinical trials in Germany and South Africa, demonstrating its safety and immunogenicity in young people. It was also successfully tested in phase IIa of a randomized clinical trial in healthy newborns in South Africa and is currently undergoing phase IIb of a study in HIV-infected newborns [52].
Second-generation rBCG ΔureC :: hly derivatives secrete cytokines to enhance immunogenicity [53] are auxotrophic for vitamin B6 [54]. In addition, the rBCG strain ΔureC :: hly ΔnuoG was obtained. It was deleted by the antiapoptotic virulence gene nuoG, which prevents the apoptosis of infected cells [55]. For mice vaccination with rBCG ΔureC :: hly ΔnuoG was safer than vaccination with unmodified BCG and more protective than rBCG ΔureC :: hly; it significantly reduced the MTB load in the lungs of mice, reduced pathological manifestations in the lungs, and enhanced immunoresponses. Transcriptome analysis of draining lymph nodes after vaccination with either rBCG ΔureC :: hly or rBCG ΔureC :: hly ΔnuoG demonstrated an earlier and stronger induction of immunoresponses than that with unmodified BCG. Thus, rBCG ΔureC :: hly ΔnuoG is a promising candidate for a vaccine with improved efficacy and safety [55].
BCG expressing perfringolysin O. The disadvatage of rBCG ΔureC :: hly is the narrow pH range of LLO activity. A BCG strain expressing perfringolysin O (Pfo), cytolysin Clostridium perfringens, was created [56]. A mutant form of this protein containing the substitution G137Q (PfoAG137Q) was created to reduce its cytotoxic effect. The mutation significantly reduced the half-life of the protein, which led to the elimination of its cytotoxicity. For the resulting rBCG strain, designated AERAS-401 (BCG1331 ΔureC ::ΩpfoAG137Q), the absence of urease activity was confirmed. The secretion of biologically active hemolysin was shown via lysis of sheep erythrocytes in the presence of a supernatant of a recombinant strain. In addition, AERAS-401 was shown to be less virulent in immunodeficient mice as compared to the parent BCG strain.
BCG Δzmp1. It was found that the removal of the zinc metalloprotease gene zmp1 weakens MTB and enhances nonspecific immunity, inflammatory, and IL-1-dependent immune mechanisms upon infection with a mutant strain. A similar deletion was obtained from BCG. In vitro and in vivo immunization experiments in mice and guinea pigs showed that the deletion of mutant BCG Δzmp1 is more immunogenic and also enhances the presentation of antigens as part of MHCII [57]. In addition, one of the Zmp1 inhibitors was able to reduce the survival of MTB in primary human macrophages [58]. Thus, BCG Δzmp1 is awaiting further evaluation and clinical trials.
rBCG-SOCS1DN. Among the various mechanisms of MTB release from the host immune system is the induction of the expression of the cytokine suppressor SOCS1 by the host cells. This suppresses cytokine signaling and disrupts JAK/STAT signaling, which is involved in innate immunity and subsequent adaptive immunity. The implementation of this mechanism by the BCG vaccine strain may be the reason for the lack of vaccine protection. A BCG strain (rBCG-SOCS1DN) that secreted a dominantly negative mutant variant of SOCS1 was obtained. It was shown that immunization with rBCG-SOCS1DN enhanced the activation of bone-marrow dendritic cells and T‑cells as compared to cells of the BCG control strain, and it increased the production of IFN-γ, TNF-α, and IL-6 cytokines. In addition, rBCG-SOCS1DN-immunized mice showed a significant decrease in the number of MTB colony-forming units (CFUs) in the lungs and spleen as compared to those in control mice immunized with BCG after infection with a highly pathogenic MTB strain. The obtained data lead to a new concept of the recombinant BCG vaccine as a tool for the immunomodulation of cytokines in host cells [59].
MTB vaccines. Attempts are being made to obtain a vaccine strain from MTB via its attenuation with the removal of the RD1 locus as a result of impaired cofactor synthesis (panCD) and amino-acid biosynthesis (leuD, lysA) [60]. The studied strains with various combinations of these mutations showed protection and immunogenicity in models with mice, guinea pigs and primates.
The most successful MTBVAC vaccine strain, MTB ΔphoP ΔfadD26, was a derivative of the MTB strain MT103. It was designed via the creation of two independent, stable genetic deletions in the phoP and fadD26 genes, which encode two main virulence factors [61]. The aforementioned phoP gene, which encode the transcription factor of the two-component virulence system PhoPR, and the gene fadD26, which is involved in the biosynthesis and export of phthiocerol dimicocerosates (PDIM), are the main genes associated with virulence of lipids of the MTB cell wall [62, 63]. In preclinical MTB evaluations, ΔphoP ΔfadD26 was comparable in safety to BCG but demonstrated better immunogenicity and protective efficacy [61, 64].
MTBVAC is currently the only live, attenuated, human pathogen–based vaccine that, after nearly 20 years of research, has successfully passed clinical trials as a prophylactic vaccine in newborns (instead of BCG) and as a prophylactic vaccine in adolescents and adults (vaccinated with BCG at birth). MTBVAC retains most of the T-cell epitopes described for TB, including the major immunodominant antigens, ESAT-6 and CFP-10 RD1. Studies have shown that MTBVAC-induced immunity to ESAT-6 and CFP-10 correlates with better protection than BCG [65].
Comparison of BCG and MTBVAC showed that the immunity associated with MTBVAC lasts longer than that with BCG with single-dose administration [66].
BCG delivery routes to the body. BCG vaccination is currently given subcutaneously, but there are other methods [67]: through mucous membranes (oral, intranasal, aerosol) or intravenously. Below, we will consider the possible impact of the vaccination route on its effectiveness.
Recent studies show a resurgence of interest in the oral administration of BCG; at one time, it was abandoned in favor of the subcutaneous due to the contamination of the vaccine with TB in Lübeck. However, some data demonstrate an increase in protective efficacy if oral administration is used together with traditional subcutaneous vaccination [68, 69].
Intranasal administration is another method of BCG delivery. This option of BCG administration to the body demonstrated sufficient efficacy in a study on mice [70, 71]. However, as compared to other pathways that also use the mucous membrane, intranasal administration has certain disadvantages. Thus, according to some reports, it is associated with a risk of the development of facial paralysis (Bell palsy) [72].
There is also aerosol administration of BCG. Thus, in macaques and guinea pigs, this pathway was shown to have greater protective properties than the subcutaneous pathway [73, 74]. A study demonstrated an increase in the immunoresponse with aerosol administration of the vaccine [75, 76]. BCG was also aerosolized in humans; this option may be promising [77].
Darra et al. studied the effect of the intravenous route of BCG administration on primates [78]. In this study, they showed the ability of BCG to provide a very high degree of protection with this delivery option: in six of ten macaques, the infection was not detected; nine of ten showed high security. Intravenous immunization induced significantly stronger T‑cell responses in the blood, spleen, and the bronchoalveolar lavage and lymph nodes of the lungs than other vaccination methods.
In summary of the data on various methods of BCG delivery into the body, it can be noted that an increase in protection against mycobacteria is observed with delivery through the mucous membranes in comparison with the currently accepted subcutaneous vaccination method; at the same time, intravenous vaccination appears to have an even stronger effect, but it is potentially more dangerous.
Use of BCG to fight other diseases. BCG vaccination is known to reduce mortality by about half in the first 6–12 months of life [79], most likely due to the stimulation of nonspecific immunity, which leads to increased resistance to respiratory diseases and sepsis in newborns [80, 81]. BCG vaccination was shown to give positive, nonspecific immune effects, leading to improved responses to other nonmycobacterial pathogens. In particular, BCG vaccination significantly increases the secretion of IL-1B, which plays an important role in antiviral immunity [82]. BCG-vaccinated mice were more resistant to smallpox virus, and they had an increased level of the production of gamma-interferon CD4+ cells [83]. The phenomenon of the immune effect after heterologous vaccination, called “trained immunity”, is supposedly caused by metabolic and epigenetic changes. It leads to the stimulation of the activity of the genetic regions encoding pro-inflammatory cytokines [84].
Due to its immunostimulating properties, BCG is a standard therapy for the prevention of relapse after surgery in the case of non-muscle-invasive bladder cancer [85]. BCG adminstration in the bladder promotes antitumor activity, probably due to stimulated recruitment of CD4+ T cells, neutrophils, and lymphocytes, as well as the activation of immune cells to eliminate cancer cells of urothelium infected with BCG [85–87]. Kanno et al. [88] obtained rBCG strains expressing Bordetella pertussis toxin (S1pt) for further use as an alternative immunotherapy for a model of bladder cancer in mice and have shown their great effectiveness in immunotherapy [88].
Current studies are aimed at the identification of clinical parameters and biomarkers that can predict an individual response to BCG therapy [89, 90]. Since side effects lead to the discontinuation of BCG therapy in some patients, BCG ΔureC :: hly is currently being tested as a replacement in phase II clinical trials.
Attempts are being made to treat melanoma with recombinant BCG strains [91]. The most successful were BCG variants expressing interleukin-2 (rBCG-IL-2) and granulocyte-macrophage colony stimulating factor (rBCG-GM-CSF).
Another possible application of BCG may be the creation of new HIV tools. Based on the vaccine strain MTBVAC described above, the recombinant strain MTBVAC.HIVA2auxo was created as a double vaccine against TB and HIV [92]. This vaccine against TB and HIV has similar protection to the parent strain MTBVAC for infection with MTB in mice and was safer than BCG and MTBVAC for mice with severe combined immunodeficiency. The MTBVAC.HIVA2auxo vaccine enhanced with the modified Ankara vaccinia virus (MVA). HIVA induced HIV-1 specific and MTB T-cell responses and HIV-1–specific CD8+ T cells.
Analysis of the current epidemiological data of COVID-19 infection revealed a correlation between BCG vaccination and a decrease in morbidity and mortality from COVID-19 worldwide [93]. It was found that countries that do not practice BCG vaccination (Italy, the Netherlands, United States) suffered more from the pandemic than countries with a universal and long-standing BCG vaccination policy. Countries with the late start of universal BCG vaccination (Iran, 1984) have higher mortality rates, which is consistent with the idea that BCG protects the vaccinated elderly population. Since BCG vaccination has been shown to provide broad protection against viral infections and sepsis [94], it is likely that the protective effect of BCG may not be directly related to protection against COVID-19. However, BCG vaccination was found to correlate with a decrease in the number of reported COVID-19 cases. This suggests that BCG may provide some protection directly from COVID-19. The widespread use of BCG vaccine in the population can reduce the number of carriers and, in combination with other measures, can slow or stop the spread of COVID-19.
A large-scale, phase III clinical study will show whether the recombinant BCG variant VPM1002 is effective against COVID-19. The study will be conducted on groups of older people and health workers, who are most at risk of the disease [95].
Thus, numerous studies show that BCG remains a promising basis for genetic modifications that can increase the effectiveness of the vaccine. In addition, the prospects for the use of BCG as an attenuated vector for various, including viral, antigens are based on the adjuvant properties of BCG, the sufficient capacity of its genome, and the intracellular localization of the pathogen. Due to its nonspecific immunotherapeutic effects, vaccination with BCG is a potentially new tool in the fight against COVID-19 and probably other infectious diseases.
FUNDING
This work was supported in part by the Russian Foundation for Basic Research (project no. 1901500149A).
COMPLIANCE WITH ETHICAL STANDARDS
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
REFERENCES
- 1.Global Tuberculosis Report, World Health Organization, Geneva, 2017.
- 2.Uplekar M., Weil D., Lonnroth K., Jaramillo E., Lienhardt C., Dias H. Lancet. 2015;385:1799–1801. doi: 10.1016/S0140-6736(15)60570-0. [DOI] [PubMed] [Google Scholar]
- 3.De Gijsel D., von Reyn C.F. Int. J. Infect. Dis. 2019;80:S6–S8. doi: 10.1016/j.ijid.2019.02.036. [DOI] [PubMed] [Google Scholar]
- 4.Lewis K., Reiling L., Guinn K., Hickey M., Smith S., Behr M., Sherman D. Bone. 2011;23:1–7. [Google Scholar]
- 5.Demangel C., Hinds J., Neyrolles O., Butcher P., Leclerc C., Cole S., Brosch R. Infect. Immun. 2006;74:88–98. doi: 10.1128/IAI.74.1.88-98.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simeone R., Bottai D., Brosch R. Curr. Opin. Microbiol. 2009;12:4–10. doi: 10.1016/j.mib.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Levi D.T., Obukhov Yu.I., Aleksandrova N.V., Volkova R.A., El’bert E.V. Al’vares, FigeroaM.V., Prokopenko, A.V., and Ludannyi, R.I., Bioprep., Profilakt., Diagn. . Lechenie. 2016;16:49–54. [Google Scholar]
- 8.Corran, P. and Griffiths, E., World Health Organization. WHO Technical Report Series no. 980, Genewa: Switzerland, 2012.
- 9.Behr M., Small P. Vaccine. 1999;17:915–922. doi: 10.1016/S0264-410X(98)00277-1. [DOI] [PubMed] [Google Scholar]
- 10.Fomukong N., Dale J., Osborn T., Grange J. J. Appl. Bacteriol. 1992;72:126–133. doi: 10.1111/j.1365-2672.1992.tb01813.x. [DOI] [PubMed] [Google Scholar]
- 11.Ahn S., Tran V., Leung A., Ng M., Li M., Liu J. Mol. Ther. 2018;26:2863–2874. doi: 10.1016/j.ymthe.2018.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun R., Skeiky Y., Izzo A., Dheenadhayalan V., Imam Z., Penn E. Vaccine. 2009;27:4412–4423. doi: 10.1016/j.vaccine.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 13.Abdallah A., Hill-Cawthorne G., Otto T., Coll F., Guerra-Assunção J., Gao G. Sci. Rep. 2015;5:1–15. doi: 10.1038/srep15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersen P., Kaufmann S. Cold Spring Harb. Perspect. Med. 2014;4:a018523. doi: 10.1101/cshperspect.a018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walters S.B., Dubnau E., Kolesnikova I., Laval F., Daffe M., Smith I. Mol. Microbiol. 2006;60:312–330. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 16.Flannagan R., Cosío G., Grinstein S. Nat. Rev. Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 17.Stanley S., Johndrow J., Manzanillo P., Cox J.S. J. Immunol. 2007;178:3143–3152. doi: 10.4049/jimmunol.178.5.3143. [DOI] [PubMed] [Google Scholar]
- 18.Van der Wel N., Hava D., Houben D., Fluitsma D., van Zon M., Pierson J., Brenner M., Peters P. Cell. 2007;129:1287–1298. doi: 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 19.Kaufmann S. Trends Immunol. 2012;33:373–379. doi: 10.1016/j.it.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Gengenbacher M., Nieuwenhuizen N.E., Kaufmann S. Curr. Opin. Immunol. 2017;47:8–16. doi: 10.1016/j.coi.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Baker J., Dechow S., Abramovitch R. Trends Microbiol. 2019;27:942–953. doi: 10.1016/j.tim.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prusa J., Zhu D., Stallings C. Pathog. Dis. 2018;76:1–13. doi: 10.1093/femspd/fty054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song H., Huff J., Janik K., Walter K., Keller C., Ehlers S. Mol. Microbiol. 2011;80:900–918. doi: 10.1111/j.1365-2958.2011.07619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukai T., Maeda Y., Tamura T., Miyamoto Y., Makino M. FEMS Immunol. Med. Microbiol. 2008;53:96–106. doi: 10.1111/j.1574-695X.2008.00407.x. [DOI] [PubMed] [Google Scholar]
- 25.Sendide K., Deghmane A., Reyrat J., Talal A., Hmama Z. Infect. Immun. 2004;72:4200–4209. doi: 10.1128/IAI.72.7.4200-4209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gengenbacher M., Kaufmann S. FEMS Microbiol. Rev. 2012;36:514–532. doi: 10.1111/j.1574-6976.2012.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saiga, H., Nieuwenhuizen, N., Gengenbacher, M., Koehler, A., Schuerer, S., Moura-Alves, P., et al., J. Infect. Dis., 2015, no. 1, pp. 1–25. [DOI] [PubMed]
- 28.Wang Y., Curry H., Zwilling B., Lafuse W. J. Immunol. 2005;174:5687–5694. doi: 10.4049/jimmunol.174.9.5687. [DOI] [PubMed] [Google Scholar]
- 29.Andersen P., Scriba T. Nat. Rev. Immunol. 2019;19:550–562. doi: 10.1038/s41577-019-0174-z. [DOI] [PubMed] [Google Scholar]
- 30.Fulton S., Reba S., Pai R., Pennini M., Torres M., Harding C., Boom W. Infect. Immun. 2004;72:2101–2110. doi: 10.1128/IAI.72.4.2101-2110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dorhoi A., Nouailles G., Jorg S., Hagens K., Heinemann E., Pradl L. Eur. J. Immunol. 2012;42:374–384. doi: 10.1002/eji.201141548. [DOI] [PubMed] [Google Scholar]
- 32.Kupz C., Er U., Staber M., Perdomo A., Dorhoi A., Brosch R., Kaufmann S. J. Clin. Invest. 2016;126:2109–2122. doi: 10.1172/JCI84978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tzelepis F., Verway M., Daoud J., Gillard J., Hassani-Ardakani K., Dunn J. J. Clin. Invest. 2015;125:752–768. doi: 10.1172/JCI77014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C., Huang D., Wang R., Shen L., Zeng G., Yao S. PLoS Pathogens. 2009;5:e1000392. doi: 10.1371/journal.ppat.1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindenstrom T., Aagaar C., Christensen D., Agger E., Andersen P. Eur. J. Immunol. 2014;44:1699–1709. doi: 10.1002/eji.201344358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu, L., Chung, A., Rosebrock, T., Ghebremichael, M., Yu, W., Grace, P., et al., Cell, vol. 167, no. 2, pp. 433–443. [DOI] [PMC free article] [PubMed]
- 37.Mavi, P.S., Singh, S., and Kumar, A., Antioxid. Redox Signal., 2019, pp. 1–59. 10.1089/ars.2019.7867
- 38.Groschel M., Sayes F., Simeone R., Majlessi L., Brosch R. Nat. Rev. Microbiol. 2016;14:677–691. doi: 10.1038/nrmicro.2016.131. [DOI] [PubMed] [Google Scholar]
- 39.Augenstreich J., Arbues A., Simeone R., Haanappel E., Wegener A., Sayes F. Cell. Microbiol. 2017;19:1–19. doi: 10.1111/cmi.12726. [DOI] [PubMed] [Google Scholar]
- 40.Tiwari, S., Casey, R., Goulding, C., Hingleywilson, S., and Jacobs, W., Microbiol. Spectr., 2019, vol. 7. 10.1128/microbiolspec [DOI] [PMC free article] [PubMed]
- 41.Collins A., Cai H., Li T., Franco L., Li X., Nair R. Cell Host Microbe. 2015;17:820–828. doi: 10.1016/j.chom.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wassermann R., Gulen M., Sala C., Perin S., Lou Y., Rybniker J. Cell Host Microbe. 2015;17:799–810. doi: 10.1016/j.chom.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 43.Bell S., MacDuff D., Kimmey J., Elie D., Olivas J. Cell Host Microbe. 2015;176:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong K., Jacobs W. Cell. Microbiol. 2011;13:1371–1384. doi: 10.1111/j.1462-5822.2011.01625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brodin P., Majlessi L., Brosch R., Smith D., Bancroft G., Clark S. J. Infect. Dis. 2004;190:115–122. doi: 10.1086/421468. [DOI] [PubMed] [Google Scholar]
- 46.Groschel M., Sayes F., Shin S., Frigui W., Pawlik A., Orgeur M. Cell Rep. 2017;18:2752–2765. doi: 10.1016/j.celrep.2017.02.057. [DOI] [PubMed] [Google Scholar]
- 47.Grode L., Ganoza C., Brohm C., Weiner J., Eisele B., Kaufmann S. Vaccine. 2013;31:1340–1348. doi: 10.1016/j.vaccine.2012.12.053. [DOI] [PubMed] [Google Scholar]
- 48.Reyrat J., Berthet F., Gicquel B. Proc. Natl. Acad. Sci. U. S. A. 1995;92:8768–8772. doi: 10.1073/pnas.92.19.8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon M. D’Arcy, HartA., and Young, P. Nature. 1980;286:79–80. doi: 10.1038/286079a0. [DOI] [PubMed] [Google Scholar]
- 50.Hamon M.A., Ribet D., Stavru F., Cossart P. Trends Microbiol. 2012;20:360–368. doi: 10.1016/j.tim.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 51.Shaughnessy L., Hoppe A., Christensen K., Swanson J. Cell. Microbiol. 2006;8:781–792. doi: 10.1111/j.1462-5822.2005.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nieuwenhuizen N., Kulkarni P., Shaligram U., Cotton M., Rentsch C., Eisele B., Grode L., Kaufmann S. Front. Immunol. 2017;8:1–9. doi: 10.3389/fimmu.2017.01147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao M., Vogelzang A., Kaiser P., Schuerer S., Kaufmann S., Gengenbacher M. PLoS One. 2013;8:1–10. doi: 10.1371/annotation/c381f4a0-efa6-41e5-8bb5-172c714510b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gengenbacher M., Vogelzang A., Schuerer S., Lazar D., Kaiser P., Kaufmann S. MBio. 2014;5:1–8. doi: 10.1128/mBio.01262-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gengenbacher M., Nieuwenhuizen N., Vogelzang A., Liu H., Kaiser P., Schuerer S. MBio. 2016;7:1–10. doi: 10.1128/mBio.00679-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun R., Skeiky Y., Izzo A., Dheenadhayalan V., Imam Z., Penn E. Vaccine. 2009;27:4412–4423. doi: 10.1016/j.vaccine.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 57.Sander P., Clark S., Petrera A., Vilaplana C., Meuli M., Selchow P. Vaccine. 2015;3:1353–1359. doi: 10.1016/j.vaccine.2015.01.058. [DOI] [PubMed] [Google Scholar]
- 58.Paolino M., Brindisi M., Vallone A., Butini S., Campiani G., Nannicini C. ChemMedChem. 2018;13:422–430. doi: 10.1002/cmdc.201700759. [DOI] [PubMed] [Google Scholar]
- 59.Mizuno S., Soma S., Inada H., Kanuma T., Matsuo K., Yasutomi Y. J. Immunol. 2019;203:188–197. doi: 10.4049/jimmunol.1800694. [DOI] [PubMed] [Google Scholar]
- 60.Sambandamurthy V., Derrick S., Jalapathy K., Chen B., Russell R., Morris S., Jacobs W. Infect. Immun. 2005;73:1196–1203. doi: 10.1128/IAI.73.2.1196-1203.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arbues A., Aguilo J., Gonzalo-Asensio J., Marinova D., Uranga S., Puentes E. Vaccine. 2013;31:4867–4873. doi: 10.1016/j.vaccine.2013.07.051. [DOI] [PubMed] [Google Scholar]
- 62.Cox J., Chess B., McNeil M., Jacobs W. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 63.Kirksey M., Tischler A., Simeone R., Hisert K., Uplekar S., Guilhot C., McKinney J. Infect. Immun. 2011;79:2829–2838. doi: 10.1128/IAI.00097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aguilo N., Uranga S., Marinova D., Monzon M., Badiola J., Martin C. Tuberculosis. 2016;96:71–74. doi: 10.1016/j.tube.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalo-Asensio J., Marinova D., Martin C., Aguilo N. Front. Immunol. 2017;8:1–8. doi: 10.3389/fimmu.2017.01803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clark, S., Lanni, F., Marinova, D., Rayner, E., Martin, C., and Williams, A., J. Infect. Dis., vol. 216, no. 5, pp. 525–533. [DOI] [PubMed]
- 67.Tanner R., Villarreal-Ramos B., Vordermeier H., McShane H. Front. Immunol. 2019;10:5–7. doi: 10.3389/fimmu.2019.01317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoft D., Brown R., Belshe R. Clin. Infect. Dis. 2000;30:217–S222. doi: 10.1086/313864. [DOI] [PubMed] [Google Scholar]
- 69.Monteiro-Maia R., de Pinho R. Mem. Inst. Oswaldo Cruz. 2014;109:838–845. doi: 10.1590/0074-0276140091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Falero-Diaz G., Challacombe S., Banerjee D., Douce G., Boyd A., Ivanyi J. Vaccine. 2000;18:3223–3229. doi: 10.1016/S0264-410X(00)00134-1. [DOI] [PubMed] [Google Scholar]
- 71.Lyadova I., Vordermeier H., Eruslanov E., Khaidukov S., Apt A., Hewinson R. Clin. Exp. Immunol. 2001;126:274–279. doi: 10.1046/j.1365-2249.2001.01667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mutsch M., Zhou W., Rhodes P., Bopp M., Chen R., Linder T., Spyr C., Steffen R. N. Engl. J. Med. 2004;350:896–903. doi: 10.1056/NEJMoa030595. [DOI] [PubMed] [Google Scholar]
- 73.Barclay W., Busey W., Dalgard D., Good R., Janicki B., Kasik J. Am. Rev. Respir. Dis. 1973;107:351–358. doi: 10.1164/arrd.1973.107.3.351. [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Contreras L., Wong Y., Muttil P., Padilla D., Sadoff J., De Rousse J. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4656–4660. doi: 10.1073/pnas.0800043105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dijkman K., Sombroek C., Vervenne R., Hofman S., Boot C., Remarque E. Nat. Med. 2019;25:255–262. doi: 10.1038/s41591-018-0319-9. [DOI] [PubMed] [Google Scholar]
- 76.Scriba T., Nemes E. Nat. Med. 2019;25:199–201. doi: 10.1038/s41591-019-0347-0. [DOI] [PubMed] [Google Scholar]
- 77.Thomas Z., McShane H. Trans. R. Soc. Trop. Med. Hyg. 2014;109:175–181. doi: 10.1093/trstmh/tru206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Darrah P., Zeppa J., Maiello P., Hackney J., Wadsworth M., Hughes T. Nature. 2020;577:95–102. doi: 10.1038/s41586-019-1817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Higgins, J., Soares-Weiser, K., Lopez-Lopez, J., Kakourou, A., Chaplin, K., Christensen, H., et al., BMJ, 2016, vol. 355. [DOI] [PMC free article] [PubMed]
- 80.Aaby P., Whittle H., Benn C. BMJ. 2012;345:1–6. doi: 10.1136/bmj.e3769. [DOI] [PubMed] [Google Scholar]
- 81.Aaby P., Kollmann T., Benn C. Nat. Immunol. 2014;15:895–899. doi: 10.1038/ni.2961. [DOI] [PubMed] [Google Scholar]
- 82.Kleinnijenhuis J., Quintin J., Preijers F., Benn C., Joosten L., Jacobs C. J. Innate Immun. 2014;6:152–158. doi: 10.1159/000355628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mathurin K., Martens G., Kornfel H., Welsh R. J. Virol. 2009;83:3528–3539. doi: 10.1128/JVI.02393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Netea, M., Joosten, L., Latz, E., Mills, K., Natoli, G., Stunnenberg, H., O’Neill, L., and Xavier, R., Science, 2016, vol. 352, no. 6284. [DOI] [PMC free article] [PubMed]
- 85.Zheng Y., Naguib Y., Dong Y., Shi Y., Bou S., Cui Z. Expert Rev. Vaccines. 2015;14:1255–1275. doi: 10.1586/14760584.2015.1068124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pichler R., Fritz J., Zavadil C., Schafer G., Culig Z., Brunner A. Oncotarget. 2016;7:39916–39930. doi: 10.18632/oncotarget.9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suttmann H., Riemensberger J., Bentien G., Schmaltz D., Stockle M., Jocham D., Bohle A., Brandau S. Cancer Res. 2006;66:8250–8257. doi: 10.1158/0008-5472.CAN-06-1416. [DOI] [PubMed] [Google Scholar]
- 88.Kanno A., Goulart C., Leite L., Pagliarone A., Nascimento I. Biomed. Res. Int. 2019;2019:9630793. doi: 10.1155/2019/9630793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Steinberg R., Thomas L., Mott S., O’Donnell M. Bl. Cancer. 2016;2:215–224. doi: 10.3233/BLC-150039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang N., Jiang G., Liu X., Na R., Wang X., Xu J. Biomed Res. Int. 2016;2016:9859021. doi: 10.1155/2016/9859021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Benitez M., Bender C., Oliveira T., Schachtschneider K., Collares T., Seixas F. Appl. Microbiol. Biotechnol. 2019;103:7903–7916. doi: 10.1007/s00253-019-10057-0. [DOI] [PubMed] [Google Scholar]
- 92.Broset E., Saubi N., Guitart N., Aguilo N., Uranga S., Kilpelainen A. Mol. Ther.—Methods Clin. Dev. 2019;13:253–264. doi: 10.1016/j.omtm.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miller, A., Reandelar, M., Fasciglione, K., Roumenova, V., Li, Y., and Otazu, G., medRxiv (preprint), 2020. 10.1101/2020.03.24.20042937
- 94.Moorlag S., Arts R., van Crevel R., Netea M. Clin. Microbiol. Infect. 2019;25:1473–1478. doi: 10.1016/j.cmi.2019.04.020. [DOI] [PubMed] [Google Scholar]
- 95.Max Planck Society, 2020. https://medicalxpress.com/ news/2020-03-vaccinebcg-immune-boost-coronavirus. html.