

Posaconazole (PCZ) is a clinically approved drug used predominantly for prophylaxis and salvage therapy of fungal infections. Here, we report its previously undescribed anti-human cytomegalovirus (HCMV) activity. By using antiviral assays, we demonstrated that PCZ, along with other azolic antifungals, has a broad anti-HCMV activity, being active against different strains, including low-passage-number clinical isolates and strains resistant to viral DNA polymerase inhibitors. Using a pharmacological approach, we identified the inhibition of human cytochrome P450 51 (hCYP51), or lanosterol 14α demethylase, a cellular target of posaconazole in infected cells, as a mechanism of anti-HCMV activity of the drug.
KEYWORDS: HCMV, antiviral, posaconazole, human CYP51, drug repurposing, synergism, antiviral agents, human cytomegalovirus
ABSTRACT
Posaconazole (PCZ) is a clinically approved drug used predominantly for prophylaxis and salvage therapy of fungal infections. Here, we report its previously undescribed anti-human cytomegalovirus (HCMV) activity. By using antiviral assays, we demonstrated that PCZ, along with other azolic antifungals, has a broad anti-HCMV activity, being active against different strains, including low-passage-number clinical isolates and strains resistant to viral DNA polymerase inhibitors. Using a pharmacological approach, we identified the inhibition of human cytochrome P450 51 (hCYP51), or lanosterol 14α demethylase, a cellular target of posaconazole in infected cells, as a mechanism of anti-HCMV activity of the drug. Indeed, hCYP51 expression was stimulated upon HCMV infection, and the inhibition of its enzymatic activity by either the lanosterol analog VFV {(R)-N-(1-(3,4′-difluoro-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide} or PCZ decreased HCMV yield and infectivity of released virus particles. Importantly, we observed that the activity of the first-line anti-HCMV drug ganciclovir was boosted tenfold by PCZ and that ganciclovir (GCV) and PCZ act synergistically in inhibiting HCMV replication. Taken together, these findings suggest that this clinically approved drug deserves further investigation in the development of host-directed antiviral strategies as a candidate anti-HCMV drug with a dual antimicrobial effect.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous beta-herpesvirus that infects from 60% to nearly 80% of the human population worldwide and establishes a life-long persistence in the host, characterized by sporadic reactivations in healthy individuals (1). HCMV is also a major opportunistic human pathogen which causes life-threatening diseases in subjects with acquired or developmental immunodeficiency, such as transplant recipients and immune-naive fetuses. Indeed, HCMV causes deafness and neurological disorders in approximately 0.1% of congenital infections (2). Moreover, HCMV has been suggested as a cofactor in vascular diseases and immune senescence (3).
To date, there is no vaccine available, and only a limited number of drugs are licensed for treatment: ganciclovir (GCV), its oral prodrug valganciclovir, foscarnet, acyclovir, its prodrug valacyclovir, cidofovir, and the recently approved letermovir (4, 5). Currently, all the available anti-HCMV drugs target virus-encoded proteins, i.e., the DNA polymerase and the terminase (5). The clinical use of these antivirals can have several drawbacks, such as an unfavorable safety profile characterized by severe acute or long-term toxicity and poor oral bioavailability (4). Moreover, no drug has been approved for the treatment of congenital infection (6), and the management of HCMV infections can be further complicated by the emergence of drug-resistant HCMV strains (7, 8). For all these reasons, there is still a strong need to develop new, safe, and effective antiviral compounds, possibly endowed with a new mechanism of action.
HCMV, like many other viruses, hijacks several cellular pathways and deeply alters many host physiological processes in order to replicate efficiently (9). Thus, the development of host-directed antiviral strategies could be an alternative or an additional approach to treat HCMV-associated diseases (10), as well as to overcome viral resistance issues. In this regard, drug repurposing can represent a powerful strategy for the identification of host pathways and factors playing a role during viral replication (11–13). Based on this rationale, we followed this strategy to identify new compounds and targets for anti-HCMV intervention through a drug screening and follow-up studies that identified several novel HCMV inhibitors, including both approved drugs and natural compounds (12, 14, 15).
Here, we report the discovery of the previously undescribed anti-HCMV activity of posaconazole (PCZ), a drug approved for the prophylaxis and salvage therapy of systemic fungal infections. Our study stems from the observation that drugs belonging to the class of azolic antifungals were identified in two previous independent drug repurposing screenings for inhibitors of HCMV replication (12, 16, 17). Starting from this observation, we initially evaluated the antiviral activity of a panel of azolic antifungals used both topically and for the treatment of invasive fungal infections. PCZ, which was not present in our original screened library, showed an interesting anti-HCMV activity; thus, we decided to characterize further its antiviral mechanism. In recent years, the antiviral properties of azolic antifungals have been an emerging topic, and PCZ, together with itraconazole, was found to have activity against both positive-stranded RNA viruses, such as picornaviruses and flaviviruses (11, 18, 19), and negative-stranded RNA viruses, such as Ebola virus and influenza virus (20, 21).
We investigated more deeply the anti-HCMV activity of PCZ and found that its antiviral activity was related to its inhibitory activity against human cytochrome P450 51 (hCYP51, or lanosterol 14α demethylase). This enzyme is the most conserved cytochrome P450, as well as the only one that catalyzes demethylation of lanosterol (22), being involved in both cholesterogenesis and synthesis of essential regulatory sterols during meiosis (23). Fungal CYP51 is the target of PCZ and other antifungals in the treatment of mycosis; however, our study suggests that human CYP51 is involved in the antiviral mechanism of PCZ against HCMV.
Our data indicate that a combination of PCZ with GCV, the gold standard for anti-HCMV therapy, greatly potentiates the antiviral effect of the latter and is synergistic in inhibiting HCMV replication in infected cells. Thus, this study provides the rationale for a possible clinical validation of the anti-HCMV activity of PCZ alone or in combination with GCV.
RESULTS
Anti-human cytomegalovirus activity of approved azolic antifungals.
In a drug repurposing campaign, we previously identified a series of molecules endowed with anti-HCMV activity, among which we noticed an overrepresentation of drugs belonging to the class of azolic antifungals (12). To extend this knowledge, a more complete panel of azolic antifungal drugs was analyzed for anti-HCMV activity (Table 1). As shown in Fig. 1A, in plaque reduction assays (PRAs), posaconazole (PCZ) and ketoconazole (KTZ) showed a dose-dependent inhibitory effect on HCMV AD169 replication in human foreskin fibroblast (HFFs), similar to what we previously reported for clotrimazole, econazole, and miconazole (12). On the other hand, under the same experimental conditions, fluconazole (FCZ), voriconazole (VCZ), and itraconazole (ITZ) did not exhibit significant anti-HCMV activity (Fig. 1B).
TABLE 1.
Antiviral activity of antifungal drugs against HCMV AD169
| Compound (abbreviation) | EC50 (μM)a | CC50 (μM)b | SIc |
|---|---|---|---|
| Clotrimazole (CTZ)d | 0.8 ± 0.6 | 40 ± 28 | 50 |
| Econazole (ECZ)d | 5.7 ± 1.7 | 85 ± 13 | 17 |
| Fluconazole (FCZ) | >25 | >250 | >10 |
| Itraconazole (ITZ) | >10 | NDe | ND |
| Ketoconazole (KTZ) | 14.4 ± 4.6 | 116 ± 6 | 8 |
| Miconazole (MCZ)d | 3.2 ± 0.8 | 55 ± 16 | 17 |
| Posaconazole (PCZ) | 3.3 ± 0.5 | >250 | >76 |
| Voriconazole (VCZ) | >25 | >250 | >10 |
| Ganciclovir (GCV) | 2.3 ± 0.6 | >500 | >217 |
Compound concentration that inhibited 50% of plaque formation, as determined by PRAs against HCMV AD169 in HFFs. Values are means ± SD of data derived from at least 3 independent experiments in duplicate. GCV was included as a positive control.
Compound concentration that produced 50% cytotoxicity, as determined by MTT assays in HFFs. Values are means ± SD of data derived from two independent experiments in duplicate.
SI, selectivity index (determined as the ratio between CC50 and EC50).
Reported in reference 12.
ND, not determined.
FIG 1.

Susceptibility of HCMV to approved azolic antifungals. Plaque reduction assays were performed in HFFs infected with HCMV and treated with different doses (from 0.1 to 25 μM) of the indicated test compounds or GCV as a control. (A) Dose-dependent inhibition of HCMV AD169 replication by PCZ and KTZ. (B) Absence of significant inhibition of HCMV AD169 replication by FCZ, ITZ, and VCZ. (ITZ could be tested only up to 10 μM due to solubility issues.) Data are means ± standard deviations (SD) from at least 3 independent experiments carried out in duplicate.
Then, to exclude the possibility that the antiviral activity of PCZ might be due to cytotoxicity, its effect on the viability of uninfected HFFs was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assays. As reported in Table 1, we found that the antiviral activity of posaconazole was not due to cytotoxicity of the target cells, since a significant toxic effect was not observed at concentrations up to 250 μM.
Broad anti-HCMV activity of posaconazole and other antifungals.
To investigate the spectrum of anti-HCMV activity of PCZ and of other antifungals, we repeated the antiviral assays with a panel of HCMV strains, including three different low-passage-number clinical isolates (TB40-UL32-EGFP, VR1814, and 388438U). As reported in Fig. 2, the anti-HCMV activity of PCZ was not dependent on the viral strain, since the 50% effective concentrations (EC50s) obtained with different HCMV strains were comparable (Table 2). Next, we evaluated the activity of PCZ against HCMV strains resistant to the available viral DNA polymerase inhibitors, as the emergence of drug resistance is an increasing cause of transplant failure associated with HCMV infections, in particular after prolonged antiviral therapy (24). PCZ fully inhibited the replication of viruses with mutations in the UL54 gene conferring cross-resistance to GCV and cidofovir or to foscarnet and acyclovir (strains 759rD100 and PFArD100, respectively) (Table 2). Anti-HCMV activity against different strains was also found for miconazole (MCZ) and econazole (ECZ) (Table 2), two other antifungal drugs which emerged as having anti-HCMV activity during the drug repurposing screening (12).
FIG 2.

Dose-dependent inhibition of the replication of indicated HCMV strains by posaconazole. Plaque reduction assays were performed in HFFs infected with the indicated HCMV strains and treated with different doses (from 0.1 to 25 μM) of PCZ. Data are means ± SD from 3 independent experiments carried out in duplicate.
TABLE 2.
Antiviral activities of antifungal drugs against different HCMV strains in different cell types
| HCMV strain | Cell type | Antiviral activity EC50 (μM)a |
|||
|---|---|---|---|---|---|
| ECZ | MCZ | PCZ | Controlb | ||
| TB40-UL32-EGFP | HFF | 4.5 ± 0.7 | 3.8 ± 0.4 | 3.5 ± 1.8 | 3.5 ± 0.7 |
| VR1814 | HFF | 3.9 ± 2.1 | 4.3 ± 1.1 | 3.5 ± 0.7 | 2.3 ± 1.3 |
| 388438U | HFF | NDc | ND | 3.2 ± 0.4 | 3.2 ± 0.8 |
| 759rD100 | HFF | 4.3 ± 1.1 | 4.5 ± 1.8 | 2.6 ± 1.2 | 75 ± 12 |
| GDGrP53 | HFF | 3.4 ± 0.9 | 2.6 ± 0.3 | 3.5 ± 1.1 | 1.9 ± 1.8 |
| PFArD100 | HFF | 4.2 ± 0.1 | 3.7 ± 0.6 | 4.8 ± 1.5 | 250 ± 18 |
| TR | HFF | ND | ND | 3.7 ± 1.3 | ND |
| TR | ARPE-19 | ND | ND | 4.2 ± 1.8 | ND |
| TR | HMVEC | ND | ND | 4.8 ± 2.0 | ND |
Compound concentration that inhibited 50% of plaque formation, as determined by PRAs. Values are means ± SD of data derived from at least 3 independent experiments in duplicate.
GCV was used as a control for all strains except for PFArD100, for which FOS was used.
ND, not determined.
Finally, the inhibitory activity of PCZ against HCMV was found not to be cell type dependent, since the EC50s measured for strain TR in epithelial (EC50 = 4.2 μM) and endothelial (EC50 = 4.8 μM) cells were in keeping with that observed in fibroblasts (EC50 = 3.7 μM) (Table 2). Moreover, since strain TR is naturally resistant to GCV and CDV, we obtained further evidence of the activity of PCZ against HCMV clinical strains that are resistant to DNA polymerase inhibitors.
Taken together, these results indicated that PCZ and other antifungals could have a mechanism of action different from that of anti-HCMV drugs targeting the viral DNA polymerase.
Effects of PCZ treatment on HCMV yield.
PCZ is routinely used as a systemic drug to prevent and treat fungal infections in immunosuppressed patients (25); thus, we decided to focus on this molecule, given that a potential dual antifungal and antiviral effect could be relevant in this clinical setting. To better characterize the anti-HCMV activity of PCZ, we also evaluated its effects on the production of virus progeny in a multicycle viral growth experiment. PCZ reduced the production of infectious HCMV progeny in a dose-dependent manner, as revealed by virus yield reduction assays (Table 3). In sum, we observed that treatment of HCMV-infected cells with PCZ after infection resulted in the inhibition of viral replication and progeny production.
TABLE 3.
Activity of posaconazole and VFV in virus yield reduction assays and in enzymatic assays in vitro
| Compound | Antiviral activity (μM) |
IC50 (μM)c | |
|---|---|---|---|
| EC50a | EC90b | ||
| PCZ | 2.2 ± 1.1 | 7.1 ± 1.1 | 8.0 ± 2.5 |
| VFV | 1.2 ± 0.9 | 10.1 ± 1.3 | 0.5 ± 0.2 |
| VCZ | NDd | ND | >100 |
Compound concentration that inhibits 50% of virus yield, as determined by titration in HFFs. Values are means ± SD of data derived from 4 independent experiments in duplicate.
Compound concentration that inhibits 90% of virus yield, as determined by titration in HFFs. Values are means ± SD of data derived from 4 independent experiments in duplicate.
Compound concentration that causes a 50% decrease in the rate of lanosterol conversion, as determined by reconstitution of the hCYP51 activity in vitro (2-min reaction). Values are means ± SD of data derived from 3 independent experiments in duplicate and calculated using GraphPad Prism 6.0 (dose response – inhibition).
ND, not determined.
Inhibition of human CYP51, a host target of posaconazole, correlates with anti-HCMV activity.
PCZ is known to interfere with different host pathways (26–29), among which are cholesterol homeostasis and synthesis, which are mediated by human CYP51, the rate-limiting enzyme in late-stage cholesterogenesis (28–30). Thus, we exploited VFV {(R)-N-(1-(3,4′-difluoro-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide)}, the most potent enzymatic inhibitor of hCYP51 identified so far (30), to investigate the involvement of hCYP51 during HCMV replication. Like other azolic compounds, VFV inhibits human CYP51 by binding in the enzyme active site and competing with the substrate lanosterol. In enzymatic assays in vitro, both VFV and PCZ inhibited the initial rate of lanosterol conversion catalyzed by purified hCYP51, with 50% inhibitory concentrations (IC50s) of 0.5 and 8 μM, respectively (Fig. 3A and Table 3). This was not observed with VCZ (IC50 >100 μM), which served as a negative control in the enzymatic assays (Fig. 3A and Table 3). When tested by PRAs, VFV had a dose-dependent inhibitory effect on HCMV replication (Fig. 3B), albeit with an EC50 higher than that of PCZ (13.3 μM versus 3.3 μM). However, given the highly hydrophobic nature of VFV (logP, 5.4), we reasoned that VFV could be poorly soluble in the semisolid medium used for PRAs; therefore, virus yield reduction assays with VFV were also performed. Indeed, VFV showed a strong and dose-dependent inhibitory effect on infectious virus production, with an EC50 in the low-micromolar range (Fig. 3C and Table 3), thus suggesting that hCYP51 enzymatic activity is required for the production of infectious HCMV progeny.
FIG 3.
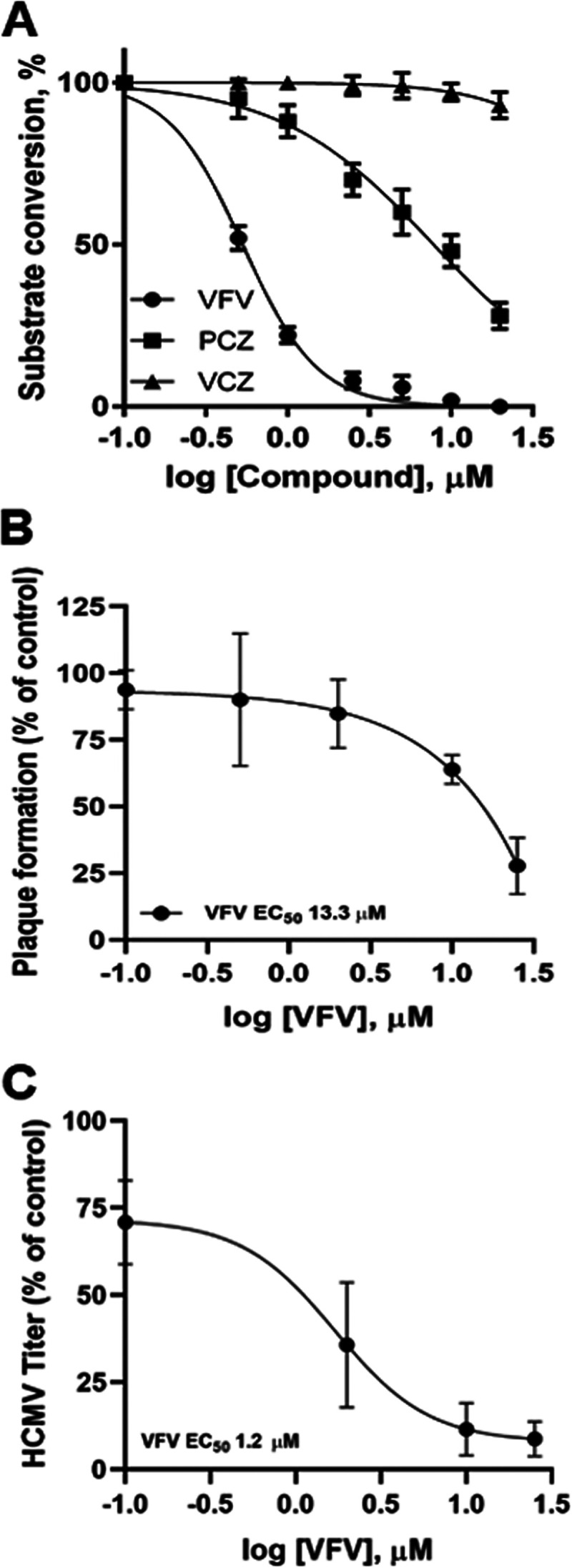
Inhibition of host hCYP51 affects HCMV replication. (A) Inhibition of enzymatic activity of purified hCYP51 by VFV, PCZ, and VCZ (2-min reaction). Data are means ± SD from 3 independent experiments carried out in duplicate. (B) Plaque reduction assays were performed in HFFs infected with HCMV AD169 and treated with different doses (from 0.1 to 25 μM) of hCYP51 inhibitor VFV. Data are means ± SD from at least 3 independent experiments carried out in duplicate. (C) Dose-dependent inhibition by VFV of virus progeny production in HCMV-infected HFFs as determined by virus yield reduction assays. Data are means ± SD from 4 independent experiments carried out in duplicate.
HCMV infection activates hCYP51 expression.
Since we observed that hCYP51 enzymatic activity is required for HCMV replication, next we investigated whether its expression could be modulated during viral infection. Indeed, under normal conditions (i.e., cells grown in regular cholesterol-containing medium), the cholesterol-biosynthetic pathway is not stimulated. To analyze the modulation of the hCYP51 promoter upon HCMV infection, we transfected permissive U-373 MG cells with a plasmid carrying a reporter gene under the control of the hCYP51 promoter and then infected them with HCMV. As shown in Fig. 4A, infection with HCMV activated the hCYP51 promoter by about 40-fold. This upregulation was not observed when the transfected cells were infected with a UV-inactivated virus, which is able to bind and enter the cells but not to express viral genes, arguing that de novo-synthesized HCMV proteins are required for hCYP51 promoter activation (Fig. 4A). Upon infection of serum-fed HFFs, an ∼2-fold increase in hCYP51 mRNA level was observed during the early phase of HCMV replication (Fig. 4B), and an accumulation of hCYP51 protein throughout the virus cycle was accordingly detected by both Western blotting (Fig. 4C and Fig. S1) and immunofluorescence analysis in living cells (Fig. S2). Altogether, these data support the view that HCMV infection activates hCYP51 gene expression.
FIG 4.

hCYP51 expression is activated during HCMV infection. (A) Activation of the hCYP51 promoter in U-373 MG cells that were mock infected or infected with either HCMV AD169 or UV-inactivated HCMV. Data are expressed in relative luciferase units (RLU), which are the luciferase units normalized to the fluorescence units derived from the expression of the cotransfected eGFP reporter gene. Data are means ± SD from 3 independent experiments carried out in duplicate. Data were analyzed by a one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison tests. ***, P ≤ 0.0001; ****, P < 0.0001. (B) Analysis of hCYP51 mRNA levels upon HCMV infection of HFFs determined by qPCR at the indicated times. The mRNA levels of UL54 were detected as a control for the progression of the infection at 24 and 48 h p.i. mRNA levels were normalized to cellular GAPDH, and gene expression was reported as relative quantification (RQ) compared to calibrator sample (mock-infected cells for hCYP51 and HCMV-infected cells at 24 h p.i. for UL54). Data are means and SD from 3 independent experiments carried out in duplicate. (C) Analysis of hCYP51 and viral protein expression during HCMV replication. Host hCYP51 protein and viral IE antigens (IEA) were detected by Western blotting both in mock-infected HFFs (M) and in HFFs infected with HCMV at an MOI of 0.5 PFU/cell at the indicated times p.i. Detection of host β-actin was used as a loading control. Molecular weights (in kilodaltons) are indicated on the left. An image from a representative experiment is shown.
Inhibition of hCYP51 enzyme during HCMV replication reduces the infectivity of viral progeny.
As shown above, both VFV and PCZ inhibited the hCYP51 enzyme, which catalyzes the rate-limiting step in late cholesterogenesis. Thus, treatment of HCMV-infected cells with these inhibitors should reduce de novo cholesterol synthesis during HCMV replication. The intracellular presence of cholesterol during HCMV infection and within the virus particle has been related to the infectivity of HCMV virions (31). We therefore hypothesized that the virus particles produced in cells treated with hCYP51 inhibitors might be less infective. To test this, we determined the particle-to-PFU ratio of cell-free HCMV released from infected HFFs treated with PCZ, VFV, or DMSO as a control, assuming that each virus particle (infectious or defective) contained one genome. Treatment of HCMV-infected cells for 120 h with either PCZ or VFV significantly reduced both the number of HCMV genomes (Fig. 5A) and the infectivity of viral progeny, with an ∼7- to 10-fold increase in particle-to-PFU ratio with respect to dimethyl sulfoxide (DMSO)-treated infected cells (Fig. 5B). Altogether, the results from these experiments indicated that hCYP51 enzymatic activity is required in the context of productive HCMV replication and could contribute to the generation of infectious HCMV particles.
FIG 5.
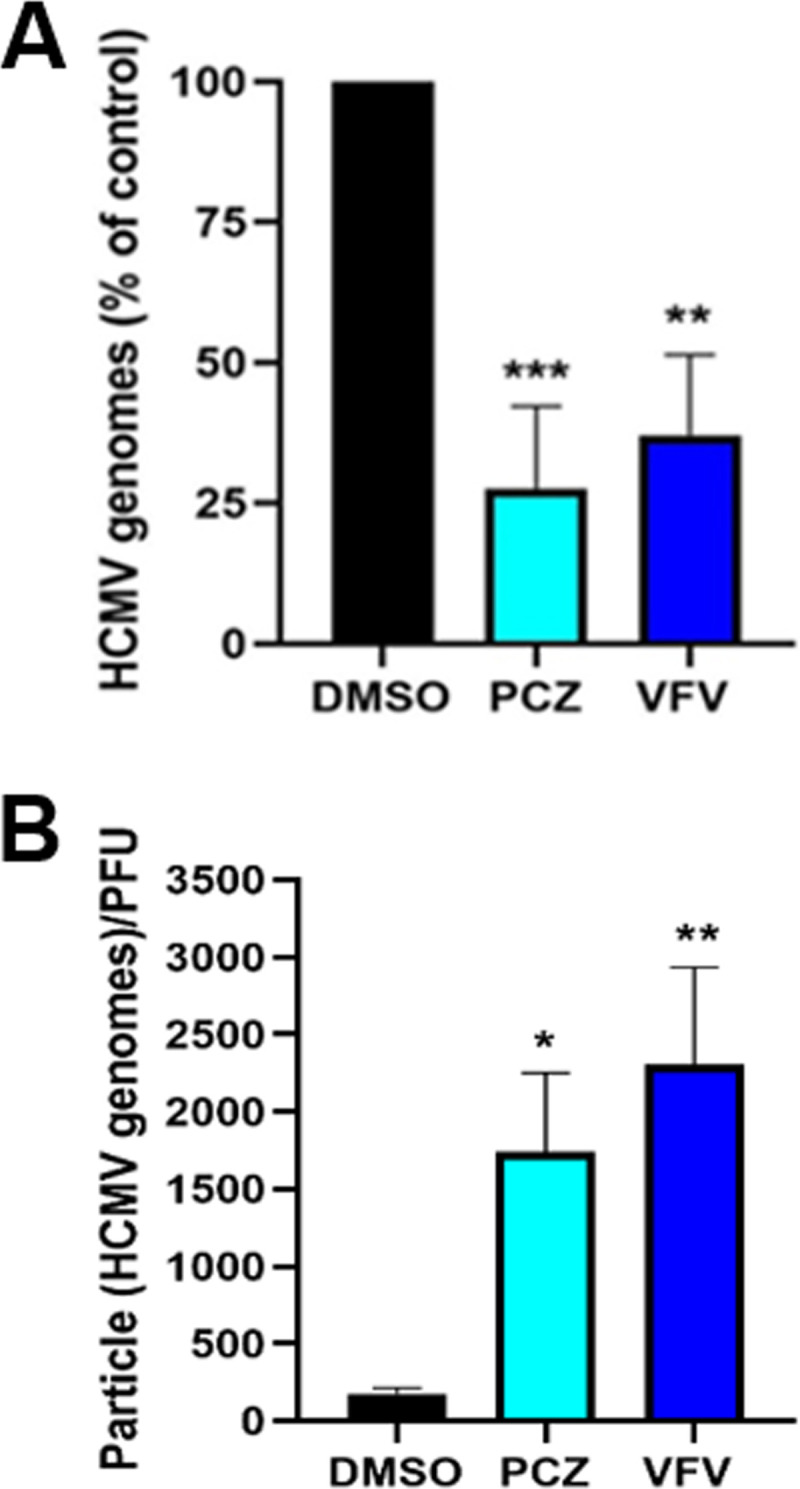
Effects of hCYP51 enzymatic activity inhibition on HCMV replication and infectivity. (A) Pharmacological inhibition of hCYP51 reduces the number of HCMV genomes. HFFs infected with HCMV at an MOI of 0.5 PFU/cell were treated with 10 μM PCZ or VFV or 0.1% DMSO as a control for 120 h. Numbers of HCMV genome copies in the supernatant of each sample were then determined by qPCR. Data are means and SD from 4 independent experiments carried out in quadruplicate. Data were analyzed by a one-way ANOVA followed by Dunnett’s multiple-comparison test. ***, P < 0.001, and **, P < 0.005, compared to the control (DMSO-treated, infected sample). (B) Pharmacological inhibition of hCYP51 reduces the infectivity of viral particles. Particle-to-PFU ratios were obtained by dividing the number of HCMV particles collected from supernatants derived from HFFs infected at an MOI of 0.5 PFU/cell and treated with test compounds (determined by qPCR) by the viral titers obtained in the same sample volume (determined by titration on fresh monolayers). Data are means and SD from 3 independent experiments carried out in quadruplicate. Data were analyzed by a one-way ANOVA followed by Dunnett’s multiple-comparison tests. *, P < 0.05, and **, P < 0.005, compared to the control (DMSO-treated, infected sample).
GCV and PCZ act synergistically against HCMV replication in infected cells.
We also used PRAs to test the antiviral efficacy of GCV in the absence or the presence of a dose of PCZ (3 μM) approximately equivalent to the mean concentration found in plasma by therapeutic drug monitoring of patients treated with PCZ (25). It is noteworthy that, in the presence of PCZ, GCV was ~10-fold more potent against HCMV than in the presence of the vehicle DMSO (EC50, 0.13 μM for the combination of GCV plus PCZ versus 1.44 μM for GCV plus DMSO) (Fig. 6). In light of these results, we investigated further the effects of the combination of PCZ with GCV against the replication of HCMV AD169 by PRAs. As reported in Table 4, the combination of GCV and PCZ resulted in antiviral synergism at all drug combinations tested, since the combination index (CI) values calculated by applying the method of Chou (32) were all <0.9 (Table 4). We did not observe any evident cell cytotoxicity when the drugs were tested in combination; thus, the synergistic effect on the reduction of viral plaque number was most likely the result of combining two drugs with different targets and mechanisms of action. Beside a reduction in the absolute number, we also observed a clear reduction in the size of the plaques when GCV was used in combination with PCZ. The calculated dose reduction indexes are reported in Table S3. Although future trials will be required to validate this observation in a clinical setting, these results suggest that a combination of GCV and PCZ might represent a new therapeutic strategy for the management of HCMV infections.
FIG 6.

A therapeutic dose of posaconazole enhances anti-HCMV activity of ganciclovir. Antiviral efficacy of GCV against HCMV AD169 in the absence (+ DMSO) or the presence (+ PCZ) of 3 μM PCZ was determined by PRA. Data are means ± SD from at least 3 independent experiments carried out in duplicate.
TABLE 4.
Analysis of the effects of the combination of GCV and PCZ against HCMV replication
| Equipotent ratio (fold of EC50a) of GCV/PCZ | CIb | Drug combination effectc |
|---|---|---|
| 0.25 | 0.625 ± 0.169 | Synergism |
| 0.5 | 0.780 ± 0.031 | Moderate synergism |
| 1 | 0.353 ± 0.278 | Synergism |
| 2 | 0.075 ± 0.039 | Very strong synergism |
| 4 | 0.064 ± 0.042 | Very strong synergism |
Fold increase of the EC50 for GCV/PCZ yielding an equipotent concentration ratio between the two combined drugs. The EC50s were determined by PRAs against HCMV AD169 in HFFs for each drug alone or in combination at concentrations from 4-fold to 0.25-fold the equipotent ratio of the drugs, using a ratio of 1:1.33, approximated from values in Table 1.
Combination index, obtained by computational analysis with Calcusyn software. Values are means ± SD of data derived from 3 independent experiments in triplicate.
Effects are defined as follows: CI < 0.1, very strong synergism; 0.1 < CI < 0.3, strong synergism; 0.3 < CI < 0.7, synergism; 0.7 < CI < 0.9, moderate synergism (32).
DISCUSSION
Our study stems from the observation of an overrepresentation of drugs belonging to the class of azolic antifungals among the molecules selected in a previous drug repurposing screening aimed at identifying new anti-HCMV compounds (12). Starting from this observation, we report for the first time that posaconazole, a drug already approved and used in both adult and pediatric immunosuppressed patients for prevention of and as salvage therapy for fungal infections, is a potent inhibitor of HCMV replication at concentrations in the low-micromolar range.
By characterizing the anti-HCMV profile of PCZ, we found that upon treatment of infected cells with PCZ and VFV, two inhibitors of hCYP51, the infectivity of the released HCMV particles is significantly reduced. Azolic antifungals have shown a polypharmacological profile mainly by affecting host cholesterol homeostasis and trafficking (26–29), in addition to being active against several unrelated viruses (11, 18–21). In human cells, PCZ is known to interfere with several pathways; for example, it interferes with cholesterol trafficking from lysosomes by inhibiting the Niemann-Pick C1 protein transporter (26), with cholesterol trafficking from the endoplasmic reticulum (ER) by inhibiting oxysterol-binding protein (OSBP), a cholesterol-shuttling protein (11, 19), and with cholesterogenesis by inhibiting hCYP51 (29, 30). However, the inhibition of OSBP, the target of PCZ in single-stranded positive RNA viruses, might be likely excluded as a possible mechanism of anti-HCMV activity of PCZ on the basis of the anti-HCMV activity also exhibited by imidazolic antifungals such as miconazole and econazole (Table 1 and Table 2), which do not bind OSBP (12, 19).
The reduced infectivity of HCMV particles released from PCZ-treated cells could be caused by the blocking of cholesterol trafficking and hCYP51 inhibition caused by the drug, leading to lower levels of available cholesterol. In fact, changes in the cholesterol uptake and efflux have been observed during HCMV infection (31, 33), and it has been reported that virion cholesterol content is crucial for the infectivity of HCMV progeny (31). Later during infection, when appropriate levels of new cholesterol molecules could be required by HCMV to produce infectious virions, inhibition of hCYP51-mediated cholesterogenesis exerted by PCZ would thus have a detrimental effect on the infectivity of the released viral progeny. However, this hypothesis remains to be tested. Nonetheless, our data suggest a role for late cholesterogenesis mediated by hCYP51 in the production of infectious HCMV virions. Indeed, we found that HCMV induces hCYP51 expression during infection. Importantly, a functional role for hCYP51 in the context of productive HCMV infection was validated by a pharmacological approach with the enzymatic inhibitors VFV and PCZ. Furthermore, the observed decrease in virus yield and in infectivity of virus particles produced upon inhibition of hCYP51 enzymatic activity supports the view that new cholesterol molecules are indeed required for the generation of infectious virus particles. Accumulation of new cholesterol molecules might contribute by conferring appropriate membrane fluidity to the enlarged cytomegalic cell, by participating in the organization of virion, or by contributing to the targeting and proper localization of viral proteins within the envelope or mature virus particle. Accordingly, in other virus models, both virion cholesterol content and effective cholesterogenesis have been reported to affect virus infectivity. Depletion of cholesterol during hepatitis B virus infection was in fact shown to induce a topological change of the large envelope protein that renders the virus noninfectious (34). Interestingly, HIV-1 Nef protein also induces hCYP51 expression to increase de novo cholesterol synthesis and enhance virion infectivity (35), and low levels of cholesterogenesis and cholesterol uptake have been related to a lower HIV transinfection ability and a slower disease progression in vivo (36).
Posaconazole peak levels (Cmax) in plasma of treated patients undergoing either prophylaxis or treatment of invasive fungal infections depend on a series of different factors, including the formulation, the diet, and the time and dosage of the administered drug (37, 51–53). During prophylaxis in patients at high risk of both fungal and HCMV infections and for therapeutic use in the treatment of mycosis, posaconazole peak levels are reported to average 1.5 to 2 μg/ml, corresponding to 2.2 to 2.9 μM (38, 39), and thus are approximately equal to or below the EC50s that we found for posaconazole against HCMV (Table 2). However, anti-HCMV activity for posaconazole within a clinically achievable range could be attained either by synergistic combination with GCV or by developing a better formulation.
We also observed that PCZ exerted anti-HCMV activity against both wild-type clinical isolates and drug-resistant viral strains in a cell type-independent manner. Importantly, we demonstrated that GCV and PCZ act synergistically in inhibiting HCMV replication in infected cells with a combination effect ranging from moderate to very strong synergism. The analysis of our data with Calcusyn software allowed the determination of the simulated dose reduction index (DRI) for both GCV and PCZ. The DRI estimates the extent to which GCV levels may be reduced when GCV is used in synergistic combination with PCZ to achieve the effect levels seen when GCV is used alone (32). For example, 90% inhibition of HCMV replication may be potentially reached by reducing the concentration of GCV and PCZ by 10- and 6-fold, respectively (Table S3), a condition that could be clinically achievable in treated patients. However, this simulation remains to be clinically tested in vivo.
In conclusion, our study suggests that repurposing of posaconazole against HCMV both alone and in combination with GCV could foster the development of new antiviral strategies against this important viral pathogen by exploiting its dual antimicrobial activity and could contribute to our understanding of how HCMV manipulates host pathways for a productive replication.
MATERIALS AND METHODS
Compounds.
Ganciclovir (GCV), foscarnet (FOS), and all the antifungal drugs were from Sigma-Aldrich. Cidofovir (CDV; Vistide) was from Gilead Sciences. VFV {(R)-N-(1-(3,4'-difluoro-[1,1'-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide)} was synthesized at Vanderbilt University as described elsewhere (40). For cellular assays, 100× stock of VFV was prepared in 25% dimethyl sulfoxide (DMSO)–34% aqueous 2-hydroxypropyl-β-cyclodextrin (vol/vol), purchased from Sigma.
Cells and viruses.
Human foreskin fibroblasts (HFFs), ARPE-19 cells, and U-373 MG cells were all from the American Type Culture Collection (ATCC) and were cultured in Dulbecco modified Eagle’s medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS; Life Technologies), 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate (both from Life Technologies). Human dermal microvascular endothelial cells (HMVECs) (CC-2543) were obtained from Clonetics and cultured in endothelial growth medium (EGM) (Clonetics). All cell cultures were maintained at 37°C in a humidified atmosphere supplemented with 5% CO2.
HCMV (strain AD169) was purchased from the ATCC. HCMV TB40E-UL32-EGFP (kindly provided by C. Sinzger, University of Ulm, Ulm, Germany) was previously described (41) as well as HCMV VR1814 (kindly provided by G. Gerna, IRCCS Policlinico San Matteo, Pavia, Italy) recovered from a cervical swab from a pregnant woman (42). HCMV 388438U clinical isolate was collected from a urine sample at the Microbiology and Virology Unit of Padua University Hospital (Italy) and was under passage 4 after primary isolation. HCMV strains resistant to antiviral drugs were obtained from the NIH AIDS Research and Reference Reagent Program (Rockville, MD) and were previously described (43). HCMV TR was reconstituted by transfecting HFFs with the corresponding TR bacterial artificial chromosome (BAC) that was prepared from the TR clinical strain, which is resistant to GCV and CDV and was isolated from an ocular specimen (44). Reconstitution of the BAC-derived TR strain in fibroblasts generated infectious virus that retains the ability to infect both endothelial and epithelial cells (45).
Plaque reduction assays.
Plaque reduction assays (PRAs) with HCMV were performed as previously described (46). Briefly, HFFs, ARPE-19 cells, and HMVECs were seeded at a density of 1.5 × 105 cells per well in 24-well plates. The next day, the cells were infected at 37°C with 80 PFU per well of the different viruses in serum-free DMEM. At 2 h postinfection (p.i.), the inocula were removed, cells were washed, and media containing various concentrations of each compound, 2% FBS, and 0.6% methylcellulose were added. After 10 days of incubation at 37°C, cell monolayers were fixed and stained with crystal violet, and viral plaques were counted.
Cytotoxicity assays.
The cytotoxicity of tested compounds was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich) method as described previously (47).
Virus yield reduction assays.
For virus yield reduction assays, HFFs were plated at 2 × 104 cells per well in 96-well plates, incubated overnight, and infected the next day with HCMV AD169 at a multiplicity of infection (MOI) of 0.1 PFU/cell. After virus adsorption for 2 h at 37°C, cells were washed and incubated with 0.2 ml of fresh medium containing 5% FBS in the absence or in the presence of test compounds. Plates were incubated for 5 days at 37°C and then subjected to one cycle of freezing and thawing. Titers were determined by transferring 0.1-ml aliquots from each well to a fresh 96-well monolayer culture of HFFs, followed by 1:5 serial dilutions across the plate. Cultures were incubated for 7 days and then fixed and stained, and the numbers of plaques were determined.
Enzymatic assays in vitro.
Recombinant human CYP51 and its redox partner NADPH-cytochrome P450 reductase (CPR) were expressed in Escherichia coli and purified as described previously (30). The standard reaction mixture contained 0.5 μM hCYP51, 1.0 μM CPR, 100 μM l-α-1,2-dilauroyl-snglycerophosphocholine, 0.4 mg/ml isocitrate dehydrogenase, and 25 mM sodium isocitrate in 50 mM potassium phosphate buffer (pH 7.2) containing 10% glycerol (vol/vol). After addition of radiolabeled [3-3H]lanosterol (∼4,000 dpm/nmol; dissolved in 45% [wt/vol] hydroxypropyl-β-cyclodextrin [HPCD]; final concentration, 50 μM) and inhibitors (concentration range, 0.1 to 20 μM), the mixture was preincubated for 30 s at 37°C in a shaking water bath; the reaction was initiated by the addition of 100 μM NADPH and stopped by extraction of the sterols with 5 ml of ethyl acetate. The extracted sterols were dried, dissolved in methanol, and analyzed by a reversed-phase high-pressure liquid chromatography (HPLC) system (Waters) equipped with a β-RAM detector (INUS Systems) using a NovaPak octadecylsilane (C18) column and a linear gradient of water-acetonitrile-methanol (1.0:4.5:4.5, vol/vol/vol) (solvent A) to CH3OH (solvent B), increasing from 0 to 100% B for 30 min at a flow rate of 1.0 ml/min. The IC50s were calculated using GraphPad Prism 6, with the percentage of lanosterol converted being plotted against inhibitor concentration and the curves fitted with nonlinear regression [log(inhibitor) versus normalized response − variable slope].
Plasmids.
The plasmid pCYP51-luc, which contains the −314/+343 human CYP51 (hCYP51) gene promoter region upstream of the luciferase reporter gene, was kindly provided by D. Rozman (Centre for Functional Genomics and Bio-Chips Institute of Biochemistry, Faculty of Medicine University of Ljubljana, Ljubljana, Slovenia) and was previously described (48). Plasmid pGAPDH-eGFP, which contains the promoter region of cellular GAPDH gene upstream of the enhanced green fluorescent protein (eGFP) gene was previously described (38) and used as a control for transfection efficiency.
Cell transfections and HCMV infection.
For the transfection/infection experiments with HCMV, U-373 MG cells were grown on 24-well plates and cotransfected using calcium phosphate (calcium phosphate transfection kit; Sigma) with 1 μg of pCYP51-luc plasmid along with 0.2 μg of pGAPDH-eGFP plasmid as a control to normalize transfection efficiency. The next day, transfected cells were either mock infected or infected with HCMV AD169 at an MOI of 0.5 PFU/cell for 2 h and then incubated with 5% FBS-containing medium. At 48 h postinfection, luciferase activity and eGFP expression were measured. For all the experiments, the values were normalized by dividing the values obtained for luciferase (LU) by the fluorescence units (FU) obtained for eGFP expression and expressed as relative luciferase units (RLU). For UV inactivation, a previously described procedure was followed (49). Briefly, HCMV diluted in serum-free DMEM was exposed for 8 min at a distance of 4 cm under a UV-light (VL-6MC; 254 nM, 6 W). Inactivation of the virus was assessed by immunofluorescence.
Quantification of gene expression.
HFFs were seeded in 6-well plates at 6 × 105 cells/well and incubated overnight at 37°C. For infection experiments, they were infected the next day with HCMV AD169 at an MOI of 1 PFU/cell for 2 h and then incubated with 5% FBS-containing medium. Total RNA was extracted from samples collected at different times p.i. using a total RNA Purification Plus kit (Norgen Biotek) according to the manufacturer’s protocol. cDNA was generated from RNA (2 μg) using random primers (Applied Biosystems) and Moloney murine leukemia virus (M-MLV) reverse transcriptase (Applied Biosystems). Quantitative PCR (qPCR) was performed with SYBR green reagent (Applied Biosystems) according to the manufacturer’s instructions on a 7900 HT Fast real-time PCR system (Applied Biosystems) using primers for hCYP51, GAPDH, and HCMV UL54 (sequences reported in Table S1). The relative changes in gene expression were calculated by the ΔΔCT method (50) using GAPDH to normalize data.
Western blotting.
For the analysis by Western blotting of the induction hCYP51 during HCMV infection, subconfluent HFFs in 6-well plates were infected with HCMV AD169 at an MOI of 0.5 PFU/cell. Whole-cell protein extracts were prepared at different times as previously described (12) and then analyzed by Western blotting with different antibodies (Table S2). Immunocomplexes were detected with the appropriate secondary anti-immunoglobulin antibodies conjugated to horseradish peroxidase (Life Technologies). Densitometry analysis was performed with ImageJ software (https://imagej.nih.gov/ij/).
Immunofluorescence and confocal microscopy analysis.
For confocal laser-scanning microscopy analysis, HFFs were infected with HCMV AD169 at an MOI of 0.25 PFU/cell. At different times p.i., cells were fixed with 4% paraformaldehyde in 1× PBS for 15 min at room temperature and then permeabilized with 0.1% Triton X-100 in PBS for 20 min at room temperature. After washing extensively with PBS, cells were incubated first with 4% FBS in PBS for 1 h at room temperature and then with different primary antibodies (listed in Table S2) diluted in 4% FBS in 1× PBS for 1 h at 37°C with shaking. Cells were then washed extensively with 4% FBS in 1× PBS and incubated with secondary fluorochrome-conjugated antibodies (listed in Table S2) for 1 h at 37°C. Nuclei were stained by incubation for 20 min with Draq5 (1:8,000 in 1× PBS). Cells were imaged using a Nikon Eclipse Ti-E microscope.
Particle-to-PFU ratio determination.
To determine the particle-to-PFU ratio of HCMV produced in the presence of test compounds, HFFs were seeded at 2 × 104 cells per well in 96-well plates, incubated overnight, and infected the next day with HCMV AD169 at an MOI of 0.5 PFU/cell. After virus adsorption for 2 h at 37°C, cells were washed and incubated with 0.2 ml of fresh medium containing 5% FBS in the presence or in the absence of test compounds. Plates were incubated for 5 days at 37°C. At the end of the incubation, 0.05 ml of supernatants was used to determine the number of virus particles that were produced under the different experimental conditions, while 0.05 ml was titrated on fresh monolayers of HFFs as previously described, to determine the number of PFU present in the same volume of supernatant. For quantitation of virus particles, 0.05 ml of supernatant was incubated with 0.2% SDS and proteinase K for 1 h at 56°C and then for 15 min at 95°C to inactivate proteinase K. Then, viral DNA was extracted with a DNA purification kit (Promega) and quantified by qPCR as described below. The particle-to-PFU ratio was determined by dividing the number of HCMV genomes by the number of PFU determined in the same volume of supernatant derived from the same sample.
Quantification of viral genomes.
To quantify the HCMV genomes in 0.05 ml of supernatants derived from the different samples collected at 120 h p.i., quantitative real-time PCR (qPCR) was performed as previously described (43). The number of viral genomes was normalized to the number of cellular β-globin gene copies. The sequences of the oligonucleotides used are listed in Table S1.
Drug combination studies.
To evaluate the combined effects of PCZ and GCV on HCMV AD169 replication, plaque reduction assays were performed as described above using 0.25×, 0.50×, 1×, 2×, and 4× EC50 for each combination of PCZ and GCV at equipotent ratio. The 2-drug combination effects were assessed using the method of Chou (32) based on mass-action law-based dynamic theory computed in CalcuSyn software, version 2.0 (Biosoft, Cambridge, United Kingdom).
Statistical analysis.
All statistical analyses were performed using GraphPad Prism, version 6.0.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by University of Padua (STARS Consolidator Grant FINDER to B. Mercorelli); by Associazione Italiana per la Ricerca sul Cancro (AIRC; grant no. IG18855 to A. Loregian); by Ministero dell’Istruzione, dell’Università e della Ricerca, Italy (PRIN 2017 no. 2017KM79NN to A. Loregian and PRIN 2017 no. 2017HWPZZZ to A. Luganini); by the British Society for Antimicrobial Chemotherapy, UK (grant BSAC-2018-0064 to A. Loregian); by the University of Turin (local research funds to G. Gribaudo and A. Luganini); by Fondazione Umberto Veronesi (to M. Celegato); and by a grant from the National Institutes of Health, USA (GM067871), to G. I. Lepesheva. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank D. Rozman, G. Pari, and M. Mach for providing plasmids, C. Sinzger and G. Gerna for providing some of the viruses used in this study, and L. Messa and T. Hargrove for experimental assistance.
B. Mercorelli, A. Luganini, G. Gribaudo, and A. Loregian have filed a provisional patent on the use of posaconazole alone and in combination with ganciclovir for the treatment of pathological conditions associated with cytomegalovirus infection.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Griffiths P, Baraniak I, Reeves M. 2015. The pathogenesis of human cytomegalovirus. J Pathol 235:288–297. doi: 10.1002/path.4437. [DOI] [PubMed] [Google Scholar]
- 2.Britt WJ. 2018. Maternal immunity and the natural history of congenital human cytomegalovirus infection. Viruses 10:405. doi: 10.3390/v10080405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klenerman P, Oxenius A. 2016. T cell responses to cytomegalovirus. Nat Rev Immunol 16:367–377. doi: 10.1038/nri.2016.38. [DOI] [PubMed] [Google Scholar]
- 4.Mercorelli B, Sinigalia E, Loregian A, Palù G. 2008. Human cytomegalovirus DNA replication: antiviral targets and drugs. Rev Med Virol 18:177–210. doi: 10.1002/rmv.558. [DOI] [PubMed] [Google Scholar]
- 5.Britt WJ, Prichard MN. 2018. New therapies for human cytomegalovirus infections. Antiviral Res 159:153–174. doi: 10.1016/j.antiviral.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meesing A, Razonable RR. 2018. New developments in the management of cytomegalovirus infection after transplantation. Drugs 78:1085–1103. doi: 10.1007/s40265-018-0943-1. [DOI] [PubMed] [Google Scholar]
- 8.Douglas CM, Barnard R, Holder D, Leavitt R, Levitan D, Maguire M, Nickle D, Teal V, Wan H, van Alewijk D, van Doorn LJ, Chou S, Strizki J. 2019. Letermovir resistance analysis in a clinical trial of cytomegalovirus prophylaxis for hematopoietic stem cell transplant recipients. J Infect Dis 221:1117–1126. doi: 10.1093/infdis/jiz577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shenk T, Alwine JC. 2014. Human cytomegalovirus: coordinating cellular stress, signaling, and metabolic pathways. Annu Rev Virol 1:355–374. doi: 10.1146/annurev-virology-031413-085425. [DOI] [PubMed] [Google Scholar]
- 10.Munger J, Bennett BD, Parikh A, Feng X-J, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol 26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strating J, van der Linden L, Albulescu L, Bigay J, Arita M, Delang L, Leyssen P, van der Schaar HM, Lanke KH, Thibaut HJ, Ulferts R, Drin G, Schlinck N, Wubbolts RW, Sever N, Head SA, Liu JO, Beachy PA, De Matteis MA, Shair MD, Olkkonen VM, Neyts J, van Kuppeveld FJ. 2015. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep 10:600–615. doi: 10.1016/j.celrep.2014.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mercorelli B, Luganini A, Nannetti G, Tabarrini O, Palù G, Gribaudo G, Loregian A. 2016. Drug repurposing approach identifies inhibitors of the prototypic viral transcription factor IE2 that block human cytomegalovirus replication. Cell Chem Biol 23:340–351. doi: 10.1016/j.chembiol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Mercorelli B, Palù G, Loregian A. 2018. Drug repurposing for viral infectious diseases: how far are we? Trends Microbiol 26:865–876. doi: 10.1016/j.tim.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mercorelli B, Luganini A, Celegato M, Palù G, Gribaudo G, Loregian A. 2018. Repurposing the clinically approved calcium antagonist manidipine dihydrochloride as a new early inhibitor of human cytomegalovirus targeting the Immediate-Early 2 (IE2) protein. Antiviral Res 150:130–136. doi: 10.1016/j.antiviral.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 15.Luganini A, Mercorelli B, Messa L, Palù G, Gribaudo G, Loregian A. 2019. The isoquinoline alkaloid berberine inhibits human cytomegalovirus replication by interfering with the viral Immediate Early-2 (IE2) protein transactivating activity. Antiviral Res 164:52–60. doi: 10.1016/j.antiviral.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Nukui M, O’Connor C, Murphy E. 2018. The natural flavonoid compound deguelin inhibits HCMV lytic replication within fibroblasts. Viruses 10:614. doi: 10.3390/v10110614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mercorelli B, Luganini A, Palù G, Gribaudo G, Loregian A. 2019. Drug repurposing campaigns for human cytomegalovirus identify a natural compound targeting the immediate-early 2 (IE2) protein: a comment on “The Natural Flavonoid Compound Deguelin Inhibits HCMV Lytic Replication within Fibroblasts.” Viruses 11:117. doi: 10.3390/v11020117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhoden E, Nix WA, Weldon WC, Selvarangan R. 2018. Antifungal azoles itraconazole and posaconazole exhibit potent in vitro antiviral activity against clinical isolates of parechovirus A3 (Picornaviridae). Antiviral Res 149:75–77. doi: 10.1016/j.antiviral.2017.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meutiawati F, Bezemer B, Strating J, Overheul GJ, Žusinaite E, van Kuppeveld FJM, van Cleef KWR, van Rij RP. 2018. Posaconazole inhibits dengue virus replication by targeting oxysterol-binding protein. Antiviral Res 157:68–79. doi: 10.1016/j.antiviral.2018.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Sun W, He S, Martínez-Romero C, Kouznetsova J, Tawa G, Xu M, Shinn P, Fisher E, Long Y, Motabar O, Yang S, Sanderson PE, Williamson PR, García-Sastre A, Qiu X, Zheng W. 2017. Synergistic drug combination . Antiviral Res 137:165–172. doi: 10.1016/j.antiviral.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schloer S, Goretzko J, Kühnl A, Brunotte L, Ludwig S, Rescher U. 2019. The clinically licensed antifungal drug itraconazole inhibits influenza virus in vitro and in vivo. Emerg Microbes Infect 8:80–93. doi: 10.1080/22221751.2018.1559709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lepesheva GI, Waterman MR. 2007. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta 1770:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Debeljak N, Fink M, Rozman D. 2003. Many facets of mammalian lanosterol 14alpha-demethylase from the evolutionarily conserved cytochrome P450 family CYP51. Arch Biochem Biophys 409:159–171. doi: 10.1016/s0003-9861(02)00418-6. [DOI] [PubMed] [Google Scholar]
- 24.Razonable RR. 2018. Drug-resistant cytomegalovirus: clinical implications of specific mutations. Curr Opin Organ Transplant 23:388–394. doi: 10.1097/MOT.0000000000000541. [DOI] [PubMed] [Google Scholar]
- 25.Clark NM, Grim SA, Lynch JP III. 2015. Posaconazole: use in the prophylaxis and treatment of fungal infections. Semin Respir Crit Care Med 36:767–785. doi: 10.1055/s-0035-1562902. [DOI] [PubMed] [Google Scholar]
- 26.Trinh MN, Lu F, Li X, Das A, Liang Q, De Brabander JK, Brown MS, Goldstein JL. 2017. Triazoles inhibit cholesterol export from lysosomes by binding to NPC1. Proc Natl Acad Sci U S A 114:89–94. doi: 10.1073/pnas.1619571114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen B, Trang V, Lee A, Williams NS, Wilson AN, Epstein EH Jr, Tang JY, Kim J. 2016. Posaconazole, a second-generation triazole antifungal drug, inhibits the Hedgehog signaling pathway and progression of basal cell carcinoma. Mol Cancer Ther 15:866–876. doi: 10.1158/1535-7163.MCT-15-0729-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamb DC, Kelly DE, Waterman MR, Stromstedt M, Rozman D, Kelly SL. 1999. Characteristics of the heterologously expressed human lanosterol 14alpha-demethylase (other names: P45014DM, CYP51, P45051) and inhibition of the purified human and Candida albicans CYP51 with azole antifungal agents. Yeast 15:755–763. doi:. [DOI] [PubMed] [Google Scholar]
- 29.Strushkevich N, Usanov SA, Park H-W. 2010. Structural basis of human CYP51 inhibition by antifungal azoles. J Mol Biol 397:1067–1078. doi: 10.1016/j.jmb.2010.01.075. [DOI] [PubMed] [Google Scholar]
- 30.Hargrove TY, Friggeri L, Wawrzak Z, Sivakumaran S, Yazlovitskaya EM, Hiebert SW, Guengerich FP, Waterman MR, Lepesheva GI. 2016. Human sterol 14alpha-demethylase as a target for anticancer chemotherapy: towards structure-aided drug design. J Lipid Res 57:1552–1563. doi: 10.1194/jlr.M069229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gudleski-O'Regan N, Greco TM, Cristea IM, Shenk T. 2012. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe 12:86–96. doi: 10.1016/j.chom.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou TC. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 33.Low H, Mukhamedova N, Cui HL, McSharry BP, Avdic S, Hoang A, Ditiatkovski M, Liu Y, Fu Y, Meikle PJ, Blomberg M, Polyzos KA, Miller WE, Religa P, Bukrinsky M, Soderberg-Naucler C, Slobedman B, Sviridov D. 2016. Cytomegalovirus restructures lipid rafts via a US28/CDC42-mediated pathway, enhancing cholesterol efflux from host cells. Cell Rep 16:186–200. doi: 10.1016/j.celrep.2016.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorobantu C, Macovei A, Lazar C, Dwek RA, Zitzmann N, Branza-Nichita N. 2011. Cholesterol depletion of hepatoma cells impairs hepatitis B virus envelopment by altering the topology of the large envelope protein. J Virol 85:13373–13383. doi: 10.1128/JVI.05423-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Y-H, Plemenitas A, Fielding CJ, Peterlin BM. 2003. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc Natl Acad Sci U S A 100:8460–8465. doi: 10.1073/pnas.1437453100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rappocciolo G, Jais M, Piazza P, Reinhart TA, Berendam SJ, Garcia-Exposito L, Gupta P, Rinaldo CR. 2014. Alterations in cholesterol metabolism restrict HIV-1 trans infection in nonprogressors. mBio 5:e01031-13. doi: 10.1128/mBio.01031-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dekkers BGJ, Bakker M, van der Elst KCM, Sturkenboom MGG, Veringa A, Span LFR, Alffenaar JC. 2016. Therapeutic drug monitoring of posaconazole: an update. Curr Fungal Infect Rep 10:51–61. doi: 10.1007/s12281-016-0255-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cornely OA, Duarte RF, Haider S, Chandrasekar P, Helfgott D, Jiménez JL, Candoni A, Raad I, Laverdiere M, Langston A, Kartsonis N, Van Iersel M, Connelly N, Waskin H. 2016. Phase 3 pharmacokinetics and safety study of a posaconazole tablet formulation in patients at risk for invasive fungal disease. J Antimicrob Chemother 71:718–726. doi: 10.1093/jac/dkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cornely OA, Robertson MN, Haider S, Grigg A, Geddes M, Aoun M, Heinz WJ, Raad I, Schanz U, Meyer RG, Hammond SP, Mullane KM, Ostermann H, Ullmann AJ, Zimmerli S, Van Iersel M, Hepler DA, Waskin H, Kartsonis NA, Maertens J. 2017. Pharmacokinetics and safety results from the Phase 3 randomized, open-label, study of intravenous posaconazole in patients at risk of invasive fungal disease. J Antimicrob Chemother 72:3406–3413. doi: 10.1093/jac/dkx263. [DOI] [PubMed] [Google Scholar]
- 40.Lepesheva G, Christov P, Sulikowski GA, Kim K. 2017. A convergent, scalable and stereoselective synthesis of azole CYP51 inhibitors. Tetrahedron Lett 58:4248–4250. doi: 10.1016/j.tetlet.2017.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sampaio KL, Cavignac Y, Stierhof YD, Sinzger C. 2005. Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular particle movements. J Virol 79:2754–2767. doi: 10.1128/JVI.79.5.2754-2767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Revello MG, Lilleri D, Zavattoni M, Stronati M, Bollani L, Middeldorp JM, Gerna G. 2001. Human cytomegalovirus immediate-early messenger RNA in blood of pregnant women with primary infection and of congenitally infected newborns. J Infect Dis 184:1078–1081. doi: 10.1086/323425. [DOI] [PubMed] [Google Scholar]
- 43.Mercorelli B, Muratore G, Sinigalia E, Tabarrini O, Biasolo MA, Cecchetti V, Palù G, Loregian A. 2009. A 6-aminoquinolone compound, WC5, with potent and selective anti-human cytomegalovirus activity. Antimicrob Agents Chemother 53:312–315. doi: 10.1128/AAC.00988-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk T. 2003. Coding potential of laboratory and clinical strains ofhuman cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cavaletto N, Luganini A, Gribaudo G. 2015. Inactivation of the human cytomegalovirus US20 gene hampers productive viral replication in endothelial cells. J Virol 89:11092–11106. doi: 10.1128/JVI.01141-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loregian A, Mercorelli B, Muratore G, Sinigalia E, Pagni S, Massari S, Gribaudo G, Gatto B, Palumbo M, Tabarrini O, Cecchetti V, Palù G. 2010. The 6-aminoquinolone WC5 inhibits human cytomegalovirus replication at an early stage by interfering with the transactivating activity of viral immediate-early 2 protein. Antimicrob Agents Chemother 54:1930–1940. doi: 10.1128/AAC.01730-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loregian A, Coen DM. 2006. Selective anti-cytomegalovirus compounds discovered by screening for inhibitors of subunit interactions of the viral polymerase. Chem Biol 13:191–200. doi: 10.1016/j.chembiol.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Halder SK, Fink M, Waterman MR, Rozman D. 2002. A cAMP-responsive element binding site is essential for sterol regulation of the human lanosterol 14alpha-demethylase gene (CYP51). Mol Endocrinol 16:1853–1863. doi: 10.1210/me.2001-0262. [DOI] [PubMed] [Google Scholar]
- 49.Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, Esclatine A. 2012. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J Virol 86:2571–2584. doi: 10.1128/JVI.05746-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real- time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 51.Kersemaekers WM, van Iersel T, Nassander U, O'Mara E, Waskin H, Caceres M, van Iersel MLPS. 2015. Pharmacokinetics and safety study of posaconazole intravenous solution administered peripherally to healthy subjects. Antimicrob Agents Chemother 59:1246–1251. doi: 10.1128/AAC.04223-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krishna G, Moton A, Ma L, Medlock MM, McLeod J. 2009. Pharmacokinetics and absorption of posaconazole oral suspension under various gastric conditions in healthy volunteers. Antimicrob Agents Chemother 53:958–966. doi: 10.1128/AAC.01034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leelawattanachai P, Montakantikul P, Nosoongnoen W, Chayakulkeeree M. 2019. Pharmacokinetic/pharmacodynamic study of posaconazole delayed-release tablet in a patient with coexisting invasive aspergillosis and mucormycosis. Ther Clin Risk Manag 15:589–595. doi: 10.2147/TCRM.S203625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.