

SUMMARY
The transmembrane domain recognition complex (TRC) targets cytoplasmic C-terminal tail-anchored (TA) proteins to their respective membranes in the endoplasmic reticulum (ER), Golgi and mitochondria. It is composed of three proteins, GET4, BAG6 and GET5. We identified an individual with compound heterozygous missense variants (p.Arg122His, p.Ile279Met) in GET4 that reduced all three TRC proteins by 70–90% in his fibroblasts, suggesting a possible defect in TA protein targeting. He presented with global developmental delay, intellectual disabilities, seizures, facial dysmorphism and delayed bone age. We found the TA protein, syntaxin 5, is poorly targeted to Golgi membranes compared to normal controls. Since GET4 regulates ER to Golgi transport, we hypothesized that such transport would be disrupted in his fibroblasts, and discovered that retrograde (but not anterograde) transport was significantly reduced. Despite reduction in the 3 TRC proteins, their mRNA levels were unchanged, suggesting increased degradation in patient fibroblasts. Treating fibroblasts with the FDA-approved proteasome inhibitor, Bortezomib (10nM), restored syntaxin 5 localization and nearly normalized the levels of all three TRC proteins. Our study identifies the first individual with GET4 mutations.
Keywords: congenital disorders of glycosylation, GET4, TRC pathway, BAG6, GET5, syntaxin 5
INTRODUCTION
The transmembrane domain recognition complex (TRC) pathway is involved in targeting tail-anchored (TA) proteins to the appropriate endoplasmic reticulum (ER), outer mitochondrial or peroxisome membranes. A C-terminal transmembrane domain and an N-terminal cytosolic region characterize TA proteins. The TRC pathway has six main components. GET1 and GET2 (CAMLG) are ER membrane receptors; GET3 is an ATPase involved in delivering TA proteins to the ER membrane; GET4 delivers TA proteins to GET3; GET5 (UBL4A) is a Ubl domain-containing protein that forms a complex with GET4; BAG6 is a BAG domain-containing protein that forms a complex with GET4/5 and SGTA is a chaperone that binds to TA proteins (Borgese and Fasana, 2011) (Figure 1A). The components of the TRC pathway also act as a triage system, wherein misfolded TA proteins that fail to insert into the target membrane are degraded through the ubiquitin proteasome system (Shao et al., 2017). Specifically, BAG6 recruits the E3 ligase RNF126 for ubiquitination and subsequent proteasome degradation of TA proteins (Rodrigo-Brenni et al., 2014).
Figure 1. Molecular data.
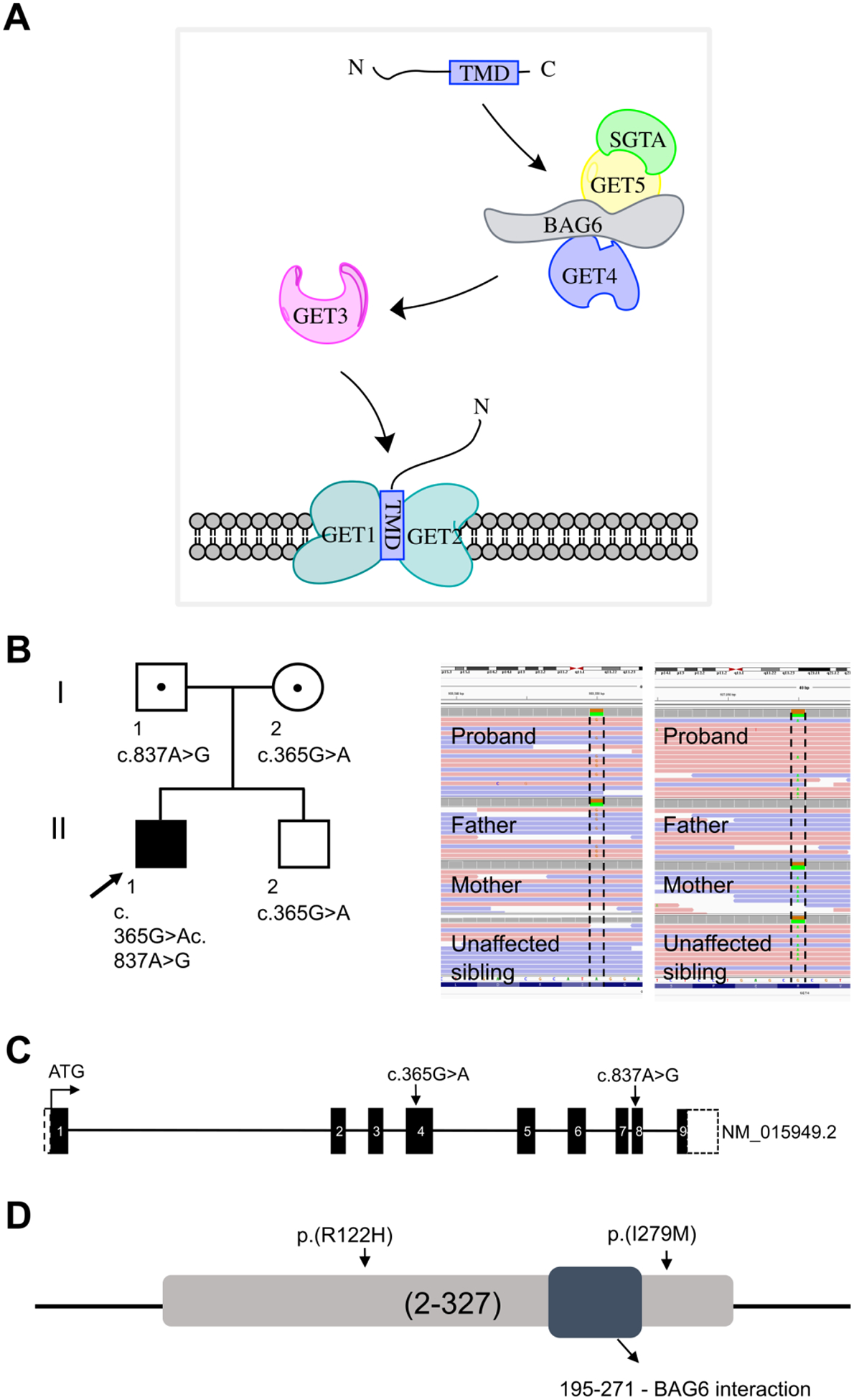
(A) Schematic representation of the TRC pathway and the components involved in insertion of TA proteins into ER membrane. (B) Family pedigree; arrow points to the proband. The Integrative Genomics Viewer (IGV) visualization of the GET4 variants. Sanger sequencing chromatogram showing biallelic mutations. The proband is a compound heterozygous for NM_015949.2 (GET4): c.365G>A; p.Arg122His (inherited from the mother) and NM_015949.2 (GET4): c.837A>G; p.Ile279Met (inherited from the father) (C) Schematic of GET4 showing the location of the variants (arrow). Each box represents the exonic regions of the gene; the black boxed are coding parts and the boxes with dashed line denote untranslated regions. (D) Schematic representation of GET4 and conservation of amino acids substituted by GET4 variants. Both variants are highly conserved across vertebrate species and are not located in the BAG6 interaction domain.
Within the TRC pathway, GET4, GET5 and BAG6 form a stable complex with each other and regulate each other’s stability (Kuwabara et al., 2015, Mock et al., 2015). The complex also forms critical interactions with other members of the TRC pathway. It interacts with the N-terminal domain of SGTA through the UBL domain of GET5. Furthermore, GET3 interacts with the complex through the N-terminal region of GET4 (Chang et al., 2010, Mariappan et al., 2010). Interaction with the complex stabilizes GET3 in a conformation suitable for “loading” TA proteins onto GET3. Thus, the GET4/5/BAG6 complex is essential for the proper functioning of the TRC pathway.
Here we present an individual with compound heterozygous mutations in GET4. Our biomolecular data indicate that expression of all members of the GET4/5/BAG6 complex is reduced in affected fibroblasts. We show that the TA client protein, syntaxin 5, is selectively mislocalized and that ER to Golgi transport is disrupted. Additionally, we show that cells treated with the proteasome inhibitor, bortezomib, substantially restore the complex.
METHODS AND MATERIALS
Evaluation of the Proband
The proband CDG-0381 (NIH designation: NIH-CDG-1101/UDP-12790) was evaluated at the National Institute of Health (NIH) under clinical protocol 14-HG-0071, “Clinical and Basic investigations into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789)” and clinical protocol 15-HG-0130 “Clinical and Genetic Evaluation of Individuals With Undiagnosed Disorders Through the Undiagnosed Diseases Network (NCT02450851)”, approved by the National Human Genome Research Institute Institutional Review Board. Written informed consent was provided by the proband’s parents.
Sample collection, processing and fibroblast culture
Peripheral blood DNA was obtained from the proband and primary dermal fibroblasts were cultured from a skin biopsy as described (Ferreira et al., 2018). Unaffected age- and gender-matched primary cell lines were obtained from Coriell Institute for Medical Research (Camden, NJ, USA) and American Type Culture Collection (ATCC, Manassas, VA, USA). GM05381, GM05565, GM09503, GM08398C, GM0407D and ATCC neonatal PCS-201–010 control fibroblast cell lines were used as controls.
Exome and genome sequencing
Whole exome sequencing was performed as previously described (Simon et al., 2017). WES for CDG-0381 was performed at the University of Washington Center for Mendelian Disorders on genomic DNA isolated from a saliva sample. Saliva genomic DNA was isolated using a DNA Genotek saliva collection kit. Details about genome sequencing can be found in supplemental methods.
Cell fractionation
Sub-cellular fractionation of fibroblasts was performed as described (Holden and Horton, 2009) with a few modifications (Tambe et al., 2019). Briefly, digitonin lysis buffer (200 μg/mL digitonin + 50 mM NaCl + 50 mM HEPES, pH 7.4) was used to obtain the cytosolic fraction. Next, NP-40 lysis buffer (0.1% NP-40 + 50 mM NaCl + 50 mM HEPES, pH 7.4) was used to obtain the membrane fraction. The remaining pellet was lysed in SDS lysis buffer (2% SDS, 62.5 mM Tris, pH 6.8, 10% glycerol) to obtain the nuclear fraction.
Western blot analysis
Western blot analysis was performed as described (Tambe et al., 2019). Equal amounts of denatured proteins were separated on a 6% or 10% (w/v) polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked and probed with the appropriate primary (rabbit anti-GET4, Abcam ab93801; mouse anti-BAG6, Santacruz Biotech sc-365928; rabbit anti-GET5, Proteintech 14253–1-AP; mouse anti-syntaxin 5, Santacruz Biotech sc-365124) and secondary (goat anti-mouse HRP, SeraCare 54500010; goat anti-rabbit HRP, Invitrogen 31446) antibodies. Western blots were developed using Pierce ECL Western Blotting Substrate (Thermo Scientific) and chemiluminescence images were obtained using ChemiDoc imaging system (BioRad). Quantitative analysis of the western blotting data was carried out using ImageJ software.
mRNA Expression Analysis
Total RNA was extracted from fibroblasts using the RNeasy Mini Kit (Qiagen, CA, USA). cDNA synthesis was performed using a High Capacity RNA to cDNA synthesis kit (Applied Biosystems) according to the manufacturer’s protocol. Quantitative RT-PCR was performed using a TaqMan gene-expression mastermix and the following TaqMan assay: GET4 (Hs00944513_g1), BAG6 (Hs00190383_m1), GET5 (Hs01006682_g1) and RPL13A (Hs04194366_g1). Expression was normalized using PRL14A and POLA4A (Hs00172187) as endogenous controls.
Brefeldin A- induced transport assays
Transport assays were performed as described (Ferreira et al., 2018). For retrograde transport assay, cell conditioned media was removed and replaced with fresh medium containing 0.25 mg/ml brefeldin A (BFA) for 0, 10, 20, 30, 40 and 50 min at room temperature. For the anterograde transport assay, cells were incubated in media containing 0.25 mg/ml brefeldin A for 1 hr at 37 °C. Next, cells were washed with DPBS, and the BFA was ‘washed out’ with pre-warmed media for 0, 20, 40, 60, 80 and 100 min at 37 °C. After the treatments, immunofluorescence was performed using Alexa Fluor 488 anti-Giantin antibodies (BioLegend, San Diego, CA, USA). Images were obtained using an Olympus IX71 microscope (Olympus Corporation, Tokyo, Japan), and processed by MetaMorph software (Molecular Devices, San Jose, CA, USA). Approximately 100 cells were counted for each time point and the percentage of cells with ER staining was determined.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 30 min at room temperature. Next, cells were permeabilized and blocked in PBS containing 1% BSA and 0.05% saponin for 30 min and stained using the appropriate primary antibody (mouse anti-syntaxin 5, Santacruz Biotech sc-365124; alexa fluor 488 anti-Giantin, Biolegend) overnight and secondary antibody (anti-mouse alexa fluor 546, Thermo Fisher) for 1 h at room temperature. Images were obtained using using a Zeiss LSM700 confocal laser-scanning microscope with a 40x oil- based objective and images were processed with LSM software ZEN Black 2012 software (Carl Zeiss Microscopy GmbH, Jena, Germany).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism software. The figure legends give details about the replicates and specific statistical analyses.
RESULTS
Clinical Findings
The proband is a 10-year-old Hispanic male, the first son of nonconsanguineous healthy parents. He was born at 41 weeks’ gestation by spontaneous vaginal delivery following an uncomplicated pregnancy, with a birth weight of 3.2 kg (10th percentile) and length of 48.3 cm. He was born with hyperextended left knee. He presented with delayed global development and was unable to sit up at 5 months of age. He babbled as an infant but eventually regressed, rolled at 18 months of age, sat at 2 years and walked with a gait trainer from the age of 3. Delayed acquisition of developmental milestones prompted extensive biochemical genetics evaluation that included the Carbohydrate Deficient Transferrin (CDT) test, which showed a modest type II glycosylation pattern lacking a single sialic acid. A glycosylation gene panel interrogation was negative for relevant mutations.
At 8 months of age he presented with tonic clonic seizures and a brain MRI showed agenesis of the corpus callosum. His seizures were poorly controlled and sometimes progressed into status epilepticus, requiring occasional emergency room visits. Between 4 and 5 years he did not have a seizure, and thereafter he had fewer than 5 seizures per year.
At the time of admission to the NIH Clinical Center at age 10, the proband weighed 18.6 kg (−4.3 SD), was 122 cm in height (−2.6 SD) and had a BMI of 12.5 (<−3 SD). He had brachycephaly and microcephaly with a head circumference of 48 cm (<−3 SD). His height was appropriate for his delayed bone age, i.e., 7 years. He presented with severe malnutrition and was discharged with the recommendation that a G-tube be placed. His face was hypotonic with bitemporal narrowing but normally placed ears, hindfoot valgus, smooth soles with underdeveloped creases and incurving of the fourth digits of the feet (Supplementary Figure 1A). He had general hypotonia, wheelchair-bound but had regular gait training. He appeared alert with eye contact and laughed but was non-verbal; he used an augmented communication device. There were no cranial nerve defects. Brain MRI showed ventriculomegaly, thin corpus callosum and moderate cortical atrophy, mild atrophy of the visual pathway, brainstem atrophy and basal ganglia volume loss. Myelination was completed, although delayed myelination had been seen up until the age of 3 (Supplementary Figure 1B). Magnetic resonance spectroscopy (MRS) showed decreased N-acetylaspartate (NAA) in the superior cerebellar vermis (Supplementary Figure 1B).
Electroencephalograms (EEG) showed frequent multifocal spikes, polyspikes, and spikes and waves in the bifrontal and frontocentral regions with poorly organized background. Nerve conduction studies showed predominantly axonal sensorimotor neuropathy. Skeletal survey revealed subluxation of the hips, scoliosis, degenerative changes of the acetabular roof, delayed bone age and low bone mineral density. Detailed clinical information and laboratory results are found in the supplementary clinical data (Supplementary Table I). Clinical lab testing failed to reveal a candidate gene or disorder, so WES was pursued.
Molecular Data
Initially, WES was performed only on the proband and revealed a potential candidate gene. However, during admission to the UDP, whole genome sequencing (WGS) was also performed on the proband, unaffected parents and a sibling to identify other potential candidates. As a result, no additional candidates were identified from clinical WGS or research reanalysis. A summary of genome analysis and the final list of variants are given in supplementary data (Supplementary Table II). Analysis of both WES and WGS results identified GET4 (NM_015949.2) as the best candidate. Clinical Sanger sequencing confirmed a maternally inherited c.365G>A (p.Arg122His) and a paternally inherited c.837A>G (pIle279Met) (Figure 1 B, C and D). Both variants are highly conserved across vertebrate species and predicted to be deleterious by SIFT, PolyPhen-2, VMM score (Gu et al., 2019) and the combined annotation dependent depletion (CADD) Phred scores. The c.365G>A (p.Arg122His) is present in the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/) at extremely low frequencies (0.00000408) and not detected in the homozygous state, while the c.837A>G (pIle279Met) is not seen.
GET4, BAG6 and GET5 protein levels are reduced in CDG-0381 fibroblasts
To establish whether the missense variants in GET4 are pathogenic, we determined their effects on GET4 protein expression and found 80–90% reduction (Figure 2A and B). We also found a corresponding reduction in GET5 and BAG6 protein levels. This result is consistent with previous studies suggesting that GET4/5/BAG6 form a stable complex and can regulate each other’s stability. Since the complex can translocate to the nucleus (Krenciute et al., 2013), we asked whether any of the three proteins is mislocalized in CDG-0381 cells. Through cell fractionation studies, we found no difference in protein localization between the CDG-0381 fibroblasts and several control lines (Figure 2A). Additionally, expression of other TRC pathway components, i.e., SGTA, GET1 and GET3, were unchanged at the protein level in CDG-0381 fibroblasts (data not shown). Collectively, these data suggest that the function of the TRC pathway might be disrupted in CDG-0381 fibroblasts.
Figure 2. GET4, BAG6 and GET5 protein levels are reduced in CDG-0381 fibroblasts.
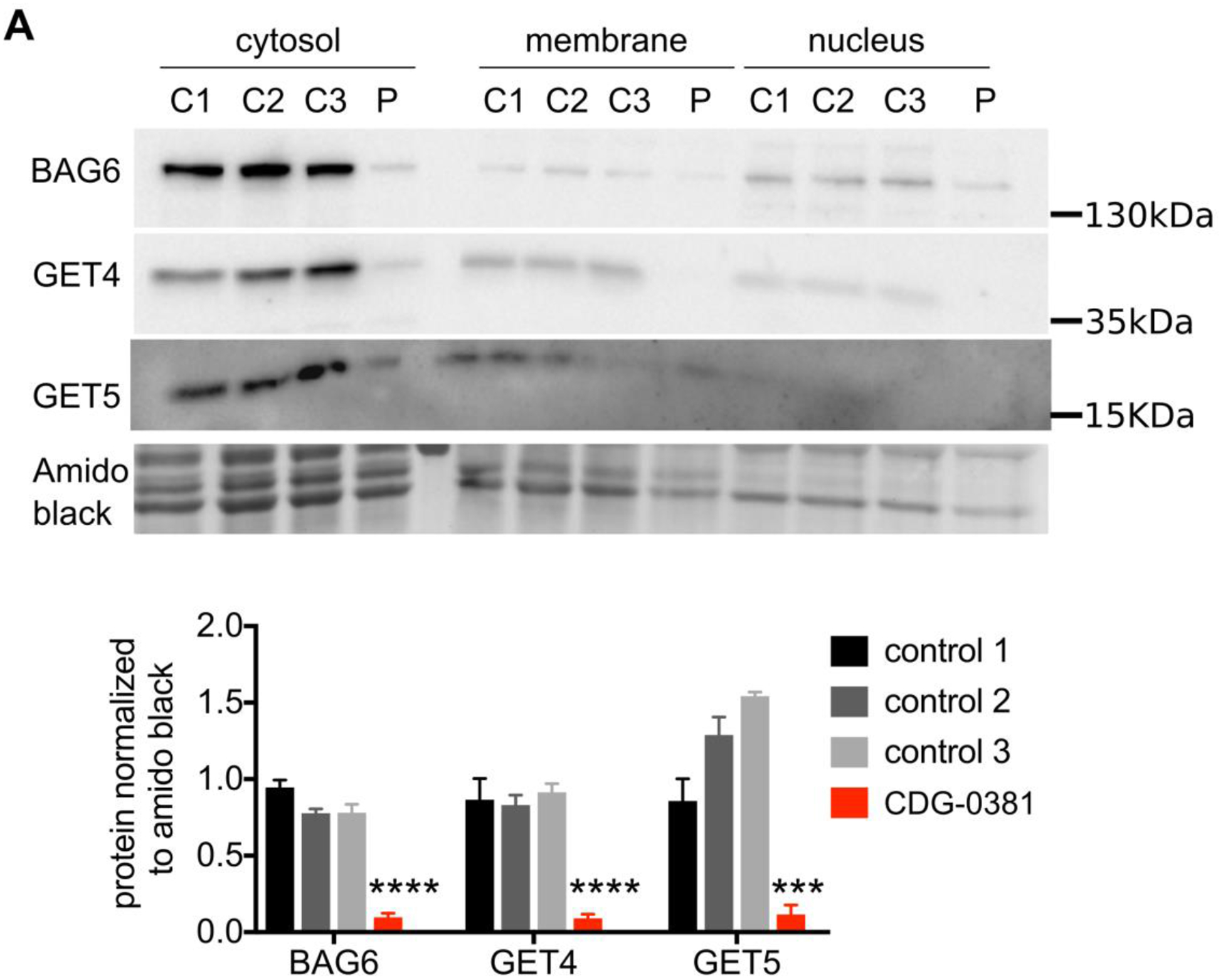
(A) Representative blots of BAG6, GET4 and GET5 protein localization in the cytosolic, membrane and nuclear fraction of three control and CDG-0381 fibroblasts. The bars indicate the molecular weight markers. (B) Analysis of protein levels of BAG6, GET4 and GET5 in the cytosolic compartment of control 1, 2 and 3 and CDG-0381 fibroblasts. The data are presented as mean ± SEM. *** p<0.001, **** p<0.0001. One-way ANOVA with Tukey’s multiple comparison test.
TA protein syntaxin 5 is mislocalized and ER to Golgi transport is disrupted
Since CDG-0381 fibroblasts lack three major components of the TRC pathway, we investigated whether TA protein targeting was disrupted as well. The Golgi-localized TA protein syntaxin 5 depends on the TRC pathway for its targeting. Both GET1 (Rivera-Monroy et al., 2016) and GET3 (Norlin et al., 2018) knockdown result in syntaxin 5 redistribution. Total protein levels of syntaxin 5 protein were unchanged in CDG-0381 by western blotting. Interestingly, cell fractionation showed 2-fold increase of syntaxin 5 in the cytosolic fraction compared to three control fibroblast lines (Figure 3A and B); this was also confirmed by immunocytochemistry (Figure 3C). Syntaxin 5 exists as a short form (Golgi localization) and a long form (Golgi and ER localization). Localization of the short form was disrupted to a greater extent than the long form. Surprisingly, another TA protein from the SNARE family, syntaxin 6, was not mislocalized in CDG-0381 fibroblasts (Supplementary Figure 2), suggesting selectivity towards TRC pathway substrates. Since syntaxin 5 plays an important role in regulating ER-Golgi transport (Dascher et al., 1994), we assessed BFA-induced anterograde and retrograde transport in three control and CDG-0381 fibroblasts. For retrograde transport, we treated control and CDG-0381 fibroblasts with 0.25 ug/mL BFA at room temperature and measured the increase in ER-localized Giantin (a Golgi marker) over time. We found that retrograde transport was significantly delayed in CDG-0381 fibroblasts (Figure 3D and E). Following BFA washout, the Golgi reassembles via anterograde transport, and this occurred similarly in control and CDG-0381 fibroblasts (data not shown). Collectively, these results identify a functional assay for GET4, GET5 and BAG6 deficiency, i.e., mislocalization of syntaxin 5 and delay of retrograde transport.
Figure 3. TA protein syntaxin 5 is mislocalized and ER to Golgi transport is disrupted.
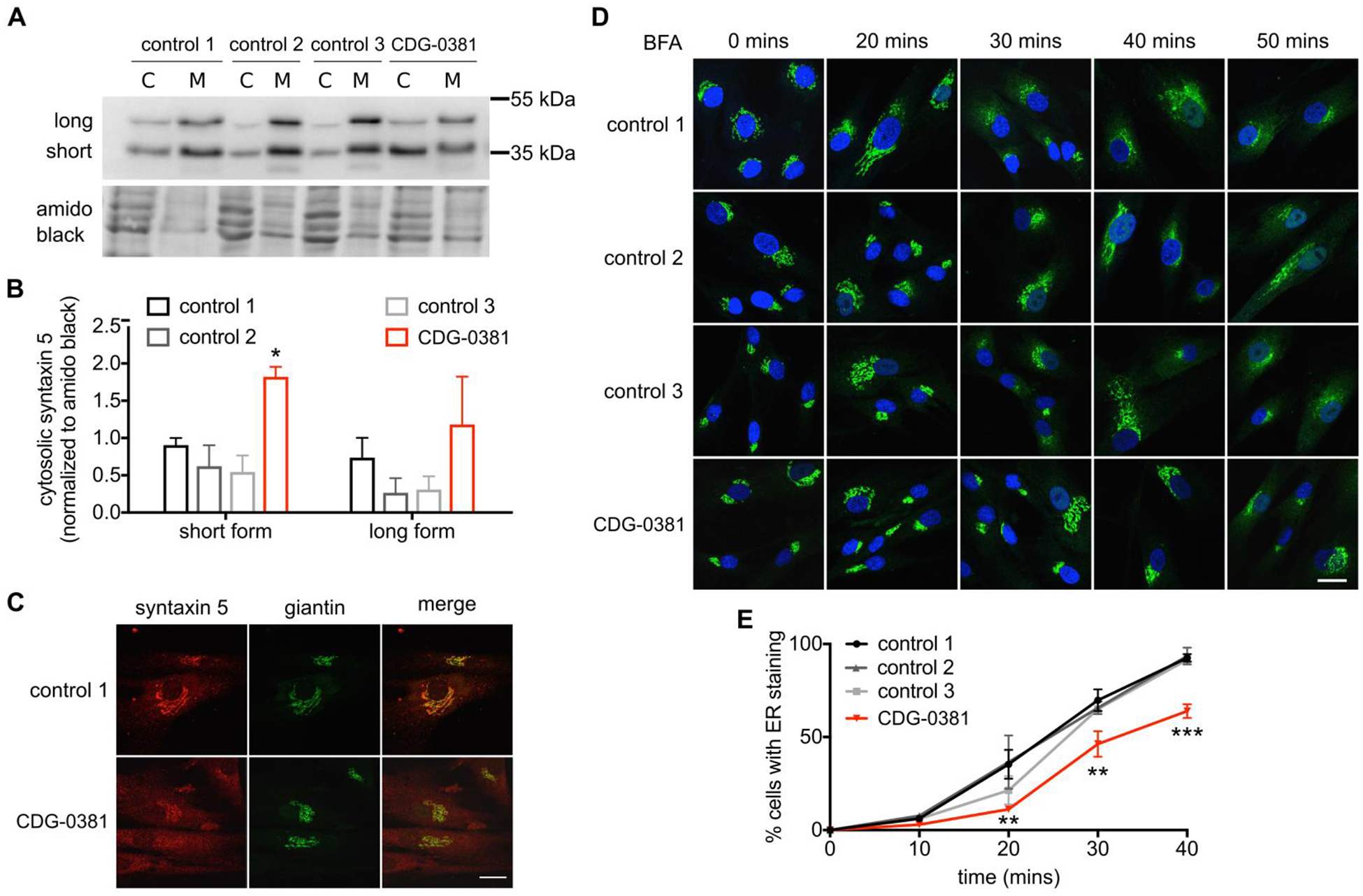
(A) Representative blot and (B) analysis of syntaxin 5 protein in cytosol and membrane fractions of control 1, 2 and 3 and CDG-0381 fibroblasts. The bars indicate the molecular weight markers. (C) A representative image showing syntaxin 5 co-localization with golgi marker giantin in control 1 and CDG-0381 fibroblasts. (D) Representative images and (E) analysis of ER localization of giantin staining in control 1, 2 and 3 and CDG-0381 fibroblasts after treatment with BFA (0.25 μg/mL) for different time points (0, 20, 30, 40 and 50 mins). Scale bar 25 μm. The data are presented as mean ± SEM. * p<0.1, ** p<0.01, *** p<0.001. Two-way ANOVA with Tukey’s multiple comparison test.
The proteasome inhibitor bortezomib restores GET4, BAG6 and GET5 levels and syntaxin 5 mislocalization
To explain the reduced GET4/5 and BAG6 protein levels in CDG-0381 fibroblasts, we analyzed mRNA levels of each by qRT PCR. Levels of GET4, GET5 and BAG6 mRNA were unchanged (Supplemetary Figure 3), suggesting increased degradation of all three proteins. To test this hypothesis, we measured the effect of proteasome inhibition on CDG-0381 fibroblasts. Treatment with 10nM bortezomib for 24 hours restored GET4/5 and BAG6 protein levels to normal (Figure 4A and B). Although bortezomib treatment increased GET4 protein (less than 2-fold) in control cells, the extent of increase was much less compared to that in CDG-0381 fibroblasts (about 5-fold). Importantly, bortezomib treatment also corrected syntaxin 5 mislocalization in CDG-0381 fibroblasts (Figure 4C, D and E). Unfortunately, treatment with bortezomib reduced BFA-induced retrograde transport in both control and CDG-0381 fibroblasts (data not shown). Hence, we could not test the effect of GET4/5/BAG6 protein restoration on ER-Golgi transport in CDG-0381 fibroblasts.
Figure 4. Proteasome inhibitor Bortezomib restores GET4, BAG6 and GET5 levels and syntaxin 5 mislocalization.
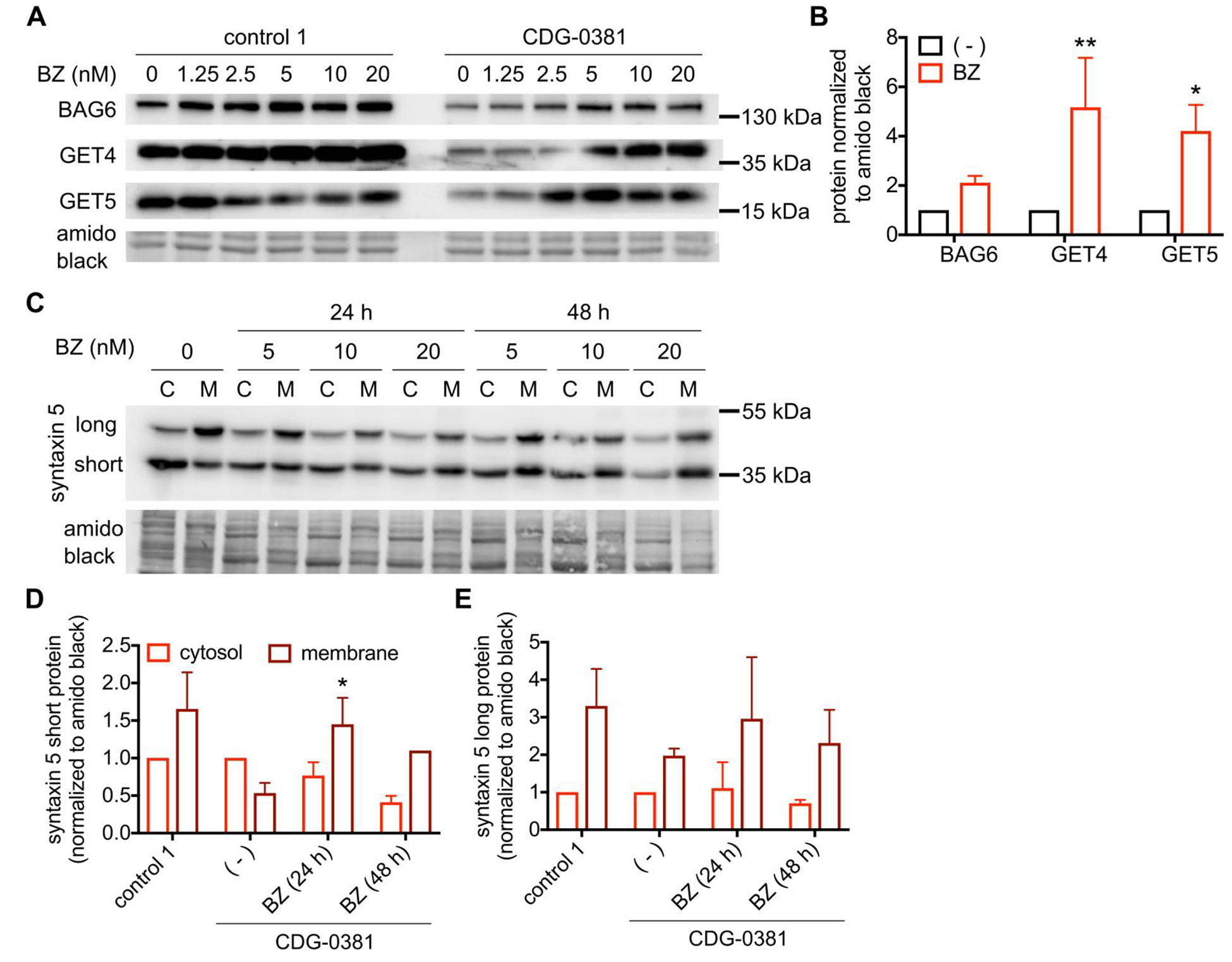
(A) Representative blots and (B) analysis of BAG6, GET4 and GET5 protein in control 1 and CDG-0381 fibroblasts after treatement with BZ (1.25, 2.5, 5, 10 and 20 nM) for 24 h. (C) Representative blots and (D and E) analysis of short and long form of syntaxin 5 protein in CDG-0381 fibroblasts after treatment with BZ (10 nM) for 24 h. The bars indicate the molecular weight markers. The data are presented as mean ± SEM. * p<0.1, ** p<0.01. Two-way ANOVA with Sidak’s multiple comparison test.
DISCUSSION
The TRC pathway is involved in targeting TA proteins to the appropriate membranes. We found that CDG-0381 fibroblasts with missense mutations in GET4 not only exhibit a loss of GET4 protein, but also have reduced levels of two other TRC pathway components, GET5 and BAG6, without changes in the mRNA levels. This can be explained by previous observations showing that GET4/5 and BAG6 form a complex and stabilize each other in the cytosol (Kuwabara et al., 2015, Mock et al., 2015). Simultaneous knockdown of GET4 and GET5 proteins reduced BAG6 protein levels. Additionally, knocking down BAG6 protein levels reduced the protein but not mRNA levels of both GET4 and GET5. Collectively, this result suggests that mutations in GET4 affect multiple components of the TRC pathway and hence likely its function.
Apart from the cytosol, the GET4/5/BAG6 complex is now known to translocate to the nucleus. In fact, GET4 interaction with BAG6 regulates its cytosol versus nuclear localization. When bound to BAG6, GET4 masks its nuclear localization sequence and increases BAG6 retention in the cytosol (Mock et al., 2017). A recent study showed that the GET4/5/BAG6 complex translocates to the nucleus and plays an important role in DNA damage response and DNA damage induced cell death, suggesting a role for the complex beyond the TA protein targeting (Krenciute et al., 2013). Several studies have linked BAG6 to cancer and autoimmune diseases (Benarroch et al., 2019). Additionally, GET5 has been shown to suppress tumor growth and metastasis in a pancreatic tumor model (Chen et al., 2019). Thus the GET4/5/BAG6 complex is involved in multiple signaling processes.
It was previously shown that loss of GET4 expression by shRNA knockdown resulted in the mislocalization of BAG6 (Wang et al., 2011). We attempted to transiently express each GET4 mutant in HELA cells to test the effects on BAG6 localization. However, this approach yielded inconsistent results, likely because, when either mutant was expressed off a plasmid, the expression was comparable to wild-type (data not shown), probably as a result of the plasmid’s strong CMV promoter. Since GET4 protein is almost absent in CDG-0381 fibroblasts, expressing the mutants off a plasmid is not physiological and hence not relevant.
We showed that the TA protein syntaxin 5, but not syntaxin 6, is mislocalized from the Golgi to the cytosol in CDG-0381 fibroblasts. This suggests selectivity toward substrates of the TCR pathway. A previous study showed that mice lacking GET1 with loss of TRC pathway function showed mislocalization of syntaxin 5 and syntaxin 6, but not sec61beta and syntaxin 8 in cardiomyocytes and hepatocytes (Rivera-Monroy et al., 2016). In another study, BAG6 siRNA resulted in mislocalization of syntaxin 6 in CHO cells (Takahashi et al., 2019). Substrate selectivity could be cell type specific, with other TA proteins affected in different cell types. Additionally, we showed retrograde but not anterograde transport was delayed in CDG-0381 fibroblasts. Syntaxin 5 has an established role in regulating ER-Golgi transport (Dascher et al., 1994). A previous study showed both syntaxin 5 and 16 were required for efficient retrograde transport in HeLa cells (Amessou et al., 2007).
Carbohydrate Deficient Transferrin (CDT) of serum samples revealed an N-linked glycosylation defect suggestive of a type II pattern missing a single sialic acid (also known as trisialo-transferrin). However, this CDT pattern has been seen in serum samples including those from mitochondrial and peroxisomal biogenesis disorders, which suggests that it could represent a secondary effect on glycosylation. Furthermore, we performed composition analysis of N-glycans from both control and CDG-0381 serum and found no clear abnormalities (data not shown). Finally, in CDG-0381 fibroblasts, we were unable to find any clear glycosylation abnormality. It is likely, then, that these N-linked glycosylation defects are a secondary effect of altered disturbed Golgi transport/function, but the specific defects are probably cell-type dependent.
We also showed that treatment with the proteasome inhibitor bortezomib restored protein levels of GET4/5 and BAG6 and also corrected syntaxin 5 mislocalization. These results confirm that GET4 mutations destabilize the GET4/5/BAG6 complex and reduce their protein levels. They also confirm that syntaxin 5 mislocalization in CDG-0381 fibroblasts is indeed a result of loss of the complex and hence TRC pathway function. Importantly, bortezomib or other proteasome inhibitors could be considered as a potential therapy for GET4/5 and BAG6 deficiency disorders. However, this FDA-approved drug, has severe side effects including gastrointestinal disturbances, thrombocytopenia and peripheral neuropathy (Field-Smith et al., 2006). Any pursuit of bortezomib therapy for these patients must consider risk vs. benefit.
In summary, our study identifies the first individual with GET4 deficiency. We show that protein levels of GET4, GET5 and BAG6 are reduced in CDG-0381 fibroblasts and that targeting of TA protein syntaxin 5 and ER-Golgi retrograde transport is disrupted. These findings contribute to further understanding of the pathogenesis of TRC pathway diseases and the characterization of individuals with mutations in GET4, GET5 and BAG6.
Supplementary Material
SYNOPSIS.
Novel pathogenic mutations in GET4 results in TRC pathway disruption and is characterized by syntaxin 5 mislocalization and delayed ER-golgi retrograde transport.
ACKNOWLEDGEMENTS
We would like to thank the families for their continued support and for providing valuable biological specimens. Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by NHGRI and NHLBI grants UM1 HG006493 and U24 HG008956. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
FUNDING DETAILS
This work was supported by grant R01DK99551 and The Rocket Fund; the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director; and the NHGRI Intramural Resaerch Program of the National Institutes of Health. Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by NHGRI and NHLBI grants UM1 HG006493 and U24 HG008956 and by the Office of the Director, NIH under Award Number S10OD021553. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
ETHICS APPROVAL AND INFORMED CONSENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study. Sanford Burnham Prebys Medical Discovery Institute (IRB-2014-038-17).
ANIMAL RIGHTS
This article does not contain any studies with animal subjects.
CONFLICT OF INTEREST
Mitali A. Tambe, Bobby G. Ng, Shino Shimada, Lynne A. Wolfe, David R. Adams, Undiagnosed Diseases Network, William A. Gahl, University of Washington Center for Mendelian Genomics, May C.V. Malicdan and Hudson H. Freeze declare that they have no conflict of interest.
REFERENCES
- AMESSOU M, FRADAGRADA A, FALGUIERES T, LORD JM, SMITH DC, ROBERTS LM, LAMAZE C & JOHANNES L 2007. Syntaxin 16 and syntaxin 5 are required for efficient retrograde transport of several exogenous and endogenous cargo proteins. J Cell Sci, 120, 1457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENARROCH R, AUSTIN JM, AHMED F & ISAACSON RL 2019. The roles of cytosolic quality control proteins, SGTA and the BAG6 complex, in disease. Adv Protein Chem Struct Biol, 114, 265–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORGESE N & FASANA E 2011. Targeting pathways of C-tail-anchored proteins. Biochim Biophys Acta, 1808, 937–46. [DOI] [PubMed] [Google Scholar]
- CHANG YW, CHUANG YC, HO YC, CHENG MY, SUN YJ, HSIAO CD & WANG C 2010. Crystal structure of Get4-Get5 complex and its interactions with Sgt2, Get3, and Ydj1. J Biol Chem, 285, 9962–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN H, LI L, HU J, ZHAO Z, JI L, CHENG C, ZHANG G, ZHANG T, LI Y, CHEN H, PAN S & SUN B 2019. UBL4A inhibits autophagy-mediated proliferation and metastasis of pancreatic ductal adenocarcinoma via targeting LAMP1. J Exp Clin Cancer Res, 38, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DASCHER C, MATTESON J & BALCH WE 1994. Syntaxin 5 regulates endoplasmic reticulum to Golgi transport. J Biol Chem, 269, 29363–6. [PubMed] [Google Scholar]
- FERREIRA CR, XIA ZJ, CLEMENT A, PARRY DA, DAVIDS M, TAYLAN F, SHARMA P, TURGEON CT, BLANCO-SANCHEZ B, NG BG, LOGAN CV, WOLFE LA, SOLOMON BD, CHO MT, DOUGLAS G, CARVALHO DR, BRATKE H, HAUG MG, PHILLIPS JB, WEGNER J, TIEMEYER M, AOKI K, UNDIAGNOSED DISEASES, N., SCOTTISH GENOME, P., NORDGREN A, HAMMARSJO A, DUKER AL, ROHENA L, HOVE HB, EK J, ADAMS D, TIFFT CJ, ONYEKWELI T, WEIXEL T, MACNAMARA E, RADTKE K, POWIS Z, EARL D, GABRIEL M, RUSSI AHS, BRICK L, KOZENKO M, THAM E, RAYMOND KM, PHILLIPS JA 3RD, TILLER GE, WILSON WG, HAMID R, MALICDAN MCV, NISHIMURA G, GRIGELIONIENE G, JACKSON A, WESTERFIELD M, BOBER MB, GAHL WA & FREEZE HH 2018. A Recurrent De Novo Heterozygous COG4 Substitution Leads to Saul-Wilson Syndrome, Disrupted Vesicular Trafficking, and Altered Proteoglycan Glycosylation. Am J Hum Genet, 103, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIELD-SMITH A, MORGAN GJ & DAVIES FE 2006. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Ther Clin Risk Manag, 2, 271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU F, WU A, GORDON MG, VLAHOS L, MACNAMARA S, BURKE E, MALICDAN MC, ADAMS DR, TIFFT CJ, TORO C, GAHL WA & MARKELLO TC 2019. A suite of automated sequence analyses reduces the number of candidate deleterious variants and reveals a difference between probands and unaffected siblings. Genet Med, 21, 1772–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLDEN P & HORTON WA 2009. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res Notes, 2, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRENCIUTE G, LIU S, YUCER N, SHI Y, ORTIZ P, LIU Q, KIM BJ, ODEJIMI AO, LENG M, QIN J & WANG Y 2013. Nuclear BAG6-UBL4A-GET4 complex mediates DNA damage signaling and cell death. J Biol Chem, 288, 20547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUWABARA N, MINAMI R, YOKOTA N, MATSUMOTO H, SENDA T, KAWAHARA H & KATO R 2015. Structure of a BAG6 (Bcl-2-associated athanogene 6)-Ubl4a (ubiquitin-like protein 4a) complex reveals a novel binding interface that functions in tail-anchored protein biogenesis. J Biol Chem, 290, 9387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARIAPPAN M, LI X, STEFANOVIC S, SHARMA A, MATEJA A, KEENAN RJ & HEGDE RS 2010. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature, 466, 1120–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOCK JY, CHARTRON JW, ZASLAVER M, XU Y, YE Y & CLEMONS WM JR. 2015. Bag6 complex contains a minimal tail-anchor-targeting module and a mock BAG domain. Proc Natl Acad Sci U S A, 112, 106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOCK JY, XU Y, YE Y & CLEMONS WM JR. 2017. Structural basis for regulation of the nucleo-cytoplasmic distribution of Bag6 by TRC35. Proc Natl Acad Sci U S A, 114, 11679–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NORLIN S, PAREKH V & EDLUND H 2018. The ATPase activity of Asna1/TRC40 is required for pancreatic progenitor cell survival. Development, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIVERA-MONROY J, MUSIOL L, UNTHAN-FECHNER K, FARKAS A, CLANCY A, COY-VERGARA J, WEILL U, GOCKEL S, LIN SY, COREY DP, KOHL T, STROBEL P, SCHULDINER M, SCHWAPPACH B & VILARDI F 2016. Mice lacking WRB reveal differential biogenesis requirements of tail-anchored proteins in vivo. Sci Rep, 6, 39464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODRIGO-BRENNI MC, GUTIERREZ E & HEGDE RS 2014. Cytosolic quality control of mislocalized proteins requires RNF126 recruitment to Bag6. Mol Cell, 55, 227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAO S, RODRIGO-BRENNI MC, KIVLEN MH & HEGDE RS 2017. Mechanistic basis for a molecular triage reaction. Science, 355, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMON MT, NG BG, FRIEDERICH MW, WANG RY, BOYER M, KIRCHER M, COLLARD R, BUCKINGHAM KJ, CHANG R, SHENDURE J, NICKERSON DA, BAMSHAD MJ, UNIVERSITY OF WASHINGTON CENTER FOR MENDELIAN, G., VAN HOVE JLK, FREEZE HH & ABDENUR JE 2017. Activation of a cryptic splice site in the mitochondrial elongation factor GFM1 causes combined OXPHOS deficiency. Mitochondrion, 34, 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKAHASHI T, MINAMI S, TSUCHIYA Y, TAJIMA K, SAKAI N, SUGA K, HISANAGA SI, OHBAYASHI N, FUKUDA M & KAWAHARA H 2019. Cytoplasmic control of Rab family small GTPases through BAG6. EMBO Rep, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAMBE MA, NG BG & FREEZE HH 2019. N-Glycanase 1 Transcriptionally Regulates Aquaporins Independent of Its Enzymatic Activity. Cell Rep, 29, 4620–4631 e4. [DOI] [PubMed] [Google Scholar]
- WANG Q, LIU Y, SOETANDYO N, BAEK K, HEGDE R & YE Y 2011. A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol Cell, 42, 758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.