

Abstract
Phosphatidylserine (PS) is a naturally occurring anionic phospholipid that is primarily located in the inner leaflet of eukaryotic cell membranes. The role of PS during apoptosis is one of the most studied biological functions of PS. Externalization of PS during apoptosis mediates an “eat me” signal for phagocytic uptake, leading to clearance of apoptotic cells and thus maintain self-tolerance by immunological ignorance. However, an emerging view is that PS exposure-mediated cellular uptake is not an immunologically silent event, but rather promoting an active tolerance towards self and foreign proteins. This biological property of PS has been exploited by parasites and viruses in order to evade immune surveillance of the host immune system. Further, this novel immune regulatory property of PS that results in tolerance induction can be harnessed for clinical applications, such as to treat autoimmune conditions and to reduce immunogenicity of therapeutic proteins. This review attempts to provide an overview of the biological functions of PS in the immune response and its potential therapeutic applications.
Keywords: Apoptosis, Phospholipids/Phosphatidylserine, Lipids, Immunology, Receptors, Immunogenicity Mitigation
1. Introduction
Eukaryotic cells display an asymmetric phospholipid distribution across their plasma membrane bilayer. Phosphatidylcholine (PC) and sphingomyelin are mainly present in the outer leaflet of a cell membrane, while phosphatidylserine (PS), phosphatidylethanolamine (PE), and phosphatidylinositol (PI) are primarily located in the inner or cytoplasmic leaflet (Chaurio et al 2009; Leventis et al 2010). PC and PE are zwitterionic phospholipids that are the most abundantly present, representing 40–50% and 25% of total phospholipids of a cell membrane, respectively (Leventis et al 2010). Despite the relatively low abundance (12% of total phospholipids), there has been extensive interest in PS due to its involvement in many biological outcomes such as blood coagulation and, most notably, clearance of apoptotic debris. This review details the role of PS exposure and immune response and how its biological properties could be harnessed for therapeutic considerations.
2. PS Asymmetry in Biological Membranes
PS is an anionic phospholipid that consists of a glycerol backbone, two fatty acid acyl chains at the sn-1 and sn-2 positions with variable length and saturation, a phosphate head group, and a polar serine head group, as seen in Figure 1. In a normal cell, PS is primarily restricted to the inner leaflet of the plasma membrane. The asymmetric distribution of PS across the cell bilayer under homeostasis is maintained by three main types of enzymes: flippases, floppases, and scramblases (Leventis et al 2010; Kay et al 2013; Marino et al 2013; Kimani et al 2014; Julian et al 2015). Flippases transport phospholipids such as PS from the outer leaflet to the inner leaflet or cytosol, while floppases translocate phospholipids from the cytosolic membrane to the external leaflet, both in an ATP-dependent manner. Scramblases differ from the unidirectional transporters in the fact that they are bidirectional, ATP-independent, and, when activated, can disrupt membrane asymmetry and promote the exposure of PS in the outer leaflet. In fact, two scramblases have been identified that contribute to the externalization of PS in the bilayer: transmembrane protein 16F (TMEM16F) and XK-related protein 8 (Xkr8), as well additional TMEM16s and Xkr proteins described as scramblases (Marino et al 2013; Kimani et al 2014; Julian et al 2015; Bevers et al 2016). TMEM16F was identified by Nagata et al. as the calcium-dependent, eight-transmembrane spanning phospholipid scramblase responsible for the PS externalization in activated platelets (Suzuki et al 2010). Xkr8 was further identified by Nagata et al. as the scramblase involved in apoptosis, in which the activation of Xkr8 by caspases resulted in increased PS exposure that were recognized as an “eat me” signal for apoptosis (Suzuki et al 2013; Suzuki et al 2014). In fact, it is believed that the externalization of PS that triggers apoptotic signaling is partially mediated by the interplay of specific Xkr8 activation and flippase (P4-ATPases) inactivation(Verhoven et al 1995; Hankins et al 2015; Segawa et al 2015). The loss of membrane asymmetry and subsequent PS externalization may trigger different signaling events that result in different biological outcomes, suggesting that not all externalized PS is functionally equivalent. PS is multifunctional, including roles in coagulation, rod cell shedding, fertilization and aopotosis(Bevers et al 2016).
FIGURE 1.
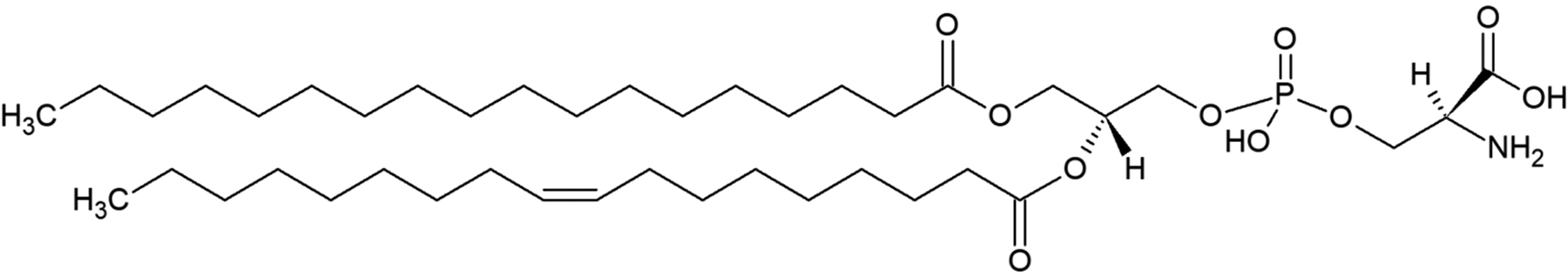
Representative structure of PS with two acyl chains, glycerol backbone, and phosphate head group.
3. Role of PS in Apoptosis
The role of PS in apoptosis, or programmed cell death, is perhaps the most extensively studied function of PS. Although apoptosis was first described by Kerr et al. in 1972, the role of PS exposure in apoptosis was initially demonstrated on apoptotic lymphocytes, in which PS externalization triggered their removal by macrophages (Fadok et al 1992; Fadok et al 1992). Apoptosis involves the rapid engulfment of the dead cell by professional phagocytes in a non-inflammatory manner in order to maintain tissue homeostasis. The apoptotic process requires several signaling events to occur that ultimately result in the clearance and removal of apoptotic cells without activation of the immune system. Apoptosis is a multi-step process that consists of “find me,” “eat me,” and “ignore me/tolerate me” signals, as summarized in Figure 2 (Munoz et al 2010). The “find me” signals secreted by the apoptotic cells stimulate a chemotactic response in order to attract and recruit phagocytes such as monocytes and macrophages into the immediate environment (Chaurio et al 2009; Munoz et al 2010; Ravichandran 2010). Potential “find me” signals have included lyso-phosphatidylcholine and sphingosine-1-phosphate (Lauber et al 2003; Mueller et al 2007; Gude et al 2008). The externalization of PS from the inner leaflet to the outer leaflet of the cell membrane provides the essential “eat me” signal that allows the phagocytes to recognize the apoptotic cell, leading to its engulfment and removal (Fadok et al 1992; Fadok et al 1992; Fadok et al 1998; Hoffmann et al 2001; Chaurio et al 2009; Munoz et al 2010; Ravichandran 2010). Finally, upon ingestion of the apoptotic cell by the phagocytes, the release of anti-inflammatory mediators such as transforming growth factor β (TGF-β), prostaglandin E2 (PGE2), and interleukin 10 (IL-10), produce the “ignore me/tolerate me” signals that help maintain the non-inflammatory environment critical for homeostasis (Fadok et al 1998; Chaurio et al 2009; Munoz et al 2010; Ravichandran 2010; Birge et al 2016).
FIGURE 2.
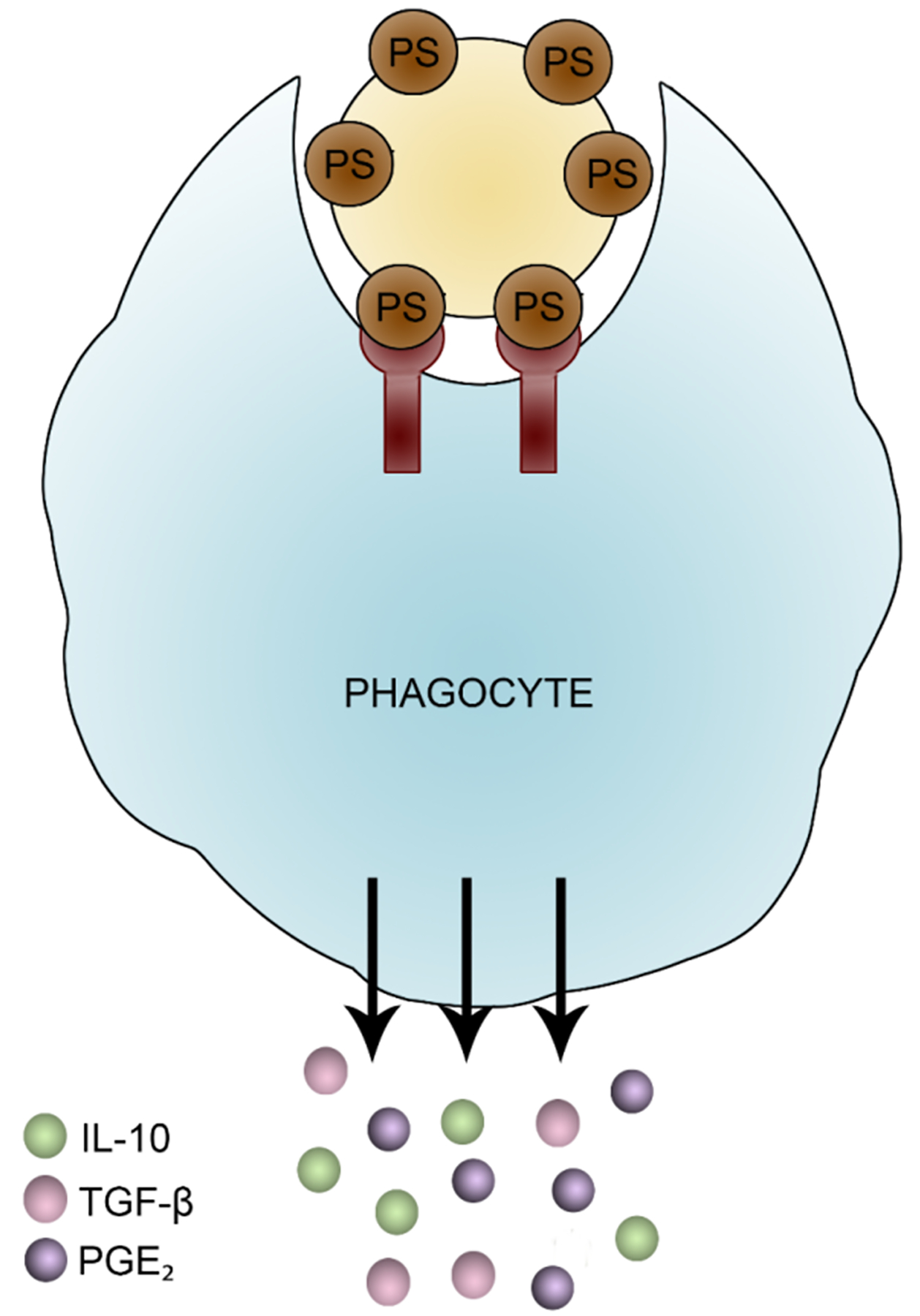
Mechanism of apoptotic cell clearance. Cells undergoing apoptosis secrete “find me” signals to attract phagocytes to the immediate vicinity, where phagocytes then recognize the apoptotic cell due to the exposure of PS that serves as an “eat me” signal. Once ingested and phagocytosed, macrophages releases “tolerate me” signals that allow for the silent clearance of cell debris.
3.1. PS Receptors Involved in Apoptosis
The recognition of PS by the phagocytes in apoptosis is mediated by the direct interactions with PS receptors and/or indirect interactions via bridging molecules. PS-specific receptors include stabilin-2, brain-specific angiogenesis inhibitor 1 (BAI1), and the T-cell immunoglobulin- and mucin (TIM) family of receptors. PS recognition can also occur by bridging molecules secreted by macrophages such as milk fat globulin-EGF-factor 8 (MFG-E8), or growth arrest-specific gene 6 (GAS6) that bind to the Axl and Tyro3 receptors to promote macrophage apoptosis(Seitz et al 2007). Both molecules can bind to the exposed PS on apoptotic cells and bridge or tether to phagocytes, allowing for clearance and removal of the apoptotic cell(Fuller et al 2008).
Stabilin-2 is a multifunctional scavenger receptor expressed on macrophages that recognizes PS in a calcium-dependent manner (Park et al 2008; Park et al 2008; Kay et al 2013). It has been reported that the EGF repeat domains in the extracellular region are the critical structures responsible for PS recognition and subsequent apoptotic engulfment. BAI1 is another important receptor that functions as an engulfment receptor for apoptotic cells (Park et al 2007; Chaurio et al 2009). It is expressed on macrophages and is able to bind to PS as well as to cardiolipin and phosphatidic acid (PA) (Park et al 2007; Kay et al 2013; Bevers et al 2016).
The TIM family of receptors include TIM-1, TIM-3, and TIM-4, have been identified as pattern recognition receptors that contain binding regions specific for PS, allowing for the direct PS-receptor interactions (Kobayashi et al 2007; Miyanishi et al 2007; Freeman et al 2010; Tietjen et al 2014; Bevers et al 2016). The TIM receptors differ in structure and expression, suggesting the distinct functions in modulating immune response. TIM-1 has been shown to be expressed on T-helper 2 (Th2) cells and serves as a co-stimulatory molecule for T cell activation. TIM-3 is expressed on T-helper 1 (Th1) cells, CD8+ T cells, and a subset of activated CD4+ T cells. Further, it has been identified on subpopulations of macrophages and dendritic cells (DCs). Engagement of TIM-3 by PS generates inhibitory signals that result in apoptosis of these cell types. The direct interaction of PS with TIM-4 is perhaps the most relevant of this family of receptors in apoptosis. TIM-4 is expressed exclusively on antigen presenting cells (APCs), such as macrophages and DCs, and is a critical mediator of apoptotic cell uptake (Kobayashi et al 2007; Miyanishi et al 2007; Freeman et al 2010). The binding site for PS is located in the extracellular IgV-like domain of the TIM-4 molecule. Structural analysis suggests a metal ion-dependent binding pocket for PS and that the binding region of TIM-4 specifically recognizes the serine head group of the PS molecule (Freeman et al 2010). The PS-TIM-4 interaction is critical for the uptake and removal of apoptotic cells, as studies have shown that blocking of this interaction by a TIM-4 antibody results in apoptotic body clearance and the development of auto-antibodies, indicating the importance of the PS-TIM-4 interplay and recognition for maintaining tolerance (Miyanishi et al 2007). In addition, PS binding to TIM-4 receptors is dependent on charge density (Kobayashi et al 2007). As the molar percentage of PS increases, the bound fraction of PS to TIM-4 increases, suggesting that PS binding to TIM-4 is additionally dependent on the surface density and expression of PS (Kerr et al 2018).
3.2. PS Structural Modifications in Apoptosis
The externalization of PS has long been the hallmark characteristic for apoptotic cell clearance. However, it is worth noting that not all PS externalization events result in efferocytosis, the engulfment of apoptotic cells, which suggests that PS exposure alone may not be sufficient for macrophage recognition (Segawa et al 2011). Studies have suggested the possibility of additional molecules that may be present to help enhance PS recognition as the “eat me” signal to allow for efficient phagocytosis and the involvement of specific structural requirements of PS for apoptosis have recently been identified (Frasch et al 2008; Frasch et al 2012; Frasch et al 2013; Kimani et al 2014; Tyurin et al 2014).
There has been extensive work investigating oxidized PS species as being the critical mediators of the “eat me” signal for efficient phagocytosis (Arroyo et al 2002; Kagan et al 2002; Matsura et al 2002). The release of reactive oxygen species (ROS) is an important contributor to apoptosis (Simon et al 2000). The oxidation of fatty acid acyl chains on PS triggers specific signals, marking the cells to undergo phagocytosis (Arroyo et al 2002; Kagan et al 2002; Borisenko et al 2004). Further, both the non-oxidized and oxidized forms of PS serve as the recognition signals for macrophages (Arroyo et al 2002; Kagan et al 2002). The administration of antibodies against oxidized phospholipids has been shown to inhibit macrophage efferocytosis, suggesting the role of oxidized phospholipids in apoptosis (Chang et al 1999; Kimani et al 2014). It has been suggested that oxidized PS may change the distribution of PS on the cell membrane, perhaps allowing for more PS exposure, or that oxidized PS may be a more potent substrate for some PS receptors, both allowing for more efficient phagocytosis (Kimani et al 2014).
Interestingly, there has been considerable literature support for the hypothesis that oxidized PS may be a better substrate for certain PS receptors. The scavenger receptor CD36 on macrophages is one of the receptors involved in apoptosis. Recognition of apoptotic cells by macrophages through CD36 occurs preferentially through interactions with oxidized PS, but not with the non-oxidized PS form (Greenberg et al 2006). Other receptors and bridging molecules that have shown to bind preferentially with oxidized PS include GAS6, MFG-E8, and BAI-1 (Borisenko et al 2004; Kimani et al 2014; Tyurin et al 2014).
The presence of a mono-acylated derivative of PS may be involved in mediating cellular signaling, as seen in Figure 3 (Frasch et al 2008). Lyso-phosphatidylserine (Lyso-PS) is thought to be generated by a deacylation reaction by the PS-specific phospholipase, PS-PLA1, and is a critical anti-inflammatory mediator (Frasch et al 2012; Kimani et al 2014). The presence of one acyl chain suggests that Lyso-PS has increased aqueous solubility and mobility to perhaps better anchor to receptors (Kay et al 2013). Work by Frasch et al. shows Lyso-PS can be generated by activated or aged apoptotic neutrophils, which then enhances efferocytosis and uptake and clearance of activated and apoptotic neutrophils(Frasch et al 2012). This is mediated by interaction of Lyso-PS with the macrophage orphan receptor G2A (Frasch et al 2008; Frasch et al 2011; Frasch et al 2012; Frasch et al 2013). Downstream signaling from G2A during efferocytosis after interaction with Lyso-PS displayed an increase in PGE2 production, an anti-inflammatory molecule that inhibits phagocytosis. (Frasch et al 2012). In addition, Lyso-PS has been shown to inhibit T cell activation (Bellini et al 1993). While the interaction between Lyso-PS and G2A has been studied in neutrophil uptake and removal, other Lyso-PS receptors have been identified, such as GPR34, Putative P2Y purinoceptor 10, and Toll-like Receptor 2 (van der Kleij et al 2002; van der Kleij et al 2003; Sugo et al 2006; Frasch et al 2012; Kitamura et al 2012; Makide et al 2013; Chung et al 2017). Although the function of the receptors and the downstream signaling of the Lyso-PS/receptor interaction is still being investigated, it is interesting to note that Lyso-PS may have a dual immune-modulatory role in certain conditions. GPR34 is present on mast cells and Lyso-PS has been shown to enhance mast cell degranulation, suggesting a pro-inflammatory response (Sugo et al 2006). Further understanding of the structural requirements of PS for efficient recognition and removal of apoptotic cells is critical in order to develop therapeutic strategies utilizing this phospholipid.
FIGURE 3.
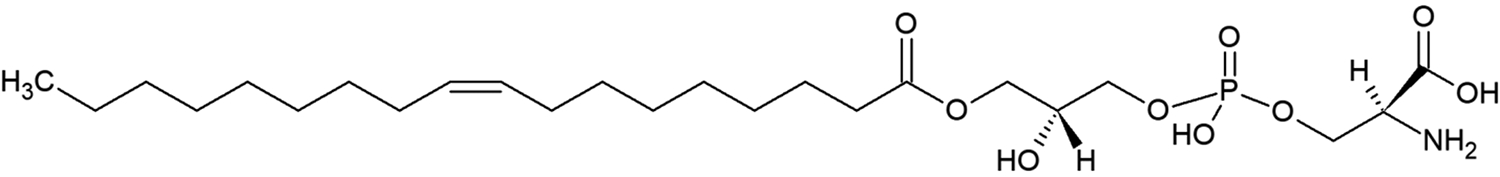
Structure of the de-acylated derivative of PS, termed Lyso-PS.
4. Therapeutic Implications of PS Exposure
Understanding of the role of PS externalization in biological events such as hemostasis and apoptosis, the mechanism of PS exposure, and the involvement of PS with specific receptors and bridging molecules to initiate specific signaling events is important to develop therapeutic applications. Knowledge of the biological functions and the impact of PS on the immune system can facilitate the development of promising therapeutic strategies in viral infections, cancer, autoimmunity, and therapeutic protein immunogenicity.
4.1. Role of PS in Parasitic/Viral Immune Evasion
The presence of PS has been identified in the envelope membrane of viral particles and exposure of PS on the surface of the parasites have been shown to contribute to their infectivity and immune evasion. Studies with the protozoon Leishmania indicated the exposure of PS on the protozoon surface and that ingestion of these parasites does not result in a pro-inflammatory response. Moreover, administration of annexin V to mask the exposed PS inhibited the infectivity of these parasites (de Freitas Balanco et al 2001). This phenomenon of apoptotic mimicry to evade the host immune system and infect target cells has also been shown for several viruses, such as HIV, Ebola virus, and Marburg virus (Birge et al 2016). In addition, parasitic schistosomes display Lyso-PS that interact with TLR2 in order to evade the host immune system (van der Kleij et al 2002). PS is exposed on the surface of viral particles either by concentrating PS within their enveloped membranes, or “cloaking” in PS-containing vesicles, either of which results in virus entry and immune evasion (Birge et al 2016). Understanding the role of PS in viral particles and the receptors and bridging molecules responsible to facilitate the entry and immune evasion of the viral particle could help develop therapeutic strategies targeting viral apoptotic mimicry. Due to the external expression of PS by these viral particles, PS blocking strategies have been investigated for host protection from viral entry and infection. Administration of bavituximab, a chimeric anti-PS monoclonal antibody, in vivo protected guinea pigs from lethal viral infection of the Pichinde virus (Soares et al 2008). Another strategy investigated is the administration of Annexin V, a naturally occurring PS binding protein. During apoptosis, it has been shown that Annexin V can inhibit phagocytosis during apoptotic cell clearance by blocking PS (Munoz et al 2007; Chaurio et al 2009). Using Annexin V in a PS blocking strategy could impair viral entry and infection. Studies by Munoz et al. demonstrated that HIV-1 infectivity in human macrophages was significantly reduced when treated with Annexin V in vitro (Munoz et al 2007). Additionally, pre-clinical studies have suggested that blocking PS with Annexin V can dysregulate tumor microenvironments and improve treatment(Birge et al 2016).
4.2. Role of PS in Tumor Microenvironment/Cancer
The presence of PS during apoptosis that facilitates the silent clearance of apoptotic cells can be exploited by tumor cells by apoptotic mimicry. The expression of PS on cancer cells can generate a local immunosuppressive environment, allowing the tumor to evade immune surveillance and detection. The exposure of PS on tumor cells may be mediated by the mildly acidic environment, the presence of ROS, and hypoxia, which can contribute to the exposure of PS on the plasma membrane (Lankry et al 2013). Within the tumor microenvironment, the presence of immature tumor vasculature, tumor-derived exosomes, and viable tumor cells contributes to the suppression of pro-inflammatory signals, due to the expression of PS (Birge et al 2016). Furthermore, the interaction of PS expressed on tumor cells with PS receptors or PS bridging molecules in the tumor microenvironment could confer the “tolerate me” signals that enhance immune escape and prevent efficient anti-tumor responses. Furthermore, exosomes isolated from patient-derived ovarian tumors and present in the ascites fluid have been shown to express PS on the surface and it has been shown that the presence of PS contributes to T cell receptor (TCR) signaling arrest. Blockade of PS with the administration of Annexin V impaired the inhibitory activity of tumor-derived exosomes (Kelleher et al 2015). Annexin V has also been investigated as a natural adjuvant in order to increase the immunogenic potential of tumor cells, ultimately enhancing anti-tumor responses (Munoz et al 2007). The identification of PS as a potential checkpoint inhibitor in the tumor environment allows for the development of PS-targeting antibodies for cancer immunotherapy. Administration of bavituximab induced M1 polarization and suppressed the progression of prostate tumors in tumor-bearing mice (Yin et al 2013). In fact, bavituximab is currently being evaluated in clinical trials as a monotherapy or in combination in several cancer indications (Chalasani et al 2015; Gerber et al 2016; Meyer et al 2017; Belzile et al 2018). Understanding the involvement of PS exposure and mediation of the immunosuppressive tumor microenvironment can facilitate the development and progress of antibody-mediated PS blockade strategies in cancer. PS-targeting therapies alone or in combination with chemotherapy, radiation, or immune checkpoint inhibitors such as anti-CTLA-4 or anti-PD1 have tremendous potential for effective tumor-specific immune responses in patients.
4.3. Role of PS in Autoimmunity
The hallmark feature of autoimmunity is the breakdown of tolerance to self-antigens and the impairment of self versus non-self recognition (Ring et al 1999). Moreover, it is widely reported that inefficient clearance of apoptotic cells contributes to auto-antibody production and autoimmunity (Cohen et al 2002; Hanayama et al 2004; Munoz et al 2010; Shao et al 2011; Julian et al 2015). The defective clearance mechanisms responsible for apoptosis could allow apoptotic cells to undergo secondary necrosis, thus enabling their cargo to be recognized as non-self. This would trigger the release of pro-inflammatory signals, instead of the “tolerate me” signals normally released under effective apoptotic conditions to maintain self-tolerance. Due to the critical function of PS as an “eat me” signal for effective recognition and clearance of apoptotic cells, impaired or defective PS signaling, either through its interaction with its receptors or bridging molecules, could contribute to the ineffective clearance of apoptotic debris that ultimately manifests in autoimmunity. The causal link between ineffective apoptotic cell removal and the onset of autoimmunity has been extensively studied in systemic lupus erythematosus (SLE) (Gaipl et al 2006; Shao et al 2011; Mahajan et al 2016). Genetic knockouts of PS receptors such as TIM-1, TIM-4, and TAM, or PS bridging molecule MGF-E8 in mice resulted in ineffective apoptotic cell clearance, auto-antibody production, and SLE-like development (Lu et al 2001; Cohen et al 2002; Hanayama et al 2004; Miyanishi et al 2007; Xiao et al 2012; Kimani et al 2014). Knowing that impaired clearance of apoptotic debris can generate auto-antibodies and autoimmune conditions and that impairment of PS signaling is partly responsible, several investigators have attempted to administer PS containing liposomes to arrest auto-antibody production and reduce the severity of disease. The ability to use PS therapeutically to treat autoimmunity is based on the ability to redirect the immune system to an immune-tolerant state and re-balance the self and non-self recognition that became lost. Studies from Pujol-Autonell et al. investigated the use of PS liposomes loaded with auto-antigens to treat autoimmune diseases such as type 1 diabetes (T1D) and multiple sclerosis (MS) (Pujol-Autonell et al 2015; Pujol-Autonell et al 2017; Rodriguez-Fernandez et al 2018). T1D is an autoimmune disease, in which the immune system recognizes insulin-producing islet cells (or pancreatic beta cells) as foreign. Studies performed by Pujol-Autonell et al. found that administration of PS liposomes with insulin peptides produced tolerogenic DCs, reduced T cell activation, reduced the incidence of diabetes and displayed a slight delay in disease onset (Pujol-Autonell et al 2015). This work was extended into experimental autoimmune encephalomyelitis (EAE), a murine form of human MS, where the investigators also demonstrated the generation of tolerogenic DCs, reduction in EAE severity, and production of Tregs (Pujol-Autonell et al 2017). Further, previous work in our laboratory have shown that pre-exposure of a myelin antigen in the presence of PS delayed disease onset, reduced disease severity, and induced Treg expression in EAE mice (Glassman et al 2018). In autoimmune conditions, understanding of PS externalization in apoptosis could be harnessed to develop PS-based therapeutic strategies that could re-establish tolerance towards self-proteins via apoptotic mimicry.
4.4. Use of PS to Mitigate Protein Immunogenicity
The role of PS exposure in apoptosis is essential for the maintenance of tissue homeostasis and tolerance to self-proteins by immunosuppressive mechanisms. Knowing that PS externalization functions as an “eat me” signal to confer immunosuppressive signals, this function PS could be exploited therapeutically to mitigate immunogenicity concerns with protein therapeutics. Despite the enormous growth of protein therapeutics in the market and in the pipeline, one of the concerns with the development of this class of compounds is the development of unwanted immune responses (Chirino et al 2004; De Groot et al 2007; Baker et al 2010). Hemophilia A (HA) is an inherited bleeding disorder that is characterized by the deficiency or dysfunction of FVIII and replacement therapy with FVIII is the first line of therapy (Lollar et al 2001; Klinge et al 2002; Aledort et al 2014). However, about 30% of severe HA patients develop neutralizing antibodies (NAbs) or inhibitors, which abrogate the activity of FVIII and potentially compromise the safety and efficacy of therapy (Lollar et al 2001; Klinge et al 2002; Aledort et al 2014). For HA patients with inhibitors, alternative therapy options include bypass agents and immune tolerance induction, in which repeated high dose of FVIII is administered (Lollar et al 2001; Astermark 2011). However, some limitations include varying success rates as well as the high cost. Therefore, the development of novel strategies to reduce immunogenicity would address an unmet clinical need.
The strategy developed in our laboratory harnesses the biologic properties of PS to form liposomes. Exploiting the known PS and FVIII interaction via the C2 domain, we first demonstrated that FVIII complexed with PS liposomes or its head group, O-phospho-L-serine (OPLS), displayed a significant decrease in inhibitor formation in HA mice (Purohit et al 2005; Ramani et al 2008). In order to investigate the mechanism of PS-mediated reduction in immune response, bone marrow-derived dendritic cells (BMDCs) were cultured and in vitro studies were performed to evaluate the impact of PS FVIII exposure on DC maturation (Gaitonde et al 2011). After treatment with PS FVIII, the expression of the co-stimulatory marker CD40 and the late maturation marker CD86 were downregulated, suggesting that PS inhibits the full maturation of DCs by skewing them towards a tolerogenic phenotype. Further studies looking at the DC and T cell interaction showed that the tolerogenic DCs generated by PS FVIII treatment displayed a reduced capacity to activate CD4+ T cells (Gaitonde et al 2011). In vitro cytokine analysis showed upregulation of regulatory cytokines, such as IL-10 and TGF-β, along with downregulation of pro-inflammatory cytokines, such as IL-17, IL-6, and TNF-α.
PS-FVIII complexes are not only less immunogenic, but also the administration of FVIII in the presence of PS liposomes presents FVIII in a tolerogenic context that could be exploited to prevent immunogenicity by desensitization and immunological tolerance. In vivo studies performed in HA mice showed that pre-exposure of PS FVIII induced hypo-responsiveness following subsequent rechallenges with free FVIII (Gaitonde et al 2013). Interestingly, control mice that were pre-exposed with FVIII and dexamethasone, a global immunosuppressant, displayed similar reductions in inhibitory titers as PS FVIII treated mice, compared with mice receiving only FVIII. However, when the dexamethasone treatment was halted and the mice were subsequently rechallenged with free FVIII, the inhibitory responses increased to comparable levels as free FVIII treated mice. In PS FVIII pre-treated mice, after the conclusion of PS desensitization and subsequent FVIII rechallenge, these mice continued to display a significant reduction in inhibitory titers. The hypo-responsiveness observed with PS treatment indicates that the PS-mediated effects extend beyond mere immunosuppression. In fact, we hypothesize that PS is able to induce immunological tolerance because of its ability to convert an immunogen to a tolerogen. The presence of PS in liposomes containing therapeutic proteins promotes uptake and reversal by additional cells that have not been observes in efferocytosis, but this uptake has enabled tolerance to be induced to therapeutic proteins.
Additional studies were performed to confirm that the PS-mediated hypo-responsiveness is antigen-specific and not immunosuppression (Gaitonde et al 2013; Ramakrishnan et al 2015). In one study, ovalbumin, an irrelevant antigen, was administered at a distal site from where PS FVIII was administered. Titer analyses showed that FVIII-specific immune responses were reduced due to the PS treatment while a robust immune response was generated towards ovalbumin. This further confirms that PS does not exert a global immunosuppressive signal, but in fact is able to induce hypo-responsiveness in an antigen-specific manner.
Now understanding that PS is able to induce hypo-responsiveness by converting an immunogen to a tolerogen, it is important to elucidate the mechanism of PS-mediated tolerance and to identify a critical cell population that mediates this effect. Further in vivo studies indicate that the expression of CD4+FoxP3+ Tregs are significantly increased in HA mice treated with PS FVIII as compared to free FVIII treatment (Gaitonde et al 2013). Tregs are a subset of CD4+ T cells that have the ability to suppress immune responses and express FoxP3 as a biomarker (Hori et al 2003). Increases and expansion of the Treg population is often used as a mechanistic marker for tolerance induction. In addition to the induction of Tregs, it has been shown that PS is able to inhibit the generation of memory B cells (Ramakrishnan et al 2015). Collectively, the mechanism of PS-mediated hypo-responsiveness is summarized in Figure 4, in which the generation of tolerogenic DCs release regulatory cytokines such as IL-10 and TGF-β that mediate the suppression of effector T cell function as well as promote the expansion of Tregs, ultimately inhibiting memory B cell generation and antibody production (Ramakrishnan et al 2015). Based on our previous studies, it is believed that, when administered with PS-antigen complexes, PS does not function as a global immunosuppressive agent. Thus, the mechanism by which PS induces antigen-specific tolerance to a protein cannot be explained by the “eat me” or “ignore me/tolerate me” signals alone. Rather, these observations are consistent with the ability of PS to induce tolerance by converting an immunogen to a tolerogen. By this property identified, PS serves to actively teach the immune system to tolerize its cargo in order to induce peripheral tolerance.
FIGURE 4.
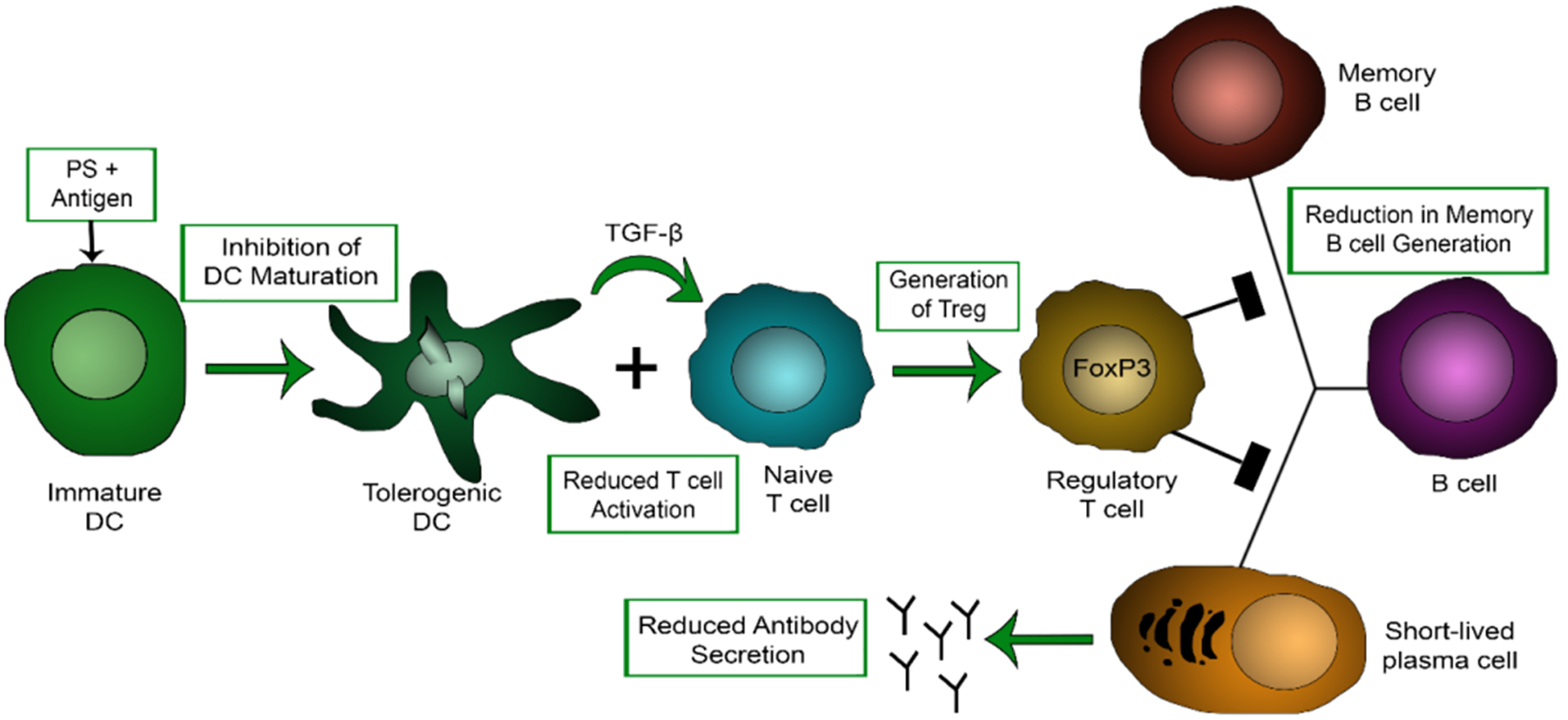
Proposed mechanism of PS-mediated tolerance induction. Administration of an antigen of interest in the presence of PS induces tolerogenic DCs, reduces T cell activation, promotes Treg expression that inhibits memory B cell generation and antibody production.
The strategy developed in our laboratory harnesses the biological properties of PS and its role during apoptosis to generate a tolerogenic PS containing nanoparticle to mitigate immunogenicity towards therapeutic proteins in part via apoptotic mimicry. While the aforementioned studies evaluated the impact of PS with FVIII as a model protein, this strategy is broadly applicable and can be extended to other antigens. For example, Pompe Disease is a lysosomal storage disorder characterized by the deficiency of acid alpha glucosidase (GAA), an enzyme responsible for the breakdown of glycogen into glucose (Raben et al 2002; Doerfler et al 2016). The deficiency in GAA results in glycogen accumulation in tissues, manifesting in skeletal muscle weakness, respiratory insufficiency, and cardiomyopathy. Despite enzyme replacement therapy with recombinant GAA, 89–100% of patients develop anti-GAA antibodies, compromising the efficacy and leaving patients with no alternative therapies for the disease (Raben et al 2002; 2006; Doerfler et al 2016). The ability of PS to induce tolerance by converting immunogenic GAA to a tolerogenic form would address this unmet clinical need. We have previously shown that subcutaneous administration of GAA in the presence of PS can induce a durable and long-lasting hypo-responsiveness in Pompe Disease mice (Schneider et al 2016; Glassman et al 2018).
Further work in our lab has shown an ability to use Lyso-PS in our nanoparticles to further reduce anti-FVIII antibody development. Biophysical characterization has shown that the incorporation of Lyso-PS forms smaller nanoparticles with an average diameter around 100 nm, compared to around a 200 nm diameter for double chain PS liposomes(Glassman et al 2018). The formation of more, smaller diameter nanoparticles also increases total PS surface area in the formulation and we have shown that PS exposure on the surface of Lyso-PS nanoparticles is higher than that of double chain PS liposomes (Unpublished Data). Administering Lyso-PS complexed with FVIII was able to reduce anti-FVIII antibody levels in an antigen specific manner, to a greater extent than double chain PS liposomes. Additionally, we have shown that Lyso-PS also binds to TIM-4 to exert its function, as blocking TIM-4 prevented Lyso-PS mediated FVIII tolerance induction(Glassman et al 2018).
The use of PS in inducing tolerance towards enzyme replacement therapies, in which no central tolerance exists, suggests a biological role that has not been discussed previously. After PS pre-exposure treatment with antigen loaded PS liposomes, rechallenge with free antigen promotes immune hypo-responsiveness and reduces antibody production. Instead of silent clearance mediated by the externalization of PS, the observation suggests that PS can induce tolerance. The broad therapeutic potential is not only to reduce immunogenicity but also to treat autoimmunity and other immune conditions.
5. CONCLUSIONS
Overall, this review discusses the biological functions of PS in apoptosis and hemostasis. Further, it discusses the implications of PS in a therapeutic setting, where PS blocking strategies are highlighted in the context of parasitic/viral immune evasion and cancer. In addition, the implications of PS is discussed in autoimmunity, where PS could be used in order to re-establish tolerance to self-antigens that was lost with the development of autoimmunity. Finally, the use of PS to mitigate immunogenicity of replacement therapies highlights a novel biological function of PS in tolerance induction that could be harnessed clinically.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health R01 HL-70227 to SVB. The authors report no conflicts of interest.
REFERENCES
- 2006. Myozyme [package insert]. Genzyme Corporation.
- Aledort L,Ljung R,Mann K,Pipe S. 2014. Factor VIII therapy for hemophilia A: current and future issues. Expert Rev Hematol 73: 373–85. [DOI] [PubMed] [Google Scholar]
- Arroyo A,Modriansky M,Serinkan FB,Bello RI,Matsura T,Jiang J,Tyurin VA,Tyurina YY,Fadeel B,Kagan VE. 2002. NADPH oxidase-dependent oxidation and externalization of phosphatidylserine during apoptosis in Me2SO-differentiated HL-60 cells. Role in phagocytic clearance. J Biol Chem 27751: 49965–75. [DOI] [PubMed] [Google Scholar]
- Astermark J. 2011. Immune tolerance induction in patients with hemophilia A. Thromb Res 127 Suppl 1: S6–9. [DOI] [PubMed] [Google Scholar]
- Baker MP,Reynolds HM,Lumicisi B,Bryson CJ. 2010. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self Nonself 14: 314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini F,Bruni A. 1993. Role of a serum phospholipase A1 in the phosphatidylserine-induced T cell inhibition. FEBS Lett 3161: 1–4. [DOI] [PubMed] [Google Scholar]
- Belzile O,Huang X,Gong J,Carlson J,Schroit AJ,Brekken RA,Freimark BD. 2018. Antibody targeting of phosphatidylserine for the detection and immunotherapy of cancer. Immunotargets Ther 7: 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevers EM,Williamson PL. 2016. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol Rev 962: 605–45. [DOI] [PubMed] [Google Scholar]
- Birge RB,Boeltz S,Kumar S,Carlson J,Wanderley J,Calianese D,Barcinski M,Brekken RA,Huang X,Hutchins JT,Freimark B,Empig C,Mercer J,Schroit AJ,Schett G,Herrmann M. 2016. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisenko GG,Iverson SL,Ahlberg S,Kagan VE,Fadeel B. 2004. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ 118: 943–5. [DOI] [PubMed] [Google Scholar]
- Chalasani P,Marron M,Roe D,Clarke K,Iannone M,Livingston RB,Shan JS,Stopeck AT. 2015. A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer Med 47: 1051–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MK,Bergmark C,Laurila A,Horkko S,Han KH,Friedman P,Dennis EA,Witztum JL. 1999. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci U S A 9611: 6353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurio RA,Janko C,Munoz LE,Frey B,Herrmann M,Gaipl US. 2009. Phospholipids: key players in apoptosis and immune regulation. Molecules 1412: 4892–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirino AJ,Ary ML,Marshall SA. 2004. Minimizing the immunogenicity of protein therapeutics. Drug Discov Today 92: 82–90. [DOI] [PubMed] [Google Scholar]
- Chung IY,Jeong KH,Mi HS. 2017. Identification of lysophosphatidylserine receptor P2Y10 as a novel G-protein coupled receptor mediating eosinophil degranulation. The Journal of Immunology 1981 Supplement: 201.14–.14. [Google Scholar]
- Cohen PL,Caricchio R,Abraham V,Camenisch TD,Jennette JC,Roubey RA,Earp HS,Matsushima G,Reap EA. 2002. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med 1961: 135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Freitas Balanco JM,Moreira ME,Bonomo A,Bozza PT,Amarante-Mendes G,Pirmez C,Barcinski MA. 2001. Apoptotic mimicry by an obligate intracellular parasite downregulates macrophage microbicidal activity. Curr Biol 1123: 1870–3. [DOI] [PubMed] [Google Scholar]
- De Groot AS,Scott DW. 2007. Immunogenicity of protein therapeutics. Trends Immunol 2811: 482–90. [DOI] [PubMed] [Google Scholar]
- Doerfler PA,Nayak S,Corti M,Morel L,Herzog RW,Byrne BJ. 2016. Targeted approaches to induce immune tolerance for Pompe disease therapy. Mol Ther Methods Clin Dev 3: 15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA,Bratton DL,Frasch SC,Warner ML,Henson PM. 1998. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ 57: 551–62. [DOI] [PubMed] [Google Scholar]
- Fadok VA,Bratton DL,Konowal A,Freed PW,Westcott JY,Henson PM. 1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 1014: 890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA,Savill JS,Haslett C,Bratton DL,Doherty DE,Campbell PA,Henson PM. 1992. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J Immunol 14912: 4029–35. [PubMed] [Google Scholar]
- Fadok VA,Voelker DR,Campbell PA,Cohen JJ,Bratton DL,Henson PM. 1992. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 1487: 2207–16. [PubMed] [Google Scholar]
- Frasch SC,Berry KZ,Fernandez-Boyanapalli R,Jin HS,Leslie C,Henson PM,Murphy RC,Bratton DL. 2008. NADPH oxidase-dependent generation of lysophosphatidylserine enhances clearance of activated and dying neutrophils via G2A. J Biol Chem 28348: 33736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch SC,Bratton DL. 2012. Emerging roles for lysophosphatidylserine in resolution of inflammation. Prog Lipid Res 513: 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch SC,Fernandez-Boyanapalli RF,Berry KA,Murphy RC,Leslie CC,Nick JA,Henson PM,Bratton DL. 2013. Neutrophils regulate tissue Neutrophilia in inflammation via the oxidant-modified lipid lysophosphatidylserine. J Biol Chem 2887: 4583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch SC,Fernandez-Boyanapalli RF,Berry KZ,Leslie CC,Bonventre JV,Murphy RC,Henson PM,Bratton DL. 2011. Signaling via macrophage G2A enhances efferocytosis of dying neutrophils by augmentation of Rac activity. J Biol Chem 28614: 12108–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ,Casasnovas JM,Umetsu DT,DeKruyff RH. 2010. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev 2351: 172–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller AD,Van Eldik LJ. 2008. MFG-E8 Regulates Microglial Phagocytosis of Apoptotic Neurons. Journal of Neuroimmune Pharmacology 34: 246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaipl US,Kuhn A,Sheriff A,Munoz LE,Franz S,Voll RE,Kalden JR,Herrmann M. 2006. Clearance of apoptotic cells in human SLE. Curr Dir Autoimmun 9: 173–87. [DOI] [PubMed] [Google Scholar]
- Gaitonde P,Peng A,Straubinger RM,Bankert RB,Balu-Iyer SV. 2011. Phosphatidylserine reduces immune response against human recombinant Factor VIII in Hemophilia A mice by regulation of dendritic cell function. Clin Immunol 1382: 135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitonde P,Ramakrishnan R,Chin J,Kelleher RJ,Bankert RB,Balu-Iyer SV. 2013. Exposure to Factor VIII Protein in the Presence of Phosphatidylserine Induces Hypo-responsiveness toward Factor VIII Challenge in Hemophilia A Mice. Journal of Biological Chemistry 28824: 17051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber DE,Spigel DR,Giorgadze D,Shtivelband M,Ponomarova OV,Shan JS,Menander KB,Belani CP. 2016. Docetaxel Combined With Bavituximab in Previously Treated, Advanced Nonsquamous Non-Small-Cell Lung Cancer. Clin Lung Cancer 173: 169–76. [DOI] [PubMed] [Google Scholar]
- Glassman FY,Balu-Iyer SV. 2018. Subcutaneous administration of Lyso-phosphatidylserine nanoparticles induces immunological tolerance towards Factor VIII in a Hemophilia A mouse model. International journal of pharmaceutics 5481: 642–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassman FY,Schneider JL,Ramakrishnan R,Dingman RK,Ramanathan M,Bankert RB,Balu-Iyer SV. 2018. Phosphatidylserine Is Not Just a Cleanup Crew but Also a Well-Meaning Teacher. J Pharm Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME,Sun M,Zhang R,Febbraio M,Silverstein R,Hazen SL. 2006. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med 20312: 2613–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gude DR,Alvarez SE,Paugh SW,Mitra P,Yu J,Griffiths R,Barbour SE,Milstien S,Spiegel S. 2008. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J 228: 2629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanayama R,Tanaka M,Miyasaka K,Aozasa K,Koike M,Uchiyama Y,Nagata S. 2004. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 3045674: 1147–50. [DOI] [PubMed] [Google Scholar]
- Hankins HM,Baldridge RD,Xu P,Graham TR. 2015. Role of Flippases, Scramblases and Transfer Proteins in Phosphatidylserine Subcellular Distribution. Traffic 161: 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann PR,deCathelineau AM,Ogden CA,Leverrier Y,Bratton DL,Daleke DL,Ridley AJ,Fadok VA,Henson PM. 2001. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol 1554: 649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S,Nomura T,Sakaguchi S. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science 2995609: 1057–61. [DOI] [PubMed] [Google Scholar]
- Julian L,Olson MF. 2015. Apoptotic membrane dynamics in health and disease. Cell Health and Cytoskeleton 7: 133–42. [Google Scholar]
- Kagan VE,Gleiss B,Tyurina YY,Tyurin VA,Elenstrom-Magnusson C,Liu SX,Serinkan FB,Arroyo A,Chandra J,Orrenius S,Fadeel B. 2002. A role for oxidative stress in apoptosis: oxidation and externalization of phosphatidylserine is required for macrophage clearance of cells undergoing Fas-mediated apoptosis. J Immunol 1691: 487–99. [DOI] [PubMed] [Google Scholar]
- Kay JG,Grinstein S. 2013. Phosphatidylserine-mediated cellular signaling. Adv Exp Med Biol 991: 177–93. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ Jr., Balu-Iyer S,Loyall J,Sacca AJ,Shenoy GN,Peng P,Iyer V,Fathallah AM,Berenson CS,Wallace PK,Tario J,Odunsi K,Bankert RB. 2015. Extracellular Vesicles Present in Human Ovarian Tumor Microenvironments Induce a Phosphatidylserine-Dependent Arrest in the T-cell Signaling Cascade. Cancer Immunol Res 311: 1269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr D,Tietjen GT,Gong Z,Tajkhorshid E,Adams EJ,Lee KYC. 2018. Sensitivity of peripheral membrane proteins to the membrane context: A case study of phosphatidylserine and the TIM proteins. Biochimica et Biophysica Acta (BBA) - Biomembranes 186010: 2126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimani SG,Geng K,Kasikara C,Kumar S,Sriram G,Wu Y,Birge RB. 2014. Contribution of Defective PS Recognition and Efferocytosis to Chronic Inflammation and Autoimmunity. Front Immunol 5: 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura H,Makide K,Shuto A,Ikubo M,Inoue A,Suzuki K,Sato Y,Nakamura S,Otani Y,Ohwada T,Aoki J. 2012. GPR34 is a receptor for lysophosphatidylserine with a fatty acid at the sn-2 position. J Biochem 1515: 511–8. [DOI] [PubMed] [Google Scholar]
- Klinge J,Ananyeva NM,Hauser CA,Saenko EL. 2002. Hemophilia A--from basic science to clinical practice. Semin Thromb Hemost 283: 309–22. [DOI] [PubMed] [Google Scholar]
- Kobayashi N,Karisola P,Peña-Cruz V,Dorfman DM,Jinushi M,Umetsu SE,Butte MJ,Nagumo H,Chernova I,Zhu B. 2007. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 276: 927–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankry D,Rovis TL,Jonjic S,Mandelboim O. 2013. The interaction between CD300a and phosphatidylserine inhibits tumor cell killing by NK cells. Eur J Immunol 438: 2151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber K,Bohn E,Krober SM,Xiao YJ,Blumenthal SG,Lindemann RK,Marini P,Wiedig C,Zobywalski A,Baksh S,Xu Y,Autenrieth IB,Schulze-Osthoff K,Belka C,Stuhler G,Wesselborg S. 2003. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 1136: 717–30. [DOI] [PubMed] [Google Scholar]
- Leventis PA,Grinstein S. 2010. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys 39: 407–27. [DOI] [PubMed] [Google Scholar]
- Lollar P,Healey JF,Barrow RT,Parker ET. 2001. Factor VIII inhibitors. Adv Exp Med Biol 489: 65–73. [DOI] [PubMed] [Google Scholar]
- Lu Q,Lemke G. 2001. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2935528: 306–11. [DOI] [PubMed] [Google Scholar]
- Mahajan A,Herrmann M,Munoz LE. 2016. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front Immunol 7: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makide K,Aoki J. 2013. GPR34 as a lysophosphatidylserine receptor. J Biochem 1534: 327–9. [DOI] [PubMed] [Google Scholar]
- Marino G,Kroemer G. 2013. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res 2311: 1247–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsura T,Serinkan BF,Jiang J,Kagan VE. 2002. Phosphatidylserine peroxidation/externalization during staurosporine-induced apoptosis in HL-60 cells. FEBS Lett 5241–3: 25–30. [DOI] [PubMed] [Google Scholar]
- Meyer J,Arriaga Y,Anandam J,Karri S,Syed S,Verma U,Abdelnaby A,Raja G,Dong Y,Beg M,Balch G. 2017. A Phase I Clinical Trial of the Phosphatidylserine-targeting Antibody Bavituximab in Combination With Radiation Therapy and Capecitabine in the Preoperative Treatment of Rectal Adenocarcinoma. Am J Clin Oncol. [DOI] [PubMed] [Google Scholar]
- Miyanishi M,Tada K,Koike M,Uchiyama Y,Kitamura T,Nagata S. 2007. Identification of Tim4 as a phosphatidylserine receptor. Nature 4507168: 435–9. [DOI] [PubMed] [Google Scholar]
- Mueller RB,Sheriff A,Gaipl US,Wesselborg S,Lauber K. 2007. Attraction of phagocytes by apoptotic cells is mediated by lysophosphatidylcholine. Autoimmunity 404: 342–4. [DOI] [PubMed] [Google Scholar]
- Munoz LE,Franz S,Pausch F,Furnrohr B,Sheriff A,Vogt B,Kern PM,Baum W,Stach C,von Laer D,Brachvogel B,Poschl E,Herrmann M,Gaipl US. 2007. The influence on the immunomodulatory effects of dying and dead cells of Annexin V. J Leukoc Biol 811: 6–14. [DOI] [PubMed] [Google Scholar]
- Munoz LE,Frey B,Pausch F,Baum W,Mueller RB,Brachvogel B,Poschl E,Rodel F,von der Mark K,Herrmann M,Gaipl US. 2007. The role of annexin A5 in the modulation of the immune response against dying and dead cells. Curr Med Chem 143: 271–7. [DOI] [PubMed] [Google Scholar]
- Munoz LE,Lauber K,Schiller M,Manfredi AA,Herrmann M. 2010. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol 65: 280–9. [DOI] [PubMed] [Google Scholar]
- Park D,Tosello-Trampont AC,Elliott MR,Lu M,Haney LB,Ma Z,Klibanov AL,Mandell JW,Ravichandran KS. 2007. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 4507168: 430–4. [DOI] [PubMed] [Google Scholar]
- Park SY,Jung MY,Kim HJ,Lee SJ,Kim SY,Lee BH,Kwon TH,Park RW,Kim IS. 2008. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ 151: 192–201. [DOI] [PubMed] [Google Scholar]
- Park SY,Kim SY,Jung MY,Bae DJ,Kim IS. 2008. Epidermal growth factor-like domain repeat of stabilin-2 recognizes phosphatidylserine during cell corpse clearance. Mol Cell Biol 2817: 5288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol-Autonell I,Mansilla MJ,Rodriguez-Fernandez S,Cano-Sarabia M,Navarro-Barriuso J,Ampudia RM,Rius A,Garcia-Jimeno S,Perna-Barrull D,Martinez-Caceres E,Maspoch D,Vives-Pi M. 2017. Liposome-based immunotherapy against autoimmune diseases: therapeutic effect on multiple sclerosis. Nanomedicine (Lond) 1211: 1231–42. [DOI] [PubMed] [Google Scholar]
- Pujol-Autonell I,Serracant-Prat A,Cano-Sarabia M,Ampudia RM,Rodriguez-Fernandez S,Sanchez A,Izquierdo C,Stratmann T,Puig-Domingo M,Maspoch D,Verdaguer J,Vives-Pi M. 2015. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PLoS One 106: e0127057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit VS,Ramani K,Sarkar R,Kazazian HH Jr.,Balasubramanian SV. 2005. Lower inhibitor development in hemophilia A mice following administration of recombinant factor VIII-O-phospho-L-serine complex. J Biol Chem 28018: 17593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N,Plotz P,Byrne BJ. 2002. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med 22: 145–66. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R,Davidowitz A,Balu-Iyer SV. 2015. Exposure of FVIII in the Presence of Phosphatidyl Serine Reduces Generation of Memory B-Cells and Induces Regulatory T-Cell-Mediated Hyporesponsiveness in Hemophilia A Mice. J Pharm Sci 1048: 2451–6. [DOI] [PubMed] [Google Scholar]
- Ramani K,Miclea RD,Purohit VS,Mager DE,Straubinger RM,Balu-Iyer SV. 2008. Phosphatidylserine containing liposomes reduce immunogenicity of recombinant human factor VIII (rFVIII) in a murine model of hemophilia A. J Pharm Sci 974: 1386–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS. 2010. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med 2079: 1807–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring GH,Lakkis FG. 1999. Breakdown of self-tolerance and the pathogenesis of autoimmunity. Semin Nephrol 191: 25–33. [PubMed] [Google Scholar]
- Rodriguez-Fernandez S,Pujol-Autonell I,Brianso F,Perna-Barrull D,Cano-Sarabia M,Garcia-Jimeno S,Villalba A,Sanchez A,Aguilera E,Vazquez F,Verdaguer J,Maspoch D,Vives-Pi M. 2018. Phosphatidylserine-Liposomes Promote Tolerogenic Features on Dendritic Cells in Human Type 1 Diabetes by Apoptotic Mimicry. Front Immunol 9: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JL,Balu-Iyer SV. 2016. Phosphatidylserine Converts Immunogenic Recombinant Human Acid Alpha-Glucosidase to a Tolerogenic Form in a Mouse Model of Pompe Disease. J Pharm Sci 10510: 3097–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K,Nagata S. 2015. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends in Cell Biology 2511: 639–50. [DOI] [PubMed] [Google Scholar]
- Segawa K,Suzuki J,Nagata S. 2011. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci U S A 10848: 19246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz HM,Camenisch TD,Lemke G,Earp HS,Matsushima GK. 2007. Macrophages and Dendritic Cells Use Different Axl/Mertk/Tyro3 Receptors in Clearance of Apoptotic Cells. The Journal of Immunology 1789: 5635–42. [DOI] [PubMed] [Google Scholar]
- Shao WH,Cohen PL. 2011. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther 131: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HU,Haj-Yehia A,Levi-Schaffer F. 2000. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 55: 415–8. [DOI] [PubMed] [Google Scholar]
- Soares MM,King SW,Thorpe PE. 2008. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat Med 1412: 1357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugo T,Tachimoto H,Chikatsu T,Murakami Y,Kikukawa Y,Sato S,Kikuchi K,Nagi T,Harada M,Ogi K,Ebisawa M,Mori M. 2006. Identification of a lysophosphatidylserine receptor on mast cells. Biochem Biophys Res Commun 3414: 1078–87. [DOI] [PubMed] [Google Scholar]
- Suzuki J,Denning DP,Imanishi E,Horvitz HR,Nagata S. 2013. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 3416144: 403–6. [DOI] [PubMed] [Google Scholar]
- Suzuki J,Imanishi E,Nagata S. 2014. Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J Biol Chem 28944: 30257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J,Umeda M,Sims PJ,Nagata S. 2010. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 4687325: 834–8. [DOI] [PubMed] [Google Scholar]
- Tietjen GT,Gong Z,Chen CH,Vargas E,Crooks JE,Cao KD,Heffern CT,Henderson JM,Meron M,Lin B,Roux B,Schlossman ML,Steck TL,Lee KY,Adams EJ. 2014. Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc Natl Acad Sci U S A 11115: E1463–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurin VA,Balasubramanian K,Winnica D,Tyurina YY,Vikulina AS,He RR,Kapralov AA,Macphee CH,Kagan VE. 2014. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ 215: 825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kleij D,Latz E,Brouwers JF,Kruize YC,Schmitz M,Kurt-Jones EA,Espevik T,de Jong EC,Kapsenberg ML,Golenbock DT,Tielens AG,Yazdanbakhsh M. 2002. A novel host-parasite lipid cross-talk. Schistosomal lyso-phosphatidylserine activates toll-like receptor 2 and affects immune polarization. J Biol Chem 27750: 48122–9. [DOI] [PubMed] [Google Scholar]
- van der Kleij D,Yazdanbakhsh M. 2003. Control of inflammatory diseases by pathogens: lipids and the immune system. Eur J Immunol 3311: 2953–63. [DOI] [PubMed] [Google Scholar]
- Verhoven B,Schlegel R,Williamson P. 1995. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. Journal of Experimental Medicine 1825: 1597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S,Brooks CR,Zhu C,Wu C,Sweere JM,Petecka S,Yeste A,Quintana FJ,Ichimura T,Sobel RA,Bonventre JV,Kuchroo VK. 2012. Defect in regulatory B-cell function and development of systemic autoimmunity in T-cell Ig mucin 1 (Tim-1) mucin domain-mutant mice. Proc Natl Acad Sci U S A 10930: 12105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y,Huang X,Lynn KD,Thorpe PE. 2013. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res 14: 256–68. [DOI] [PubMed] [Google Scholar]