

Abstract
The enhancement of nuclear relaxation rates due to the interaction with a paramagnetic center (known as Paramagnetic Relaxation Enhancement) is a powerful source of structural and dynamics information, widely used in structural biology. However, many signals affected by the hyperfine interaction relax faster than the evolution periods of common NMR experiments and therefore they are broadened beyond detection. This gives rise to a so-called blind sphere around the paramagnetic center, which is a major limitation in the use of PREs. Reducing the blind sphere is extremely important in paramagnetic metalloproteins. The identification, characterization, and proper structural restraining of the first coordination sphere of the metal ion(s) and its immediate neighboring regions is key to understand their biological function. The novel HSQC scheme we propose here, that we termed R2-weighted, HSQC-AP, achieves this aim by detecting signals that escaped detection in a conventional HSQC experiment and provides fully reliable R2 values in the range of 1H R2 rates ca. 50–400 s−1. Independently on the type of paramagnetic center and on the size of the molecule, this experiment decreases the radius of the blind sphere and increases the number of detectable PREs. Here, we report the validation of this approach for the case of PioC, a small protein containing a high potential 4Fe-4S cluster in the reduced [Fe4S4]2+ form. The blind sphere was contracted to a minimal extent, enabling the measurement of R2 rates for the cluster coordinating residues.
Keywords: Paramagnetic NMR, Iron sulfur proteins, Pulse sequences, NMR based structural restraints, Transverse relaxation, Paramagnetic relaxation enhancement
Introduction
The hyperfine interaction between electron and nuclear spins gives rise to additional contributions to chemical shifts and nuclear relaxation, both of which can be used as a source of structural restraints (Piccioli and Turano 2015; Turner et al. 1998). Nowadays, NMR solution structures of paramagnetic macromolecules are obtained by a combination of conventional restraints (Ab et al. 2006; Mori et al. 2008), such as NOE and residual dipolar couplings, and of paramagnetic-based restraints (Arnesano et al. 2006; Clore 2015; Kudhair, et al. 2280; Parigi et al. 2019; Pintacuda et al. 2007). Depending on which paramagnet is present in the system, different combinations of contact shifts, pseudocontact shifts, paramagnetic relaxation enhancements and cross correlation rates can be used (Koehler and Meiler 2011; Clore and Iwahara 2009; Kateb and Piccioli 2003; Pintacuda 2004). However, since about two decades, many groups have promoted the use of paramagnetism-based NMR restraints to study also diamagnetic proteins: metal binding tags have been used as spin-labels and provide various restraints capable to complement the NMR information available on a native, diamagnetic, derivative (Iwahara et al. 2004; Miao 2019; Matei and Gronenborn 2016; Liu et al. 2014; Nitsche and Otting 2017; Joss and Haussinger 2019). NMR of paramagnetic systems is not anymore a playground reserved to scientists working with inorganic or bio-inorganic systems, but a tool for a larger community of structural biologists with many potential applications (Tang et al. 2006; Softley et al. 2020).
Very recently, we have shown that structural restraints derived from Paramagnetic Relaxation Enhancements (PRE) can be used as the unique source of restraints for the structure calculation of small metalloproteins (Trindade et al. 2020b). A key point for a successful PRE-only approach is the availability of many accurate relaxation rate values measured throughout the entire protein, including the close proximity of the paramagnetic center, where nuclear spins are strongly affected by paramagnetism. When hyperfine shifted signals are well isolated outside the bulk diamagnetic envelope, they can be characterized using 1D experiments and relaxation based filters (Inubushi and Becker 1983), although usually only a few signals, arising from the side chains of metal-bound residues, can be identified via this approach (Hansen and Led 2006; Sato et al. 2003; Brancaccio 2017). Paramagnetic relaxation depends on γ2 of the observed nucleus, therefore 13C or 15N direct detection (Arnesano 2003; Kolczak et al. 1999; Lin et al. 2003) have been successful as an efficient alternative to 1H detected experiments, not only for assignment purposes (Bermel 2005; Machonkin et al. 2002; Bertini et al. 2005b) but also for the obtainment of PREs. However, the 15 N HSQC experiment still remains the “easiest” molecular fingerprint, therefore methods to exploit the use of 1HN T1 and T2 rates are welcome.
To date, relaxation-based restraints are indeed obtained via 1H T1 and T2 measurements from 15N HSQC-type experiments (Donaldson 2001; Iwahara et al. 2007). However, many signals affected by the hyperfine interaction relax faster than the evolution periods used in these experiments; therefore, they will be broadened beyond detection or, when they are detected, their intensity decay cannot be properly sampled. Thus, we need to design novel pulse sequences to measure R2 and R1 rates of resonances that escape detection in conventional HSQC experiments. For longitudinal 1H relaxation rates, we have shown that an inversion recovery filtered 15N HSQC experiment acquired in antiphase (15N IR-HSQC-AP) (Ciofi-Baffoni et al. 2014) is very effective for the identification of signals severely affected by paramagnetism, and also for obtaining R1 rates faster than those measurable in established experiments. However, it has been reported that 1H R2 values are less susceptible to internal motions and cross relaxation than 1H R1 values (Iwahara et al. 2004; Iwahara and Clore 2010). Therefore, we are interested to develop an experiment to obtain accurate measurements of 1H transverse relaxation also in the close proximity of a paramagnetic center.
The experiment we present here has been customized for the case of the HiPIP (High Potential Iron-sulfur Protein) PioC from Rhodopseudomonas palustris TIE-1 (Bird, et al. 2014). PioC has 54 amino acids, contains the typical HiPIP binding motif CXXCXnCXmC and it is the smallest HiPIP isolated so far. Due to the high reduction potential (E0 = + 450 mV vs SHE) of the [Fe4S4]3+/[Fe4S4]2+ redox pair (both oxidation states are paramagnetic), the protein is stable in the reduced [Fe4S4]2+ state. The paramagnetic 1H NMR spectrum of PioC is very similar to the NMR spectra of other HiPIPs in the reduced state (Bertini 1992), thus indicating that the electronic relaxation time (τe) in PioC must be similar to previously studied HiPIPs (Banci et al. 2018). However, PioC is the smallest HiPIP isolated so far and about 60% of the protein is affected by paramagnetic relaxation. Counter-intuitively, the small size of the protein makes it more difficult to study, because paramagnetic effects are active in the majority of the protein, scalar and dipolar couplings are quenched and a number of HN signals are not observable in a conventional HSQC experiment (Cheng and Markley 1995; Machonkin et al. 2005; Lin 2009). Throughout this article we will first review why experimental approaches that are effective to study proteins containing metal bindings tags are not equally efficient for native metalloproteins; then we will discuss how implementation of this novel HSQC scheme allows the measurement of relaxation rate values in a range 50–400 s−1. Rates in this range were once very difficult to measure reliably, however they are necessary to reduce the blind sphere enabling the characterization, and proper structural restraining of the first coordination sphere of the metal ion(s) and its immediate surrounding residues.
Materials and methods
Protein expression and purification
PioC was expressed and purified as previously reported (Bird et al. 2014). Uniformly 15N labeled samples of PioC were produced and the expression and purification protocol was identical throughout except in the addition of ammonium sulfate (15N2, 99%) in the M9 minimal media. BL21 DE3 cells were double transformed with pET32h, a plasmid containing the construct thioredoxin–6xHis–thrombin cleavage site–PioC, and with pDB1281, a plasmid that carries the machinery for the assembly of iron-sulfur clusters. Cells were grown in Luria–Bertani (LB) medium supplemented with 100 mg*dm−3 ampicillin and 35 mg*dm−3 chloramphenicol until the OD600nm of 0.6 where they were induced with 1.0 mM arabinose and 20 μM FeCl3 and 200 μM cysteine were added. Cells were again incubated until the OD600nm of 1 and then harvested and washed in M9 minimal media salts before resuspension in M9 minimal media. Once re-suspended, cells were incubated for one hour before induction with 0.5 mM IPTG. After 4 h cells were harvested by centrifugation and disrupted using a French Press at 1000 psi. The lysate was ultra-centrifuged at 204,709×g for 90 min at 4 °C to remove cell membranes and debris and the supernatant was dialyzed overnight against 50 mM potassium phosphate buffer pH 5.8 with 300 mM NaCl before injection in a His-trap affinity column (GE Healthcare). The fraction containing Histag-PioC eluted with 250 mM imidazole and was incubated overnight with Thrombin (GE Healthcare) for digestion. The final purified PioC (His-tag free) was then concentrated from the flow through of a 2nd passage through the His-trap column using an Amicon Ultra Centrifugal Filter (Millipore) with a 3 kDa cutoff. The purity of PioC was confirmed by SDS-PAGE with Blue Safe staining (NzyTech) and by UV–Visible spectroscopy.
NMR spectroscopy
Measurements of 1H R2 transverse relaxation rates were carried out using 11.7 T Bruker AVANCE 500 equipped with a triple resonance, inverse detection, cryoprobe (TXI). 1HN R2 measurements were obtained from a series of R2-weighted 15 N-HSQC-AP experiments, developed throughout this article. For each experiment, 256 scans were collected over 256 increments. Acquisition time and recycle delay were 47.1 ms and 150 ms. A series of sixteen R2-weighted 15N-HSQC-AP experiments was recorded, using INEPT transfer periods of 0.2 ms, 0.4 ms, 0.6 ms, 0.8 ms, 1.2 ms, 1.6 ms, 2.0 ms, 2.4 ms, 2.8 ms, 3.2 ms, 4.0 ms, 4.8 ms, 5.6 ms, 6.4 ms, 8.0 ms, 10.0 ms. Total experimental time was about 58 h. 1HN R2 measurements have been obtained also with the established approach (Donaldson 2001), in which a relaxation delay T is inserted into the INEPT building block of an in-phase 15 N HSQC experiment. For each experiment, 32 scans were collected over 156 increments, using acquisition time and recycle delay of 47.1 ms and 4 s, respectively. A series of fourteen15N-HSQC-IP experiments was recorded using relaxation delays T of 13.3 ms, 17.3 ms, 21.3 ms, 33.3 ms, 45.3 ms, 57.3 ms, 69.3 ms, 81.3 ms, 93.3 ms, 117.3 ms, 141.3 ms, 165.3 ms, 205.3 ms and 245.3 ms. A 1400 μs selective 1HN inversion pulse was used for 3JHNHα decoupling. Total experimental time was about 80 h. Both series have been performed using 256 × 1024 data point matrices, over spectral windows of 80.0 ppm × 21.7 ppm. Squared cosine weighting functions and apodization were used in both dimensions prior to FT, spectra dimensions was 512 × 2048 data points. Peak intensities were used to calculate R2 values, using the equations described in the results section. All relaxation data were analyzed using the Bruker Topspin Dynamics Center.
NMR assignment of PioC
The backbone NMR assignment of PioC (Trindade et al. 2020a) has been published on Biomolecular NMR Assignment and deposited in the BMRB data bank (ID 34487).
Results
State of the art: an overview
The state-of-the-art approach for 1H R2 relaxation is to use a 15N HSQC experiment and insert a relaxation period within the INEPT block to measure HN rates (Donaldson 2001). Relaxation rates R2 can then be obtained either by collecting, for each HN signal, a complete decay curve or by measuring R2 rates from a two time-point measurement, thereby enabling the direct determination of R2 values and their associated errors without any fitting procedure (Iwahara et al. 2007). For non-deuterated proteins, band selective 1H 180° pulses should be used to avoid the evolution of 3JHNHα coupling during the relaxation period. The paramagnetic contribution to the overall nuclear relaxation is given (Bertini et al. 2016) by Eq. (1)
| 1 |
when R2dia and R2obs values can be measured with high precision and accuracy (Iwahara et al. 2007), then very small values of paramagnetic relaxation enhancement R2para can be obtained by the difference of the two measured values. This has been exploited by attaching a metal binding peptide, such as ATCUN or HHP at the N-terminus site of a diamagnetic protein, as shown in Fig. 1a (Donaldson 2001). Metal binding peptides containing ions with long electronic correlation times, such as, for instance, Cu2+ or Mn2+, provide paramagnetic relaxation enhancements that can be measured, in the case of Cu2+ ion, for all protons located approximately 10–30 Å apart from the metal center (Donaldson 2001; Harford and Sarkar 1997; Jensen et al. 2004; Keizers and Ubbink 2011). The strength of this approach is that, within the above range, both R2obs and R2dia can be experimentally measured with high accuracy and R2para as small as 0.5 s−1 can be obtained, thus allowing the use of Paramagnetic Relaxation Enhancement (PRE, hereafter) for long metal-to-proton distance restraints. Protein–protein interaction surfaces and catalytic centers typically fall within this range, therefore one could obtain additional structural information for protein–protein/protein–ligand interactions, structure refinement, etc. (Battiste and Wagner 2000; Hass and Ubbink 2014; Cetiner 2019; Anthis and Clore 2015; Spronk 2018). On the other hand, signals at less than 12 Å from the copper(II) ion will experience R2para larger than 50 s−1, that will give rise to signals that, in the R2 experiment, will be very weak and eventually broadened beyond detection. The loss of information typically occurs in a protein region where no catalytic reactions or biochemical events occur, since the metal binding peptide is attached far from the protein core, usually at the N-term site. In metalloproteins, however, the topology of the system is completely different: the metal center(s) and its first coordination sphere always constitute the core region for the protein function. Structural biologists are therefore interested to obtain detailed information in the close proximity of the metal center(s), where the biochemically relevant events occur. Assuming the same paramagnetic relaxation enhancement of the previously described situation, we face a loss of information in the most interesting part of the protein. Such blind region (Balayssac et al. 2006) is, indeed, always the core region of a metalloprotein (Fig. 1B). In this frame, reducing the blind sphere around the paramagnetic center(s) by measuring relaxation rates of nuclear spins that are most affected by paramagnetism becomes extremely important.
Fig. 1.

Measurable paramagnetic relaxation enhancements vs metal-to-proton distances. a Assuming a 10 Å blind sphere and a 10–30 Å sphere where PREs can be measured, the use of a metal binding tag at the N-term site of Ubiquitin (Donaldson 2001) gives measurable PRE for about 90% of the protein. b When the same relaxation enhancement parameters is considered for a small metalloprotein such as HiPIP (Trindade et al. 2020b), more than 70% of the protein would fall in the blind sphere. c Different electronic correlation times (τe) provide different dimensions for the regions affected by PREs. Assuming 50 s1 and 0.5 s−1 as upper and lower limits for the detection of PREs, the simulated behavior of paramagnetic centers with different τe is shown
The replacement of Cu(II), i.e. the metal ion used in the case of Ubiquitin shown in Fig. 1a, with another metal ion will change the radius of the sphere where paramagnetic relaxation enhancement is effective and measurable, but the above consideration will remain: the metal center is always surrounded by a blind sphere, where signals are broadened beyond detection, and by an outer sphere, in which PREs can be measured and factorized. The situation is described in Fig. 1c, where we considered 50 s−1 as upper limit value to obtain reliable R2 measurements and 0.5 s−1 as lower limit for the precision of the measurement. At 500 MHz, considering a protein of small size and neglecting contact relaxation, R2 is dominated by the Solomon equation (Solomon 1955) which, in turn, depends on the electronic relaxation time. For τe = 5*10–9 s, which is an estimate for the electronic relaxation time of non-blue Cu2+ chromophores, 1H signals at less than 11 Å are predicted to be unobservable while those at more than 24 Å are almost unaffected by paramagnetism. Different cases may occur for iron ions: for a high spin (S = 2) Fe2+, with typical τe = 5*10–12 s, the above limits would be, respectively, 8 Å and 17 Å, while for a low spin (S = 1/2) Fe3+, with τe = 1*10–12 s, they would be 4 Å and 9 Å, respectively. Different electronic relaxation times, together with other experimental parameters, such as magnetic field strength and protein size will give rise to different radii for the blind sphere and for the sphere where PREs are measurable. Also the replacement of 1H with 13C or 15 N nuclei reduces both spheres and provides an alternate route to obtain PREs (Mateos et al. 2019). To extend the range of application of PREs in 1H detected experiments it is necessary, on the one hand, to increase the accuracy of experimental methods and observe smaller PRE values and, on the other hand, to measure with reliability R2 values larger than those achievable with the existing experimental approaches.
As extensively studied in the past, [Fe4S4]2+ clusters in proteins have an electronic ground state S = 0 due to the antiferromagnetic coupling (Phillips et al. 1970; Mouesca and Lamotte 1998; Bertini et al. 1992). However, each iron ion is formally in the oxidation state Fe2.5+ and the system is paramagnetic at room temperature due to the population of the excited energy levels of the electron spin ladder (Banci et al. 1990). The distribution of population among the spin levels depends on the extent of the antiferromagnetic coupling constant(s) operative in the [Fe4S4]2+ cluster (Blondin and Girerd 1990). Therefore, the choice of the parameters to input in the Solomon equation to predict the behavior of R2 vs H-Fe distance in [Fe4S4]2+ clusters is not obvious (Bertini et al. 1997): if we consider that each iron ion will relax with the same electronic relaxation time of a High Spin Fe2+ ion (τe = 5*10–12), and that paramagnetism at room temperature arises from the population of the first excited state of the electron spin energy ladder, characterized by S = 1, we obtain, as shown in Fig. 1C, a behavior somehow intermediate between the two cases considered here for iron ions, with expected values for the blind sphere and for the PRE sphere of about 6 Å and 13 Å. This makes PioC an ideal test case for the optimization of sequences aiming at measuring R2 rates in the proximity of the cluster.
Pulse sequence description
Here we propose a novel pulse sequence, shown in Fig. 2a, in which the relaxation delay is embedded within the INEPT evolution, the refocusing INEPT is removed and signal is acquired as antiphase doublet as soon as the HyNz magnetization is created by the last 1H 90° pulse. We called the experiment 1H R2-weighted 15N-HSQC-AP because the INEPT period, typically 1/(2JHN), is replaced here by a variable relaxation delay T. In this very simple sequence, 1H transverse relaxation is active only during the delay T, when the 2HxNz coherence evolves from Hy as HxNzsin(πJHNT). The relaxation rate of the 2HxNz antiphase term is a combination of 15 N R1 and 1H R2 relaxation rates; paramagnetic relaxation rates have a γ2 dependence from the observed nucleus (Solomon 1955; Bertini 1986), therefore the contribution of 15N R1 can be neglected and the observed rates are fully due to 1H R2 relaxation (Iwahara et al. 2007). Also cross correlation between 1H Curie Spin Relaxation and HN dipole–dipole relaxation is not contributing to the observed rates (Pintacuda et al. 2003; Mori et al. 2010). In order to sample 1H relaxation rates also at T values of a few μs, we avoid using pulsed field gradients, generally applied during the INEPT period. The delay T can then be arrayed from zero to 10 ms to sample the evolution of Hy → 2HxNz coherence transfer for half a sinusoidal period (1/J), during which 1H R2 relaxation is active. To avoid signal losses due to 1H R2 relaxation, the inverse INEPT block is removed and the 2HyNz coherence, created by the two 90° pulses at the end of 15N evolution, is acquired in antiphase without 15N decoupling. Removing the 15N decoupling also rules out duty cycle problems: without decoupling, one can safely use very short recycle delays in order to increase S/N of fast relaxing signals and to suppress water signal via progressive saturation (Camponeschi et al. 2019). This may affect the observed relaxation rates of HN signals; however, when the observed values in Eq. (1) are dominated by R2para, water saturation should not play a significant role. Another very important feature of this sequence is the suitability for cryo-probes, because there are no risks associated to coil heating due to an excess of RF power (Helms and Satterlee 2013). The use of a refocusing INEPT and 15N decoupling during acquisition is indeed a severe limiting factor for the recycle delay that, by no means, could have been as short as we used in our experiments.
Fig. 2.

The 1H R2 weighted 15N HSQC-AP pulse experiment. a Pulse sequence used for the experiment. Hard 90° and 180° pulses are used for both 1H and 15N channels, using phase x when undefined. Phase cycling: φ1 = x, −x, y, −y; φ2 = 2(y), 2(x); φ3 = 2(x), 2(−x); φ4 = 4(x), 4(−x); φrec = x, −x, −x, x, −x, x, x, −x. PFG gradients of 200 μs were used, with 100 μs for gradient recovery. Standard parameters to measure fast relaxing nuclei as follows: aq = 47 ms, T = ranging from 200 μs to 10 ms, recycle delay = 150 ms. b Comparison between different R2 relaxation building blocks. Considering the relaxation building block shown on the left panel (Donaldson 2001; Iwahara et al. 2007), using τa = 2.65 ms and T/4 = 1.8 ms as shortest possible delay, the exponential decay can be measured from about 12.5 ms. Signals with R2 values over 70 s−1 will be lost or detected at very weak intensities for few points. In the R2-weighted building block shown in the right panel, they could be easily measured for a sufficient number of time delays in the range 0–10 ms, provided signals have enough S/N. The time scales of the two curves highlight the complementarity of the two experiments
The R2-weighted HSQC-AP is essentially the simplest possible scheme for measuring 1H R2 relaxation with an HSQC-type experiment. Figure 2b shows the features of the R2-weighted building block with respect to the relaxation building block commonly used (Donaldson 2001). For the latter, the relaxation delay T must accommodate the selective 1H 180° pulse, the Pulsed Field Gradients, the INEPT transfer period 2τa. The shortest possible value of the relaxation delay T is set to about 12 ms, indeed this is why very fast relaxing signals are not observed with this approach. The removal of the 1H 180° selective pulse and shorter gradients allow one to decrease this value, even though it can’t be below 6.5 ms. As shown in Fig. 2b, for an R2 rate of 70 s−1 signals will be already at 50% of the initial intensity at the first time point of the series, while those exceeding 150 s−1 will be beyond detection after two time points of the R2 series, for all these situations the exponential decay could not be properly analyzed. On the other hand, the R2-weighted INEPT building block is highly complementary to this approach, because it will monitor the evolution of signal intensities during the first 10 ms of the relaxation recovery, starting from T period as small as 100 μs, therefore being able to monitor also very fast relaxing signals. Another interesting feature of the R2-weighted HSQC is that fast and slow relaxing resonances will be measured with comparable precision.
As expected, the most representative spectra of the two R2 series are rather different. In Fig. 3a, the first time point of the exponential decay of the in-phase experiment is superimposed to the spectrum of the R2-weighted HSQC-AP series, recorded with T = 2.4 ms. Several signals, such as those of residues 22, 27–28, 36–37, 47–51, are indeed observable only in the R2-weighted HSQC-AP, while other residues are only barely visible in the first point of the exponential decay (e.g. residues 25 and 35). Fast relaxing signals are much better observed in the R2 weighted experiment, although signals are much broader than in the in-phase experiment, because they have been acquired as HN antiphase doublets. Like homonuclear 1H and 13C cases, a dispersion phase mode of the antiphase doublets produces the sum of the two dispersive components of opposite phase, thus giving rise to a pseudo-singlet with the maximum of signal intensity (Turner 1993; Bertini et al. 1994, 2005a). As an example, Fig. 3b shows selected rows for His7 and Asn27. In the case of His7, which has a negligible R2para contribution, we observe that, when the signal linewidth is smaller than the splitting of the HN doublet, the typical pattern of an antiphase doublet phased in dispersion mode is observed; when signals are broader than the scalar coupling constant, like the case of Asn27 (R2 = 221 s−1), the doublet splitting is lost and the dispersive component of the doublet give rise to a well pseudo observable pseudo singlet. The removal of the inverse INEPT prevents transverse relaxation to be operative before 1H detection. Indeed, an additional refocusing would prevent the observation of signals characterized by T2 shorter than the INEPT period.
Fig. 3.
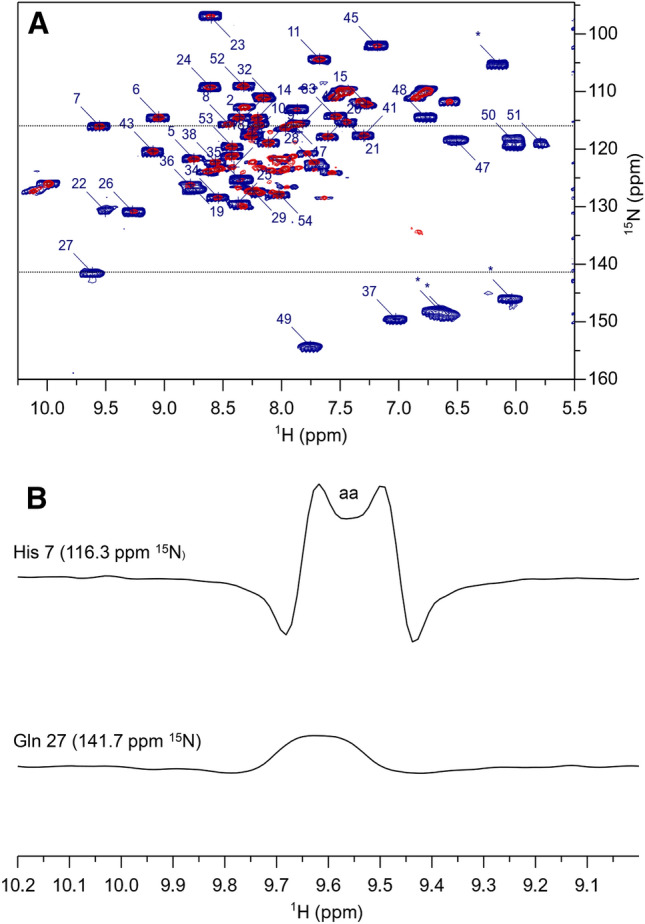
a 15N HSQC spectra of PioC obtained with an R2-weighted 15N-HSQC-AP experiment collected with T = 2.4 ms (blue) and the first point of the R2 series collected with the standard experiment with an overall relaxation delay = 12.5 ms (red). Experiments were recorded using a 500 MHz NEO-Avance Bruker spectrometer equipped with Triple resonances inverse cryoprobe (CP-TXI). R2-weighted 15N-HSQC-AP was recorded with 256 scans each fid using an overall recycle delay of 200 ms, the in-phase experiment with 16 scans each fid and 4 s as recycle delay. Folded peaks are marked with an asterisk. b Expanded plot of selected rows of R2-weighted 15 N-HSQC-AP, corresponding to His7 and Asn27
Fitting of R2 values. The intensity of observed signals in the R2-weighted HSQC-AP can be analyzed according to:
| 2 |
where T is the variable delay. In principle, a three-parameter fitting will provide values for I0, J and R2, respectively representing the initial signal intensity, the 1JHN scalar coupling constant, and the 1H R2 relaxation rate. However, the interplay between the two parameters J and R2 is such that a three parameters fitting of the buildup curves tends either to over-estimate J and then compensate it with an under-evaluation of R2 values or the other way around (smaller J and higher R2 values). We therefore used a two-parameter fitting for Eq. (2), with constant J values. As shown in Fig. 4a, for J values in the range 92–96 Hz, i.e. the range of admissible values for 1JHN scalar coupling at 500 MHz (residual dipolar couplings are expected to be negligible at 500 MHz for a protein of this size containing a [Fe4S4]2+ cluster), the deviations among calculated R2 values were much smaller than the errors observed in each two-parameters fitting. We therefore decided to consider values and uncertainties taken from the two-parameter fitting obtained using J = 94 Hz as a fixed value. Some representative build-up curves are reported in Fig. 4b. The R2-weighted, 15 N-HSQC-AP experiment is able to measure R2 values for all HN signals of the protein, including those belonging to residues of the iron-bound cysteines, to residues H-bonded to cluster sulfide ions, and those spatially close to the 4Fe-4S cluster although not in direct electronic contact with the prosthetic group.
Fig. 4.

Peak intensities in the R2-weighted 15N-HSQC-AP experiment. Relative intensity are expressed with respect to I0 term of Eq. (2). a Buildup curves obtained with a two-parameter fitting of Eq. (2) using fixed J values. Fitted intensities are those of the R2-weighted 15N-HSQC-AP experiment for the case of Cys22 HN signal. When J was varied from 92 to 96 Hz, the interplay between J and R2 provide essentially the same best fitting curve, with R2 values in the range 308–319 s−1. The uncertainty of R2 due to the admissible values of J is smaller than the standard error (± 19.6 s−1) of each individual fitting. b Experimental build-up curves of some selected signals. Curve fitting using a two-parameter fit and J = 94 Hz give R2 values as indicated in Figure
Data evaluation and assessment
The R2 values obtained with the R2-weighted HSQC-AP experiments are summarized in Table 1, together with those measured using the standard sequence. A data assessment is reported in Fig. 5, where, for both series, the value of the relative error (ΔR2/R2) vs R2 is shown. When R2 values are lower than 45 s−1, the accuracy of the sequence based on exponential decay and in-phase acquisition is much higher than that of the R2-weighted-HSQC-AP. Indeed, when INEPT (2τa) and R2 relaxation (T) evolve into separate building blocks, a single exponential decay is measured and several methods allow one to measure R2 values with very good precision and accuracy (Iwahara et al. 2007). However, in the range 45–80 s−1 the relative errors become larger than those obtained with the R2-weighted-AP and, above 80 s−1, R2 values are measurable only (with the experimental conditions we used to perform the experiments) with the R2-weighted-AP sequence, up to R2 rates as large as 310 s−1. This indicates that, for signals with R2 rates larger than ca. 50 s−1, the R2-weighted-AP sequence should be the preferred method to measure relaxation rates, while the standard approach should be used in all other cases. Because all backbone HN signals of PioC have been identified and assigned and this was the fastest rate among them, it is not possible to set the upper limit threshold of the R2-weighted-HSQC-AP experiment. In principle (see Fig. 2b), one should be able to measure R2 values up to 400–500 s−1, provided that very broad signals do not fall within a crowded spectral region.
Table 1.
500 MHz, 298 K, observed transverse relaxation rates for PioC HN amide protons
| δ1H (ppm) | δ15 N (ppm) | residue | R2 (s−1) AP | error (s−1) AP | R2 (s−1) IP | error (s−1) AP | |
|---|---|---|---|---|---|---|---|
| VAL | 1 | ||||||
| 8.26 | 117.2 | THR | 2 | 11.1 | 0.4 | ||
| 8.42 | 123.7 | LYS | 3 | 15.4 | 1.3 | ||
| 8.43 | 121.8 | LYS | 4 | 23.0 | 6.9 | 19.1 | 1.5 |
| 8.76 | 122.3 | ALA | 5 | 25.8 | 7.3 | 23.4 | 0.8 |
| 9.08 | 115.2 | SER | 6 | 38.8 | 5.2 | 17.8 | 0.6 |
| 9.64 | 116.3 | HIS | 7 | 37.8 | 4.4 | 21.3 | 0.8 |
| 8.49 | 116.5 | LYS | 8 | 52.9 | 19.1 | 32.8 | 2.2 |
| 8.13 | 119.5 | ASP | 9 | 35.3 | 5.3 | 16.9 | 0.7 |
| 8.23 | 118.2 | ALA | 10 | 34.6 | 4.3 | 19.6 | 0.8 |
| 7.69 | 105.0 | GLY | 11 | 46.8 | 3.9 | 20.0 | 1.4 |
| 8.19 | 116.3 | TYR | 12 | 36.6 | 3.8 | 29.9 | 1.5 |
| 8.67 | 124.5 | GLN | 13 | 54.3 | 3.8 | 46.4 | 4.1 |
| 8.22 | 115.2 | GLU | 14 | 19.4 | 1.1 | ||
| 7.29 | 112.8 | SER | 15 | 21.9 | 5.8 | 21.6 | 1.0 |
| PRO | 16 | ||||||
| 7.92 | 116.4 | ASN | 17 | 31.2 | 4.4 | 22.8 | 1.1 |
| 8.17 | 111.6 | GLY | 18 | 43.5 | 5.2 | 25.1 | 1.5 |
| 8.78 | 126.9 | ALA | 19 | 52.6 | 12.0 | 44.4 | 2.8 |
| 7.62 | 118.4 | LYS | 20 | 41.8 | 10.9 | 17.8 | 0.8 |
| 7.47 | 115.9 | ARG | 21 | 38.0 | 2.8 | 44.4 | 3.1 |
| 9.47 | 131.4 | CYS | 22 | 313.6 | 19.6 | n.o | |
| 8.62 | 97.5 | GLY | 23 | 60.3 | 6.2 | 48.8 | 6.3 |
| 8.62 | 109.8 | THR | 24 | 45.9 | 4.1 | 42.0 | 2.8 |
| 8.36 | 130.0 | CYS | 25 | 65.0 | 5.1 | 44.1 | 7.5 |
| 9.28 | 131.4 | ARG | 26 | 47.9 | 4.9 | 55.5 | 5.3 |
| 9.62 | 141.7 | GLN | 27 | 221.0 | 15.1 | n.o | |
| 7.72 | 123.4 | PHE | 28 | 172.3 | 8.4 | n.o | |
| 8.19 | 128.2 | ARG | 29 | 39.2 | 5.2 | 44.9 | 3.0 |
| PRO | 30 | ||||||
| PRO | 31 | ||||||
| 8.15 | 111.8 | SER | 32 | 43.5 | 5.2 | 25.1 | 1.5 |
| 7.55 | 114.9 | SER | 33 | 43.4 | 4.3 | 24.2 | 0.8 |
| 8.55 | 129.0 | CYS | 34 | 37.5 | 3.9 | 42.5 | 3.7 |
| 8.36 | 126.0 | ILE | 35 | 84.5 | 5.9 | 65.6 | 26 |
| 8.75 | 127.6 | THR | 36 | 115.0 | 12.3 | n.o | |
| 7.00 | 149.9 | VAL | 37 | 161.5 | 10.7 | n.o | |
| 8.61 | 122.8 | GLU | 38 | 43.0 | 4.0 | 24.2 | 0.8 |
| 8.43 | 115.7 | SER | 39 | 35.2 | 8.3 | 19.7 | 1.0 |
| PRO | 40 | ||||||
| 7.28 | 118.2 | ILE | 41 | 25.4 | 7.5 | 27.7 | 1.3 |
| 7.84 | 116.4 | SER | 42 | 36.7 | 3.8 | 42.8 | 2.4 |
| 9.12 | 121.2 | GLU | 43 | 24.1 | 3.4 | 19.2 | 0.5 |
| 7.98 | 116.8 | ASN | 44 | 25.7 | 5.2 | 27.4 | 1.5 |
| 7.20 | 102.6 | GLY | 45 | 55.5 | 3.8 | 46.2 | 6.5 |
| 7.86 | 113.7 | TRP | 46 | 73.1 | 5.0 | 54.8 | 14.8 |
| 6.47 | 118.9 | CYS | 47 | 182.7 | 11.0 | n.o | |
| 6.79 | 115.2 | ARG | 48 | 57.9 | 5.3 | 74.0 | 28.6 |
| 7.74 | 154.7 | LEU | 49 | 168.4 | 12.4 | n.o | |
| 6.02 | 119.5 | TYR | 50 | 56.6 | 14.3 | n.o | |
| 5.46 | 119.0 | ALA | 51 | 216.6 | 42.0 | n.o | |
| 8.33 | 109.7 | GLY | 52 | 39.4 | 4.4 | 23.0 | 1.3 |
| 8.43 | 120.2 | LYS | 53 | 24.9 | 5.0 | 18.0 | 0.9 |
| 8.04 | 128.5 | ALA | 54 | 7.7 | 0.3 | ||
| 8.53 | 124,0 | TRP sc | 46 | 53.1 | 4.2 | 33.9 | 2.1 |
Columns 5–6 refer to values observed with the R2-weighted HSQC-AP experiment, columns 7–8 to values observed with an in-phase HSQC experiment with a single exponential T2 decay prior to INEPT
Fig. 5.
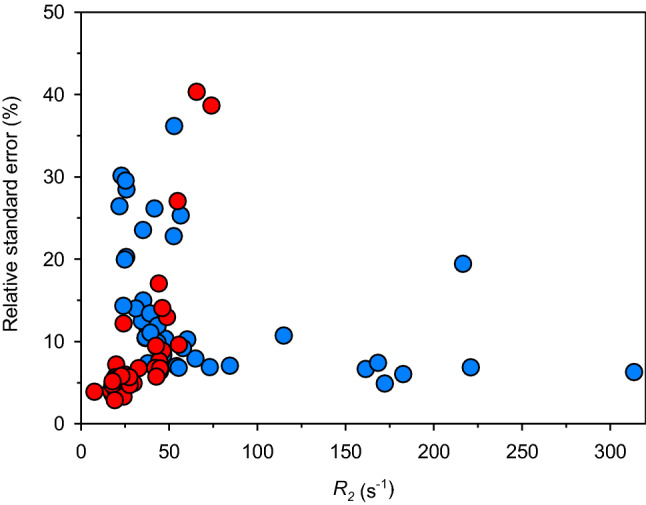
Transverse relaxation rates percentage errors (ΔR2/R2) obtained with the two series of experiments discussed in Fig. 2: (red) in-phase detected HSQC with relaxation delay preceding INEPT; (blue) R2-weighted HSQC-AP. The recycle delays for the two series were 4 s and 200 ms, respectively. For R2 values under 40 s−1, data from the in-phase experiment have much smaller errors than those from the R2-weighted HSQC-AP experiment. For R2 values over 50 s−1, the R2-weighted-HSQC-AP has much better performances. For R2 values above 80 s−1, only the R2-weighted-HSQC-AP provided reliable R2 measurements
The poor agreement between the two sets of values obtained with the two experiments and reported in Table 1 also deserves a comment. Indeed, the R2-weighted HSQC-AP experiment has been optimized to measure R2 rates of fast relaxing signals, which are the target of this experiment. To this end, we have used, for the R2-weighted HSQC-AP, recycle delays that are a factor 20 shorter than those used in the in-phase sequence (200 ms vs 4 s). Due to the fast repetition of the experiment, signals with R2 < 35 s−1 suffer from partial saturation. As a matter of speculation, it would always be possible to perform the R2-weighted HSQC-AP experiment with longer recycle delays to properly fit R2 values of slower relaxing signals. However, the obvious complementarity between the two experiments, given by the time scale of the recoveries measurable with the two experiments, and the intrinsic higher precision of the in phase experiment provided by the single exponential dependence, suggest that a combination of two R2 measurements, respectively optimized for the quantification of small and large PREs would be, by far, the most efficient approach.
Conclusions
In summary, the R2-weighted HSQC-AP experiment, proposed here, is the simplest experiment for R2 relaxation, in which 1H magnetization is kept along the transverse plane only during the relaxation delay and t2 acquisition and all periods of JHN evolution/refocusing are removed. This simplified scheme not only detects signals that escape detection in a conventional HSQC experiment, but measures R2 values that are fully reliable, in the range of rates 50–400 s−1. This is extremely important for metalloproteins, where the first coordination sphere of the metal ion(s) and its immediate neighboring regions can be identified, characterized and, finally, restrained into NMR structure calculations only when it is possible to obtain paramagnetism-based structural restraints such as PREs. The small HiPIP protein PioC is a challenging and significant example of the application of this experiment, where this objective was achieved and the R2 could be measured for all four cysteines coordinating the paramagnetic cluster. Finally, it is worth noting that this approach can be useful in all other cases where R2 is larger than 50 s−1: conformation dynamics and exchange phenomena often increase relaxation rates above this threshold also in diamagnetic systems, thus extending the potential applications of this approach.
Acknowledgments
Open access funding provided by Università degli Studi di Firenze within the CRUI-CARE Agreement. This work benefited from access to CERM/CIRMMP, the Instruct-ERIC Italy centre. Financial support was provided by European EC Horizon2020 TIMB3 (Project 810856) Instruct-ERIC (PID 4509). This article is based upon work from COST Action CA15133, supported by COST (European Cooperation in Science and Technology). Fondazione Ente Cassa di Risparmio di Firenze (CRF 2016 0985) is acknowledged for providing fellowship to MI. Project LISBOA-01–0145-FEDER-007660 (Microbiologia Molecular, Estrutural e Celular) funded by FEDER funds through COMPETE2020—Programa Operacional Competitividade e Internacionalização (POCI), Fundação para a Ciência e a Tecnologia (FCT) Portugal Grant PD/BD/135187/2017 to IBT.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Availability of data and material:
Complete protein NMR Assignment reported in BMRB ID-34487. Pulse sequences and NMR data sets available upon request.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ricardo O. Louro, Email: louro@itqb.unl.pt
Mario Piccioli, Email: piccioli@cerm.unifi.it.
References
- Ab E, Atkinson AR, Banci L, Bertini I, Ciofi-Baffoni S, Brunner K, Diercks T, Doetsch V, Engelke F, Folkers G, Griesinger C, Gronwald W, Gunther H, Habeck M, de Jong R, Kalbitzer HR, Kieffer B, Leeflang BR, Loss S, Luchinat C, Marquardsen T, Moskau D, Neidig KP, Nilges M, Piccioli M, Pierattelli R, Rieping W, Schippmann T, Schwalbe H, Trave G, Trenner JM, Wohnert J, Zweckstetter M, Kaptein R. NMR in Structural Proteomics. Acta Crystallogr D Biol Crystallogr. 2006;62:1150–1161. doi: 10.1107/S0907444906032070. [DOI] [PubMed] [Google Scholar]
- Anthis NJ, Clore GM. Visualizing transient dark states by NMR spectroscopy. Q Rev Biophys. 2015;48:35–116. doi: 10.1017/S0033583514000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnesano F, et al. A strategy for the NMR characterization of type II copper(II) proteins: the case of the copper trafficking protein CopC from Pseudomonas syringae. J Am Chem Soc. 2003;125:7200–7208. doi: 10.1021/ja034112c. [DOI] [PubMed] [Google Scholar]
- Arnesano F, Banci L, Piccioli M. NMR structures of paramagnetic metalloproteins. Q Rev Biophys. 2006;38:167–219. doi: 10.1017/S0033583506004161. [DOI] [PubMed] [Google Scholar]
- Balayssac S, Jiménez B, Piccioli M. Assignment strategy for fast relaxing signals: complete aminoacid identification in thulium substituted calbindin D9k. J Biomol NMR. 2006;34:63–73. doi: 10.1007/s10858-005-5359-z. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Luchinat C. The 1H NMR parameters of magnetically coupled dimers - the Fe2S2 proteins as an example. Struct Bonding. 1990;72:113–135. [Google Scholar]
- Banci L, Camponeschi F, Ciofi-Baffoni S, Piccioli M. The NMR contribution to protein-protein networking in Fe-S protein maturation. J Biol Inorg Chem. 2018;23:687–687. doi: 10.1007/s00775-018-1552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste JL, Wagner G. Utilization of site-directed spin labelling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited Nuclear Overhauser Effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- Bermel W, et al. Complete assignment of heteronuclear protein resonances by protonless NMR spectroscopy. Angew Chem Int Ed. 2005;44:3089–3092. doi: 10.1002/anie.200461794. [DOI] [PubMed] [Google Scholar]
- Bertini I, et al. Identification of the iron ions of HiPIP from Chromatium vinosum within the protein frame through 2D NMR experiments. J Am Chem Soc. 1992;114:3332–3340. [Google Scholar]
- Bertini I, Capozzi F, Luchinat C, Piccioli M, VicensOliver M. NMR is a unique and necessary step in the investigation of iron-sulfur proteins: the HiPIP from R. gelatinosus as an example. Inorg Chim Acta. 1992;198–200:483–491. [Google Scholar]
- Bertini I, Donaire A, Luchinat C, Rosato A. Paramagnetic relaxation as a tool for solution structure determination: Clostridium pasterianum ferredoxin as an example. Proteins Struct Funct Genet. 1997;29:348–358. [PubMed] [Google Scholar]
- Bertini I, Jiménez B, Piccioli M. 13C direct detected experiments: optimisation to paramagnetic signals. J Magn Reson. 2005;174:125–132. doi: 10.1016/j.jmr.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Bertini I, Jiménez B, Piccioli M, Poggi L. Asymmetry in C−C COSY spectra provides information on ligand geometry in paramagnetic proteins. J Am Chem Soc. 2005;127(35):12216–12217. doi: 10.1021/ja051058m. [DOI] [PubMed] [Google Scholar]
- Bertini I, Luchinat C, Parigi G, Ravera E. NMR of paramagnetic molecules. Amsterdam: Elsevier; 2016. [Google Scholar]
- Bertini I, Luchinat C, Piccioli M, Tarchi D. COSY spectra of paramagnetic macromolecules, observability, scalar effects, cross correlation effects, relaxation allowed coherence transfer. Concepts Magn Reson. 1994;6:307–335. [Google Scholar]
- Bertini I, Luhat C. NMR of paramagnetic molecules in biological systems. Menlo Park: Benjamin/Cummings; 1986. [Google Scholar]
- Bird LJ, et al. Nonredundant roles for cytochrome c2 and two high-potential iron-sulfur proteins in the photoferrotroph Rhodopseudomonas palustris TIE-1. J Bacteriol. 2014;196(4):850–858. doi: 10.1128/JB.00843-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondin G, Girerd J-J. Interplay of electron exchange and electron transfer in metal polynuclear complexes in proteins or chemical models. Chem Rev. 1990;90:1359–1376. [Google Scholar]
- Brancaccio D, et al. [4Fe-4S] Cluster Assembly in Mitochondria and Its Impairment by Copper. J Am Chem Soc. 2017;139:719–730. doi: 10.1021/jacs.6b09567. [DOI] [PubMed] [Google Scholar]
- Camponeschi F, Muzzioli R, Ciofi-Baffoni S, Piccioli M, Banci L. Paramagnetic (1)H NMR Spectroscopy to Investigate the Catalytic Mechanism of Radical S-Adenosylmethionine Enzymes. J Mol Biol. 2019;431:4514–4522. doi: 10.1016/j.jmb.2019.08.018. [DOI] [PubMed] [Google Scholar]
- Cetiner EC, et al. Paramagnetic-iterative relaxation matrix approach: extracting PRE-restraints from NOESY spectra for 3D structure elucidation of biomolecules. J Biomol NMR. 2019;73:699–712. doi: 10.1007/s10858-019-00282-0. [DOI] [PubMed] [Google Scholar]
- Cheng H, Markley JL. NMR spectroscopic studies of paramagnetic proteins: iron-sulfur proteins. Annu Rev Biophys Biomol Struct. 1995;24:209–237. doi: 10.1146/annurev.bb.24.060195.001233. [DOI] [PubMed] [Google Scholar]
- Ciofi-Baffoni S, Gallo A, Muzzioli R, Piccioli M. The IR-N-15-HSQC-AP experiment: a new tool for NMR spectroscopy of paramagnetic molecules. J Biomol NMR. 2014;58:123–128. doi: 10.1007/s10858-013-9810-2. [DOI] [PubMed] [Google Scholar]
- Clore GM. Practical aspects of paramagnetic relaxation enhancement in biological macromolecules. Methods Enzymol. 2015;564:485–497. doi: 10.1016/bs.mie.2015.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson LW, et al. Structural characterization of proteins with an attached ATCUN Motif by paramagnetic relaxation enhancement NMR spectroscopy. J Am Chem Soc. 2001;123:9843–9847. doi: 10.1021/ja011241p. [DOI] [PubMed] [Google Scholar]
- Hansen DF, Led JJ. Determination of the geometric structure of the metal site in a blue copper protein by paramagnetic NMR. Proc Natl Acad Sci USA. 2006;103:1738–1743. doi: 10.1073/pnas.0507179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harford C, Sarkar B. Amino Terminal Cu(II)- and Ni(II)-Binding (ATCUN) Motif of Proteins and Peptides: Metal Binding, DNA Cleavage, and Other Properties. Acc Chem Res. 1997;30:123–130. [Google Scholar]
- Hass MAS, Ubbink M. Structure determination of protein-protein complexes with long-range anisotropic paramagnetic NMR restraints. Curr Opin Struct Biol. 2014;24:45–53. doi: 10.1016/j.sbi.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Helms G, Satterlee JD. Keeping PASE with WEFT: SHWEFT-PASE pulse sequences for H-1 NMR spectra of highly paramagnetic molecules. Magn Reson Chem. 2013;51:222–229. doi: 10.1002/mrc.3929. [DOI] [PubMed] [Google Scholar]
- Inubushi T, Becker ED. Efficient detection of paramagnetically shifted NMR resonances by optimizing the WEFT pulse sequence. J Magn Reson. 1983;51:128–133. [Google Scholar]
- Iwahara J, Clore GM. Structure-independent analysis of the breadth of the positional distribution of disordered groups in macromolecules from order parameters for long, variable-length vectors using NMR paramagnetic relaxation enhancement. J Am Chem Soc. 2010;132:13346–13356. doi: 10.1021/ja1048187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara J, Schwieters CD, Clore GM. Ensemble approach for NMR structure refinement against H-1 paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule. J Am Chem Soc. 2004;126:5879–5896. doi: 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]
- Iwahara J, Tang C, Clore GM. Practical aspects of 1H transverse paramagnetic relaxation enhancement measurements on macromolecules. J Magn Reson. 2007;184:185–195. doi: 10.1016/j.jmr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MR, Lauritzen C, Dahl SW, Pedersen J, Led JJ. Binding ability of a HHP-tagged protein towards Ni2+ studied by paramagnetic NMR relaxation: the possibility of obtaining long-range structure information. J Biomol NMR. 2004;29:175–185. doi: 10.1023/B:JNMR.0000019251.09648.c4. [DOI] [PubMed] [Google Scholar]
- Joss D, Haussinger D. Design and applications of lanthanide chelating tags for pseudocontact shift NMR spectroscopy with biomacromolecules. Prog Nucl Magn Reson Spectrosc. 2019;114–115:284–312. doi: 10.1016/j.pnmrs.2019.08.002. [DOI] [PubMed] [Google Scholar]
- Kateb F, Piccioli M. New routes to the detection of relaxation allowed coherence transfer in paramagnetic molecules. J Am Chem Soc. 2003;125:14978–14979. doi: 10.1021/ja038101v. [DOI] [PubMed] [Google Scholar]
- Keizers PHJ, Ubbink M. Paramagnetic tagging for protein structure and dynamics analysis. Prog Nucl Magn Reson Spectrosc. 2011;58:88–96. doi: 10.1016/j.pnmrs.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Koehler J, Meiler J. Expanding the utility of NMR restraints with paramagnetic compounds: background and practical aspects. Prog Nucl Magn Reson Spectrosc. 2011;59:360–389. doi: 10.1016/j.pnmrs.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolczak U, Salgado J, Siegal G, Saraste M, Canters GW. Paramagnetic NMR studies of blue and purple copper proteins. Biospectroscopy. 1999;5:S19–S32. doi: 10.1002/(SICI)1520-6343(1999)5:5+<S19::AID-BSPY3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kudhair BK, et al. Structure of a Wbl protein and implications for NO sensing by M. tuberculosis. Nat Commun. 2017;8:2280. doi: 10.1038/s41467-017-02418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin IJ, et al. Hyperfine-shifted 13C and 15N NMR signals from Clostridium pasteurianum Rubredoxin: extensive assignments and quantum chemical verification. J Am Chem Soc. 2009;131:15555–15563. doi: 10.1021/ja905928x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin I, Gebel EB, Machonkin TE, Westler WM, Markley JL. Correlation bewtween hydrogen bond lenghts and reduction potentials in Clostridium Pasteurianum Rubredoxin. J Am Chem Soc. 2003;125:1464–1465. doi: 10.1021/ja028710n. [DOI] [PubMed] [Google Scholar]
- Liu W-M, Overhand M, Ubbink M. The application of paramagnetic lanthanoid ions in NMR spectroscopy on proteins. Coord Chem Rev. 2014;273–274:2–12. [Google Scholar]
- Machonkin TE, Westler WM, Markley JL. 13C–13C 2D NMR: a novel strategy for the study of paramagnetic proteins with slow electronic relaxation times. J Am Chem Soc. 2002;124:3204–3205. doi: 10.1021/ja017733j. [DOI] [PubMed] [Google Scholar]
- Machonkin TE, Westler WM, Markley JL. Paramagnetic NMR spectroscopy and density functional calculations in the analysis of the geometric and electronic structures of iron-sulfur proteins. Inorg Chem. 2005;44:779–797. doi: 10.1021/ic048624j. [DOI] [PubMed] [Google Scholar]
- Madl T, Felli IC, Bertini I, Sattler M. Structural analysis of protein interfaces from 13C direct-detect paramagnetic relaxation enhancements. J Am Chem Soc. 2010;132:7285–7287. doi: 10.1021/ja1014508. [DOI] [PubMed] [Google Scholar]
- Matei E, Gronenborn AM. (19)F paramagnetic relaxation enhancement: a valuable tool for distance measurements in proteins. Angew Chem Int Ed Engl. 2016;55:150–154. doi: 10.1002/anie.201508464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos B, Konrat R, Pierattelli R, Felli IC. NMR characterization of long-range contacts in intrinsically disordered proteins from paramagnetic relaxation enhancement in C-13 direct-detection experiments. ChemBioChem. 2019;20:335–339. doi: 10.1002/cbic.201800539. [DOI] [PubMed] [Google Scholar]
- Miao Q, et al. A double-armed, hydrophilic transition metal complex as a paramagnetic NMR Probe. Angew Chem Int Ed Engl. 2019;58:13093–13100. doi: 10.1002/anie.201906049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Jiménez B, Piccioli M, Battistoni A, Sette M. The solution structure of the monomeric copper, zinc superoxide dismutase from Salmonella enterica: structural insights to understand the evolution toward the dimeric structure. Biochemistry. 2008;47:12954–12963. doi: 10.1021/bi801252e. [DOI] [PubMed] [Google Scholar]
- Mori M, Kateb F, Bodenhausen G, Piccioli M, Abergel D. Towards structural dynamics: protein motions viewed by chemical shift modulations and direct detection of C'N multiple-quantum relaxation. J Am Chem Soc. 2010;132:3594–3600. doi: 10.1021/ja9103556. [DOI] [PubMed] [Google Scholar]
- Mouesca J-M, Lamotte B. Iron-Sulfur clusters and their electronic and magnetic properties. Coord Chem Rev. 1998;178–180:1573–1614. [Google Scholar]
- Nitsche C, Otting G. Pseudocontact shifts in biomolecular NMR using paramagnetic metal tags. Prog Nucl Magn Reson Spectrosc. 2017;98–99:20–49. doi: 10.1016/j.pnmrs.2016.11.001. [DOI] [PubMed] [Google Scholar]
- Parigi G, Ravera E, Luchinat C. Magnetic susceptibility and paramagnetism-based NMR. Prog Nucl Magn Reson Spectrosc. 2019;114–115:211–236. doi: 10.1016/j.pnmrs.2019.06.003. [DOI] [PubMed] [Google Scholar]
- Phillips WD, Poe M, McDonald CC, Bartsch RG. Proc Natl Acad Sci USA. 1970;67:682–682. doi: 10.1073/pnas.67.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccioli M, Turano P. Transient iron coordination sites in proteins: exploiting the dual nature of paramagnetic NMR. Coord Chem Rev. 2015;284:313–328. [Google Scholar]
- Pintacuda G, et al. Fast structure /based assignment of 15N HSQC spectra of selectively 15N labeled paramagnetic proteins. J Am Chem Soc. 2004;126:2963–2970. doi: 10.1021/ja039339m. [DOI] [PubMed] [Google Scholar]
- Pintacuda G, Hohenthanner K, Otting G, Muller N. Angular dependence of dipole-dipole-Curie-spin cross-correlation effects in high-spin and low-spin paramagnetic myoglobin. J Biomol NMR. 2003;27:115–132. doi: 10.1023/a:1024926126239. [DOI] [PubMed] [Google Scholar]
- Pintacuda G, John M, Su XC, Otting G. NMR structure determination of protein-ligand complexes by lanthanide labeling. Acc Chem Res. 2007;40:206–212. doi: 10.1021/ar050087z. [DOI] [PubMed] [Google Scholar]
- Sato K, Kohzuma T, Dennison C. Active-site structure and electron-transfer reactivity of plastocyanins. J Am Chem Soc. 2003;125:2101–2112. doi: 10.1021/ja021005u. [DOI] [PubMed] [Google Scholar]
- Siegal G, Selenko P. Cells, drugs and NMR. J Magn Reson. 2019;306:202–212. doi: 10.1016/j.jmr.2019.07.018. [DOI] [PubMed] [Google Scholar]
- Softley CA, Bostock MJ, Popowicz GM, Sattler M. Paramagnetic NMR in drug discovery. J Biomol NMR. 2020;74:287–309. doi: 10.1007/s10858-020-00322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon I. Relaxation processes in a system of two spins. Phys Rev. 1955;99:559–565. [Google Scholar]
- Spronk C, et al. Structure and dynamics of Helicobacter pylori nickel-chaperone HypA: an integrated approach using NMR spectroscopy, functional assays and computational tools. J Biol Inorg Chem. 2018;23:1309–1330. doi: 10.1007/s00775-018-1616-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Iwahara J, Clore GM. Visualization of transient encounter complexes in protein-protein association. Nature. 2006;444:383–386. doi: 10.1038/nature05201. [DOI] [PubMed] [Google Scholar]
- Trindade IB, Invernici M, Cantini F, Louro RO, Piccioli M. 1H, 13C and 15N assignment of the paramagnetic high potential iron-sulfur protein (HiPIP) PioC from Rhodopseudomonas palustris TIE-1. Biomol NMR Assign. 2020 doi: 10.1007/s12104-020-09947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trindade I, Invernici M, Cantini F, Louro RO, Piccioli M (2020b) NOE-less protein NMR structures-an alternative approach in Highly Paramagnetic Systems. arXiv: 2002.01228 [DOI] [PubMed]
- Turner DL. Optimization of COSY and related methods. Applications to 1H NMR of horse ferricytochrome c. J Magn Reson Ser A. 1993;104:197–202. [Google Scholar]
- Turner DL, Brennan L, Chamberlin SG, Louro RO, Xavier AV. Determination of solution structures of paramagnetic proteins by NMR. Eur Biophys J. 1998;27:367–375. doi: 10.1007/s002490050144. [DOI] [PubMed] [Google Scholar]