

Summary
Entamoeba histolytica, a protozoan parasite in the lumen of the human large intestine, occasionally spreads to the liver and induces amebic liver abscesses (ALAs). Upon infection with E. histolytica, high levels of type 2 cytokines are induced in the liver early after infection. However, the sources and functions of these initial type 2 cytokines in ALA formation remain unclear. In this study, we examined the roles of group 2 innate lymphoid cells (ILC2s) in ALA formation. Hepatic ILC2 numbers were significantly increased and they produced robust levels of IL-5. The in vivo transfer of ILC2s into Rag2−/−common γ chain (γc)−/− KO mice aggravated ALA formation accompanied by eosinophilia and neutrophilia. Furthermore, IL-33-deficient mice and IL-5-neutralized mice had less ALA formations. These results suggest that ILC2s contribute to exacerbating the pathogenesis of ALA by producing early type 2 cytokines and promoting the accumulation of eosinophils and neutrophils in the liver.
Subject Areas: Immunology, Microbiology, Microbiology Parasite
Graphical Abstract
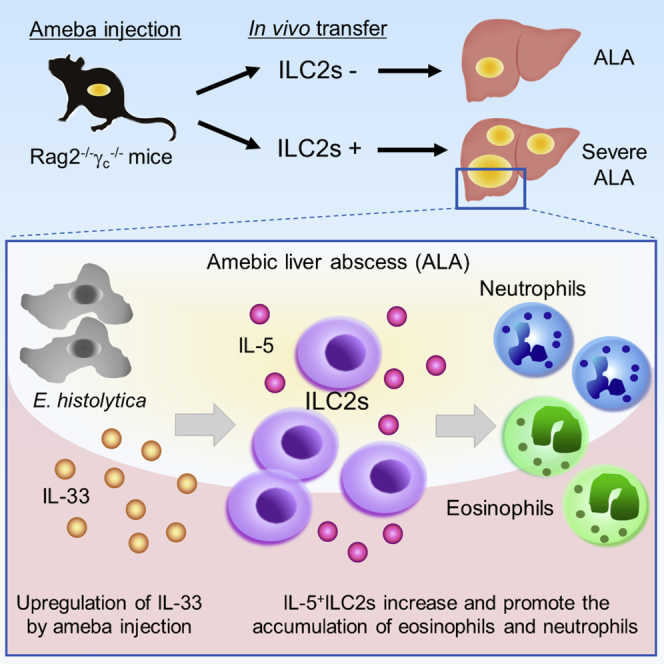
Highlights
-
•
ILC2s exacerbate ALA by promoting the accumulation of eosinophils and neutrophils
-
•
Hepatic ILC2s are increased and the main source of IL-5 in the early phase of ALA
-
•
Hepatic ILC2s localize with IL-33+ cells in the inflammatory areas of ALA
-
•
IL-33 is a trigger of ILC2-mediated ALA formation
Immunology; Microbiology; Microbiology Parasite
Introduction
In developing countries where the social infrastructure and hygienic environment are poor, pathogenic parasites often contaminate water and food and cause intestinal infectious diseases in humans. Amebiasis is caused by infection with Entamoeba histolytica, an enteric protozoan parasite, and it remains as a leading parasitic disease globally, affecting about 50 million people and causing 55,500 deaths each year (Kotloff et al., 2013; Murray et al., 2012). Occasionally, E. histolytica invades the intestinal mucosa causing amebic colitis and then spreads to extra-intestinal organs, especially the liver through the portal circulation, resulting in amebic liver abscess (ALA) formation (Hamano et al., 2013; Haque et al., 2003; Ravdin, 1988). Approximately 80% of patients with ALA develop symptoms within 2–4 weeks including fever and abdominal pain, which, if not treated adequately, can cause the progressive focal destruction of liver tissues (Prakash and Bhimji, 2017). However, the factors associated with severe and chronic ALA are unknown.
IFN-γ was critically important in controlling the early formation of ALA in a mouse model of ALA induced by the injection of E. histolytica via the liver parenchyma (Lotter et al., 2006; Seydel et al., 2000). In vitro studies demonstrated that neutrophils and macrophages, including Kupffer cells, were activated to kill E. histolytica by an IFN-γ-dependent mechanism in humans and mice (Denis and Chadee, 1989a, 1989b; Ghadirian and Salimi, 1993; Salata et al., 1987). Thus, IFN-γ activates innate immune cells and prevents severe ALA formation early after E. histolytica translocation and invasion.
It is generally accepted that type 1 responses characterized by IFN-γ-dependent mechanism are counteracted by type 2 responses. Type 2 immune responses (IL-4, IL-5, and IL-13) were induced simultaneously with IFN-γ in a mouse model of amebic colitis (Houpt et al., 2002). A hamster model of ALA produced IL-5 and IL-13 at the early phase after the direct injection of E. histolytica into the hepatic parenchyma (Cervantes-Rebolledo et al., 2009). Thus far, the source of IL-5 and IL-13 and their roles in ALA formation remain unclear.
Group 2 innate lymphoid cells (ILC2s), a new subset of innate immune cells, produce abundant levels of type 2 cytokines including IL-5, IL-13, and IL-9 in response to epithelial stress signals such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP). Large amounts of type 2 cytokines secreted by ILC2s are responsible for the initiation of type 2 immune responses. Furthermore, ILC2s had a critical role in the progress of allergic lung inflammation (Halim et al., 2012; Motomura et al., 2014) and the rapid expulsion of intestinal helminths (Moro et al., 2010; Neill et al., 2010). IL-5 secreted by ILC2s induces eosinophil accumulation, and IL-13 upregulates mucin production into the gut to expel enteric nematodes such as Nippostrongylus brasiliensis. However, it is unclear whether ILC2s and its production of type 2 cytokines are protective against protozoan infection or promote the pathogenesis induced by protozoans. Thus, we hypothesized that ILC2s are the main source of early type 2 cytokines in the liver during amebic infection.
Here we report that the numbers of ILC2s and type 2 cytokine production were significantly increased during severe ALA formation in Rag2 KO mice. In vivo depletion of ILC2s in Rag2 KO mice controlled ALA formation without any difference in IFN-γ production, and in vivo ILC2-transfer experiments showed that ILC2s were responsible for severe ALA formation by promoting the accumulation of eosinophils and neutrophils in the liver. Our results demonstrate that ILC2s are the main source of type 2 cytokines in the early phase of ALA and contribute to exacerbating the pathogenesis of ALA.
Results
Increased Numbers of ILC2s and Upregulated Type 2 Cytokines in Rag2 KO Livers after E. histolytica Inoculation via the Intraportal Vein
To elucidate whether ILC2s are a source of type 2 cytokines in the early phase of ALA and the role and function of ILC2s in ALA formation, Rag2 knockout (KO) mice, deficient for T cells and B cells, were inoculated with E. histolytica, and the kinetics of ILC2s and type 2 cytokine production were monitored. Previous studies injected E. histolytica directly into liver parenchyma to mimic ALA (Campbell et al., 1999; Cieslak et al., 1992); however, the injection procedure and injection itself may lead to the release of danger signals such as IL-33. As an alternative method, we induced a natural infection type of ALA by inoculating ameba into the portal vein without damaging the liver parenchyma. To mimic ALA in mice, we infected C57BL/6 (WT) and Rag2 KO mice with 2 × 105 E. histolytica via the portal vein without damaging the liver. We used Rag2 KO mice, which are deficient for T and B cells, to specifically investigate the role of ILCs in ALA formation without influence from T and B cells and their production of cytokines. The direct inoculation of E. histolytica into a portal vein, which mimics the natural route of metastasis of ameba, led to ALA formation in WT and Rag2 KO mice. Figures 1A and S1A show ALA formation as white spots as a gross observation in WT and Rag2 KO livers at day 4 after E. histolytica injection. Amebic abscesses were accompanied by the necrosis of hepatocytes and accumulation of leukocytes around amebae in WT and Rag2 KO mice (Figures 1B and S1B). The area and number of abscess in the liver were similar between WT and Rag2 KO mice, suggesting that T and B cells are dispensable for the early phase of ALA formation (Figures 1C and 1D). As Rag2 KO and WT mice developed ALA via the intra-portal injection of ameba, we measured the mRNA expression of type 2 cytokines during ALA formation to examine the involvement of ILC2s. Hepatic IL-5, but not IL-13, mRNA expression was significantly upregulated in WT and Rag2 KO mice on day 4 (Figures 1E and 1F). ILC2s are activated by the epithelial cell-derived cytokines IL-25, IL-33, and TSLP and produce a large amount of IL-5 and IL-13 in the lungs and intestine upon infection with the helminth Nippostrongylus brasiliensis (Moro et al., 2010; Neill et al., 2010). We examined the expression of epithelial cell-derived cytokines in the liver. E. histolytica injection into WT and Rag2 KO mice enhanced IL-33 mRNA expression on day 4 (Figure 1G); however, IL-25 was not detected (data not shown). Next, we examined the kinetics of ILC2s in the livers of WT and Rag2 KO mice. The hepatic ILC2s were identified as GATA3+, Thy1.2+, CD45+, lineage−, T1/ST2+ cell population. The absolute number of ILC2s in the liver was significantly increased on day 4 in WT and Rag2 KO mice after E. histolytica injection (Figures 1H and S2). IL-17RB+, KLRG1+, T1/ST2-, CD45+, lineage− cells, which is inflammatory ILC2s, were not detected in this model, suggesting that the hepatic ILC2s that increased during ALA formation were tissue-resident ILC2s. During ALA formation, other innate immune cells in the liver, such as eosinophils and neutrophils were increased and showed similar kinetics in both WT and Rag2 KO mice, whereas NK cells were not significantly increased in either WT or Rag2 KO mice (Figures S3A–S3C). The proportion of neutrophils in blood was significantly increased, whereas the blood eosinophils were significantly decreased in Rag2 KO mice on day 4 compared with uninfected mice (Figures S3D and S3E). The number of ILC2s was increased in the livers of Rag2 KO mice accompanied by the upregulation of type 2 cytokines at the early phase of ALA formation similar to that in WT mice. This indicated that the portal vein injection of E. histolytica to Rag2 KO mice is a suitable model of ALA to assess the role of ILC2s in the liver.
Figure 1.

ILC2s Increase in the Livers of WT and Rag2 KO Mice during ALA Formation via Intra-Portal Vein Injection with 2 × 105E. histolytica
(A) Macro observation of ALA formation in livers from WT and Rag2 KO mice on day 4. Arrows show a representative abscess in the liver. See also Figure S1A.
(B) Histology of liver tissues from WT and Rag2 KO mice stained with H&E on day 4. Representative figures are shown. Original magnification: × 2.5 and scale bar, 1 mm. See also Figure S1B.
(C and D). The area (C) and number (D) of ALA formed in livers on day 4 (n = 6 per group).
(E–G) Expression of mRNA encoding IL-5 (E), IL-13 (F), and IL-33 (G) in liver tissues on days 4 and 7 (n = 3 per time point in each group).
(H) Absolute number of hepatic ILC2s on days 0 (uninfected), 4, and 7 (n = 3 per time point in each group). Hepatic lymphocytes were stained with markers for ILC2 (lineage− CD45+ Gata3+ Thy1.2+ T1/ST2+ population) by flow cytometry. Absolute numbers were counted by multiplying the percentage of ILC2s by the absolute number of hepatic lymphocytes. Statistically significant differences between day 0 and indicated time points in each group are indicated with p values (∗p < 0.05, ∗∗p < 0.01, ANOVA). Each point shows the mean ± standard deviation (SD). Data are representative of three independent experiments. See also Figures S2, S3, and S7.
Hepatic ILC2s Produce High Levels of IL-5 in WT and Rag2 KO Mice during ALA Formation
To clarify whether ILC2s are a major source of IL-5 and IL-13 in the liver early after E. histolytica infection, we isolated hepatic lymphocytes on days 0 (naive), 4, and 7 after E. histolytica injection and monitored IL-5 and IL-13-producing ILC2s by intracellular staining. Importantly, hepatic ILC2s spontaneously produced high levels of IL-5 in the setting cultured in the medium alone in WT and Rag2 KO mice on day 4 after E. histolytica injection (Figures 2A and S4). The number of hepatic IL-5+ ILC2s were markedly increased on day 4 in WT and Rag2 KO mice (Figure 2B). To examine hepatic ILC2s, we stimulated hepatic lymphocytes with PMA and ionomycin followed by staining with mAbs to ILC2 markers as well as IL-5 and IL-13 on days 4 and 7 after E. histolytica injection. In WT and Rag2 KO mice, ILC2s were a major producer of IL-5 in response to PMA and ionomycin (Figures 2C, 2D, and S4). However, IL-13-producing ILC2s were rare in the liver during ALA formation (Figure S4). These results indicated that hepatic ILC2s are a principal source of early IL-5 during ALA formation in WT and Rag2 KO mice.
Figure 2.

ILC2s Spontaneously Produce High IL-5 Levels in the Livers of WT and Rag2 KO Mice at the Early Phase of ALA Formation
The proportion (A and C) and absolute number (B and D) of IL-5-producing ILC2s in livers from WT and Rag2 KO mice on days 0 (naive), 4, and 7 (n = 3 per time point in each group). Hepatic lymphocytes were stimulated with PMA and ionomycin (C and D; PMA + ion) or were unstimulated (A and B; none stimulation).
Statistically significant differences between day 0 and indicated time points in each group are indicated with p values (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ANOVA). Each point shows the mean ± standard deviation (SD). Data are representative of at least three independent experiments. See also Figure S4.
ALA Development Is Controlled by the In Vivo Depletion of ILC2s
It is unclear whether ILC2s exacerbate ALA formation or protect mice from ALA. To clarify the roles of ILC2s in the development of ALA, we depleted ILC2s in the liver by the administration of anti-mouse (α) CD25 mAb into Rag2 KO mice, which lack T and B cells, every 2 days from 3 days before E. histolytica injection. Because activated T cells, especially regulatory T (Treg) cells and Th2 cells, also express CD25 on their surface, the in vivo administration of αCD25 mAb into Rag2 KO mice is a suitable method to deplete ILCs alone and produce an in vivo setting where the presence or absence of ILCs are the major difference between the two groups. We confirmed the substantial reduction of ILC2s in the Rag2 KO liver, whereas there was no difference in the proportion of blood eosinophils, neutrophils, and the absolute number of other immune cells such as NK cells, ILC1s, resident Kupffer cells (KCs), and transient inflammatory monocyte-derived KCs after two doses administration of αCD25 mAb (Figures 3A and S5). IL-17RB+ KLRG1+ T1/ST2- CD45+ lineage− inflammatory ILC2 was not detected in the liver treated with αCD25 mAb (data not shown). The number of hepatic ILC2s on day 4 after injection was significantly decreased by αCD25 mAb treatment (Figure 3B). We observed the liver histology on day 4 after E. histolytica injection. ILC2-depleted Rag2 KO mice reduced ALA formation compared with control Rag2 KO mice treated with isotype-matched mAb (Figure 3C). The area and number of ALA were not detectable in ILC2-depleted Rag2 KO (Figures 3D and 3E). Thus, ALA was controlled by the in vivo depletion of ILC2s in Rag2 KO mice, indicating ILC2s are responsible for ALA formation.
Figure 3.

ALA Development Is Better Controlled by the In Vivo Depletion of ILC2s in Rag2 KO Mice
(A) Confirmation of in vivo ILC2 depletion in the livers of Rag2 KO mice treated with αCD25 mAb or control rat IgG1 by flow cytometry. Hepatic ILC2s were detected after two administrations of αCD25 mAb prior to E. histolytica injection. The numbers indicate the percentage of cells in corresponding quadrants.
(B) The absolute number of hepatic ILC2s on day 4 after E. histolytica injection.
(C) Histology of liver tissues stained with H&E on day 4. Representative figures are shown. Original magnification: ×5. Scale bar, 500 μm. Arrows show a representative abscess in the liver.
(D and E) The area (D) and number (E) of ALA formed in the liver sections on day 4. The plots in (D) show each area of all ALA observed in three ameba-injected mice. N.D. indicates no detection of abscesses.
(F) ELISA analysis of IFN-γ production by hepatic lymphocytes from ILC2-depleted Rag2 KO mice on day 4. NS indicates no statistical difference.
(G and H) The absolute numbers of eosinophils (G; MHC class II− SiglecF+ CD11b+ cells), neutrophils (H; CD11b+ Gr-1+ F4/80− MHC class II− SiglecF− cells) on day 4.
Statistically significant differences between control rat IgG1 treated and ILC2-depleted Rag2 KO mice are indicated with p values (∗p < 0.05, unpaired 2-tailed Student’s t test). Each point shows the mean ± standard deviation (SD). Data are representative of at least three independent experiments. See also Figure S5.
ILC2 Depletion Rescues Rag2 KO Mice from Severe ALA without Upregulating IFN-γ Production
IFN-γ has a protective role in innate immunity against ALA in SCID mice (Seydel et al., 2000). As IFN-mediated type 1 and type 2 immune responses are mutually regulated, ILC2s might exacerbate ALA by regulating innate IFN-γ production in vivo. To examine the impact of ILC2 depletion on IFN-γ production in the liver, we assessed the ability of hepatic lymphocytes to produce IFN-γ in Rag2 KO with or without ILC2s on day 4 after E. histolytica injection. There was no difference in IFN-γ production between ILC2-depleted and control Rag2 KO mice (Figure 3F). This result shows that the control of severe ALA formation observed in ILC2-depleted Rag2 KO mice was not due to the upregulation of IFN-γ production. ILC2-depleted Rag2 KO mice also had significantly fewer eosinophils and neutrophils in the liver compared with control IgG-treated Rag2 KO mice (Figures 3G and 3H). Thus, ILC2s might exacerbate ALA by increasing the numbers of eosinophils and neutrophils in the liver.
ILC2s Directly Exacerbate the Pathogenesis of ALA Accompanied by Eosinophilia and Neutrophilia
To elucidate whether ILC2s contribute directly to the pathogenesis of ALA, we adoptively transferred ILC2s into Rag2−/−γc−/− mice, which lack ILCs, NK cells, T cells, and B cells. Transferred ILC2s were originally isolated from the mesentery of naive WT mice and possessed the ability to produce IL-5 and IL-13 under IL-33 stimulation (Figures S6A and S6B). Overall, 2 × 106 non-stimulated ILC2s were intravenously transferred into Rag2−/−γc−/− mice 1 day before the injection of 2 × 105 E. histolytica. The transferred ILC2s were confirmed to be in the liver on day 4 after E. histolytica injection (Figure S6C). Interestingly, ILC2-transferred Rag2−/−γc−/− mice developed severe ALA associated with hemorrhagic necrosis in the central part of ALA compared with non-transferred Rag2−/−γc−/− mice on day 4 after E. histolytica injection (Figure 4A). The ILC2 transfer significantly increased the area and number of ALA (Figures 4B and 4C). In this setting, we examined the accumulation of eosinophils and neutrophils and found they were significantly increased in the livers of ILC2-transferred Rag2−/−γc−/− mice on day 4 (Figures 4D and 4E). These results suggest that ILC2s exacerbate the pathogenesis of ALA accompanied by eosinophilia and neutrophilia.
Figure 4.

ILC2-Transferred Rag2−/−γc−/− KO Mice Develop Severe ALA Accompanied by the Accumulation of Eosinophils and Neutrophils in the Liver
Naive ILC2s (2 × 106) were transferred into Rag2−/−γc−/− (DKO) mice 1 day before E. histolytica injection
(A) Histology of liver tissues stained with H&E on day 4 after E. histolytica injection. Representative figures are shown. Original magnification: ×2.5. Scale bar, 1 mm. Each group consisted of three mice.
(B and C) The area (B) and number (C) of ALA formed in liver sections from DKO mice with or without ILC2 transfer on day 4 (∗p < 0.05, unpaired 2-tailed Student’s t test, n = 3 per group).
(D and E) The absolute numbers of eosinophils (D) and neutrophils (E) in livers from DKO with or without ILC2 transfer on day 4 (n = 3 per group).
Statistically significant differences between ILC2-transferred and non-transferred DKO mice are indicated with p value (∗∗p < 0.01, ∗∗∗p < 0.001, ANOVA). Each point shows the mean ± standard deviation (SD). Data are representative of at least two independent experiments. See also Figure S6.
IL-33 Is a Trigger of ILC2-Mediated ALA Formation
Next, we investigated how ILC2s are activated to exacerbate the pathogenesis of ALA after ameba injection. As IL-33, but not IL-25, mRNA expression was profoundly upregulated in the liver (Figure 1G), we clarified whether IL-33 is crucial for the activation of ILC2s, which exacerbate ALA. IL-33GFP/GFP mice, which lack IL-33 protein expression, showed reduced ALA formation compared with WT mice on day 4 after E. histolytica injection (Figures 5A and 5C). In IL-33-deficient mice, large ALA (>0.2 mm2) were not observed, whereas they were scattered throughout the liver in WT mice (Figure 5B). To examine the localization of ILC2s in the liver in detail, prior to the pathogen exposure and during acute E. histolytica infection, immunofluorescent staining of frozen tissue sections was performed. As shown in Figure 6A, in both naive WT and Rag2 KO mice, some ILC2s identified by strong red signal (T1/ST2) and blue signal (Gata3) were only detected in portal triad area in liver lobule. It is known that WT liver contains tissue-resident Tregs, which expressed Gata3 and T1/ST2 (Popovic et al., 2017). However, the similar strong Gata3 high, T1/ST2 high signal and localization of ILC2s were observed in both WT and Rag2 KO mice, indicating the Gata3 and T1/ST2 double-positive cells were not tissue-resident liver Tregs. In infected liver on day 7 post infection, ILC2s were observed within the necrotic lesions of ALA and surrounding inflammatory areas filled with IL-33+ cells in green signal (Figures 6B–6D). In both uninfected and infected phases, the close physical juxtaposition of ILC2 and IL-33+ cell is well revealed. It should be noted that the strong red signal from ILC2 merges to green of IL-33+ cell to generate yellow signal shown in detail region of Figures 6C and 6D. It is still unclear so far what type of cells produce IL-33 in this model. But, given that IL-33 is a key activator of ILC2s, it is possible that ILC2s recruited to the lesion are the critical trigger cell in the pathogenesis of ALA.
Figure 5.

IL-33-Deficient Mice Show Less ALA Formation Compared with WT Mice
IL-33GFP/GFP (IL-33-deficient) mice were injected with 2 × 105E. histolytica.
(A) Histology of liver tissues stained with H&E on day 4. Representative figures are shown. Original magnification: ×2.5. Scale bar, 1 mm.
(B and C) The area (B) and number (C) of ALA formed in liver sections from WT (n = 6) and IL-33-deficient mice (n = 3) on day 4. ∗∗p < 0.01, unpaired 2-tailed Student’s t test.
Figure 6.

Localization of ILC2s and IL-33+ Cells in the Liver after Ameba Injection
(A) The liver sections from uninfected WT and Rag2 KO mice were stained with anti-T1/ST2 (red), anti-Gata3 (blue), and anti-CD31 (green) mAbs.
(B) Immunofluorescent staining with isotype control Ab and DAPI (white).
(C) The liver section from uninfected WT mice was stained for ILC2s and IL-33+ cells with anti-T1/ST2 (red), anti-Gata3 (blue), and anti-IL-33 (green) mAbs.
(D) The liver section from infected WT mice was stained for ILC2s and IL-33+ cells using anti-T1/ST2 (red), Gata3 (blue), and IL-33 (green) mAbs. Scale bar, 50 μm. Yellow dotted lines show the necrotic lesion of ALA. Numbers in white dotted squares show further detail on the right images as detail 1 and 2. Results are representative of three sections from three mice examined.
IL-5 Leads to the Development of ALA Accompanied by Eosinophil Accumulation
To clarify whether IL-5 directly contributes to the pathogenesis of ALA, we analyzed ALA formation in Rag2 KO mice after neutralizing IL-5 with anti-IL-5 mAb. ALA formation was reduced in anti-IL-5-treated mice (Figure 7A). The area and number of ALA were markedly decreased in anti-IL-5-treated Rag2 KO mice compared with WT mice (Figures 7A–7C). Eosinophil accumulation in the liver was significantly decreased in anti-IL-5-treated Rag2 KO mice compared with rat IgG1 treated Rag2 KO mice (Figure 7D). However, in this setting, there was no difference in the absolute number of ILC2s and neutrophils in the livers between groups (Figures 7E and 7F). These results suggest that IL-5 produced by ILC2s induces eosinophil accumulation, which then exacerbates ALA.
Figure 7.

ALA Formation Is Regulated by IL-5 with the Accumulation of Eosinophils in Rag2 KO Mice
Rag2 KO mice were intraperitoneally administered 250 μg/mouse anti-(α) IL-5 mAb every 2 days from 3 days before E. histolytica injection (day −3, −1, 1, and 3).
(A) Histology of liver tissues stained with H&E on day 4 after E. histolytica injection. Representative figures are shown. Original magnification: ×2.5. Scale bar, 1 mm.
(B and C) The area (B) and number (C) of ALA formed in liver sections from Rag2 KO mice treated with αIL-5 mAb or control rat IgG1 on day 4 (∗p < 0.05, unpaired 2-tailed Student’s t test, n = 6 per group). The plots in (B) show each area of all ALA observed in six mice per group.
(D–F) The absolute numbers of eosinophils (D), hepatic ILC2s (E) and neutrophils (F) on day 4 (n = 3 per group). Statistically significant differences between isotype control Ab treated-Rag2 KO and IL-5 depleted-Rag2 KO mice are indicated with p value (∗p < 0.05, unpaired two-tailed Student’s t test). Each point shows the mean ± standard deviation (SD). Data are representative of at least three independent experiments.
Discussion
In this study, ILC2s were identified as the main source of type 2 cytokines in early ALA formation. Furthermore, type 2 cytokine-producing ILC2s exacerbated the pathology of ALA formation in mice injected with ameba via the portal vein.
To specifically investigate the role of innate immune cells in ALA formation, we used WT and Rag2 KO mice, which are deficient for T and B cells. In WT and Rag2 KO mice, similar levels of ALA formation were observed in the early phase and the number of hepatic ILC2s producing high levels of IL-5 was significantly increased on day 4 after E. histolytica injection. This implies that innate immune cells, in particular, ILC2s, but not T and B cells, are strongly involved in the pathogenesis of ALA.
ILC2s are tissue-resident cells that expand locally and produce abundant IL-5 and IL-13 in non-lymphoid organs such as the lungs and small intestine, and the mesenteric lymph nodes during the acute phase of Nippostrongylus brasiliensis infection (Gasteiger et al., 2015; Moro et al., 2016). On day 7 after E. histolytica injection, the number of ILC2s and IL-5-producing ILC2s were diminished in the liver. As ALA formation peaks on day 4 and injected mice can survive with recovery from ALA in this experimental model, the decrease of the number and function of ILC2s on day 7 may correlate with the recovery from ALA. After E. histolytica injection, IL-13 mRNA expression was augmented in the livers of WT, but not Rag2 KO, mice on day 7. We did not detect a significant increase in IL-13-producing ILC2s in the livers of WT and Rag2 KO mice on day 4 even after ex vivo stimulation with PMA and ionomycin. It is possible from these results that IL-13 production is dependent on conventional T cells after ameba infection.
IFN-γ is critical for clearing ameba from the liver and suppressing the function and proliferation of ILC2s (Mchedlidze et al., 2016; Moro et al., 2016). In ILC2-depleted Rag2 KO mice, levels of IFN-γ production were similar to that in WT mice. Thus, IFN-γ production in the liver was not affected by ILC2 depletion, suggesting that ILC2s exacerbate the pathology of ALA without changing the production of IFN-γ in the microenvironment.
How IL-5 is involved in the pathogenesis of ALA is currently unclear. In this study, we neutralized IL-5 in vivo to clearly show that IL-5 exacerbated ALA progression. IL-5 produced by ILC2s induced the survival and proliferation of eosinophils, which lead to eosinophilia (Nussbaum et al., 2013). In a susceptible mouse model of ALA, severe abscesses were mainly mediated by eosinophils that strongly infiltrated into the abscess (Estrada-Villasenor et al., 2007), indicating that ILC2-produced IL-5 directly induces eosinophil infiltration that helps to form severe ALA around E. histolytica.
In this study, we focused on the accumulation of eosinophils and neutrophils during ILC2-mediated ALA formation. The kinetics of eosinophils and neutrophils in the blood were inconsistent with those of liver eosinophils and neutrophils, suggesting that the size and composition of the ALA do not simply reflect the levels of neutrophils and eosinophils in the blood. Although systemic changes are induced after ameba injection, the pathology observed during ALA is not the simple reflection of such systemic changes. Other immune cells activated by ILC2s may also be involved in the pathogenesis of ALA. It was reported that IL-5 increased the number of alternatively activated macrophages and fibroblasts (Reiman et al., 2006). In a hamster model of ALA caused by E. nuttalli, which is highly prevalent in macaques, alternatively activated macrophages were increased in the liver in the late stage of abscess progression (Guan et al., 2018). Thus, alternatively activated macrophages augmented by IL-5 might contribute to the pathology of ALA caused by E. histolytica. We need to clarify further the involvement of alternatively activated macrophages in ILC2-mediated ALA.
The adoptive transfer of naive ILC2s into Rag2−/−γc−/− KO mice clearly showed that ILC2s exacerbated the pathogenesis of ALA accompanied by the augmented accumulation of neutrophils. Recently it was reported that ILC2 subpopulation exhibited the conversion into IL-17-producing cells in response to IL-1β and IL-23 (Bernink et al., 2019; Hochdorfer et al., 2019). As IL-17 induces the migration of neutrophils into the site of infection, ILC2 plasticity into IL-17-producing cells may provide a possibility to increase the accumulation of neutrophils in the liver. A previous study reported that IL-33 inoculated into the ears of mice induced inflammatory psoriatic lesions accompanied by the recruitment of neutrophils into the ear after phorbol ester challenge. Recruited neutrophils produced large amounts of the chemokine KC (CXCL1) in the inflammatory ear lesions (Lefrancais et al., 2012). Because IL-33-deficient mice showed less ALA formation, IL-33 is likely responsible for the initiation of ALA formation. We noted that there was a trend of the co-localization of ILC2 and IL-33+ cell in the liver from naive and infected mice. As hepatic ILC2s infiltrated around ALA accompanied by the interaction with a lot of IL-33+ cells, suggesting a possibility that IL-33 activates ILC2s and induces the accumulation of neutrophils, which then produce chemokines and/or other inflammatory factors involved in liver inflammation and subsequently severe ALA. However, to date, it is unclear whether neutrophils are responsible for ALA formation in this setting and whether ILC2s activated by IL-33 are associated with the function of neutrophils. Further investigations are required to reveal the relevance of ILC2s and neutrophils to ALA.
A number of studies about the immune response against protozoan parasitic diseases such as leishmaniasis, malaria, and toxoplasmosis have revealed the role of type 1 immunity in controlling the infection. Regarding type 2 immunity in protozoan infection, a few studies reported that type 2 immunity provided the susceptibility to leishmanial infection and protected against cerebral malaria via IL-33-mediated ILC2, M2 macrophages, and Tregs (Besnard et al., 2015). Our study has revealed the involvement of ILC2s during a protozoan infection and the contributions of ILC2s in the pathogenesis of liver abscesses caused by E. histolytica. To date, few studies have investigated the function of ILC2s in the liver. One study reported that ILC2s exacerbated Con A-induced hepatitis in mice, and another clarified the association between increased ILC2s and human liver fibrosis (Gonzalez-Polo et al., 2019; Zhang et al., 2019). Consistent with previous studies, we clearly demonstrated that ILC2s exacerbate ALA caused by E. histolytica.
In conclusion, we showed that ILC2s are a major source of early type 2 cytokines, such as IL-5, during ALA formation and that they contribute to the exacerbation of ALA accompanied by the accumulation of eosinophils and neutrophils. Furthermore, IL-33-deficient and IL-5-neutralized mice revealed the mechanism of ILC2-mediated ALA formation in which IL-5-producing ILC2s activated by IL-33 exacerbated ALA. Further studies are required to clarify how ILC2s induce severe ALA via eosinophils and neutrophils and/or other immune cells and whether IL-5-producing ILC2s have functional relevance to neutrophil accumulation in the pathogenesis of ALA.
Limitations of the Study
In this study, we indicated that ILC2s contribute to the pathogenesis of ALA at the early phase of the disease using a mouse model inoculated with E. histolytica via the hepatic portal vein. The present animal model of ALA may provide a concern about the physiological relevance about the portal vein inoculation. However, there is no way for the natural infection model via oral to develop ALA so far, much less a translocation model of E. histolytica trophozoites to the liver from the intestine. To mimic the translocation of ameba for the natural occurring of ALA, we used the portal vein inoculation model in which we can avoid the damage of liver parenchyma that may lead to the production of IL-33 and other alarmins. ILC2s can produce abundant type 2 cytokines, but the number and proportion of ILC2s are much lower compared with T cell at steady state. To monitor the kinetics and role of ILC2s, we used Rag2 KO mice (T cell-deficient mice) to exclude type 2 responses by Th2 cell. We confirmed that both WT (under the presence of T cells) and Rag2 KO mice developed a similar level of ALA formation on day 4. This result clearly indicated that T cell does not influence the development and pathogenesis of ALA at least at this time point. Although we suggest ILC2s exacerbate amebic liver abscess in mice, we should consider the balance between ILC2s and T cell in immunocompetent mice. In flow cytometry analysis, we could not exclude dead cells by the specific staining owing to the limitation of detectors in the flow cytometer. As hepatic ILC2s do not have a specific marker, the identification of hepatic ILC2s needs staining of a number of surface and intracellular markers. Since our study made it a priority to stain these markers for detecting hepatic ILC2s, a detector of flow cytometer did not remain for dead cell staining. Instead of the specific staining for live/ dead cell discrimination, dead cells were distinguished by gating on the FSC/SSC plot. ILC2 depletion in Rag2 KO mice rescued ALA formation without any difference in IFN-γ production on day 4. It remains the possibility that IFN-γ production by ILC1s may occur much earlier time point than on day 4. We could not examine it owing to the restriction of outing by COVID-19 pandemic. So far, the roles and functions of ILC1s in ALA formation remain unclear. We need further investigations to reveal the relevance of ILC1s and ILC2s at the early time point of ALA.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shinjiro Hamano (shinjiro@nagasaki-u.ac.jp).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The published article includes all data generated or analyzed during this study. Any additional information will be available from the lead contact upon request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors are grateful to Megumi Hamasaki and Hiromi Oda for breeding, genotyping, maintenance, and care of the mice used in this study. We also thank Fumie Hara for technical assistance. This study was conducted at the Joint Usage/Research Center on Tropical Disease, Institute of Tropical Medicine, Nagasaki University, Japan. This study was supported by a Grant-in-Aid for Young Scientists (B) JP16K19118 to R.N. from the Japan Society for the Promotion of Science (JSPS), a Grant-in-Aid for Scientific Research on Priority Areas from MEXT (21022037 to S.H.), Grants-in-Aid for International Scientific Research (B) from JSPS (23406009 and 26305013 to S.H.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Author Contributions
R.N. designed this study, analyzed the data, and wrote the manuscript. R.N. and S.D. maintained amebae and performed the experiments. A.Y. supported and performed the immunofluorescence of the liver. T.M. supported culture of amebae and performed antibody administration for revised experiments. S.K. and K.M. provided protocols for ILC2 analysis and in vitro culture of ILC2s and data interpretation. M.K. supported the analysis of real-time PCR data and advised the manuscript. M.S. made histological sections of the liver. S.H., K.M., and S.K. participated in the preparation and the edition of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101544.
Contributor Information
Risa Nakamura, Email: risa-n@nagasaki-u.ac.jp.
Shinjiro Hamano, Email: shinjiro@nagasaki-u.ac.jp.
Supplemental Information
References
- Bernink J.H., Ohne Y., Teunissen M.B.M., Wang J., Wu J., Krabbendam L., Guntermann C., Volckmann R., Koster J., Van Tol S. c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nat. Immunol. 2019;20:992–1003. doi: 10.1038/s41590-019-0423-0. [DOI] [PubMed] [Google Scholar]
- Besnard A.G., Guabiraba R., Niedbala W., Palomo J., Reverchon F., Shaw T.N., Couper K.N., Ryffel B., Liew F.Y. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog. 2015;11:e1004607. doi: 10.1371/journal.ppat.1004607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell D., Gaucher D., Chadee K. Serum from Entamoeba histolytica-infected gerbils selectively suppresses T cell proliferation by inhibiting interleukin-2 production. J. Infect. Dis. 1999;179:1495–1501. doi: 10.1086/314781. [DOI] [PubMed] [Google Scholar]
- Cervantes-Rebolledo C., Moreno-Mendoza N., Morales-Montor J., De La Torre P., Laclette J.P., Carrero J.C. Gonadectomy inhibits development of experimental amoebic liver abscess in hamsters through downregulation of the inflammatory immune response. Parasite Immunol. 2009;31:447–456. doi: 10.1111/j.1365-3024.2009.01120.x. [DOI] [PubMed] [Google Scholar]
- Cieslak P.R., Virgin H.W.T., Stanley S.L., Jr. A severe combined immunodeficient (SCID) mouse model for infection with Entamoeba histolytica. J. Exp. Med. 1992;176:1605–1609. doi: 10.1084/jem.176.6.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis M., Chadee K. Cytokine activation of murine macrophages for in vitro killing of Entamoeba histolytica trophozoites. Infect. Immun. 1989;57:1750–1756. doi: 10.1128/iai.57.6.1750-1756.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis M., Chadee K. Human neutrophils activated by interferon-gamma and tumour necrosis factor-alpha kill Entamoeba histolytica trophozoites in vitro. J. Leukoc. Biol. 1989;46:270–274. doi: 10.1002/jlb.46.3.270. [DOI] [PubMed] [Google Scholar]
- Estrada-Villasenor E., Morales-Montor J., Rodriguez-Dorantes M., Ramos-Martinez E., Nequiz-Avendano M., Ostoa-Saloma P. IL-6 KO mice develop experimental amoebic liver infection with eosinophilia. J. Parasitol. 2007;93:1424–1428. doi: 10.1645/GE-1223.1. [DOI] [PubMed] [Google Scholar]
- Gasteiger G., Fan X., Dikiy S., Lee S.Y., Rudensky A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–985. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadirian E., Salimi A. In vitro effect of recombinant interferon gamma in combination with LPS on amoebicidal activity of murine Kupffer cells. Immunobiology. 1993;188:203–219. doi: 10.1016/S0171-2985(11)80498-5. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Polo V., Pucci-Molineris M., Cervera V., Gambaro S., Yantorno S.E., Descalzi V., Tiribelli C., Gondolesi G.E., Meier D. Group 2 innate lymphoid cells exhibit progressively higher levels of activation during worsening of liver fibrosis. Ann. Hepatol. 2019;18:366–372. doi: 10.1016/j.aohep.2018.12.001. [DOI] [PubMed] [Google Scholar]
- Guan Y., Feng M., Min X., Zhou H., Fu Y., Tachibana H., Cheng X. Characteristics of inflammatory reactions during development of liver abscess in hamsters inoculated with Entamoeba nuttalli. PLoS Negl. Trop. Dis. 2018;12:e0006216. doi: 10.1371/journal.pntd.0006216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim T.Y., Krauss R.H., Sun A.C., Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36:451–463. doi: 10.1016/j.immuni.2011.12.020. [DOI] [PubMed] [Google Scholar]
- Hamano S., Cherry J., Demmlar-Harrison J.G., Kaplan S., Steinbach J.W., Hotez J.P. Elsevier; 2013. Amebiasis. Feigin and Cherrys's Textbook of Pediatric Infectious Diseases. [Google Scholar]
- Haque R., Huston C.D., Hughes M., Houpt E., Petri W.A., Jr. Amebiasis. N. Engl. J. Med. 2003;348:1565–1573. doi: 10.1056/NEJMra022710. [DOI] [PubMed] [Google Scholar]
- Hochdorfer T., Winkler C., Pardali K., Mjosberg J. Expression of c-Kit discriminates between two functionally distinct subsets of human type 2 innate lymphoid cells. Eur. J. Immunol. 2019;49:884–893. doi: 10.1002/eji.201848006. [DOI] [PubMed] [Google Scholar]
- Houpt E.R., Glembocki D.J., Obrig T.G., Moskaluk C.A., Lockhart L.A., Wright R.L., Seaner R.M., Keepers T.R., Wilkins T.D., Petri W.A., Jr. The mouse model of amebic colitis reveals mouse strain susceptibility to infection and exacerbation of disease by CD4+ T cells. J. Immunol. 2002;169:4496–4503. doi: 10.4049/jimmunol.169.8.4496. [DOI] [PubMed] [Google Scholar]
- Kotloff K.L., Nataro J.P., Blackwelder W.C., Nasrin D., Farag T.H., Panchalingam S., Wu Y., Sow S.O., Sur D., Breiman R.F. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- Lefrancais E., Roga S., Gautier V., Gonzalez-De-Peredo A., Monsarrat B., Girard J.P., Cayrol C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. U S A. 2012;109:1673–1678. doi: 10.1073/pnas.1115884109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotter H., Jacobs T., Gaworski I., Tannich E. Sexual dimorphism in the control of amebic liver abscess in a mouse model of disease. Infect. Immun. 2006;74:118–124. doi: 10.1128/IAI.74.1.118-124.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchedlidze T., Kindermann M., Neves A.T., Voehringer D., Neurath M.F., Wirtz S. IL-27 suppresses type 2 immune responses in vivo via direct effects on group 2 innate lymphoid cells. Mucosal Immunol. 2016;9:1384–1394. doi: 10.1038/mi.2016.20. [DOI] [PubMed] [Google Scholar]
- Moro K., Kabata H., Tanabe M., Koga S., Takeno N., Mochizuki M., Fukunaga K., Asano K., Betsuyaku T., Koyasu S. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat. Immunol. 2016;17:76–86. doi: 10.1038/ni.3309. [DOI] [PubMed] [Google Scholar]
- Moro K., Yamada T., Tanabe M., Takeuchi T., Ikawa T., Kawamoto H., Furusawa J., Ohtani M., Fujii H., Koyasu S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463:540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- Motomura Y., Morita H., Moro K., Nakae S., Artis D., Endo T.A., Kuroki Y., Ohara O., Koyasu S., Kubo M. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity. 2014;40:758–771. doi: 10.1016/j.immuni.2014.04.013. [DOI] [PubMed] [Google Scholar]
- Murray C.J., Vos T., Lozano R., Naghavi M., Flaxman A.D., Michaud C., Ezzati M., Shibuya K., Salomon J.A., Abdalla S. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- Neill D.R., Wong S.H., Bellosi A., Flynn R.J., Daly M., Langford T.K., Bucks C., Kane C.M., Fallon P.G., Pannell R. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum J.C., Van Dyken S.J., Von Moltke J., Cheng L.E., Mohapatra A., Molofsky A.B., Thornton E.E., Krummel M.F., Chawla A., Liang H.E., Locksley R.M. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502:245–248. doi: 10.1038/nature12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic B., Golemac M., Podlech J., Zeleznjak J., Bilic-Zulle L., Lukic M.L., Cicin-Sain L., Reddehase M.J., Sparwasser T., Krmpotic A., Jonjic S. IL-33/ST2 pathway drives regulatory T cell dependent suppression of liver damage upon cytomegalovirus infection. PLoS Pathog. 2017;13:e1006345. doi: 10.1371/journal.ppat.1006345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash V., Bhimji S.S. StatPearls Publishing; 2017. Abscess, Amebic Liver. [PubMed] [Google Scholar]
- Ravdin J.I. Human infection by Entamoeba histolytica. In: RAVDIN J.I., editor. Amebiasis. New York. Edinburgh: Churchill Livingstone; 1988. pp. 594–613. [Google Scholar]
- Reiman R.M., Thompson R.W., Feng C.G., Hari D., Knight R., Cheever A.W., Rosenberg H.F., Wynn T.A. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect. Immun. 2006;74:1471–1479. doi: 10.1128/IAI.74.3.1471-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salata R.A., Murray H.W., Rubin B.Y., Ravdin J.I. The role of gamma interferon in the generation of human macrophages cytotoxic for Entamoeba histolytica trophozoites. Am. J. Trop. Med. Hyg. 1987;37:72–78. doi: 10.4269/ajtmh.1987.37.72. [DOI] [PubMed] [Google Scholar]
- Seydel K.B., Smith S.J., Stanley S.L., Jr. Innate immunity to amebic liver abscess is dependent on gamma interferon and nitric oxide in a murine model of disease. Infect. Immun. 2000;68:400–402. doi: 10.1128/iai.68.1.400-402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Qi C., Li L., Hua S., Zheng F., Gong F., Fang M. CD8(+) T cell/IL-33/ILC2 axis exacerbates the liver injury in Con A-induced hepatitis in T cell-transferred Rag2-deficient mice. Inflamm. Res. 2019;68:75–91. doi: 10.1007/s00011-018-1197-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated or analyzed during this study. Any additional information will be available from the lead contact upon request.