

Keywords: diet, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, transcriptome
Abstract
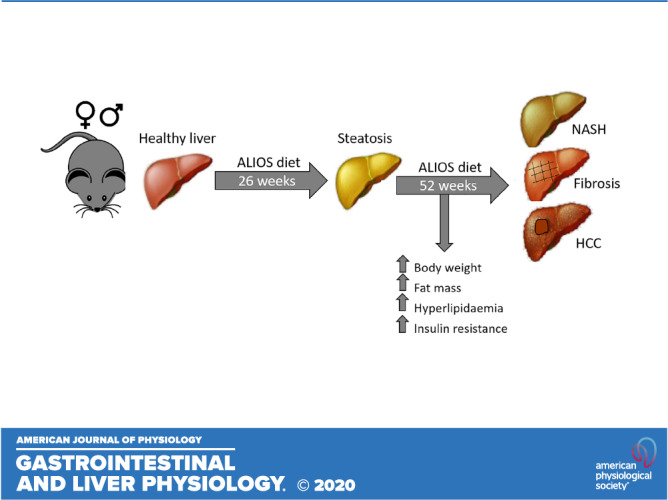
The pathogenesis of nonalcoholic fatty liver disease (NAFLD) and the progression to nonalcoholic steatohepatitis (NASH) and increased risk of hepatocellular carcinoma remain poorly understood. Additionally, there is increasing recognition of the extrahepatic manifestations associated with NAFLD and NASH. We demonstrate that intervention with the American lifestyle-induced obesity syndrome (ALIOS) diet in male and female mice recapitulates many of the clinical and transcriptomic features of human NAFLD and NASH. Male and female C57BL/6N mice were fed either normal chow (NC) or ALIOS from 11 to 52 wk and underwent comprehensive metabolic analysis throughout the duration of the study. From 26 wk, ALIOS-fed mice developed features of hepatic steatosis, inflammation, and fibrosis. ALIOS-fed mice also had an increased incidence of hepatic tumors at 52 wk compared with those fed NC. Hepatic transcriptomic analysis revealed alterations in multiple genes associated with inflammation and tissue repair in ALIOS-fed mice. Ingenuity Pathway Analysis confirmed dysregulation of metabolic pathways as well as those associated with liver disease and cancer. In parallel the development of a robust hepatic phenotype, ALIOS-fed mice displayed many of the extrahepatic manifestations of NAFLD, including hyperlipidemia, increased fat mass, sarcopenia, and insulin resistance. The ALIOS diet in mice recapitulates many of the clinical features of NAFLD and, therefore, represents a robust and reproducible model for investigating the pathogenesis of NAFLD and its progression.
NEW & NOTEWORTHY Nonalcoholic fatty liver disease (NAFLD) affects 30% of the general population and can progress to nonalcoholic steatohepatitis (NASH) and potentially hepatocellular carcinoma. Preclinical models rely on mouse models that often display hepatic characteristics of NAFLD but rarely progress to NASH and seldom depict the multisystem effects of the disease. We have conducted comprehensive metabolic analysis of both male and female mice consuming a Western diet of trans fats and sugar, focusing on both their hepatic phenotype and extrahepatic manifestations.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome and is the most common form of liver disease in the Western world (2, 25). NAFLD currently affects ~30% of the normal population and rises to 80% in patients with obesity and type 2 diabetes (24, 25). It is a spectrum disease, ranging from simple steatosis through to the necroinflammatory disease nonalcoholic steatohepatitis (NASH). Development of NASH subsequently increases the risk of fibrosis, cirrhosis, and eventually hepatocellular carcinoma (HCC; Refs. 51, 52). Recent evidence supports the concept that NAFLD is a multisystemic condition impacting on a variety of organs and systems (5, 48) and is associated with multiple extrahepatic clinical features, including insulin resistance, hyperlipidemia, and sarcopenia.
Preclinical mouse models of NAFLD and NASH typically rely on genetic manipulation, hepatic cytotoxic injury, or formulated dietary extremes (11, 16, 29). Although some of these models exhibit the histological features of NAFLD, they rarely progress to NASH or HCC and, therefore, do not reflect the mechanisms of the human disease. Commonly used genetic models of obesity, the leptin-deficient (ob/ob) mouse and leptin receptor-deficient (db/db) mouse, have excess hepatic fat deposition but do not develop NASH or HCC (1, 11, 36), most likely because leptin is involved in regulating inflammation and fibrosis. In addition, models using high-fat diet (HFD; 60% fat) cause simple steatosis but do not progress to NASH or develop hepatic injury (11, 34). Fructose has been shown to be a driver of hepatic de novo lipogenesis (43); however, fructose-only dietary interventions often fail to induce dyslipidemia, hepatic steatosis, and inflammation (28, 41). Previous studies have utilized “fast food” diets (7, 22), which contain 40% fat (12% from saturated fat) with the addition of fructose in drinking water; although these studies progress to characteristic NASH, they seldom highlight the extrahepatic features of the disease, and sexual dimorphism has not been explored. Alternative models use hepatic toxins to drive liver injury. These commonly include carbon tetrachloride with which animals develop hepatic histological features of NASH and fibrosis from as early as 8 wk, but often do not present with other clinical features of NASH, such as weight gain and insulin resistance (47). Diethylnitrosamine is able to induce HCC but does so without the progression from NAFLD and NASH (19). Furthermore, the use of genetic manipulation in combination with toxins and HFD in generating models of NAFLD poses questions as to the relevance and similarity to human NAFLD and NASH.
The American lifestyle-induced obesity syndrome (ALIOS) mouse model is a dietary intervention based on the nutritional content of commonly consumed fast foods of the Western world (46). Mice are fed high-fat chow (45%), including trans fats, with high-fructose corn syrup added to the drinking water; animals become obese and insulin resistant and develop hepatic steatosis with a necroinflammatory response (10, 46). When aged to 12 mo, mice fed ALIOS also develop NASH-driven HCCs (10). To date, studies using ALIOS have only treated male mice; therefore, the effects of ALIOS on female metabolism is entirely unexplored. In addition, with the increasing evidence suggesting the importance of the extrahepatic impact of NAFLD, models that can accurately replicate a clinical condition are likely to be more highly informative, with respect to natural history but also the potential impact of intervention. In the published literature, studies have largely focused on the hepatic phenotype associated with the ALIOS diet, and its multisystem impact has not been evaluated in detail.
We have, therefore, conducted a comprehensive metabolic analysis of both male and female mice consuming either an ALIOS diet or standard chow from 11 to 52 wk of age, focusing on both their hepatic phenotype and extrahepatic manifestations.
METHODS
Mouse husbandry.
Male and female C57BL/6NTac mice were kept and studied in accordance with United Kingdom Home Office legislation and local ethical guidelines issued by the Medical Research Council (Responsibility in the Use of Animals in Medical Research, July 1993; Home Office license 30/3146). All procedures were conducted in accordance with the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 (SI 4 2012/3039). Mice were kept under controlled light (light 7 AM to 7 PM, dark 7 PM to 7 AM), temperature (21 ± 2°C), and humidity (55 ± 10%) conditions. They had free access to water (9–13 ppm chlorine) and were fed ad libitum on a commercial diet (SDS Rat and Mouse No.3 Breeding diet, RM3; Special Diet Services, Witham, Essex, United Kingdom) until 10 wk of age when they were then transferred to a control (NC; SDS Rat and Mouse No.3 Breeding diet, RM3) or ALIOS diet [D06031302, Research Diets; and TD.06415 with hydrogenated vegetable fats, Envigo (45% fat, of which 30% is trans fat) with 55% fructose-45% glucose in drinking water] for 26 or 52 wk before culling by cervical dislocation and tissue analysis.
Experimental design.
Cohorts of male and female mice were bred for longitudinal metabolic phenotyping tests (26-wk cohort: n = 12 NC males, n = 15 ALIOS males, n = 15 NC females, n = 15 ALIOS females; 52-wk cohort: n = 17 NC males, n = 12 ALIOS males, n = 15 NC females, n = 15 ALIOS females). Mice were housed in single-sex groups across multiple litters and were not randomized into groups. Animal identifiers (IDs) and diets were recorded on the cages and were not blinded to the operator carrying out the animal procedure, although subsequent tests only included animal ID information. Sample size estimates were based on previous experience of mouse models in which relevant traits were measured (9, 10, 38).
Body weight and composition.
Body weight was measured weekly in the morning using average weights (in grams) calculated by Adventure Pro balances (OHAUS). Fat and lean masses were assessed by dual-energy X-ray absorptiometry (DXA) at 17, 25, 39, and 51 wk of age.
Calorimetry.
Calorimetric data were collected in a PhenoMaster system (TSE Systems) at 14, 25, 36, and 48 wk of age. Mice were kept under controlled light (light 7 AM to 7 PM, dark 7 PM to 7 AM), temperature (21 ± 2°C), and humidity (55 ± 10%) conditions. They had free access to water (9–13 ppm chlorine) and were fed ad libitum. O2 consumption (V̇o2), CO2 production (V̇co2), and respiratory exchange ratio (V̇co2/V̇o2, an estimate of fuel usage) were calculated and recorded electronically over 12 h for each mouse. Total locomotor activity (measured by x-, y-, and z-axis infrared beam breaks) and diet consumption were also recorded electronically for each mouse. Data were collected at three to four time points each hour.
Intraperitoneal glucose tolerance test.
Glucose tolerance was assessed at 15, 25, 37, and 49 wk of age. Mice were fasted overnight for 16 h and then injected intraperitoneally with 20% glucose solution (2 g glucose/kg body wt; Sigma, Dorset, United Kingdom). Blood was sampled from the tail vein, and glucose concentration was measured at t = 0, 15, 30, 60, and 120 min (AlphaTRAK; Abbott). Insulin concentrations at t = 0, 60, and 120 min were determined by ELISA (Crystal Chem). The homeostatic model assessment of insulin resistance (HOMA-IR) index was calculated as fasting glucose (in millimoles per liter) times fasting insulin (in microunits per milliliter)/22.5 (13).
Intraperitoneal insulin tolerance test.
Insulin tolerance was assessed at 25, 37, and 49 wk. Mice were fasted for 4–5 h and then injected intraperitoneally with insulin at a concentration of 0.75 or 1.25 IU/kg for females and males, respectively (Hypurin Bovine Insulin). Blood was sampled from the tail vein, and glucose concentration was measured at t = 0, 15, 30, 45, and 90 min (AlphaTRAK).
Blood biochemistry.
At termination, mice were anesthetized with isoflurane, and blood was collected via retroorbital bleed. Samples were kept on ice and then centrifuged for 10 min at 8,000 g at room temperature. Alkaline phosphatase, alanine aminotransferase (ALT), aspartate aminotransferase (AST), cholesterol, high-density lipoprotein (HDL), low-density lipoprotein (LDL), triglycerides, free fatty acids, and bilirubin were determined from plasma on an AU680 Clinical Chemistry Analyzer (Beckman Coulter, High Wycombe, United Kingdom). Creatinine was analyzed using a colorimetric kit appropriate for mouse serum (Cayman Chemical, Cambridge, United Kingdom).
Tissue biochemistry.
Hepatic triacylglycerol content was measured on an AU480 Clinical Chemistry Analyzer from a homogenate of frozen liver tissue (100 mg in 500 µL of PBS/0.1% Triton X-100).
Tissue histology.
Liver tissue was fixed in 4% (vol/vol) buffered formaldehyde, samples were subsequently paraffin-embedded, and 5 μm sections were prepared on a microtome. Sections were stained with hematoxylin-eosin and viewed at ×200 magnification. Inflammation score was determined by counting the number of inflammatory foci in five fields of view over three sections from each mouse liver (20, 23), and the average was scored as follows: no foci = 0, less than two per field of view = 1, two to four per field of view = 2, more than four per field of view = 3. A focus was determined as a cluster of five or more inflammatory cells. Percentage of hepatic fibrosis was determined by staining three sections from each mouse liver with Sirius Red and quantifying percentage of positive staining over six fields of view by ImageJ (NIH, Bethesda, MD; https://imagej.nih.gov/ij/).
Immunohistochemistry.
Immunohistochemistry was performed on wax-embedded liver sections (5 µm). Briefly, sections were dewaxed and rehydrated before incubation with antibodies against glutamine synthetase (5 µg/mL; Millipore, Watford, United Kingdom) and Sox9 (1 µg/mL; Millipore) after heat-induced antigen retrieval. Bound primary antibody was detected using a peroxidase-conjugated secondary antibody (Dako) with visualization using 3,3-diaminobenzidine (SigmaFast; Sigma). For negative control samples, nonimmune goat serum replaced primary antibodies.
Protein extraction and immunoblotting.
Total protein was extracted from whole liver tissue using radioimmunoprecipitation assay buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0; Sigma), with protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Loughborough, United Kingdom). Protein concentrations were measured using a BCA protein quantification kit according to the manufacturer’s protocol (Thermo Fisher Scientific). Primary collagen type 1 (Col1a1; Cell Signaling Technology, Leiden, The Netherlands) and secondary antibodies from Dako (Agilent Technologies, Santa Clara, CA) were used at dilutions of 1:1,000 and 1:2,000, respectively. Bands were visualized with ECL (Pierce Thermo Fisher Scientific) and ChemiDoc XRS+ imager (Bio-Rad, Watford, United Kingdom). Bands were quantified by densitometry and normalized to total protein using ImageJ.
RNA sequencing.
Total liver RNA was extracted using TRI reagent (Sigma). Concentration was determined spectrophotometrically at 260 nm optical density on a NanoDrop spectrophotometer (Thermo Fisher Scientific, Hemel Hempstead, United Kingdom) and quality checked (QCed) on a 2100 Bioanalyzer system (Agilent Technologies, Stockport, United Kingdom). Only samples with RNA integrity numbers >7 were used for analysis. cDNA was generated from total RNA using first oligo(dT) and subsequently random priming using the TruSeq Stranded mRNA HT Sample Prep Kit for Illumina (according to manufacturer’s instructions). The prepared libraries were QCed and multiplexed, followed by pair-end sequencing (75 bp) over one lane of a NextSeq 75 SR flow cell (Illumina, Cambridge, United Kingdom) to a total depth of 130 million read pairs on the Illumina NextSeq 500 platform. Reads were mapped with Stampy (31) on default settings with GRCm38/mm10 as genome reference and BAM files merged using BamTools. Gene level read counts for all protein-coding RNA transcripts present in refGene mm10 were quantified in a strand-specific manner using featureCounts from the Subread package version 1.6.0. Differential expression analysis was performed using DESeq2 (30). Differentially expressed genes (DEGs) were reported for q ≤ 0.05 and fold change of 2 or q ≤ 0.1. Statistical significance was determined by unpaired parametric t test, and differentially regulated genes were defined by a false discovery rate (Benjamini–Hochberg method) adjusted P value <1%. The online bioinformatics tools Metascape (https://metascape.org/) and Enrichr (http://amp.pharm.mssm.edu/Enrichr/) were used for enrichment analysis of the DEGs. Ingenuity Pathway Analysis (IPA; Ingenuity Systems) was used to examine biological functions and disease and functional relationships between gene networks.
Bioinformatic data analysis.
RNA-sequencing (RNA-Seq) data were downloaded from the National Center for Biotechnology Information (GSE126848). Gene counts (GSE126848_Gene_counts_raw.txt) and sample identification were determined from the series matrix (GSE126848_series_matrix.txt). The clinical characteristics of patients are described in Supplemental Table S1 (all supplemental material is available at https://doi.org/10.6084/m9.figshare.12666860). Gene symbols were converted from Ensembl ID using org.Hs.eg.db version 3.11.4. Differential gene expression was determined using edgeR version 3.11. As the human NASH data set contained 12 male samples and 4 female samples, RNA-Seq libraries from female samples were removed. All remaining samples were included (normal weight, obese, NAFLD, and NASH), low counts were removed (counts per minute > 0.25 in 2 libraries), and differential expression for NASH versus normal weight samples was determined, this gave 3,152 downregulated and 3,491 upregulated genes (using edgeR glmLRT). Differentially expressed genes from male mice (ALIOS vs. normal chow, 2,153 downregulated and 2,865 upregulated) were used to convert to human symbols. Mouse gene symbols (5,018) were converted using the package biomaRt (version 2.440). Four thousand seven hundred one genes were matched between human and mouse. NASH-regulated genes were then compared with ALIOS-regulated genes. A list of 2,052 genes was identified as overlapping, with significance determined using Fisher exact test (a Venn diagram was produced, using the ggVennDiagram R package). The overlapping genes were used for pathway analysis and plotted on heat maps (using ggplot2). The top 30 significant genes (sorted by false discovery rate) in the NASH data set were extracted. Log counts per million were used for each heat map, and each data set (mouse and human) was scaled separately before plotting.
Statistics.
Data are presented as means ± standard error. Data analysis was performed using GraphPad Prism software (GraphPad Software, La Jolla, CA) and considered statistically significant at P < 0.05. Normality was assessed using the Shapiro–Wilk test. Two-tailed, unpaired t tests were used to compare differences in the mean between diets within each sex. Mann–Whitney tests were used with data sets of nonparametric distributions. For data collected across time, repeated-measures two-way analysis of variance (ANOVA) was used.
RESULTS
Male and female mice fed ALIOS have progressive weight gain and increased fat mass.
Weight gain was greater in both male and female ALIOS-fed mice, and body weight continued to rise throughout the study, from the onset of ALIOS at 11 wk until 52 wk (Fig. 1, A and B). From 15 wk onward, DXA analysis confirmed that ALIOS-fed mice of both sexes had an increase in fat mass compared with normal chow controls (Fig. 1, C and D). Interestingly, at 15 and 37 wk, female mice also displayed a decrease in lean mass. There was no change in lean mass in the male mice throughout the duration of the study.
Fig. 1.

The American lifestyle-induced obesity syndrome (ALIOS) diet increased body weight (BW) and fat mass in male and female mice from 11 to 52 wk. Body weight curves for male [normal chow (NC), n = 29; ALIOS, n = 27; A] and female NC (n = 30)- and ALIOS (n = 30; B)-fed mice and dual-energy X-ray absorptiometry-determined lean and fat mass in male (NC, n = 17; ALIOS, n = 12; C) and female (NC, n = 15; ALIOS, n = 15; D) mice from 15 to 52 wk (w) are shown. Changes in adipose depot mass in male (E) and female (F) NC- and ALIOS-fed mice at 26 and 52 wk are also shown. BAT, brown adipose tissue; GF, gonadal fat; Mes, mesenteric fat; PR, perirenal fat; Subcut, subcutaneous fat. Data are expressed as means ± SE. 26 wk: NC males, n = 12; ALIOS males, n = 15; NC females, n = 15; ALIOS females, n = 15. 52 wk: NC males, n = 17; ALIOS males, n = 12; NC females, n = 15; ALIOS females, n = 15. *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from mice fed NC at each time point.
At 26 wk, male ALIOS-fed mice had increased fat pad weights across all depots compared with NC controls. However, by 52 wk, gonadal fat was the only enlarged depot (Fig. 1E). ALIOS-fed female mice had increased fat depots at both 26 and 52 wk compared with NC controls (Fig. 1F). Consistent with increased lipid storage, ALIOS-fed male and female mice had increased serum levels of total, HDL, and LDL cholesterol from 25 wk onward (Table 1). Circulating levels of triacylglycerols and free fatty acids were significantly decreased in ALIOS-fed mice compared with NC controls, at 52 wk and from 25 wk in males and females, respectively (Table 1). In addition, data from metabolic cage experiments revealed that male mice were more sedentary than females and that ALIOS-fed mice had reduced total activity compared with NC controls (Supplemental Tables S2 and S3). As expected, respiratory exchange ratios were reduced in male and female ALIOS-fed mice, indicating increased fat catabolism for energy production (Supplemental Tables S2 and S3).
Table 1.
Circulating serum levels of lipids
| 16 wk |
25 wk |
37 wk |
52 wk |
|||||
|---|---|---|---|---|---|---|---|---|
| NC | ALIOS | NC | ALIOS | NC | ALIOS | NC | ALIOS | |
| Males, mmol/L | ||||||||
| Total cholesterol | 2.6 ± 0.1 | 3.9 ± 0.2‡ | 2.1 ± 0.1 | 4.1 ± 0.2§ | 2.4 ± 0.1 | 4.6 ± 0.3§ | 2.5 ± 0.2 | 5.8 ± 0.4§ |
| HDL | 1.8 ± 0.1 | 2.7 ± 0.1§ | 1.5 ± 0.1 | 2.9 ± 0.1§ | 1.7 ± 0.1 | 3.1 ± 0.2§ | 1.7 ± 0.1 | 3.7 ± 0.4§ |
| LDL | 0.59 ± 0.02 | 0.9 ± 0.1‡ | 0.43 ± 0.03 | 0.9 ± 0.1§ | 0.6 ± 0.1 | 1.3 ± 0.2§ | 0.5 ± 0.1 | 1.8 ± 0.2§ |
| TAG | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.3 ± 0.1 | 1.1 ± 0.1 | 0.94 ± 0.04 | 0.67 ± 0.04§ |
| Free fatty acids | 1.7 ± 0.1 | 1.5 ± 0.1 | 0.9 ± 0.1 | 0.81 ± 0.05 | 1.9 ± 0.1 | 1.8 ± 0.1 | 0.8 ± 0.1 | 0.67 ± 0.04* |
| Females, mmol/L | ||||||||
| Total cholesterol | 2.0 ± 0.1 | 2.4 ± 0.2* | 2.0 ± 0.1 | 2.8 ± 0.2‡ | 1.7 ± 0.1 | 2.9 ± 0.2‡ | 1.7 ± 0.1 | 3.2 ± 0.4‡ |
| HDL | 1.3 ± 0.1 | 1.5 ± 0.1 | 1.4 ± 0.1 | 2.0 ± 0.1‡ | 1.2 ± 0.1 | 2.1 ± 0.1‡ | 1.2 ± 0.1 | 2.1 ± 0.2‡ |
| LDL | 0.48 ± 0.03 | 0.57 ± 0.04 | 0.45 ± 0.02 | 0.61 ± 0.04‡ | 0.43 ± 0.01 | 0.71 ± 0.08† | 0.36 ± 0.02 | 0.74 ± 0.13‡ |
| TAG | 0.8 ± 0.1 | 1.0 ± 0.1 | 0.65 ± 0.03 | 0.55 ± 0.02* | 0.97 ± 0.02 | 0.71 ± 0.04‡ | 0.81 ± 0.03 | 0.55 ± 0.03† |
| Free fatty acids | 1.7 ± 0.1 | 1.7 ± 0.1 | 0.81 ± 0.04 | 0.64 ± 0.04† | 1.95 ± 0.15 | 1.37 ± 0.07‡ | 0.97 ± 0.06 | 0.69 ± 0.04† |
Values are means ± SE. n = 12–15 Mice in each group. ALIOS, American lifestyle-induced obesity syndrome; TAG, triacylglycerol. Significantly different from normal chow (NC) at same age,
P < 0.05,
P < 0.01,
P < 0.001, and
P < 0.0001.
At 26 wk, kidney size was reduced in both male and female mice, and quadricep weight decreased in female mice only (Supplemental Fig. S1). By 52 wk, male ALIOS-fed mice had decreased heart, quadricep, and testes mass compared with those fed NC, alongside an increase in spleen mass (Supplemental Fig. S1A). Serum creatinine levels were elevated in male ALIOS-fed mice compared with NC controls at 52 wk (Table 2). Female ALIOS-fed mice mirrored the changes in quadricep and spleen mass seen in the males and had a reduction in kidney mass (Supplemental Fig. S1B) and increase in serum creatinine (Table 2).
Table 2.
Circulating serum levels of renal and liver biochemistry
| 16 wk |
25 wk |
37 wk |
52 wk |
|||||
|---|---|---|---|---|---|---|---|---|
| NC | ALIOS | NC | ALIOS | NC | ALIOS | NC | ALIOS | |
| Males | ||||||||
| Creatinine, mg/dL | N/A | 0.69 ± 0.06 | 0.81 ± 0.11 | N/A | 0.67 ± 0.08 | 0.96 ± 0.08* | ||
| ALP, U/L | 76.6 ± 3.3 | 72.6 ± 4.0 | 64.9 ± 2.0 | 75.2 ± 7.6 | 70.1 ± 2.9 | 88.7 ± 12.1 | 66.2 ± 3.1 | 118.5 ± 11.0§ |
| ALT, U/L | 42.1 ± 1.9 | 83.7 ± 23.9 | 33.0 ± 2.6 | 99.9 ± 16.4† | 34.4 ± 2.9 | 238.0 ± 82.4† | 38.0 ± 4.0 | 291.2 ± 47.4§ |
| AST, U/L | 93.7 ± 6.3 | 126.3 ± 22.1 | 69.5 ± 8.1 | 135.9 ± 17.0† | 72.4 ± 7.9 | 250.7 ± 80.5* | 72.4 ± 4.6 | 346.3 ± 50.8§ |
| AST-to-ALT ratio | 2.2 ± 0.1 | 1.7 ± 0.1* | 2.2 ± 0.3 | 1.6 ± 0.2 | 2.2 ± 0.3 | 1.1 ± 0.1† | 2.0 ± 0.1 | 1.2 ± 0.1§ |
| Total bilirubin, µmol/L | 3.7 ± 0.5 | 2.9 ± 0.4 | 1.9 ± 0.1 | 1.8 ± 0.1 | 2.2 ± 0.2 | 2.1 ± 0.1 | 1.9 ± 0.1 | 1.9 ± 0.1 |
| Females | ||||||||
| Creatinine, mg/dL | N/A | 0.62 ± 0.07 | 0.65 ± 0.08 | N/A | 0.64 ± 0.07 | 0.91 ± 0.08 | ||
| ALP, U/L | 138.9 ± 11.3 | 120.9 ± 8.1 | 105.1 ± 3.6 | 108.4 ± 4.4 | 122.7 ± 15.1 | 142.3 ± 9.8 | 130.5 ± 7.5 | 138.5 ± 13.7 |
| ALT, U/L | 43.9 ± 7.2 | 46.7 ± 6.8 | 28.5 ± 1.4 | 136.3 ± 32.8‡ | 33.0 ± 1.7 | 226.3 ± 54.4‡ | 37.7 ± 4.1 | 310.4 ± 41.5‡ |
| AST, U/L | 114.9 ± 20.5 | 125.9 ± 11.0 | 68.3 ± 3.6 | 251.7 ± 47.0‡ | 83.7 ± 6.4 | 338.5 ± 55.4‡ | 101.3 ± 8.6 | 501.3 ± 45.3‡ |
| AST-to-ALT ratio | 2.6 ± 0.1 | 2.8 ± 0.2 | 2.4 ± 0.1 | 2.2 ± 0.2 | 2.5 ± 0.1 | 1.7 ± 0.1‡ | 2.8 ± 0.2 | 1.7 ± 0.1‡ |
| Total bilirubin, µmol/L | 3.2 ± 0.6 | 4.4 ± 0.4 | 1.7 ± 0.1 | 1.9 ± 0.1 | 2.3 ± 0.2 | 2.1 ± 0.2 | 1.7 ± 0.1 | 2.0 ± 0.1* |
Values are means ± SE. n = 12–15 Mice in each group. Creatinine, n = 10 mice in each group. ALIOS, American lifestyle-induced obesity syndrome; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; N/A, serum not analyzed at this time point. Significantly different from normal chow (NC) at same age,
P < 0.05,
P < 0.01,
P < 0.001, and
P < 0.0001.
ALIOS-fed mice have normal glucose tolerance but are insulin resistant.
There was no difference in glucose tolerance in ALIOS-fed male mice throughout the duration of the study (Fig. 2A). In females, glucose tolerance was impaired at 25 and 37 wk in ALIOS mice, although by 49 wk, there was no difference in comparison with NC-fed animals (Fig. 2B). However, the ALIOS diet increased serum insulin levels in response to the glucose bolus, in both male and female mice, compared with NC controls, consistent with the development of insulin resistance (Fig. 2, E and F, and Supplemental Fig. S2).
Fig. 2.

American lifestyle-induced obesity syndrome (ALIOS) diet-fed mice have normal glucose tolerance. Intraperitoneal glucose tolerance tests (ipGTTs) were performed on normal chow (NC)- and ALIOS-fed male (A) and female (B) mice at 15, 25, 37, and 49 wk. ipGTTs are represented as areas under the curve (AUCs) in male (C) and female (D) NC- and ALIOS-fed mice at 15, 25, 37, and 49 wk (w). AUCs of serum insulin collected at t = 0, 60, and 120 min during the ipGTT in male (E) and female (F) mice are shown. Data are expressed as means ± SE. n = 7 mice in each group at each time point; *P < 0.05, **P < 0.01.
Further evidence of insulin resistance was obtained from insulin tolerance testing; in both male and female ALIOS-fed mice from 37 wk onward, there was an impaired glycemic response to intraperitoneal insulin injection (Fig. 3, A and B, and Supplemental Fig. S3). There was no change in insulin sensitivity at 25 wk in ALIOS-fed mice of either sex. The HOMA-IR was increased in both male and female ALIOS-fed mice from 15 wk onward (Fig. 3, C and D).
Fig. 3.

Male and female American lifestyle-induced obesity syndrome (ALIOS) diet-fed mice are insulin resistant. Intraperitoneal insulin tolerance tests (ipITTs) are represented as areas under the curve (AUCs) in male (A) and female (B) normal chow (NC)- and ALIOS-fed mice at 25, 37, and 50 wk (w). Homeostatic model assessment of insulin resistance (HOMA-IR) of male (C) and female (D) ALIOS- and NC-fed mice at 15, 25, 37, and 49 wk. Data are expressed as means ± SE. n = 7 Mice in each group at each time point; *P < 0.05, **P < 0.01, ***P < 0.001.
ALIOS drives hepatic steatosis and inflammation.
Liver to body weight mass was increased in both male and female ALIOS-fed mice compared with NC at 26 and 52 wk (Fig. 4, A and B). Additionally, hepatic triglyceride was elevated in ALIOS-fed mice of both sexes at 26 wk and in male mice at 52 wk (Fig. 4, C and D). Histological examination of all ALIOS livers identified steatosis, with evidence of macro- and microvesicular lipid droplets and ballooning of hepatocytes (Fig. 4, E and F). ALIOS-fed male and female mice had altered expression of genes associated with lipid metabolism (Srebf1, Elovl3, and Lpl; Fig. 4, G and H) and insulin signaling (Irs1, G6pc, Glut4, and Pck1; Fig. 4, I and J).
Fig. 4.

American lifestyle-induced obesity syndrome (ALIOS) diet drives hepatic steatosis. Liver mass of male (A) and female (B) and hepatic triacylglycerol (TAG) content in male (C) and female (D) normal chow (NC)- and ALIOS-fed mice at 26 and 52 wk (w; 26 wk: NC males, n = 12; ALIOS males, n = 15; NC females, n = 15; ALIOS females, n = 15; 52 wk: NC males, n = 17; ALIOS males, n = 12; NC females, n = 15; ALIOS females, n = 15) are shown. Representative hematoxylin-eosin images are from male mice at 52 wk fed NC (E) and ALIOS (F) depicting macro- and microvesicular lipid droplets. Data are expressed as means ± SE. Scale bar is 25 µm. ALIOS-fed male and female mice had altered expression of genes associated with lipid metabolism (G, males; H, females) and insulin signaling (I, males; J, females). n = 8 Mice in each group. Genes are expressed as fold changes in log10 compared with NC. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. BW, body weight.
At 26 wk, male ALIOS-fed mice displayed no change in histologically determined inflammation score; however, this was increased by 52 wk (Fig. 5A). In contrast, female mice fed ALIOS had an increased inflammation score from 26 wk that persisted to 52 wk (Fig. 5B). ALIOS-fed male and female mice had increased hepatic expression of genes involved in inflammation, including macrophage infiltration (Tnf, Ccl2, Cd68, and f4/80; Fig. 5, C and D), compared with NC-fed mice at 52 wk.
Fig. 5.

Mice fed American lifestyle-induced obesity syndrome (ALIOS) diet have increased hepatic inflammation. Histologically determined inflammation scores of male (A) and female (B) normal chow (NC)- and ALIOS-fed mice at 26 wk (w; NC males, n = 12; ALIOS males, n = 15; NC females, n = 15; ALIOS females, n = 15) and 52 wk (NC males, n = 17; ALIOS males, n = 12; NC females, n = 15; ALIOS females, n = 15) are shown. Data are expressed as means ± SE. Both male (C) and female (D) mice had increased hepatic expression of genes involved in inflammation, including macrophage infiltration compared with NC-fed mice at 52 wk. n = 8 Mice in each group; *P < 0.05, ***P < 0.001, ****P < 0.0001.
Male ALIOS-fed mice have increased incidence of fibrosis and HCCs.
ALIOS-fed mice of both sexes had elevated serum levels of ALT and AST from 26 wk onward and continued to rise throughout the duration of the study, suggesting hepatocyte damage and the presence of fibrosis (Table 2); the AST-to-ALT ratio was decreased in ALIOS-fed mice compared with controls, which is suggestive of NAFLD. Serum levels of bilirubin, however, were unchanged in ALIOS-fed mice compared with NC controls (Table 2). Hepatic fibrosis percentage was increased in male ALIOS-fed mice compared with NC controls at 26 and 52 wk (Fig. 6A), whereas in female ALIOS-fed mice percentage fibrosis was only increased at 52 wk (Fig. 6B). Both ALIOS-fed male and female mice had increased hepatic expression of genes associated with cell adhesion (Col1a1, Col1a2, Dpt, and Lum; Fig. 6, C and D) as well as increased protein levels of Col1a1 (Fig. 6, E and F).
Fig. 6.

American lifestyle-induced obesity syndrome (ALIOS) diet drives hepatic fibrosis in aged mice. Percentage fibrosis determined by Sirius Red staining in male (A) and female (B) normal chow (NC)- and ALIOS-fed mice at 26 wk (w; NC males, n = 12; ALIOS males, n = 15; NC females, n = 15; ALIOS females, n = 15) and 52 wk (NC males, n = 17; ALIOS males, n = 12; NC females, n = 15; ALIOS females, n = 15) and hepatic expression of genes associated with cell adhesion in male (C) and female (D) ALIOS-fed mice (n = 8 mice in each group; genes are expressed as fold changes in log10 compared with NC) are shown. Western blotting of Col1a1 in male (E) and female (F) NC- and ALIOS-fed mice at 52 wk (n = 10 mice in each group) was performed. Data are expressed as means ± SE; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Representative images depict Sirius Red staining in male NC (G) and ALIOS (H) mice at 52 wk. I: hematoxylin-eosin staining of a potential hepatocellular carcinoma (HCC) from a male ALIOS-fed mouse at 52 wk (T: tumor) highlighting (J) the compressed border of cells (asterisk) and multinucleic cells (arrow). An example is given of positive Sox9 (K) and glutamine synthetase (L) labeling in an HCC from an ALIOS-fed male mouse at 52 wk.
Advanced fibrotic disease increases the risk of HCC; male mice fed ALIOS had an increased frequency of macroscopic liver growths (NC: 5.8%, ALIOS: 25%; P < 0.05). There was no evidence of liver lesions in female ALIOS-fed mice. The lesions were associated with compressed adjacent nontumor tissue, although there appeared to be no invasion of blood vessels or surrounding liver tissue (Fig. 6I). Histological assessment revealed that the lesions were composed of atypical hepatocytes with increased nuclear-to-cytoplasmic ratios as well as some multinucleated cells (Fig. 6J). As there was no obvious evidence of invasion to confirm malignancy, characterization was performed using HCC markers commonly used in mice and humans. Sox9 is a marker of hepatic stem cell activation previously implicated in tumor pathogenesis (18, 37), and labeling was positive in the nucleus of the atypical hepatocytes of only 1/3 lesions (Fig. 6K). Glutamine synthetase (GS) has previously been used to determine well-differentiated HCCs from premalignant tumors in human liver (10, 37, 49). Diffuse GS was also only present in 2/3 ALIOS lesions (Fig. 6L).
ALIOS diet alters the hepatic transcriptome in male and female mice.
Clustering of NC and ALIOS of the top 100 DEGs revealed 2 distinct groups between NC and ALIOS with almost no overlapping (Fig. 7, A and B). In male mice, 5,018 genes were differentially expressed between NC and ALIOS: 2,153 were downregulated and 2,865 were upregulated (Table 3). Within the top 10 upregulated genes, 3 were associated with the major histocompatibility complex (MHC). Gene ontology analysis highlighted that the most upregulated genes were associated with reorganization of cellular structures and collagen binding as well as inflammatory and immune response (Fig. 7C). The downregulated genes were mostly clustered to biological pathways involved in metabolism and protein processing (Fig. 7D). In line with these findings, the top diseases and functions captured in IPA included cancer, organismal injury, and endocrine disorders (Table 4). The top toxicology lists by IPA included liver necrosis and hepatic fibrosis as well as nuclear factor erythroid 2-related factor 2-mediated oxidative stress response.
Fig. 7.

Hierarchical clustering analysis of the top 100 differentially expressed genes from male (A) and female (B) normal chow (NC)- and American lifestyle-induced obesity syndrome (ALIOS) diet-fed mice at 52 wk. Data are presented in a heat-map format in which NC and ALIOS are separated into columns and genes are in rows. Red corresponds to genes that are upregulated in ALIOS compared with NC, and blue corresponds to those that are downregulated. Enriched gene pathways are of ALIOS-fed mice. Upregulated (C) and downregulated (D) gene ontology pathways in male mice and upregulated (E) and downregulated (F) pathways in females are shown. Pathways are ranked by P values.
Table 3.
The 10 most up- and downregulated genes in NC vs. ALIOS liver tissue at 52 wk
| Gene | Description | Log Fold | FDR |
|---|---|---|---|
| Males | |||
| Clec7a | C-type lectin domain family 7 | 2.3128 | 7.98 × 10−33 |
| Mmp12 | Matrix metallopeptidase 12 | 4.1723 | 7.04 × 10−31 |
| H2-Ab1 | Histocompatibility 2, class II antigen A, beta 1 | 1.9017 | 4.10 × 10−29 |
| Cx3cr1 | Chemokine (C-X3-C motif) receptor 1 | 2.8669 | 1.24 × 10−28 |
| H2-Aa | Histocompatibility 2, class II antigen A, alpha | 2.1164 | 2.10 × 10−28 |
| Tmem86a | Transmembrane protein 86A | 1.7757 | 4.03 × 10−28 |
| Col1a1 | Collagen, type I, alpha 1 | 3.5204 | 1.72 × 10−26 |
| Ephb2 | Eph receptor B2 | 4.7443 | 2.27 × 10−25 |
| Cd63 | CD63 antigen | 3.0092 | 3.45 × 10−25 |
| H2-Eb1 | Histocompatibility 2, class II antigen E beta | 1.9343 | 1.11 × 10−24 |
| Ces2a | Carboxylesterase 2A | −1.9190 | 7.04 × 10−31 |
| Ces1b | Carboxylesterase 1B | −1.3651 | 6.89 × 10−23 |
| Tnfaip8l1 | Tumor necrosis factor, alpha-induced protein 8-like 1 | −1.0119 | 1.99 × 10−17 |
| Scarb2 | Scavenger receptor class B, member 2 | −0.7034 | 3.34 × 10−14 |
| Retsat | Retinol saturase (all trans retinol 13,14 reductase) | −1.0527 | 6.13 × 10−14 |
| Marf1 | Meiosis regulator and mRNA stability 1 | −0.7292 | 2.89 × 10−13 |
| Tuba4a | Tubulin, alpha 4A | −0.9631 | 3.80 × 10−13 |
| Hectd1 | HECT domain E3 ubiquitin protein ligase 1 | −0.6121 | 9.50 × 10−13 |
| Angptl4 | Angiopoietin-like 4 | −1.0041 | 1.27 × 10−12 |
| Pxmp4 | Peroxisomal membrane protein 4 | −0.9919 | 1.46 × 10−12 |
| Females | |||
| Wfdc2 | WAP four-disulfide core domain 2 | 2.6371 | 1.97 × 10−40 |
| Uap1l1 | UDP-N-acetylglucosamine pyrophosphorylase 1-like 1 | 2.6742 | 6.04 × 10−39 |
| Ifi27l2b | Interferon, alpha-inducible protein 27 like 2B | 2.8447 | 2.95 × 10−37 |
| Ly6d | Lymphocyte antigen 6 complex, locus D | 3.6763 | 7.04 × 10−29 |
| Clec7a | C-type lectin domain family 7, member a | 2.4672 | 4.90 × 10−28 |
| Mmp12 | Matrix metallopeptidase 12 | 5.0590 | 5.69 × 10−28 |
| Ms4a6d | Membrane-spanning 4-domains, subfamily A, member 6D | 2.0780 | 7.87 × 10−28 |
| Hcar2 | Hydroxycarboxylic acid receptor 2 | 2.5398 | 1.12 × 10−27 |
| Lpl | Lipoprotein lipase | 2.5220 | 1.16 × 10−27 |
| Osbpl3 | Oxysterol binding protein-like 3 | 3.5456 | 2.10 × 10−27 |
| Abhd6 | Abhydrolase domain containing 6 | −1.1633 | 6.92 × 10−27 |
| Gm3787 | Predicted gene 3787* | −2.6100 | 1.97 × 10−22 |
| Ces2a | Carboxylesterase 2A | −1.4585 | 6.56 × 10−19 |
| Mttp | Microsomal triglyceride transfer protein | −0.8436 | 2.59 × 10−17 |
| Cyp2c23 | Cytochrome P-450, family 2, subfamily c, polypeptide 23 | −1.6328 | 1.10 × 10−16 |
| Avpr1a | Arginine vasopressin receptor 1A | −1.5448 | 2.88 × 10−16 |
| Fam234b | Family with sequence similarity 234, member B | −1.2050 | 9.72 × 10−16 |
| Sult5a1 | Sulfotransferase family 5A, member 1 | −2.6802 | 1.79 × 10−15 |
| Cyp4a10 | Cytochrome P-450, family 4, subfamily a, polypeptide 10 | −1.2606 | 2.61 × 10−14 |
| Sult3a2 | Sulfotransferase family 3A, member 2 | −3.7032 | 9.57 × 10−14 |
ALIOS, American lifestyle-induced obesity syndrome; FDR, false discovery rate; NC, normal chow.
Function of predicted genes unknown in current annotation.
Table 4.
IPA of differentiated genes in male and female ALIOS-fed mice at 52 wk
| Biological Function | P Value | Genes, n |
|---|---|---|
| Males | ||
| Diseases and disorders | ||
| Cancer | 1.04 × 10−14 to 1.69 × 10−88 | 4,093 |
| Organismal injury and abnormalities | 1.04 × 10−14 to 1.69 × 10−88 | 4,206 |
| Endocrine system disorders | 1.03 × 10−27 to 2.92 × 10−70 | 3,316 |
| Gastrointestinal disease | 6.12 × 10−15 to 9.14 × 10−57 | 3,682 |
| Inflammatory response | 9.02 × 10−15 to 7.51 × 10−54 | 1,364 |
| Molecular and cellular functions | ||
| Cell death and survival | 7.61 × 10−15 to 2.49 × 10−72 | 1,622 |
| Cellular movement | 7.21 × 10−15 to 6.10 × 10−68 | 1,216 |
| Cellular compromise | 5.16 × 10−19 to 7.51 × 10−54 | 354 |
| Cell-to-cell signaling and interaction | 1.07 × 10−14 to 6.98 × 10−41 | 953 |
| Lipid metabolism | 1.10 × 10−14 to 1.40 × 10−39 | 797 |
| Top toxicology list | Overlap (ratio) | |
| NRF2-mediated oxidative stress response | 2.83 × 10−15 | 42.1% (101/240) |
| Liver necrosis/cell death | 6.98 × 10−15 | 38.5% (124/322) |
| Renal necrosis/cell death | 1.40 × 10−14 | 33.3% (191/573) |
| Hepatic fibrosis | 1.41 × 10−13 | 51.4% (57/111) |
| Xenobiotic metabolism signaling | 1.19 × 10−11 | 35.2% (123/349) |
| Females | ||
| Diseases and disorders | ||
| Cancer | 8.17 × 10−15 to 2.35 × 10−78 | 3,434 |
| Organismal injury and abnormalities | 1.86 × 10−14 to 2.35 × 10−78 | 3,528 |
| Endocrine system disorders | 1.94 × 10−36 to 3.14 × 10−64 | 2,815 |
| Inflammatory response | 1.29 × 10−14 to 7.07 × 10−56 | 1,187 |
| Gastrointestinal disease | 1.09 × 10−14 to 3.97 × 10−51 | 3,122 |
| Molecular and cellular functions | ||
| Cell death and survival | 1.35 × 10−14 to 7.31 × 10−58 | 1,375 |
| Cellular movement | 1.32 × 10−14 to 2.23 × 10−55 | 999 |
| Cellular compromise | 1.35 × 10−14 to 4.09 × 10−55 | 374 |
| Cellular function and maintenance | 2.32 × 10−15 to 1.81 × 10−50 | 1,237 |
| Cell-to-cell signaling and interaction | 1.75 × 10−14 to 1.79 × 10−41 | 707 |
| Top toxicology list | Overlap (ratio) | |
| Hepatic fibrosis | 2.00 × 10−16 | 50.5% (56/111) |
| Renal necrosis/cell death | 5.35 × 10−13 | 28.4% (163/573) |
| Liver necrosis/cell death | 1.11 × 10−12 | 32.6% (105/322) |
| Increases liver steatosis | 3.83 × 10−12 | 45.0% (49/109) |
Data represent the number of genes up- or downregulated in American lifestyle-induced obesity syndrome (ALIOS) diet-fed mice at 52 wk. Biological functions, molecular functions, and top toxicology lists were assigned using findings extracted from literature and stored in Ingenuity Pathway Analysis (IPA). P values were determined by IPA software using Fisher exact test and determine the probability that the pathway or function assigned is explained by chance alone. The percentage overlap and ratio were calculated from the number of observed genes compared with the number of known genes for that category in the Ingenuity Knowledge Base. NRF2, nuclear factor erythroid 2-related factor 2.
In livers of ALIOS-fed female mice, a total of 4,221 genes showed differential expression: 2,350 genes were up- and 1,871 were downregulated (Table 3). The most upregulated genes were associated exclusively with inflammatory processes (Fig. 7E), whereas downregulated genes, as in male mice, clustered to metabolic processes (Fig. 7F). Similarly, the top diseases and functions identified by IPA included cancer, injury, and endocrine disorders (Table 4). The top toxicology lists as determined by IPA included hepatic fibrosis, liver necrosis, and steatosis.
To identify DEGs associated with NASH in humans, published RNA-Seq data from liver biopsies of patients with NASH were reanalyzed alongside biopsies from patients with normal body weight. A total of 4,558 human DEGs were identified. Comparative analysis revealed that 2,052 (22.5%) genes were shared by the human NASH patients and male ALIOS-fed mice (Fig. 8A). Out of the 2,052 common genes, 1,018 were upregulated, and 1,034 were downregulated. The clustering of the top 30 overlapped genes revealed similarities between livers of ALIOS-fed mice and human NASH liver biopsies (Fig. 8B). Genes associated with lipid metabolism (Fig. 8C; LPL), insulin signaling (G6PC and PCK1), inflammation (TNFA1P3, CCL2, and CXCR4), and cell adhesion (DPT, LUM, and COL1A1) were among those that were altered in both human NASH and ALIOS-fed male mice. Additionally, gene ontology analysis revealed that the top 100 shared genes were enriched in pathways associated with immune response, metabolic processes, and cell migration (Fig. 8D), confirming that most of the genes and pathways conserved between the ALIOS mouse model and human NASH are associated with inflammation and fibrosis.
Fig. 8.

A: American lifestyle-induced obesity syndrome (ALIOS) diet-fed male mice and human patients with nonalcoholic steatohepatitis (NASH) share 22.5% of differentially expressed genes (DEGs). Numbers in Venn diagram represent genes. B: hierarchical clustering analysis of the top 30 DEGs from human NASH and ALIOS-fed male mice. Values are z-scores. Overlapping DEGs are associated with metabolism, inflammation, and cell adhesion [genes are expressed as fold changes in log10 compared with normal chow (NC); C], and gene ontology of the top 100 overlapping DEGs shows associations with alterations in metabolic pathways, immune response, and cell adhesion (D). Pathways are ranked by P values.
DISCUSSION
NAFLD is rapidly becoming the most common cause for liver transplantation. However, there are aspects of its pathology that remain poorly understood, and informative preclinical models that accurately reflect the clinical condition, particularly its natural history and progression, can provide valuable mechanistic insight. In this study, we have shown that male and female mice fed the ALIOS diet for 52 wk develop a classic and reproducible hepatic phenotype (including elevated liver chemistry, histological features of NASH, and the development of HCC). However, we also have shown that they develop many of the extrahepatic features associated with NAFLD, including increased fat mass, sedentary behavior, abnormal circulating lipid profiles, insulin resistance, and sarcopenia. Kidney weights were reduced and serum creatinine levels were elevated in ALIOS-fed mice. Recent reports have highlighted the association between NAFLD and renal dysfunction in humans (32, 44). Furthermore, we have highlighted the differential gene expressions in ALIOS-fed mice compared with NC controls that may prove highly informative in enhancing our understanding of the pathogenesis of NAFLD.
Male and female ALIOS-fed mice had increased body weight from as early as 11 wk, driven by increased fat mass. Of note, body weights in female ALIOS-fed mice continue to increase at 52 wk and do not plateau, contrasting with male ALIOS-fed mice. It is interesting to speculate that this sexually dimorphic trajectory might extend to the liver in that the females might also develop HCCs as seen in the males, if left for >1 yr. Unfortunately, we did not extend our observations beyond 1 yr in either sex. Detailed body composition analysis has not been undertaken in this model previously, and we have been able to show increased fat depot mass, with additional evidence for reduced skeletal muscle (quadricep mass). This finding mirrors the sarcopenia that is associated with NAFLD (8); Koo et al. (21) reported that sarcopenia was present in 17.9 and 35% of patients with NAFLD and NASH, respectively, and was associated with significant fibrosis and insulin resistance (21). Previous studies have demonstrated abnormal glucose handling and insulin resistance in mice fed the ALIOS diet (10, 46), and we have now shown that this persists throughout the duration of the intervention (at least to 52 wk). Total (including both LDL and HDL) cholesterol became elevated soon after the commencement of the diet, although circulating triglyceride levels were lower in both male and female mice on the ALIOS diet at 52 wk. It is possible that this may reflect impaired hepatic lipid export, therefore, contributing to increased hepatic triglyceride accumulation.
The liver phenotype that we have observed is similar to that which has been described previously (10, 46), and we have now extended the detailed phenotyping to 52-wk duration. At 26 wk, steatosis is predominately periportal, but, by 52 wk, macro- and microvesicle steatosis has extended to the centrilobular region. The development of microvesicular steatosis may be linked to increased disease progression; in humans, microvesicular steatosis from liver biopsies correlated positively with increased NASH diagnosis and advanced fibrosis (45). By 52 wk, there was clear evidence of hepatic fibrosis in both male and female mice. In male mice, the ALIOS diet was associated with an increased incidence of liver tumors as we (10) have shown previously. However, female mice appeared to be completely protected from this. Male predisposition to HCC is well described (15, 17, 35), and in this regard, the ALIOS model appears to replicate clinical findings. The atypical hepatocytes seen in the liver lesions from ALIOS-fed male mice are a recognized feature of human hepatic tumors. Unfortunately, there is no established panel of murine HCC markers that are comparable with human (40); the use of immunohistochemical markers was variable across different lesions from different mice. Glutamine synthetase is a target gene of β-catenin, and its overexpression is associated with mutations of β-catenin and/or activation of its pathway. In mice, hepatocellular tumors express differing levels of GS depending on the type of mutation within the neoplasm (40).
Unbiased transcriptomic analysis provides a powerful tool with which to interrogate the processes that drive NAFLD to the more advanced disease stages. Our analysis demonstrated increased mRNA expression of proinflammatory cytokines (Tnf, Ccl2, and Ccl3) in male and female ALIOS-fed mice as well as markers of macrophage (Cd68, Cd40, and F4/80) and Kupffer cell infiltration. Endorsing these observations, a direct comparison of the transcriptome from ALIOS-fed male mice with publicly available RNA-Seq data from biopsies of patients with NASH highlights a significant overlap of genes associated with NAFLD and NASH. Moylan et al. (33) reported a 64-gene profile of upregulated genes in severe NAFLD in humans, which included genes associated with inflammation, cell adhesion, and liver progenitor cells. These pathways are believed to be crucial in the progression from steatosis to NASH as well as to the development of HCC (11, 12), further validating the ALIOS diet as a good model of the full NAFL spectrum of disease. In addition, the top pathways highlighted from IPA mirror those seen in patients with severe NAFLD (33), including cancer.
Of the most upregulated genes in ALIOS-fed mice, two are common between males and females, and both genes have key roles in driving the inflammatory phenotype. Clec7a encodes membrane receptors that play a role in the innate immune response. Activation of Clec7a leads to production of the transcription factor NF-κβ (14), which induces synthesis of inflammatory cytokines such as TNF, IL-6, or IL-2 (6), suggesting that Clec7a is key contributor to the inflammatory profile of the liver in NAFLD. Mmp12 is predominately expressed by macrophages, and in human adipose tissue, Mmp12 expression correlates positively with macrophage infiltration, inflammation, and insulin resistance (26). Mmp12 expression also correlates positively with arterial stiffening in mice (27), suggesting that the hepatic macrophage filtration in ALIOS-fed mice further drives progression of the disease. Among the most strongly downregulated genes in ALIOS-fed mice was Ces2A, encoding a hepatic serine hydrolase. In humans and mice, obesity decreases the activity of Ces2, which leads to hepatic dyslipidemia (39). Indeed, normal expression of Ces2 contributes to suppression of hepatic inflammation, improving adiposity and glucose tolerance (39); downregulation in the ALIOS-fed mice may, therefore, be a key driver of NAFLD progression. In males, three genes associated with MHC class II were also upregulated. Previous reports have indicated disease susceptibility is strongly influenced by the MHC class II pathway; increased expression of MHC class II-related genes is associated with increased hepatic fibrosis in response to toxic insults and hepatocyte damage (4, 42), suggesting the progression of NASH may be due to antigen presentation through MHC, particularly in males.
Previous studies using fast food diets have also highlighted the characteristics of NASH (7, 22). However, these diets do not contain trans fats, which Tetri et al. (46) suggested are the main driver of hepatic injury to promote fibrotic disease and its potential progression to HCCs. The use of trans fats has recently been phased out from the food industry, due to their impact on metabolic disease. The ALIOS diet aimed to generate a rodent model that replicated the clinical characteristics of human NAFLD and NASH, and, therefore, trans fats were used to drive an adverse liver phenotype rather than recapitulate current human diets.
Previous fast-food-diet studies have focused primarily on the hepatic phenotype and have also detailed mitochondrial dysfunction in NASH. The ALIOS study did not include a fructose-only cohort as the aim was to fully characterize a diet that induces steatosis, inflammation, and fibrosis. The role of fructose in hepatic lipid accumulation has been well characterized; however, recent studies using fructose-only interventions have previously failed to induce hepatic steatosis and inflammation (28, 41). The combination of adverse diets and different genetic backgrounds have also made substantial contributions to preclinical NASH models (3, 11, 50). The ALIOS model has investigated a dietary driver of NAFLD and NASH, and it is plausible this may behave differently on different genetic backgrounds, but this is beyond the scope of this study. Additionally, previous preclinical NASH mouse models have primarily neglected to analyze females and their response to these altered diets. The current ALIOS study has extended the comprehensive metabolic analysis to 1 yr and detailed the extrahepatic phenotype of the syndrome, as well as full transcriptomic analysis, in both male and female mice.
In conclusion, we have provided the most comprehensive, longitudinal assessment of the ALIOS diet, both with regard to its hepatic phenotype and its extrahepatic manifestations. The ALIOS diet closely recapitulates many of the features of clinical NAFLD, and our transcriptomic analysis has revealed many common pathways that are shared between clinical samples and the ALIOS intervention. The ALIOS model, therefore, represents a robust and reproducible tool to further understand the complex nature of NAFLD and its progression to the most advanced stages, including NASH and HCC.
GRANTS
This study was supported by Medical Research Council (programme grant to J. W. Tomlinson, ref. MR/P011462/1; project grant to R. D. Cox, ref. MC_U142661184), National Institute for Health Research Oxford Biomedical Research Centre (principal investigator award to J. W. Tomlinson), and an Oxford Brookes University Nigel Groome PhD Studentship (studentship award to A. Arvaniti, principle investigator L. L. Gathercole).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.E.H. and J.W.T. conceived and designed research; S.E.H., A.A., and L.L.G. performed experiments; S.E.H. and T.M.P. analyzed data; S.E.H., T.M.P., and J.W.T. interpreted results of experiments; S.E.H. prepared figures; S.E.H. drafted manuscript; S.E.H., R.D.C., L.L.G., and J.W.T. edited and revised manuscript; S.E.H., T.M.P., A.A., R.D.C., L.L.G., and J.W.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Phenotyping team at the Mary Lyon Centre, Medical Research Council Harwell Institute for the breeding and husbandry of the mice and for conducting the phenotyping assessments. We also thank the Oxford Genomics Centre at the Wellcome Centre for Human Genetics (funded by Wellcome Trust Grant Reference 203141/Z/16/Z) for the generation and initial processing of sequencing data.
REFERENCES
- 1.Abe N, Kato S, Tsuchida T, Sugimoto K, Saito R, Verschuren L, Kleemann R, Oka K. Longitudinal characterization of diet-induced genetic murine models of non-alcoholic steatohepatitis with metabolic, histological, and transcriptomic hallmarks of human patients. Biol Open 8: bio041251, 2019. doi: 10.1242/bio.041251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong MJ, Houlihan DD, Bentham L, Shaw JC, Cramb R, Olliff S, Gill PS, Neuberger JM, Lilford RJ, Newsome PN. Presence and severity of non-alcoholic fatty liver disease in a large prospective primary care cohort. J Hepatol 56: 234–240, 2012. doi: 10.1016/j.jhep.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 3.Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min HK, Mirshahi F, Bedossa P, Sun X, Hoshida Y, Koduru SV, Contaifer D Jr, Warncke UO, Wijesinghe DS, Sanyal AJ. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 65: 579–588, 2016. doi: 10.1016/j.jhep.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba Y, Doi K. MHC class II-related genes expression in porcine-serum-induced rat hepatic fibrosis. Exp Mol Pathol 77: 214–221, 2004. doi: 10.1016/j.yexmp.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Ballestri S, Mantovani A, Nascimbeni F, Lugari S, Lonardo A. Extra-hepatic manifestations and complications of nonalcoholic fatty liver disease. Future Med Chem 11: 2171–2192, 2019. doi: 10.4155/fmc-2019-0003. [DOI] [PubMed] [Google Scholar]
- 6.Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol 6: 33–43, 2006. doi: 10.1038/nri1745. [DOI] [PubMed] [Google Scholar]
- 7.Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, Gores G. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 301: G825–G834, 2011. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung GE, Kim MJ, Yim JY, Kim JS, Yoon JW. Sarcopenia is significantly associated with presence and severity of nonalcoholic fatty liver disease. J Obes Metab Syndr 28: 129–138, 2019. doi: 10.7570/jomes.2019.28.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dowman JK, Hopkins LJ, Reynolds GM, Armstrong MJ, Nasiri M, Nikolaou N, van Houten EL, Visser JA, Morgan SA, Lavery GG, Oprescu A, Hübscher SG, Newsome PN, Tomlinson JW. Loss of 5α-reductase type 1 accelerates the development of hepatic steatosis but protects against hepatocellular carcinoma in male mice. Endocrinology 154: 4536–4547, 2013. doi: 10.1210/en.2013-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowman JK, Hopkins LJ, Reynolds GM, Nikolaou N, Armstrong MJ, Shaw JC, Houlihan DD, Lalor PF, Tomlinson JW, Hübscher SG, Newsome PN. Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high-fat/fructose diet and sedentary lifestyle. Am J Pathol 184: 1550–1561, 2014. doi: 10.1016/j.ajpath.2014.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical models for studying NASH-driven HCC: how useful are they? Cell Metab 29: 18–26, 2019. doi: 10.1016/j.cmet.2018.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Font-Burgada J, Sun B, Karin M. Obesity and cancer: the oil that feeds the flame. Cell Metab 23: 48–62, 2016. doi: 10.1016/j.cmet.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 13.Fraulob JC, Ogg-Diamantino R, Fernandes-Santos C, Aguila MB, Mandarim-de-Lacerda CA. A mouse model of metabolic syndrome: insulin resistance, fatty liver and non-alcoholic fatty pancreas disease (NAFPD) in C57BL/6 mice fed a high fat diet. J Clin Biochem Nutr 46: 212–223, 2010. doi: 10.3164/jcbn.09-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med 197: 1107–1117, 2003. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ioannou GN, Green P, Lowy E, Mun EJ, Berry K. Differences in hepatocellular carcinoma risk, predictors and trends over time according to etiology of cirrhosis. PLoS One 13: e0204412, 2018. doi: 10.1371/journal.pone.0204412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kammoun HL, Allen TL, Henstridge DC, Kraakman MJ, Peijs L, Rose-John S, Febbraio MA. Over-expressing the soluble gp130-Fc does not ameliorate methionine and choline deficient diet-induced non alcoholic steatohepatitis in mice. PLoS One 12: e0179099, 2017. doi: 10.1371/journal.pone.0179099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, Li L, Desiderio R, Thrift AP, Asch SM, Chu J, El-Serag HB. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 155: 1828–1837.e2, 2018. doi: 10.1053/j.gastro.2018.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawai T, Yasuchika K, Ishii T, Miyauchi Y, Kojima H, Yamaoka R, Katayama H, Yoshitoshi EY, Ogiso S, Kita S, Yasuda K, Fukumitsu K, Komori J, Hatano E, Kawaguchi Y, Uemoto S. SOX9 is a novel cancer stem cell marker surrogated by osteopontin in human hepatocellular carcinoma. Sci Rep 6: 30489, 2016. doi: 10.1038/srep30489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kishida N, Matsuda S, Itano O, Shinoda M, Kitago M, Yagi H, Abe Y, Hibi T, Masugi Y, Aiura K, Sakamoto M, Kitagawa Y. Development of a novel mouse model of hepatocellular carcinoma with nonalcoholic steatohepatitis using a high-fat, choline-deficient diet and intraperitoneal injection of diethylnitrosamine. BMC Gastroenterol 16: 61, 2016. doi: 10.1186/s12876-016-0477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321, 2005. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 21.Koo BK, Kim D, Joo SK, Kim JH, Chang MS, Kim BG, Lee KL, Kim W. Sarcopenia is an independent risk factor for non-alcoholic steatohepatitis and significant fibrosis. J Hepatol 66: 123–131, 2017. doi: 10.1016/j.jhep.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 22.Krishnan A, Abdullah TS, Mounajjed T, Hartono S, McConico A, White T, LeBrasseur N, Lanza I, Nair S, Gores G, Charlton M. A longitudinal study of whole body, tissue, and cellular physiology in a mouse model of fibrosing NASH with high fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 312: G666–G680, 2017. doi: 10.1152/ajpgi.00213.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larner DP, Morgan SA, Gathercole LL, Doig CL, Guest P, Weston C, Hazeldine J, Tomlinson JW, Stewart PM, Lavery GG. Male 11β-HSD1 knockout mice fed trans-fats and fructose are not protected from metabolic syndrome or nonalcoholic fatty liver disease. Endocrinology 157: 3493–3504, 2016. doi: 10.1210/en.2016-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E, Koteish A, Brancati FL, Clark JM. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol 178: 38–45, 2013. doi: 10.1093/aje/kws448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le MH, Devaki P, Ha NB, Jun DW, Te HS, Cheung RC, Nguyen MH. Prevalence of non-alcoholic fatty liver disease and risk factors for advanced fibrosis and mortality in the United States. PLoS One 12: e0173499, 2017. doi: 10.1371/journal.pone.0173499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JT, Pamir N, Liu NC, Kirk EA, Averill MM, Becker L, Larson I, Hagman DK, Foster-Schubert KE, van Yserloo B, Bornfeldt KE, LeBoeuf RC, Kratz M, Heinecke JW. Macrophage metalloelastase (MMP12) regulates adipose tissue expansion, insulin sensitivity, and expression of inducible nitric oxide synthase. Endocrinology 155: 3409–3420, 2014. doi: 10.1210/en.2014-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu SL, Bae YH, Yu C, Monslow J, Hawthorne EA, Castagnino P, Branchetti E, Ferrari G, Damrauer SM, Puré E, Assoian RK. Matrix metalloproteinase-12 is an essential mediator of acute and chronic arterial stiffening. Sci Rep 5: 17189, 2015. doi: 10.1038/srep17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu XJ, Duan NN, Liu C, Niu C, Liu XP, Wu J. Characterization of a murine nonalcoholic steatohepatitis model induced by high fat high calorie diet plus fructose and glucose in drinking water. Lab Invest 98: 1184–1199, 2018. doi: 10.1038/s41374-018-0074-z. [DOI] [PubMed] [Google Scholar]
- 29.Lo L, McLennan SV, Williams PF, Bonner J, Chowdhury S, McCaughan GW, Gorrell MD, Yue DK, Twigg SM. Diabetes is a progression factor for hepatic fibrosis in a high fat fed mouse obesity model of non-alcoholic steatohepatitis. J Hepatol 55: 435–444, 2011. doi: 10.1016/j.jhep.2010.10.039. [DOI] [PubMed] [Google Scholar]
- 30.Love MI, Anders S, Kim V, Huber W. RNA-Seq workflow: gene-level exploratory analysis and differential expression. F1000 Res 4: 1070, 2015. doi: 10.12688/f1000research.7035.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res 21: 936–939, 2011. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantovani A, Turino T, Lando MG, Gjini K, Byrne CD, Zusi C, Ravaioli F, Colecchia A, Maffeis C, Salvagno G, Lippi G, Bonora E, Targher G. Screening for non-alcoholic fatty liver disease using liver stiffness measurement and its association with chronic kidney disease and cardiovascular complications in patients with type 2 diabetes. Diabetes Metab S1262-3636(19)30184-3, 2019. doi: 10.1016/j.diabet.2019.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, Murphy SK, Ashley-Koch AE, Choi SS, Michelotti GA, Hampton DD, Chen Y, Tillmann HL, Hauser MA, Abdelmalek MF, Diehl AM. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 59: 471–482, 2014. doi: 10.1002/hep.26661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, Koike K, Kaufman RJ, Karin M. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26: 331–343, 2014. doi: 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Natri HM, Wilson MA, Buetow KH. Distinct molecular etiologies of male and female hepatocellular carcinoma. BMC Cancer 19: 951, 2019. doi: 10.1186/s12885-019-6167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140: 197–208, 2010. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park YN. Update on precursor and early lesions of hepatocellular carcinomas. Arch Pathol Lab Med 135: 704–715, 2011. doi: 10.1043/2010-0524-RA.1. [DOI] [PubMed] [Google Scholar]
- 38.Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, Seckl JR, Mullins JJ. Metabolic syndrome without obesity: hepatic overexpression of 11β-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci USA 101: 7088–7093, 2004. doi: 10.1073/pnas.0305524101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruby MA, Massart J, Hunerdosse DM, Schönke M, Correia JC, Louie SM, Ruas JL, Näslund E, Nomura DK, Zierath JR. Human carboxylesterase 2 reverses obesity-induced diacylglycerol accumulation and glucose intolerance. Cell Reports 18: 636–646, 2017. doi: 10.1016/j.celrep.2016.12.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salleng KJ, Revetta FL, Deane NG, Washington MK. The applicability of a human immunohistochemical panel to mouse models of hepatocellular neoplasia. Comp Med 65: 398–408, 2015. [PMC free article] [PubMed] [Google Scholar]
- 41.Savari F, Mard SA, Badavi M, Rezaie A, Gharib-Naseri MK. A new method to induce nonalcoholic steatohepatitis (NASH) in mice. BMC Gastroenterol 19: 125, 2019. doi: 10.1186/s12876-019-1041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singer JB, Lewitzky S, Leroy E, Yang F, Zhao X, Klickstein L, Wright TM, Meyer J, Paulding CA. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet 42: 711–714, 2010. doi: 10.1038/ng.632. [DOI] [PubMed] [Google Scholar]
- 43.Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci 61: 1282–1293, 2016. doi: 10.1007/s10620-016-4054-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun DQ, Ye FZ, Kani HT, Yang JR, Zheng KI, Zhang HY, Targher G, Byrne CD, Chen YP, Yuan WJ, Yilmaz Y, Zheng MH. Higher liver stiffness scores are associated with early kidney dysfunction in patients with histologically proven non-cirrhotic NAFLD. Diabetes Metab S1262-3636(19)30183-1, 2019. doi: 10.1016/j.diabet.2019.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Tandra S, Yeh MM, Brunt EM, Vuppalanchi R, Cummings OW, Ünalp-Arida A, Wilson LA, Chalasani N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J Hepatol 55: 654–659, 2011. doi: 10.1016/j.jhep.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol 295: G987–G995, 2008. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y, Friedman SL. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 69: 385–395, 2018. [Erratum in J Hepatol 69: 988, 2018.] doi: 10.1016/j.jhep.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.VanWagner LB, Rinella ME. Extrahepatic manifestations of nonalcoholic fatty liver disease. Curr Hepatol Rep 15: 75–85, 2016. doi: 10.1007/s11901-016-0295-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weber A, Boger R, Vick B, Urbanik T, Haybaeck J, Zoller S, Teufel A, Krammer PH, Opferman JT, Galle PR, Schuchmann M, Heikenwalder M, Schulze-Bergkamen H. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 51: 1226–1236, 2010. doi: 10.1002/hep.23479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei G, An P, Vaid KA, Nasser I, Huang P, Tan L, Zhao S, Schuppan D, Popov YV. Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am J Physiol Gastrointest Liver Physiol 318: G174–G188, 2020. doi: 10.1152/ajpgi.00041.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang JD, Ahmed F, Mara KC, Addissie BD, Allen AM, Gores GJ, Roberts LR. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology 71: 907–916, 2020. doi: 10.1002/hep.30858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang X. NAFLD related-HCC: the relationship with metabolic disorders. Adv Exp Med Biol 1061: 55–62, 2018. doi: 10.1007/978-981-10-8684-7_5. [DOI] [PubMed] [Google Scholar]