

Abstract
Carbonic anhydrase (CAII) binds to the electrogenic basolateral Na+- cotransporter (NBCe1) and facilitates reabsorption across the proximal tubule. However, whether the inhibition of CAII with acetazolamide (ACTZ) alters NBCe1 activity and interferes with the ammoniagenesis pathway remains elusive. To address this issue, we compared the renal adaptation of rats treated with ACTZ to NH4Cl loading for up to 2 wk. The results indicated that ACTZ-treated rats exhibited a sustained metabolic acidosis for up to 2 wk, whereas in NH4Cl-loaded rats, metabolic acidosis was corrected within 2 wk of treatment. excretion increased by 10-fold in NH4Cl-loaded rats but only slightly (1.7-fold) in ACTZ-treated rats during the first week despite a similar degree of acidosis. Immunoblot experiments showed that the protein abundance of glutaminase (4-fold), glutamate dehydrogenase (6-fold), and SN1 (8-fold) increased significantly in NH4Cl-loaded rats but remained unchanged in ACTZ-treated rats. Na+/H+ exchanger 3 and NBCe1 proteins were upregulated in response to NH4Cl loading but not ACTZ treatment and were rather sharply downregulated after 2 wk of ACTZ treatment. ACTZ causes renal wasting and induces metabolic acidosis but inhibits the upregulation of glutamine transporter and ammoniagenic enzymes and thus suppresses ammonia synthesis and secretion in the proximal tubule, which prevented the correction of acidosis. This effect is likely mediated through the inhibition of the CA-NBCe1 metabolon complex, which results in cell alkalinization. During chronic ACTZ treatment, the downregulation of both NBCe1 and Na+/H+ exchanger 3, along with the inhibition of ammoniagenesis and generation, contributes to the maintenance of metabolic acidosis.
Keywords: ammonium, carbonic anhydrase, electrogenic basolateral sodium-bicarbonate cotransporter, glutaminase, proximal tubule
INTRODUCTION
The kidney plays an important role in the control of acid-base homeostasis by regulating net acid excretion in the urine (23, 31, 70). Several processes are involved in the maintenance of blood pH within a narrow physiological range. These processes include H+ secretion/ reabsorption along the nephron, ammoniagenesis, which produces and generates from glutamine metabolism, and excretion of titratable acids represented mainly by phosphoric acid, which provides additional buffer allowing the increase of net acid excretion in the urine (17, 23, 31). These processes operate mainly in the distal nephron and proximal tubule, both of which are equipped with appropriate membrane transporters and enzymes necessary for transepithelial reabsorption and glutamine transport and metabolism (1, 10, 16, 21, 45, 64). Moreover, the proximal tubule can adapt to an acid insult and ensuing metabolic acidosis by increasing the expression and activity of many of these transport pathways and enzymes, which increase ammonia ( + NH3) synthesis and generation and reabsorption and ultimately contribute to increased net acid excretion and correction of metabolic acidosis (10, 16, 34, 64, 70). Upregulation of the glutamine transporter SNAT3/SN1 and ammoniagenic enzymes [i.e., glutaminase (GA) and glutamate dehydrogenase (GDH)] in response to an acid stress is mediated mainly via intracellular acidic pH, per se (15, 56, 59, 62).
The reabsorption of in the proximal tubule requires H+ secretion via both H+-ATPase and Na+/H+ exchanger 3 (NHE3) in the brush-border membrane, membrane-bound as well as intracellular carbonic anhydrases (CAs), which facilitate the dehydration/hydration of carbonic acid and thus allow a rapid transfer of CO2 to the cell, and reabsorption via the electrogenic basolateral Na+- cotransporter (NBCe1) (10, 16, 23, 31). This process contributes to the reabsorption of a significant amount of fluid (Na+, , Cl−, and water) in the proximal tubule. Because of this important role in fluid reabsorption, CAs have been targeted with inhibitors such as acetazolamide (ACTZ), which works as a proximal diuretic. Indeed, ACTZ is commonly used to treat several clinical conditions, including metabolic alkalosis induced by the use of loop diuretics or mechanical ventilation in patients with chronic obstructive pulmonary disease, intraocular pressure or glaucoma, and conditions associated with volume overload such as congestive heart failure, and to prevent or reduce the symptoms of altitude sickness (25, 35, 41, 48, 67). In most of these conditions, however, the action of ACTZ to induce fluid loss is associated with significant wasting in the urine and the development of metabolic acidosis.
The mechanism by which ACTZ inhibits reabsorption in the proximal tubule has evolved in recent years (53). Indeed, recent studies have demonstrated that CA inhibitors cause intracellular alkalinization in the microperfused kidney proximal tubule (66). Furthermore, these studies demonstrated that basolateral CA plays an essential role in the reabsorption of and fluid in in the proximal tubule (66). Subsequent studies have shown that intracellular CAII binds to NBCe1 in a metabolon complex (CA-NBCe1) and facilitates Na+ and transport in mouse proximal tubule cells (22, 54) and oocytes (57). These studies indicated that in addition to inactivation of apical membrane-bound CAIV and inhibition of CO2 transfer into the cells, CA inhibitors also indirectly inhibit NBCe1-mediated transport across the basolateral membrane and likely contribute to the alkalinization of proximal tubule cells (66). It is worth noting that the interaction of CAs with the transport proteins is not specific to NBCe1, but CAII was also shown to interact with many other transport proteins and increase their activities, including Cl−/ anion exchanger 1 (61, 68), and downregulated in adenoma or DRA (60), NHE3 (32), and monocarboxylate transporter (29).
Many decades ago, the effects of ACTZ on renal ammoniagenesis was the subject of many investigations. Studies have shown that ACTZ-induced bicarbonaturia was associated with a significant reduction in urinary excretion in both humans and experimental animals. Acute intravenous infusion of ACTZ to patients with liver cirrhosis is associated with the shift of ammonia from alkaline urine to renal venous blood without affecting its production in the proximal tubule (47). However, acute ACTZ treatment causes a significant reduction in urinary ammonium excretion in both rats (51) and dogs (55). The investigators used clearance studies and isolated perfused kidneys and showed that the reduction in excretion by acute ACTZ treatment resulted from the inhibition of GDH activity in dogs (55) and suppression of the activity of GA (7, 51), γ-glutamyltransferase (51), and γ-glutamyltranspeptidase (65) in rats. However, the role of these enzymes in ACTZ-induced ammoniagenesis inhibition was not confirmed in a direct in vitro study using rat kidney homogenates (14). A subsequent study by Tannen and Ross (63) investigated the effects of ACTZ on renal ammoniagenesis in rat kidneys and showed a significant reduction in ammonia production in isolated perfused kidneys of control rats as well as an inhibition of ammoniagenesis in cortical tubules isolated from both control and acidotic rats. Chronic administration of CA inhibitors caused a significant metabolic acidosis that was not associated with increased urinary ammonium excretion in either humans (42) or experimental animals (38, 58).
Thus, in light of these observations, our objective was to demonstrate whether chronic inhibition of the CA-NBCe1 metabolon complex by ACTZ alters ammonia metabolism in proximal tubule cells in the presence of concomitant cell alkalinization and significant systemic metabolic acidosis. To address this question, rats were placed in metabolic cages and treated with ACTZ or its vehicle. Chronic effects of ACTZ on systemic acid-base composition, urinary excretion of ammonium, and expression of acid-base transporters, glutamine transporters, and ammoniagenic enzymes in the kidney cortex were studied. The results of these experiments were compared with those obtained in rats subjected to metabolic acidosis using NH4Cl loading versus their controls.
MATERIALS AND METHODS
The experiments performed in the present studies were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati. Male Sprague-Dawley rats (250–360 g) were housed individually in metabolic cages with free access to rat chow and distilled water and maintained in a temperature-controlled room regulated on a 12:12-h light-dark cycle for the duration of the experiment. After 3 days of acclimation to metabolic cages, rats were subjected to the following treatments.
Animal Treatments
Study 1.
acute actz treatment.
Because the plasma half-life of ACTZ is 4–8 h (25, 53), we examined the renal effects of two doses. Accordingly, rats were injected subcutaneously around 1:00 PM with 100 or 200 mg/kg body wt or vehicle (n = 5 rats in each group). Urine volume was measured, and urine samples were collected at 4 and 24 h after the injection of each group. In addition to urine volume, we measured urine pH, urine osmolality, and urine electrolyte levels to assess the renal response to ACTZ versus vehicle (Table 1).
Table 1.
ACTZ effects on urine output, urine pH, urine osmolality, and electrolyte excretion
| Urine Output | Urine pH | Urine Osmolality | Na+ | K+ | Cl− | |
|---|---|---|---|---|---|---|
| Vehicle | ||||||
| 4 h | 0.83 ± 0.15 | 6.58 ± 0.19 | 2,015 ± 68 | 0.044 ± 0.02 | 0.17 ± 0.04 | 0.11 ± 0.02 |
| 24 h | 13.0 ± 0.87 | 7.80 ± 0.12 | 1,485 ± 100 | 1.420 ± 0.06 | 2.59 ± 0.04 | 2.04 ± 0.05 |
| ACTZ (100 mg/kg) | ||||||
| 4 h | 9.0 ± 0.67a | 8.46 ± 0.12b | 568 ± 24a | 0.97 ± 0.09a | 1.13 ± 0.09b | 0.37 ± 0.04c |
| 24 h | 14.0 ± 0.64 | 7.06 ± 0.10 | 1,006 ± 142d | 1.07 ± 0.11 | 2.12 ± 0.25 | 1.56 ± 0.17e |
| ACTZ (200 mg/kg) | ||||||
| 4 h | 11.0 ± 0.64a | 8.81 ± 0.09b | 665 ± 22b | 1.30 ± 0.12b | 1.24 ± 0.08a | 0.54 ± 0.05a |
| 24 h | 22.0 ± 1.32f | 8.27 ± 0.06b | 781 ± 36f | 1.60 ± 0.11 | 2.41 ± 0.11 | 1.14 ± 0.09f |
Data are means ± SE; n = 5 rats in each group. Urine output was measured as mL/indicated time, urine pH as pH unit, urine osmolality as mosm/kgH2O, and electrolyte excretion (Na+, K+, and Cl−) as mmol/indicated time. ACTZ, acetazolamide.
P < 0.001 vs. 4 h of vehicle;
P < 0.01 vs. 4 h of vehicle;
P < 0.02 vs. 4 h of vehicle;
P < 0.05 vs. 24 h of vehicle;
P < 0.02 vs. 24 h of vehicle;
P < 0.001 vs. 24 h of vehicle.
Study 2.
chronic actz treatment.
A group of 11 rats received daily injections of 200 mg/kg body wt ACTZ subcutaneously and were euthanized at 6 days (n = 6) or 2 wk (n = 5) after the beginning of the treatment. ACTZ was prepared daily as a suspension in a solution containing 1× DMSO + 4× sunflower oil (ACTZ-V1)1. Another group of rats received daily injections of DMSO + sunflower oil (vehicle 1) and were euthanized after 6 days (n = 5) or 2 wk (n = 5) of treatment. All rats were fed rodent chow and distilled water ad libitum for the duration of the experiment.
chronic nh4cl loading-induced metabolic acidosis.
NH4Cl loading is extensively used to induce metabolic acidosis in both human and experimental animals (4, 6, 18, 27, 49). Accordingly, rats were subjected to either NH4Cl loading (280 mM) in the drinking water (n = 10) or control (no treatment, n = 5). NH4Cl-loaded rats were then euthanized at 6 days (n = 5) or 2 wk (n = 5) after the initiation of the treatment. Control rats (n = 5) were euthanized 10 days after the beginning of the experiment. Both groups were fed rodent chow and distilled water for the duration of the experiment.
Study 3.
The above studies revealed a significant increase in urinary excretion in vehicle-treated rats. To confirm the inhibitory effect of ACTZ on excretion, the study 2 was repeated using a different solution containing 1× DMSO + 4× 1,2-propanediol (vehicle 2) to dissolve ACTZ (ACTZ-V2)1. Rats were injected daily with freshly prepared ACTZ (200 mg/kg body wt) and vehicle 2 and euthanized after 6 days (n = 5 in each group) or 2 wk (n = 5 in each group) of treatment. All rats were fed rodent chow and distilled water ad libitum for the duration of the experiment.
Data Collection and Urine and Blood Chemistry Analysis
Food intake, water intake, and urine volume were measured daily for the first 6 days and on day 14 of the treatment. Collected urine samples were used to measure urine , urine pH, and electrolyte (Na+, K+, and Cl−) excretion. Animals were anesthetized using 50 mg pentobarbital sodium, blood was collected from the heart, and the kidneys were rapidly removed and placed in ice-cold Hank’s solution. Cortex tissues were then dissected, snap frozen in liquid nitrogen, and stored at −80°C for protein isolation. Blood chemistry was assessed using an i-STAT1 analyzer with EC8+ cartridges (Abbott Laboratories, Abbott Park, IL). Urine Na+, K+, and Cl− were determined using Easylyte Plus Na+/K+/Cl− analyzer (MEDICA, Bedford, MA), and urine osmolality was measured using the Advanced Micro osmometer (model 3300). Urine concentration was measured using the phenol/sodium hypochlorite method described by Berthelot and previously used in our laboratory (1).
Isolation of Homogenates and Membrane Fractions From the Renal Cortex
The expression of transport proteins and ammoniagenic enzymes was examined in membrane fractions and homogenates, respectively, isolated from the kidney cortex of rats from study 2 treated with ACTZ versus vehicle 1 (DMSO + sunflower oil). Accordingly, homogenates (all cell debris resulting from homogenization before any centrifugation) and total membrane fractions containing plasma membrane proteins were prepared as previously described (1, 4, 6, 18). Briefly, cortex samples were homogenized in ice-cold isolation solution (250 mM sucrose and 10 mM triethnolamine, pH 7.6) supplemented with protease inhibitors (0.1 mg/mL phenazine methylsulfonyl fluoride and 1 mg/mL leupeptin) using a Polytron homogenizer. Immediately after homogenization, aliquots (homogenates) were saved and the remaining samples were centrifuged at low speed (1,000 g) for 10 min at 4°C to remove nuclei and cell debris. The resulting supernatants were spun at 150,000 g for 90 min at 4°C. Pellets containing the plasma membrane were suspended in isolation solution containing protease inhibitors. The total protein concentration was measured using a BCA kit, and membrane fractions were solubilized at 60°C for 20 min in Laemmli sample buffer.
Electrophoresis and Immunoblot Analysis
The procedure was performed as previously described in our laboratory (1, 4, 6, 18). Briefly, solubilized proteins (homogenates or membrane proteins) were size fractionated on an 8% vertical polyacrylamide minigel under denaturing conditions. The separated proteins were electrophoretically transferred to nitrocellulose membranes using a Bio-Rad transfer apparatus (Bio-Rad Laboratories, Hercules, CA). Membranes were then blocked with 5% milk proteins for 30 min and probed with affinity-purified primary specific antibodies against the proteins of interest. After an overnight incubation at 4°C, membranes were washed in Tris-buffered saline (NaCl/KCl) with Tween 20 and then incubated with the corresponding secondary antibody for 1 h. Membranes were washed again with Tris-buffered saline with Tween 20, and the site of antigen-antibody complexation on the nitrocellulose membranes was visualized using the chemiluminescence method and captured on light-sensitive imaging film (Kodak). Membranes were then stripped and reprobed with β-actin antibody as a control for the equity of protein loading. Bands corresponding to the proteins of interest were quantitated by densitometric analysis using a scanner (HP Officejet 4500, Hewlett Packard) and UN-SCAN-IT gel software (Silk Scientific, Orem, UT), and the results are expressed as percentages of control.
Antibodies
Generation of SNAT3/SN1 antibody-peptide-derived polyclonal antibody specific to kidney basolateral Na+-dependent glutamine transporter SNAT3 or SN1 encoded by Slc38a3 was generated using commercial services (Biomatik, Cambridge, ON, Canada). Two peptides corresponding to rat SNAT3 amino acid residues 2–18 (EIPRQTEMVELVPNGKHC) and amino acid residues 41–55 (CGEGKGFLQQSSSKE) were synthesized, conjugated, and used to develop a polyclonal antibody using tow rabbits. Both peptides were injected simultaneously into each rabbit, and the ELISA titer was done for each immunizing peptide and for each rabbit. The test yielded an ELISA titer of >1:100,000. Antisera were affinity purified by covalently immobilizing the immunizing peptides on purification columns. The specificity of the SNAT3 antibody was demonstrated by competitive inhibition of the antibody by the immunizing peptides (Fig. 3).
Fig. 3.
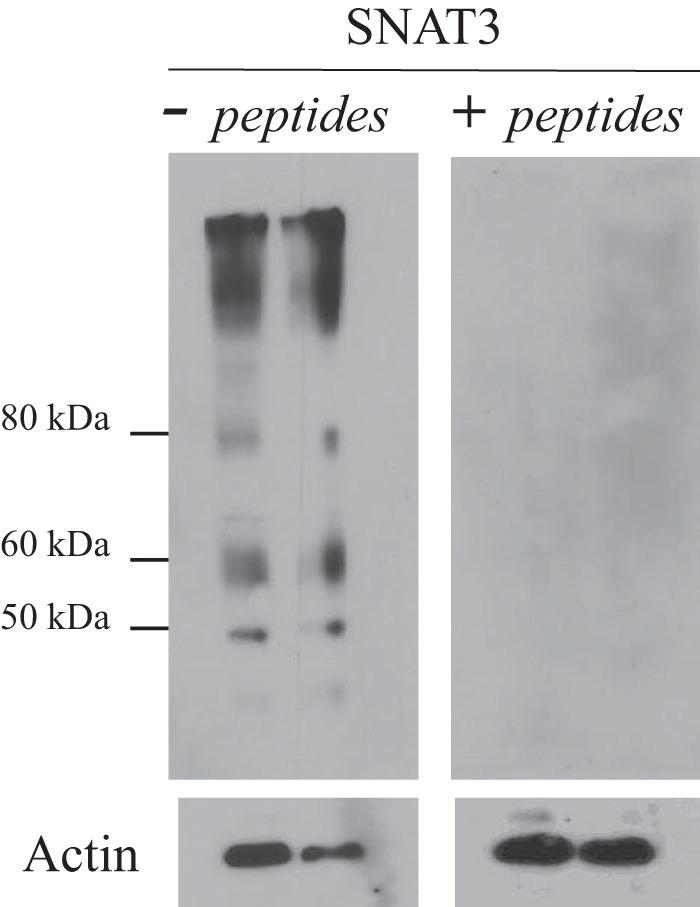
Specificity of the SNAT3 antibody. Homogenates of kidneys harvested from acidotic rats were used for immunoblot analysis. After protein transfer, PVDF membranes were incubated with untreated SNAT3 antibody (left) or preadsorbed antibody with the immunogenic peptides (right). Actin was used as a control for protein loading in the gel. Membrane proteins (25 μg) from kidney homogenates of different rats were loaded per lane.
Phosphate-dependent GA polyclonal antibody was a generous gift from Dr. Norman P. Curthoys (Colorado State University). Rabbit anti-GDH antibody was purchased from Rockland Immunochemicals (Limerick, PA). Rabbit anti-glutamine synthetase or glutamate-ammonia ligase antibody was purchased from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal anti-NHE3 antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). Rabbit polyclonal anti-Na+- cotransporter antibody was purchased from ProteinTech Group (Rosemont, IL). Rabbit polyclonal anti-NaPi-IIa antibody was generated and previously used in our laboratory (11, 69).
Materials
Polyacrylamide minigel cassettes for the NOVEX apparatus, SuperSignal substrate for chemiluminescence, membrane stripping buffer (Restore Western blot solution), BCA kit, and donkey anti-rabbit secondary antibodies were purchased from Thermo Scientific (Rockford, IL). The paper blotting and light-sensitive imaging film (Kodak) were purchased from Midwest Scientific (St. Louis, MO). PVDF nitrocellulose membranes was purchased from Bio-Rad Laboratories. β-Actin and mouse anti-goat secondary antibodies were purchased from Santa Cruz Biotechnology. All other chemicals, including ACTZ, were purchased from Sigma-Aldrich.
Statistical Analysis
Data are expressed as absolute values or percentages of control. Results are presented as means ± SE. Statistical significance between control and experimental groups was determined by a Student’s unpaired t test or one-way ANOVA as needed using SigmaPlot software 13.0. P < 0.05 was considered significant.
RESULTS
Dose-Response and Time-Course Effects of ACTZ on Urine Composition
The data from noncumulative urine samples shown in Table 1 indicate that both doses of ACTZ caused significant diuresis and reduced urine osmolality and bicarbonaturia as shown by alkaline urine pH within the first 4 h of treatment compared with vehicle (Table 1). The effects of a higher dose of 200 mg/kg were exacerbated for urine volume and urine pH or remained rather unchanged for urine osmolality within the next 24 h (Table 1). However, the effects of ACTZ at the lower dose of 100 mg/kg were either dissipated or significantly corrected within 24 h after the treatment compared with vehicle (Table 1).
Urinary excretion of Na+, K+, and Cl− increased significantly in response to both doses within the first 4 h of the treatment. During this early time period, ACTZ is potentially natriuretic, as Na+ excretion increased by 22- and 30-fold, K+ excretion increased by 6.7- and 7-fold, and Cl− excretion increased by ~3.4- and ~5-fold in response to 100 and 200 mg/kg body wt, respectively, compared with vehicle (Table 1). During the following 24 h, excretion of Na+ and K+ in response to both doses of ACTZ was not significantly different from vehicle (Table 1), whereas Cl− excretion was reduced in a dose-dependent manner, compared with vehicle (Table 1), indicating an increase in Cl− retention by the kidney.
These results indicate that ACTZ at 200 mg/kg body wt is more efficient in causing a sustained inhibition of NaHCO3 and water reabsorption in the proximal tubule. Hence, we chose to conduct the following studies with a single daily injection of ACTZ at 200 mg/kg ACTZ.
Effect of Chronic ACTZ Treatment on Blood Composition
The results of measured blood electrolytes and acid-base components were not different between ACTZ studies. Hence, the results shown in Table 2 are pooled data from both studies 2 and 3 where rats were injected with ACTZ dissolved in two different vehicles. The results indicate that rats treated with ACTZ exhibited significant hyperchloremic metabolic acidosis, as indicated by increased Cl− concentration and reduced total CO2 and blood pH, respectively, within the first 6 days of the treatment compared with vehicle-treated group. These effects were sustained for up to 2 wk of treatment. ACTZ treatment did not alter Na+, K+, and blood urea nitrogen levels for the duration of the experiment. A mild, albeit significant, reduction in blood glucose levels (nonfasting state) was observed in ACTZ-treated animals versus vehicle-treated animals (Table 2).
Table 2.
Blood composition
| Na+, mM | K+, mM | Cl−, mM | Total CO2, mM | pH | Glucose, mg/dL | Blood Urea Nitrogen, mg/dL | |
|---|---|---|---|---|---|---|---|
| Vehicle | 139 ± 0.33 | 4.98 ± 0.16 | 96 ± 1.61 | 33 ± 0.22 | 7.41 ± 0.01 | 170 ± 2.02 | 18 ± 0.22 |
| P value | NS | NS | <0.001* | <0.001* | <0.001* | <0.001* | NS |
| ACTZ, 6 days | 137 ± 0.95 | 4.6 ± 0.10 | 108 ± 0.63 | 21 ± 0.54 | 7.33 ± 0.01 | 139 ± 4.74 | 19 ± 0.52 |
| P value | NS | NS | <0.004* | <0.001* | <0.001* | <0.001* | NS |
| ACTZ, 2 wk | 138 ± 0.88 | 4.74 ± 0.09 | 107 ± 0.65 | 22 ± 1.04 | 7.33 ± 0.01 | 139 ± 2.23 | 19 ± 0.47 |
Data are means ± SE; n = 20 rats for vehicle, n = 11 for 6 days of acetazolamide (ACTZ) treatement, and n = 10 for 2 wk of ACTZ treatment. The results are pooled data from studies 2 and 3. NS, not significant.
Compared with vehicle.
Urinary Electrolyte Excretion and Food Intake in ACTZ Versus NH4Cl Loading
NH4Cl loading.
The results showed a slight but significant reduction in food intake by day 6 (P < 0.05 vs. control), which recovered toward the control level by day 14 of NH4Cl loading (Table 3). Na+ excretion was also significantly reduced by day 6 (P < 0.04 vs. control) and returned to the control level by day 14 of NH4Cl loading (Table 3). As expected, Cl− excretion was increased drastically in NH4Cl-loaded rats, whereas K+ excretion remained unchanged throughout the experiment (Table 3). A detailed study related to the renal handling of Na+ during NH4Cl loading-induced metabolic acidosis in rats was previously performed and reported by our laboratory (18).
Table 3.
Food intake and urinary electrolyte excretion
| NH4Cl Loading |
ACTZ-V2 |
|||||
|---|---|---|---|---|---|---|
| Control | 6 days | 14 days | V2 | 6 days | 14 days | |
| Food intake, g/24 h | 20 ± 0.82 | 17 ± 0.44* | 19 ± 0.33 | 17 ± 0.55 | 18 ± 0.25 | 18 ± 0.64 |
| Na+ excretion, mmol/24 h | 1.82 ± 0.11 | 1.54 ± 0.04† | 1.88 ± 0.68 | 1.37 ± 0.07 | 1.69 ± 0.08* | 1.84 ± 0.07† |
| K+ excretion, mmol/24 h | 3.23 ± 0.19 | 3.03 ± 0.06 | 3.42 ± 1.02 | 3.16 ± 0.16 | 3.37 ± 0.16 | 4.07 ± 0.12* |
| Cl− excretion, mmol/24 h | 2.55 ± 0.10 | 9.06 ± 0.43‡ | 10.71 ± 0.40‡§ | 2.13 ± 0.10 | 3.02 ± 0.11† | 3.41 ± 0.18† |
Data are means ± SE; n = 5 rats in each group except for the vehicle 2 (V2) group, where n = 10. Data shown for V2 are pooled data from 6 and 14 days of V2 treatment. ACTZ, acetazolamide.
P < 0.05 vs. control or vehicle 2;
P < 0.04 vs. control or vehicle 2;
P < 0.001 vs. control;
P < 0.01 vs. 6 days.
ACTZ treatment.
As expected, ACTZ-treated rats exhibited significant natriuresis after 6 days (P < 0.05 vs. pooled vehicle 2), which increased further after 14 days (P < 0.04 vs. pooled vehicle 2) of treatment (Table 3). A significant increase in K+ excretion was shown in rats treated with ACTZ for 14 days (P < 0.05 vs. pooled vehicle 2) but not after 6 days of treatment (Table 3). Cl− excretion increased significantly after 6 days (P < 0.04) and further increased after 14 days (P < 0.04) of ACTZ treatment compared with pooled vehicle 2 (Table 3). The increased Cl− excretion is likely due to downregulation of pendrin, the apical Cl−/ exchanger in type B intercalated cells, by the ensuing metabolic acidosis (50).
Food intake was slightly, but significantly, decreased (17 ± 0.50 g/day, n = 6) in ACTZ-treated animals at 6 days versus vehicle 1-treated animals (22 ± 0.95, n = 10, P < 0.02) but was not significantly altered in animals treated with ACTZ for 2 wk (20 ± 1.40 g/day, n = 5, P > 0.05) compared with vehicle 1-treated animals (n = 10). Unlike ACTZ suspended in vehicle 1, ACTZ dissolved in vehicle 2 did not alter food intake throughout the duration of the treatment (Table 3).
Blood and Cl− Levels in ACTZ Treatment Versus NH4Cl Loading
In a time-course study, we compared the changes in blood levels of and Cl− between ACTZ treatment and the classical model of NH4Cl loading-induced metabolic acidosis. The composition of ACTZ vehicles did not significantly alter the effect of ACTZ on blood electrolytes and acid-base composition, as discussed above. Hence, in Fig. 1, we pooled serum Cl− concentration and serum concentration data obtained from both ACTZ studies. The results shown in Fig. 1 indicate that ACTZ-treated animals exhibited significant hyperchloremic metabolic acidosis, as shown by the increase in blood Cl− (Fig. 1A) and decreased (Fig. 1B) levels, respectively, within the first 6 days of treatment compared with vehicle (time 0). Changes in these parameters persisted for up to 2 wk of ACTZ treatment (Fig. 1, A and B) compared with vehicle (time 0). NH4Cl-loaded animals exhibited a similar degree of hyperchloremic (Fig. 1C) and metabolic acidosis (Fig. 1D) within the first 6 days of treatment compared with control animals (time 0). However, both Cl− and were corrected to near control levels within the last 8 days of NH4Cl loading (Fig. 1, C and D). At this time point, blood levels remained slightly, albeit significantly, below the control level (P < 0.002; Fig. 1D).
Fig. 1.

Time-course effects of acetazolamide (ACTZ) and NH4Cl loading on blood Cl− and levels. Rats were placed in metabolic cages with free access to rodent chow and distilled water and randomly divided into different groups. Rats were treated with ACTZ (n = 10) or its vehicle (n = 10) compared with rats subjected to NH4Cl loading (n = 10) or control (n = 5) and were euthanized after 6 or 14 days. A and B: time course of blood concentration in rats treated with ACTZ (A) or loaded with NH4Cl (B) compared with pooled vehicle [vehicle 1/vehicle 2 (V1/V2)]-treated or control rats, respectively. C and D: time course of blood Cl− concentration in rats treated with ACTZ (C) or loaded with NH4Cl (D) compared with pooled vehicle-treated (n = 10) or control rats, respectively. As shown, hyperchloremic metabolic acidosis was corrected within 2 wk in NH4Cl-loaded but not ACTZ-treated rats. *P < 0.002 vs. vehicle or control; **P < 0.03 vs. vehicle or control; §P < 0.01 vs. 6 days; ¶P < 0.001 vs. vehicle.
Urinary Excretion in ACTZ-Treated Versus NH4Cl-Loaded Animals
The time-course data shown in Fig. 2A demonstrated a sharp and gradual increase in urinary excretion2 in NH4Cl-loaded rats during the first 3 days and plateaued thereafter at higher levels for the duration of the treatment. However, the curve of excretion was shifted sharply to the right in ACTZ-treated rats, with a significant inhibition of urinary excretion over 14 days of ACTZ treatment whether ACTZ was dissolved in vehicle 2 or suspended in vehicle 1 (Fig. 2, A and B). The presence of sunflower oil in vehicle 1 likely altered ammonia metabolism and increased excretion in rats treated with ACTZ dissolved in vehicle 1 (compare vehicle 1 with vehicle 2 and control in Fig. 2B). As clearly shown in Fig. 2C, the magnitude of the increase in excretion was sharply reduced in ACTZ-treated (~1.7-fold) versus NH4Cl-loaded (11-fold) animals (Fig. 2B). This inhibition occurred despite a similar degree of metabolic acidosis within the first 6 days of treatment ( concentration: 19 ± 0.75 vs. 20 ± 1.10 mM in ACTZ vs. NH4Cl treatment, respectively; Fig. 1, B and D).
Fig. 2.

Time course of urinary excretion in acetazolamide (ACTZ) treatment versus NH4Cl loading. A: time course of urinary excretion in rats loaded with NH4Cl versus rats treated with ACTZ suspended in vehicle 1 (ACTZ-V1) or ACTZ dissolved in vehicle 2 (ACTZ-V2). *P < 0.03 vs. baseline (time 0); £P < 0.01 vs. ACTZ-V2. B: urinary excretion was measured in urine collected 6 or 14 days after treatment of rats with ACTZ-V1 or ACTZ-V2 or loaded with NH4Cl compared with their vehicles or control, respectively. @P < 0.001 vs. vehicle 2 or control; ¥P < 0.05 vs. vehicle 1 or vehicle 2; #P < 0.04 vs. 6 days; ¶P < 0.0001 vs. control. C: fold increase in excretion on days 6 and 14 in each treatment. excretion increased by ~11-fold in NH4Cl-loaded rats but only by ~2-fold in ACTZ-treated rats despite a similar degree of metabolic acidosis during the first 6 days of treatment. #P < 0.04 vs. 6 days; §P < 0.001 vs. the same period of ACTZ treatment. D: time course of urine pH in rats loaded with NH4Cl and rats treated with ACTZ dissolved in vehicle (ACTZ-V2). **P < 0.01 vs. baseline (time 0); *P < 0.05 vs. baseline (time 0) or vs. vehicle 2. n = 5–10 rats in each group as indicated above.
The results shown in Fig. 2D demonstrate the time course of urinary pH changes over the course of 6 days of treatment. As shown, the results obtained from study 2 indicate an initial sharp increase in urine pH within the first 24 h of ACTZ treatment, which gradually decreased after that but remained slightly, albeit significantly, higher than the baseline level or compared with urine pH of vehicle-treated rats (Fig. 2D). In contrast, NH4Cl-loaded rats exhibited a significant reduction in urine pH, which remained low for the duration of the treatment (Fig. 2D). Urine pH of control rats remained unchanged for the duration of the experiment (data not shown).
Adaptation of Glutamine Transporter SNAT3 and Ammoniagenic Enzymes to ACTZ- Versus NH4Cl Loading-Induced Metabolic Acidosis
Next, we examined the abundance of proteins involved in the ammoniagenesis process in the proximal tubule. These include glutamine transporter SNAT3 and glutamine-metabolizing enzymes such as phosphate-dependent GA (PGA), GDH, and glutamine synthetase (GS). SNAT3, PGA, and GDH were upregulated (15, 56, 59, 62), whereas GS was downregulated (36, 71), in response to metabolic acidosis.
Characterization of SNAT3 antibody.
We characterized new rabbit polyclonal antibodies against SNAT3 generated against two synthetic peptides corresponding to a fragment of SNAT3 peptide as described in materials and methods. Figure 3 shows immunoblots loaded with membrane proteins isolated from whole kidney homogenates harvested from rats subjected to NH4Cl loading-induced metabolic acidosis and probed with SNAT3 antibody. As shown, the antibody detected several bands (Fig. 3, left), all of which were undetectable when the antibody was preadsorbed with excess immunogenic peptides before the immunoblot analysis (Fig. 3, right). This suggests that the antibody specifically recognized SNAT3 protein in the kidney homogenates. In our expression studies, we focused on the protein abundance of SNAT3 represented by the bands between 50 and 60 kDa as previously studied by many laboratories, including ours (1), using different antibodies (8, 12, 13, 26, 40, 59).
Six days of treatment with NH4Cl loading or ACTZ.
The results shown in Fig. 4 indicate that NH4Cl loading-induced metabolic acidosis for 6 days, as expected, caused a sharp upregulation in the protein abundance of SNAT3 (30- and 50-fold increase in both bands, P < 0.001 and P < 0.0001, respectively; Fig. 4, A and B), PGA (6-fold, P < 0.001; Fig. 4, A and C), and GDH (164%, P < 0.001; Fig. 4, A and C) compared with control (n = 5 rats in each group). The protein abundance of GS was not altered (P > 0.05; Fig. 4, A and C) in response to NH4Cl loading compared with control (n = 5 rats in each group). Interestingly, ACTZ treatment for 6 days caused only a slight but significant upregulation of SNAT3 protein (60-kDa band, P < 0.002, n = 5; Fig. 4, D and E) but did not alter the protein abundance of PGA (P > 0.05, n = 5; Fig. 4, D and F) or GDH (P > 0.05, n = 5; Fig. 4, D and F), despite a degree of metabolic acidosis that was similar to that induced by NH4Cl loading (6 days; Fig. 1, A and B). However, a slight but significant decrease in the protein abundance of GS was observed in rats treated with ACTZ for 6 days (P < 0.02; Fig. 4, D and F) compared with vehicle (n = 5 rats in each group).
Fig. 4.

Adaptation of glutamine transporter and ammoniagenic enzymes in the kidney cortex of rats treated with acetazolamide (ACTZ) or NH4Cl loading for 6 days. A and D: tissue homogenates (enzymes) and membrane fractions (SNAT3) were isolated from the kidney cortex of rats treated with NH4Cl loading (A) or ACTZ (D) versus their respective control or vehicle treatment and were used for immunoblot analysis using specific antibodies for each protein. B, C, E, and F: corresponding average means ± SE of the densitometry analysis of SNAT3 (B and E) and ammoniagenic enzymes (C and F) in response to NH4Cl loading versus control (B and C) or in ACTZ-treated versus vehicle-treated rats (E and F). The abundance of these proteins was normalized to actin used for the control of gel loading. As shown, SNAT3, phosphate-dependent glutaminase (PGA), and glutamate dehydrogenase (GDH) were sharply upregulated in NH4Cl-loaded rats but not in ACTZ-treated rats. n = 5 rats in each group. Each lane was loaded with 10−30 μg protein from the kidney cortex. Note that the exposure time for SNAT3 in vehicle versus ACTZ (D) was much higher than that in control versus 6 days of NH4Cl loading (A). GS, glutamine synthetase; NS, not significant.
Two weeks of treatment with NH4Cl loading or ACTZ.
Over 2 wk of treatment, rats subjected to NH4Cl loading exhibited a significant increase in the protein abundance of SNAT3 (~2- and ~4-fold increase in both bands, P < 0.001 and P < 0.01, respectively; Fig. 5, A and B), PGA (~3-fold increase, P < 0.0001; Fig. 5, A and C), and GDH (+ 50%, P < 0.01; Fig. 5, A and C) compared with control (n = 5 rats in each group). However, the magnitude of the effect was significantly less than that induced over 6 days of treatment. Like in 6 days, the protein abundance of GS was not altered (P > 0.05; Fig. 5, A and C) by 2 wk of NH4Cl loading versus control (n = 5 rats in each group). In response to 2 wk of ACTZ treatment, the protein abundance of SNAT3 was not altered (P > 0.05 for either band; Fig. 5, D and E), whereas PGA (+47%, P < 0.004; Fig. 5, D and F) and GDH (+26%, P < 0.03; Fig. 5, D and F) increased slightly but significantly compared with vehicle group (n = 5 rats in each group). The abundance of GS protein did not change (P > 0.05; Fig. 5, D and F) in response to 2 wk of ACTZ treatment versus vehicle (n = 5 rats in each group).
Fig. 5.

Adaptation of glutamine transporter and ammoniagenic enzymes in the kidney cortex of rats treated with acetazolamide (ACTZ) or NH4Cl loading for 2 wk. A and D: tissue homogenates (enzymes) and membrane fractions (SNAT3) were isolated from the kidney cortex of rats treated with NH4Cl loading (A) or ACTZ (D) versus their respective control or vehicle treatment and were used for immunoblot analysis using specific antibodies for each protein. B, C, E, and F: corresponding average means ± SE of the densitometry analysis of SNAT3 (B and E) and ammoniagenic enzymes (C and F) in response to NH4Cl loading versus control (B and C) or in ACTZ-treated versus vehicle-treated rats (E and F). The abundance of these proteins was normalized to actin used for the control of gel loading. As shown, SNAT3 was sharply upregulated in NH4Cl-loaded rats but not ACTZ-treated rats for 2 wk. Phosphate-dependent glutaminase (PGA) and glutamate dehydrogenase (GDH) were slightly, but significantly, upregulated in ACTZ-treated rats for 2 wk. The magnitude of PGA induction was greater in NH4Cl loading versus ACTZ treatment. n = 5 rats in each group. Each lane was loaded with 10−30 μg protein from the kidney cortex. Note that the exposure time for SNAT3 in vehicle versus ACTZ (D) was much higher than that in control versus 6 days of NH4Cl loading (A). GS, glutamine synthetase; NS, not significant.
The densitometry analysis data of SNAT3 and ammoniagenic enzymes in response to ACTZ and NH4Cl loading versus their vehicle or control (Figs. 4 and 5, B, C, E, and F) are also shown in Table 4. The control and vehicle-treated groups were pooled (control/vehicle) for each time, and the statistical analysis between control/vehicle, ACTZ, and NH4Cl loading for each time period was done using one-way ANOVA. This allowed a direct comparison of the effects of ACTZ versus NH4Cl loading on SNAT3 and ammoniagenic enzymes (Table 4).
Table 4.
Densitometry analysis of SNAT3 and the enzyme-to-actin ratio expressed as a percentage of vehicle or control
| SNAT3 |
Phosphate-Dependent Glutaminase | Glutamate Dehydrogenase | Glutamine Synthetase | ||
|---|---|---|---|---|---|
| 60 kDa | 50 kDa | ||||
| Vehicle/control, 6 days | 100 ± 15 | 100 ± 20 | 100 ± 13 | 100 ± 8 | 100 ± 7 |
| 6 days of ACTZ | 193 ± 17a | 120 ± 9 | 96 ± 7 | 89 ± 9 | 64 ± 10a |
| 6 days of acidosis | 2,687 ± 462b | 4,729 ± 265b | 582 ± 84c | 210 ± 17d | 121 ± 5d |
| Vehicle/control, 2 wk | 100 ± 12 | 100 ± 7 | 100 ± 10 | 100 ± 9 | 100 ± 7 |
| 2 wk of ACTZ | 118 ± 14 | 107 ± 16 | 147 ± 11e | 126 ± 6f | 100 ± 8 |
| 2 wk of acidosis | 293 ± 60g | 483 ± 57g | 394 ± 29g | 157 ± 16 | 91 ± 8 |
Data are means ± SE; n = 10 rats for pooled vehicle and control groups for each time period and n = 5 rats for other groups. Data are also shown in Figs. 4 and 5.
P < 0.03 vs. vehicle/control;
P < 0.001 vs. 6 days of acetazolamide (ACTZ) treatment;
P < 0.02 vs. 6 days of ACTZ treatment;
P < 0.05 vs. 6 days of ACTZ treatment;
P < 0.02 vs. 2 wk of vehicle/control treatment;
P < 0.05 vs. 2 wk of vehicle/control treatment;
P < 0.05 vs. 2 wk of ACTZ treatment. One-way ANOVA was used for comparison between groups.
We should mention that although urinary excretion was increased by ~0.5 mmol/day in rats injected with vehicle 1 (DMSO + sunflower oil) versus those injected with vehicle 2 (DMSO + 1,2-propanediol), the expression of SNAT3 and ammoniagenic enzymes was not different between these two vehicles.
Adaptation of Proximal Tubule NHE3, NBC, and NaPi-IIa to ACTZ- Versus NH4Cl Loading-Induced Metabolic Acidosis
NH4Cl loading-induced metabolic acidosis for 6 days caused a significant increase in the protein abundance of the apical Na+/H+ exchanger NHE3 (+77%, P < 0.02; Fig. 6, A and B) and the basolateral Na+- cotransporter NBCE1 or NBCe-1 (+48%, P < 0.03; Fig. 6, A and B) but did not alter protein expression of apical Na+-dependent phosphate cotransporter NaPi-IIa (P > 0.05; Fig. 6, A and B) compared with control animals (n = 5 rats in each group). Interestingly, none of these transporters was affected in animals subjected to 6 days of ACTZ (P > 0.05; Fig. 6, C and D) versus vehicle treatment (n = 5 rats in each group), despite the presence of significant metabolic acidosis (Fig. 1A).
Fig. 6.

Response of acid-base transporters [Na+/H+ exchanger 3 (NHE3) and electrogenic basolateral Na+- cotransporter (NBCe1)] and phosphate transporter (NaPi-IIa) to acetazolamide (ACTZ) or NH4Cl loading for 6 days. A and C: membrane fractions were isolated from the kidney cortex of rats treated with NH4Cl loading (A) or ACTZ (C) versus their respective control or vehicle treatment and were used for immunoblot analysis using specific antibodies for each protein. B and D: corresponding average means ± SE of the densitometry analysis of transport proteins in response to NH4Cl loading versus control (B) or in ACTZ-treated versus vehicle-treated rats (D). The abundance of these proteins was normalized to actin used for the control of gel loading. As shown, NHE3 and NBCe1 proteins were significantly upregulated in NH4Cl-loaded but not ACTZ-treated rats despite the same degree of acidosis. n = 5 rats in each group. Each lane was loaded with 40 μg protein from the kidney cortex. NS, not significant.
After 2 wk of NH4Cl loading, the abundance of NHE3 protein returned toward the baseline level, whereas the protein abundance of NBCe1 was significantly reduced (P < 0.03; Fig. 7, A and B) compared with control (n = 5 rats in each group). In contrast, the protein abundance of NaPi-IIa was significantly upregulated in response to 2 wk of NH4Cl loading (+54%, P < 0.02; Fig. 7, A and B) versus control (n = 5 rats in each group). However, 2 wk of ACTZ treatment resulted in a sharp reduction in the abundance of both NHE3 (−46%, P < 0.002) and NBCe1 (−76%, P < 0.001) proteins (Fig. 7, C and D) compared with the vehicle-treated group (n = 5 rats in each group). In contrast, the protein abundance of NaPi-IIa was rather significantly increased (+60%, P < 0.01; Fig. 7, C and D) in 2-wk ACTZ- versus vehicle-treated rats (n = 5 rats in each group).
Fig. 7.

Response of acid-base transporters [Na+/H+ exchanger 3 (NHE3) and electrogenic basolateral Na+- cotransporter (NBCe1)] and phosphate transporter (NaPi-IIa) to acetazolamide (ACTZ) or NH4Cl loading for 2 wk. A and C: membrane fractions were isolated from the kidney cortex of rats treated with NH4Cl loading (A) or ACTZ (C) versus their respective control or vehicle treatment and were used for immunoblot analysis using specific antibodies for each protein. B and D: corresponding average means ± SE of the densitometry analysis of transport proteins in response to NH4Cl loading versus control (B) or in ACTZ-treated versus vehicle-treated rats (D). The abundance of these proteins was normalized to actin used for the control of gel loading. As shown, 2 wk of ACTZ caused a sharp downregulation of both NHE3 and NBCe1 but increased the abundance of NaPi-IIa. Except for NHE3, similar findings were seen in NH4Cl-loaded rats with normal acid-base status. n = 5 rats in each group. Each lane was loaded with 40 μg protein from the kidney cortex. NS, not significant.
DISCUSSION
Previous studies in both humans and experimental animals have demonstrated that acute inhibition of CA causes a reduction in urinary excretion despite significant bicarbonaturia (7, 47, 51, 55, 63, 65) and that chronic administration of CA inhibitors causes significant metabolic acidosis that is not associated with increased renal ammoniagenesis (38, 48, 58). The present studies demonstrate, in a reproducible manner, that chronic ACTZ treatment is associated with the development of significant hyperchloremic metabolic acidosis in rats that is maintained for as long as the animals are treated (Fig. 1). This observation is in sharp contrast with the conventional understanding of renal adaptation to chronic metabolic acidosis (CMA). In fact, our present studies show that when hyperchloremic metabolic acidosis is induced in rats using the classical model of NH4Cl loading, this disorder is corrected within the second week despite continuation of NH4Cl loading (Fig. 1). The stimulation of ammoniagenesis and increased ammonia ( + NH3) excretion is the key component of increased urinary net acid excretion during renal adaptation to chronic metabolic acidosis. The stimulation of ammoniagenesis during CMA results from increased glutamine catabolism via upregulation of ammoniagenic mitochondrial enzymes (PGA and GDH) and upregulation of the cytosolic gluconeogenic enzyme phosphoenolpyruvate carboxykinase, which produces glucose and in the proximal tubule (15, 16, 43, 44, 59, 70, 71). This process correlates with upregulation of both glutamine transporters (SNAT3 or SN1) and acid-base transporters including NHE3 and NBCe1 in the proximal tubule (34, 52, 59). The upregulation of NHE3 drives secretion in the tubular fluid (28), whereas the increased expression and activity of NBCe1 allow the transfer of newly generated in the bloodstream. The bulk of secreted in the proximal tubule is reabsorbed in the medullary thick ascending limb via Na+-K+-2Cl− cotransporters (3, 5, 20) accumulated in the medullary interstitium, secreted in the collecting duct via transporter Rh family B and family C glycoproteins, and ultimately excreted in the urine (30, 70, 71). In addition to excretion, the increased titratable acids, represented mainly by phosphate ions, contribute to the increased net acid excretion during CMA. In this regard, the expression and/or activity of phosphate transporter NaPi-IIa has been shown to be altered in the proximal tubule in response to acid loading (46).
Our data indicate that ACTZ treatment is associated with a rapid increase in wasting in the urine (Table 1), which ultimately leads to the development of hyperchloremic metabolic acidosis, as indicated by the reduction in concentration and increased serum Cl− levels (Table 2 and Fig. 1). When we compared the effects of ACTZ to NH4Cl loading on blood acid-base composition, we observed that ACTZ treatment generated significant metabolic acidosis within 6 days, which was maintained for the duration (2 wk) of the treatment (Fig. 1). NH4Cl loading caused the same degree of acidosis during the first week but was corrected within the second week of treatment (Fig. 1) despite continuous NH4Cl loading. This clearly indicates an impairment in the mechanisms responsible for increased urinary net acid excretion and renal adaptation of metabolic acidosis when the latter is induced by ACTZ. Indeed, our results show that the magnitude of urinary excretion is significantly diminished in ACTZ-treated versus NH4Cl-loaded animals (Fig. 2). To gain insight into the molecular mechanisms responsible for the inhibition of ammonia synthesis in ACTZ-induced acidosis, we examined the abundance of proteins involved in glutamine transport and metabolism in the proximal tubule of ACTZ-treated versus NH4Cl-loaded animals. Surprisingly, our results showed a significant upregulation in the protein abundance of glutamine transporter SNAT3 and ammoniagenic enzymes PGA and GDH in NH4Cl-loaded but not ACTZ-treated animals (Fig. 4 and Table 4), despite the same degree of metabolic acidosis associated with these treatments. During the 2 wk of treatments, SNAT3 and PGA/GDH were still significantly upregulated in NH4Cl-loaded rats, with only a slight, but significant, increase in PGA and GDH expression observed in ACTZ-treated rats (Fig. 5). It should be noted that the magnitude of the stimulation of expression of both ammoniagenic proteins (SNAT3, PGA, and GDH) and acid-base transporters (NHE3 and NBCe1) was reduced after 2 wk versus 6 days of NH4Cl loading (Figs. 4, 5, 6, and 7). In fact, expression of NBCe1 was significantly reduced by 43% after 2 wk of NH4Cl loading versus control (Fig. 7, A and B). Interestingly, the rate of excretion remained elevated and unchanged during the entire period of NH4Cl loading (Fig. 2B), indicating that the activity of ammoniagenic proteins, NHE3, and NBCe1, likely remained elevated despite changes in their protein expression levels.
With regard to the adaptation of the proximal tubule acid-base transporters, the protein abundance of NHE3 and NBCe1 increased significantly during the first week of NH4Cl loading (Fig. 6). The upregulation of NHE3 is in agreement with previous findings (reviewed in 34), whereas the upregulation of NBCe1 is supported by the increase in NBCe1 activity in response to metabolic acidosis previously reported by others (52). During the second week of NH4Cl loading, which is associated with correction of metabolic acidosis (Fig. 1), NHE3 expression returned to baseline level (Fig. 7), whereas the abundance of NBCe1 was slightly, but significantly, reduced (−43%) below the control level (Fig. 7). However, NHE3 and NBCe1 expression did not change during the first week of ACTZ treatment (Fig. 6) despite the presence of significant metabolic acidosis (Fig. 1), but their protein abundance was sharply reduced during the second week of ACTZ treatment (Fig. 7). Although the mechanism mediating the downregulation of these proteins after 2 wk of ACTZ is unclear, it must be noted that this effect is specific to NHE3 and NBCe1, as phosphate transporter NaPi-IIa protein is rather significantly upregulated (Fig. 7).
Hence, taken all together, our results indicate that ACTZ causes metabolic acidosis in two phases. First is a generation phase during which ACTZ inhibits the reabsorption of filtered in the proximal tubule and causes wasting in the urine, as indicated by the immediate alkalinization of urine pH (Table 1). This is followed by a maintenance phase during which ACTZ not only inhibits ammoniagenesis process, which is supposed to generate new molecules and provide ammonia buffer for net acid excretion, but also inhibits the adaptation of both NHE3 and NBCe1 to ensuing metabolic acidosis during the first week with subsequent sharp downregulation of their protein abundance during the second week of treatment (Figs. 6 and 7).
The molecular mechanism underlying the adaptation of glutamine transporter SNAT3 and ammoniagenic enzymes (GA and GDH) to metabolic acidosis has been extensively explored. Studies have shown that acidic pH, per se, is responsible for the upregulation of both PGA and GDH in response to metabolic acidosis. The intracellular acidic pH activates cellular mechanisms, which lead to the activation of gene transcription as well as the increased mRNA stability of both PGA and GDH (15, 56, 62). The stability of the mRNA encoding for these enzymes is increased in response to acidosis via the activation of pH-response element (AU-rich sequence) present in the 3′UTR of their mRNA transcripts (15, 56, 62). Subsequent studies reported that the upregulation of glutamine transporter SNAT3/SN1 in response to metabolic acidosis involves an acidic pH-responsive element (similar AU-rich region present in PGA and GDH mRNA transcripts) present in the 3′UTR of SNAT3 mRNA transcript (59).
As described in the introduction, ACTZ inhibits the activity of CAs and causes wasting in the urine, which results in metabolic acidosis. However, ACTZ was also shown to suppress the activation of basolateral Na+: cotransporter (NBCe1) by inhibiting CAII and causing cell alkalinization (22, 54). It should be noted that the inhibition of CAIV (bound to both apical and basolateral membranes) by ACTZ and its effects on apical H+ secretion and basolateral transport, with resulting effects on intracellular pH, can also affect ammonia metabolism in the proximal tubule cells. Moreover, other mechanisms independent of changes in NBCe1 activity may contribute to ACTZ-induced cell alkalinization and inhibition of ammoniagenesis in the proximal tubule cells. Nevertheless, in our working model depicted in Fig. 8, we propose that ACTZ directly inhibits CAII and CAIV, which then blocks the stimulation of NBCe1 by CAII and CAIV, and indirectly inhibits NBCe1 activity in the proximal tubule. At this time, it is unclear whether ACTZ-CA complex prevents the binding of CA to NBCe1 (as shown in Fig. 8) or if ACTZ binds to CA-NBCe1 metabolon and inhibits NBCe1 activation by CA. Either of these mechanisms contributes to the intracellular alkalinization, which prevents the activation of cytosolic proteins and their binding to pH-responsive elements in SNAT3, PGA, and GDH, and inhibits the upregulation of these proteins, therefore preventing the stimulation of ammoniagenesis despite the presence of systemic acidosis in ACTZ-treated animals (Fig. 8). Alternatively, possible changes in the intracellular calcium levels secondary to changes in cell pH and/or variations in membrane potential resulting from altered NBCe1 activity could trigger other signaling pathway(s), which contribute to the inhibition of SNAT3 and/or ammoniagenic enzymes in the presence of ACTZ.
Fig. 8.

Proposed working model for acetazolamide (ACTZ)-induced inhibition of ammoniagenesis and maintenance of metabolic acidosis. In the absence of ACTZ (left), carbonic anhydrase (CA) binds to the electrogenic basolateral Na+- cotransporter (NBCe1) and increases its activity; during an acid insult (NH4Cl loading), the ensuing acidosis acidifies proximal tubule (PT) cells and subsequently stimulates every cellular component involved in the ammoniagenesis process, which leads to increased synthesis and generation. The increased excretion combined with increased delivery to the bloodstream leads to the correction of metabolic acidosis. In the presence of ACTZ (right), the latter binds to the Na+- cotransporter and suppresses its activity and causes to accumulate and alkalinize PT cells. Alkaline intracellular pH (pHi) prevents the stimulation of ammoniagenesis and prevents the correction of metabolic acidosis. Long-term ACTZ treatment also downregulates the protein abundance of both Na+/H+ exchanger 3 (NHE3) and Na+- cotransporter. Both of these events contribute to the maintenance on metabolic acidosis during ACTZ treatment. GA, glutaminase; GDH, glutamate dehydrogenase; PEPCK, phosphoenolpyruvate carboxykinase.
Nevertheless, the activity of NBCe1 in the proximal tubule appears to play an important role in the stimulation of ammoniagenesis in response to an acid insult. This is supported by the observation that patients bearing missense mutations in the gene SLC4A4 encoding for NBCe1 develop proximal renal tubular acidosis with inappropriately lower urinary ammonia excretion (2, 9, 33, 37). This phenomenon was confirmed in experimental animal studies, which showed that mice with NBCe1 deletion exhibited a significant decrease in ammonia synthesis and excretion despite the presence of significant metabolic acidosis and intact proximal tubular structure (19, 24). It should be noted that the inhibition of CA and transporters by ACTZ is not specific to the proximal tubule, and hence it could also affect NH4+ transport in the downstream nephron segments. For instance, the inhibition of CA in medullary thick ascending limb (mTAL) would likely alter NH4+ reabsorption by decreasing the activity of the basolateral NBCn1, which was reported to play an indirect role in NH4+ reabsorption in the mTAL (71). In the collecting duct, the inhibition of AE1 (61) and CA by ACTZ could affect H+ production and secretion, which in turn could interfere with NH4+ secretion by type A intercalated cells (71). We should emphasize that our present experiments were conducted in male rats, and whether the conclusions drawn from these studies can be extrapolated to female gender is uncertain.
In addition to NBCe1, CAII was also shown to physically interact with the apical Na+/H+ exchanger NHE3 and increase its activity (32). However, the maximal interaction occurs only at alkaline pH (32), whereas the maximal activity of NHE3 is reached at acidic pH. Hence, the physiologic implication of this interaction is less clear. Moreover, studies showed that ammonia synthesis and excretion were not altered in mice with proximal tubule-specific deletion of NHE3 (39). Hence, it is unlikely that ACTZ inhibits NH4+ synthesis and secretion as a result of NHE3 inhibition, at least, during the first week of ACTZ treatment.
In conclusion, ACTZ causes renal wasting and induces metabolic acidosis but inhibits the stimulation of ammoniagenesis in the proximal tubule. ACTZ prevented the upregulation of glutamine transporter SNAT3 and ammoniagenic enzymes (GA and GDH) as well as acid-base transporters (NHE3 and NBCe1) despite a significant acidemia. This effect is likely mediated through the inhibition of the activity of CA-NBCe1 metabolon complex and resulting cell alkalinization. The downregulation of both NBCe1 and NHE3 along with the inhibition of ammoniagenesis and generation contribute to the maintenance of metabolic acidosis during chronic ACTZ treatment.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-083582 and academic development funds from the College of Medicine and Department of Internal Medicine, University of Cincinnati, and Dialysis Clinic (C-4095) to H. Amlal.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.A. and H.A. conceived and designed research; P.A., S.A., and H.A. performed experiments; P.A., S.A., C.V.T., and H.A. analyzed data; P.A., S.A., C.V.T., and H.A. interpreted results of experiments; P.A. and H.A. prepared figures; H.A. drafted manuscript; P.A., S.A., C.V.T., and H.A. edited and revised manuscript; P.A., S.A., C.V.T., and H.A. approved final version of manuscript.
Footnotes
ACTZ-V1 refers to ACTZ dissolved in vehicle 1 (1× DMSO + 4× sunflower oil). ACTZ-V2 refers to ACTZ dissolved in vehicle 2 (1× DMSO + 4× 1,2-propanediol). Vehicle refers to vehicle 1.
The ammonia excretion rates do not always equal ammonia production (ammoniagenesis) rates. To actually measure renal ammonia production rates is difficult and involves measuring the rates of ammonia excretion into the urine and the rates at which ammonia leaves the kidneys via the renal veins. There was a correlation between the protein levels of SNAT3 and ammoniagenic enzymes and ammonium excretion rates, which suggests that differences in the rates of renal ammoniagenesis were likely to have played a role in differences in rates of urinary ammonia excretion.
REFERENCES
- 1.Abu Hossain S, Chaudhry FA, Zahedi K, Siddiqui F, Amlal H. Cellular and molecular basis of increased ammoniagenesis in potassium deprivation. Am J Physiol Renal Physiol 301: F969–F978, 2011. doi: 10.1152/ajprenal.00010.2011. [DOI] [PubMed] [Google Scholar]
- 2.Alper SL. Familial renal tubular acidosis. J Nephrol 23, Suppl 16: S57–S76, 2010. [PubMed] [Google Scholar]
- 3.Amlal H, Paillard M, Bichara M. NH4+ transport pathways in cells of medullary thick ascending limb of rat kidney. conductance and K+/(H+) antiport. J Biol Chem 269: 21962–21971, 1994. [PubMed] [Google Scholar]
- 4.Amlal H, Sheriff S, Soleimani M. Upregulation of collecting duct aquaporin-2 by metabolic acidosis: role of vasopressin. Am J Physiol Cell Physiol 286: C1019–C1030, 2004. doi: 10.1152/ajpcell.00394.2003. [DOI] [PubMed] [Google Scholar]
- 5.Attmane-Elakeb A, Mount DB, Sibella V, Vernimmen C, Hebert SC, Bichara M. Stimulation by in vivo and in vitro metabolic acidosis of expression of rBSC-1, the Na+-K+()-2Cl− cotransporter of the rat medullary thick ascending limb. J Biol Chem 273: 33681–33691, 1998. doi: 10.1074/jbc.273.50.33681. [DOI] [PubMed] [Google Scholar]
- 6.Balkovetz DF, Chumley P, Amlal H. Downregulation of claudin-2 expression in renal epithelial cells by metabolic acidosis. Am J Physiol Renal Physiol 297: F604–F611, 2009. doi: 10.1152/ajprenal.00043.2009. [DOI] [PubMed] [Google Scholar]
- 7.Beaton JR. The inhibition by acetazoleamide of renal phosphate-activated glutaminase in rats. Can J Biochem Physiol 39: 663–669, 1961. doi: 10.1139/o61-067. [DOI] [PubMed] [Google Scholar]
- 8.Bignon Y, Pinelli L, Frachon N, Lahuna O, Figueres L, Houillier P, Lourdel S, Teulon J, Paulais M. Defective bicarbonate reabsorption in Kir4.2 potassium channel deficient mice impairs acid-base balance and ammonia excretion. Kidney Int 97: 304–315, 2020. doi: 10.1016/j.kint.2019.09.028. [DOI] [PubMed] [Google Scholar]
- 9.Brenes LG, Sanchez MI. Impaired urinary ammonium excretion in patients with isolated proximal renal tubular acidosis. J Am Soc Nephrol 4: 1073–1078, 1993. [DOI] [PubMed] [Google Scholar]
- 10.Brown D, Wagner CA. Molecular mechanisms of acid-base sensing by the kidney. J Am Soc Nephrol 23: 774–780, 2012. doi: 10.1681/ASN.2012010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burris D, Webster R, Sheriff S, Faroqui R, Levi M, Hawse JR, Amlal H. Estrogen directly and specifically downregulates NaPi-IIa through the activation of both estrogen receptor isoforms (ERα and ERβ) in rat kidney proximal tubule. Am J Physiol Renal Physiol 308: F522–F534, 2015. doi: 10.1152/ajprenal.00386.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busque SM, Wagner CA. Potassium restriction, high protein intake, and metabolic acidosis increase expression of the glutamine transporter SNAT3 (Slc38a3) in mouse kidney. Am J Physiol Renal Physiol 297: F440–F450, 2009. doi: 10.1152/ajprenal.90318.2008. [DOI] [PubMed] [Google Scholar]
- 13.Bürki R, Mohebbi N, Bettoni C, Wang X, Serra AL, Wagner CA. Impaired expression of key molecules of ammoniagenesis underlies renal acidosis in a rat model of chronic kidney disease. Nephrol Dial Transplant 30: 770–781, 2015. doi: 10.1093/ndt/gfu384. [DOI] [PubMed] [Google Scholar]
- 14.Chapman SK, Hoover MS. Acetazolamide and renal ammoniagenesis. Am J Physiol Renal Physiol 234: F235–F237, 1978. [DOI] [PubMed] [Google Scholar]
- 15.Curthoys NP, Gstraunthaler G. Mechanism of increased renal gene expression during metabolic acidosis. Am J Physiol Renal Physiol 281: F381–F390, 2001. doi: 10.1152/ajprenal.2001.281.3.F381. [DOI] [PubMed] [Google Scholar]
- 16.Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol 9: 1627–1638, 2014. doi: 10.2215/CJN.10391012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elkinton JR, Huth EJ, Webster GD Jr, McCance RA. The renal excretion of hydrogen ion in renal tubular acidosis. I. Quantitative assessment of the response to ammonium chloride as an acid load. Am J Med 29: 554–575, 1960. doi: 10.1016/0002-9343(60)90090-5. [DOI] [PubMed] [Google Scholar]
- 18.Faroqui S, Sheriff S, Amlal H. Metabolic acidosis has dual effects on sodium handling by rat kidney. Am J Physiol Renal Physiol 291: F322–F331, 2006. doi: 10.1152/ajprenal.00338.2005. [DOI] [PubMed] [Google Scholar]
- 19.Gawenis LR, Bradford EM, Prasad V, Lorenz JN, Simpson JE, Clarke LL, Woo AL, Grisham C, Sanford LP, Doetschman T, Miller ML, Shull GE. Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/ cotransporter. J Biol Chem 282: 9042–9052, 2007. doi: 10.1074/jbc.M607041200. [DOI] [PubMed] [Google Scholar]
- 20.Good DW. Adaptation of and transport in rat MTAL: effects of chronic metabolic acidosis and Na+ intake. Am J Physiol Renal Physiol 258: F1345–F1353, 1990. doi: 10.1152/ajprenal.1990.258.5.F1345. [DOI] [PubMed] [Google Scholar]
- 21.Good DW, Burg MB. Ammonia production by individual segments of the rat nephron. J Clin Invest 73: 602–610, 1984. doi: 10.1172/JCI111250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross E, Pushkin A, Abuladze N, Fedotoff O, Kurtz I. Regulation of the sodium bicarbonate cotransporter kNBC1 function: role of Asp986, Asp988 and kNBC1-carbonic anhydrase II binding. J Physiol 544: 679–685, 2002. doi: 10.1113/jphysiol.2002.029777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamm LL, Nakhoul N, Hering-Smith KS. Acid-base homeostasis. Clin J Am Soc Nephrol 10: 2232–2242, 2015. doi: 10.2215/CJN.07400715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Handlogten ME, Osis G, Lee HW, Romero MF, Verlander JW, Weiner ID. NBCe1 expression is required for normal renal ammonia metabolism. Am J Physiol Renal Physiol 309: F658–F666, 2015. doi: 10.1152/ajprenal.00219.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heming N, Urien S, Faisy C. Acetazolamide: a second wind for a respiratory stimulant in the intensive care unit? Crit Care 16: 318, 2012. doi: 10.1186/cc11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karinch AM, Lin CM, Wolfgang CL, Pan M, Souba WW. Regulation of expression of the SN1 transporter during renal adaptation to chronic metabolic acidosis in rats. Am J Physiol Renal Physiol 283: F1011–F1019, 2002. doi: 10.1152/ajprenal.00106.2002. [DOI] [PubMed] [Google Scholar]
- 27.Kim HY, Han JS, Jeon US, Joo KW, Earm JH, Ahn C, Kim S, Lee JS, Kim GH. Clinical significance of the fractional excretion of anions in metabolic acidosis. Clin Nephrol 55: 448–452, 2001. [PubMed] [Google Scholar]
- 28.Kinsella JL, Aronson PS. Interaction of and Li+ with the renal microvillus membrane Na+-H+ exchanger. Am J Physiol Cell Physiol 241: C220–C226, 1981. doi: 10.1152/ajpcell.1981.241.5.C220. [DOI] [PubMed] [Google Scholar]
- 29.Klier M, Andes FT, Deitmer JW, Becker HM. Intracellular and extracellular carbonic anhydrases cooperate non-enzymatically to enhance activity of monocarboxylate transporters. J Biol Chem 289: 2765–2775, 2014. doi: 10.1074/jbc.M113.537043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knepper MA, Packer R, Good DW. Ammonium transport in the kidney. Physiol Rev 69: 179–249, 1989. doi: 10.1152/physrev.1989.69.1.179. [DOI] [PubMed] [Google Scholar]
- 31.Koeppen BM. The kidney and acid-base regulation. Adv Physiol Educ 33: 275–281, 2009. doi: 10.1152/advan.00054.2009. [DOI] [PubMed] [Google Scholar]
- 32.Krishnan D, Liu L, Wiebe SA, Casey JR, Cordat E, Alexander RT. Carbonic anhydrase II binds to and increases the activity of the epithelial sodium-proton exchanger, NHE3. Am J Physiol Renal Physiol 309: F383–F392, 2015. doi: 10.1152/ajprenal.00464.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurtz I, Zhu Q. Structure, function, and regulation of the SLC4 NBCe1 transporter and its role in causing proximal renal tubular acidosis. Curr Opin Nephrol Hypertens 22: 572–583, 2013. doi: 10.1097/MNH.0b013e328363ff43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laghmani K, Preisig PA, Moe OW, Yanagisawa M, Alpern RJ. Endothelin-1/endothelin-B receptor-mediated increases in NHE3 activity in chronic metabolic acidosis. J Clin Invest 107: 1563–1569, 2001. doi: 10.1172/JCI11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leaf DE, Goldfarb DS. Mechanisms of action of acetazolamide in the prophylaxis and treatment of acute mountain sickness. J Appl Physiol 102: 1313–1322, 2007. doi: 10.1152/japplphysiol.01572.2005. [DOI] [PubMed] [Google Scholar]
- 36.Lee HW, Osis G, Handlogten ME, Lamers WH, Chaudhry FA, Verlander JW, Weiner ID. Proximal tubule-specific glutamine synthetase deletion alters basal and acidosis-stimulated ammonia metabolism. Am J Physiol Renal Physiol 310: F1229–F1242, 2016. doi: 10.1152/ajprenal.00547.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lemann J Jr, Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int 58: 1267–1277, 2000. doi: 10.1046/j.1523-1755.2000.00282.x. [DOI] [PubMed] [Google Scholar]
- 38.Leonard E, Orloff J. Regulation of ammonia excretion in the rat. Am J Physiol 182: 131–138, 1955. doi: 10.1152/ajplegacy.1955.182.1.131. [DOI] [PubMed] [Google Scholar]
- 39.Li HC, Du Z, Barone S, Rubera I, McDonough AA, Tauc M, Zahedi K, Wang T, Soleimani M. Proximal tubule specific knockout of the Na+/H+ exchanger NHE3: effects on bicarbonate absorption and ammonium excretion. J Mol Med (Berl) 91: 951–963, 2013. doi: 10.1007/s00109-013-1015-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lister A, Bourgeois S, Imenez Silva PH, Rubio-Aliaga I, Marbet P, Walsh J, Shelton LM, Keller B, Verrey F, Devuyst O, Giesbertz P, Daniel H, Goldring CE, Copple IM, Wagner CA, Odermatt A. NRF2 regulates the glutamine transporter Slc38a3 (SNAT3) in kidney in response to metabolic acidosis. Sci Rep 8: 5629, 2018. doi: 10.1038/s41598-018-24000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.López C, Alcaraz AJ, Toledo B, Cortejoso L, Gil-Ruiz MA. Acetazolamide therapy for metabolic alkalosis in pediatric intensive care patients. Pediatr Crit Care Med 17: e551–e558, 2016. doi: 10.1097/PCC.0000000000000971. [DOI] [PubMed] [Google Scholar]
- 42.Nadell J, Kalinsky H. The effects of the carbonic anhydrase inhibitor 6063 on electrolytes and acid-base balance in two normal subjects and two patients with respiratory acidosis. J Clin Invest 32: 622–629, 1953. doi: 10.1172/JCI102773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagami GT. Ammonia production and secretion by S3 proximal tubule segments from acidotic mice: role of ANG II. Am J Physiol Renal Physiol 287: F707–F712, 2004. doi: 10.1152/ajprenal.00189.2003. [DOI] [PubMed] [Google Scholar]
- 44.Nagami GT, Chang JA, Plato ME, Santamaria R. Acid loading in vivo and low pH in culture increase angiotensin receptor expression: enhanced ammoniagenic response to angiotensin II. Am J Physiol Renal Physiol 295: F1864–F1870, 2008. doi: 10.1152/ajprenal.90410.2008. [DOI] [PubMed] [Google Scholar]
- 45.Nonoguchi H, Uchida S, Shiigai T, Endou H. Effect of chronic metabolic acidosis on ammonia production from l-glutamine in microdissected rat nephron segments. Pflugers Arch 403: 229–235, 1985. doi: 10.1007/BF00583592. [DOI] [PubMed] [Google Scholar]
- 46.Nowik M, Picard N, Stange G, Capuano P, Tenenhouse HS, Biber J, Murer H, Wagner CA. Renal phosphaturia during metabolic acidosis revisited: molecular mechanisms for decreased renal phosphate reabsorption. Pflugers Arch 457: 539–549, 2008. doi: 10.1007/s00424-008-0530-5. [DOI] [PubMed] [Google Scholar]
- 47.Owen EE, Tyor MP, Flanagan JF, Berry JN. The kidney as a source of blood ammonia in patients with liver disease: the effect of acetazolamide. J Clin Invest 39: 288–294, 1960. doi: 10.1172/JCI104039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 19: 199–229, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Pathare G, Dhayat N, Mohebbi N, Wagner CA, Cheval L, Neuhaus TJ, Fuster DG. Acute regulated expression of pendrin in human urinary exosomes. Pflugers Arch 470: 427–438, 2018. doi: 10.1007/s00424-017-2049-0. [DOI] [PubMed] [Google Scholar]
- 50.Petrovic S, Wang Z, Ma L, Soleimani M. Regulation of the apical Cl−/ exchanger pendrin in rat cortical collecting duct in metabolic acidosis. Am J Physiol Renal Physiol 284: F103–F112, 2003. doi: 10.1152/ajprenal.00205.2002. [DOI] [PubMed] [Google Scholar]
- 51.Phenix P, Welbourne TC. Renal glutaminases: diamox inhibition of glutamyltransferase. Am J Physiol 228: 1269–1275, 1975. doi: 10.1152/ajplegacy.1975.228.4.1269. [DOI] [PubMed] [Google Scholar]
- 52.Preisig PA, Alpern RJ. Chronic metabolic acidosis causes an adaptation in the apical membrane Na/H antiporter and basolateral membrane Na(HCO3)3 symporter in the rat proximal convoluted tubule. J Clin Invest 82: 1445–1453, 1988. doi: 10.1172/JCI113750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purkerson JM, Schwartz GJ. The role of carbonic anhydrases in renal physiology. Kidney Int 71: 103–115, 2007. doi: 10.1038/sj.ki.5002020. [DOI] [PubMed] [Google Scholar]
- 54.Pushkin A, Abuladze N, Gross E, Newman D, Tatishchev S, Lee I, Fedotoff O, Bondar G, Azimov R, Ngyuen M, Kurtz I. Molecular mechanism of kNBC1-carbonic anhydrase II interaction in proximal tubule cells. J Physiol 559: 55–65, 2004. doi: 10.1113/jphysiol.2004.065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Risquez A, Hernandez W, Preuss HG. Effects of acetazolamide on renal ammoniagenesis in dogs. Kidney Blood Press Res 2: 205–213, 1979. doi: 10.1159/000172710. [DOI] [Google Scholar]
- 56.Schroeder JM, Liu W, Curthoys NP. pH-responsive stabilization of glutamate dehydrogenase mRNA in LLC-PK1-F+ cells. Am J Physiol Renal Physiol 285: F258–F265, 2003. doi: 10.1152/ajprenal.00422.2002. [DOI] [PubMed] [Google Scholar]
- 57.Schueler C, Becker HM, McKenna R, Deitmer JW. Transport activity of the sodium bicarbonate cotransporter NBCe1 is enhanced by different isoforms of carbonic anhydrase. PLoS One 6: e27167, 2011. doi: 10.1371/journal.pone.0027167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seldin DW, Rector FC Jr, Teng HC. Effects of prolonged administration of diamox on excretion of acid and carbonic anhydrase and glutaminase activities in the kidney. Am J Physiol 189: 551–556, 1957. doi: 10.1152/ajplegacy.1957.189.3.551. [DOI] [PubMed] [Google Scholar]
- 59.Solbu TT, Boulland JL, Zahid W, Lyamouri Bredahl MK, Amiry-Moghaddam M, Storm-Mathisen J, Roberg BA, Chaudhry FA. Induction and targeting of the glutamine transporter SN1 to the basolateral membranes of cortical kidney tubule cells during chronic metabolic acidosis suggest a role in pH regulation. J Am Soc Nephrol 16: 869–877, 2005. doi: 10.1681/ASN.2004060433. [DOI] [PubMed] [Google Scholar]
- 60.Sterling D, Brown NJ, Supuran CT, Casey JR. The functional and physical relationship between the DRA bicarbonate transporter and carbonic anhydrase II. Am J Physiol Cell Physiol 283: C1522–C1529, 2002. doi: 10.1152/ajpcell.00115.2002. [DOI] [PubMed] [Google Scholar]
- 61.Sterling D, Reithmeier RA, Casey JR. A transport metabolon. Functional interaction of carbonic anhydrase II and chloride/bicarbonate exchangers. J Biol Chem 276: 47886–47894, 2001. doi: 10.1074/jbc.M105959200. [DOI] [PubMed] [Google Scholar]
- 62.Tang A, Curthoys NP. Identification of ζ-crystallin/NADPH:quinone reductase as a renal glutaminase mRNA pH response element-binding protein. J Biol Chem 276: 21375–21380, 2001. doi: 10.1074/jbc.M101941200. [DOI] [PubMed] [Google Scholar]
- 63.Tannen RL, Ross BD. The impact of acetazolamide on renal ammoniagenesis and gluconeogenesis. J Lab Clin Med 102: 536–542, 1983. [PubMed] [Google Scholar]
- 64.Taylor L, Curthoys NP. Glutamine metabolism: role in acid-base balance*. Biochem Mol Biol Educ 32: 291–304, 2004. doi: 10.1002/bmb.2004.494032050388. [DOI] [PubMed] [Google Scholar]
- 65.Thornley-Brown D, Dass PD, Welbourne TC. Acetazolamide inhibition of renal gamma-glutamyl transpeptidase. Biochem Pharmacol 31: 3347–3352, 1982. doi: 10.1016/0006-2952(82)90610-4. [DOI] [PubMed] [Google Scholar]
- 66.Tsuruoka S, Swenson ER, Petrovic S, Fujimura A, Schwartz GJ. Role of basolateral carbonic anhydrase in proximal tubular fluid and bicarbonate absorption. Am J Physiol Renal Physiol 280: F146–F154, 2001. doi: 10.1152/ajprenal.2001.280.1.F146. [DOI] [PubMed] [Google Scholar]
- 67.Van Berkel MA, Elefritz JL. Evaluating off-label uses of acetazolamide. Am J Health Syst Pharm 75: 524–531, 2018. doi: 10.2146/ajhp170279. [DOI] [PubMed] [Google Scholar]
- 68.Vince JW, Reithmeier RA. Identification of the carbonic anhydrase II binding site in the Cl−/ anion exchanger AE1. Biochemistry 39: 5527–5533, 2000. doi: 10.1021/bi992564p. [DOI] [PubMed] [Google Scholar]
- 69.Webster R, Sheriff S, Faroqui R, Siddiqui F, Hawse JR, Amlal H. Klotho/fibroblast growth factor 23- and PTH-independent estrogen receptor-α-mediated direct downregulation of NaPi-IIa by estrogen in the mouse kidney. Am J Physiol Renal Physiol 311: F249–F259, 2016. doi: 10.1152/ajprenal.00542.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weiner ID, Verlander JW. Ammonia transporters and their role in acid-base balance. Physiol Rev 97: 465–494, 2017. doi: 10.1152/physrev.00011.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weiner ID, Verlander JW. Emerging features of ammonia metabolism and transport in acid-base balance. Semin Nephrol 39: 394–405, 2019. doi: 10.1016/j.semnephrol.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]