

To the Editor: Here we introduce Philosopher (https://philosopher.nesvilab.org), a free, open-source, versatile, and robust data analysis toolkit designed to bring easy access to a powerful and comprehensive set of computational tools for shotgun proteomics data analysis.
Computational analysis is a central component of any modern experiment, and mass spectrometry-based proteomics is no exception. As technologies continue to rapidly advance with respect to throughput and sensitivity, bioinformatics tools must keep pace with large-scale experiments. While existing proteomics tools, such as the Trans-Proteomic Pipeline (TPP)1, MaxQuant2, and PeptideShaker3 are capable of performing high-quality analyses, all require installation and are dependent upon specific operating systems, libraries, and other software. Managing these tools can be a daunting task, even for research groups with substantial bioinformatics expertise. This is particularly true when experiments demand high performance configurations such as GNU/Linux clusters or cloud computing. To address this challenge, we initially built and deployed Docker containers with different applications for proteomics, which in part inspired the creation of the BioContainers resource for different bioinformatics fields4. Though efficient for packing and sharing resources, we found that chaining different applications with custom implementation of established algorithms in a transparent and dependency-free way was still a challenge for containerization. The Philosopher toolkit integrates high-performance algorithms and existing tools (Fig. 1), and is a dependency-free, fast, and comprehensive proteomics pipeline, able to rapidly process even the most complex proteomics data sets with efficient resource management.
Figure 1. Philosopher provides easy access to a complete suite of computational proteomics tools.
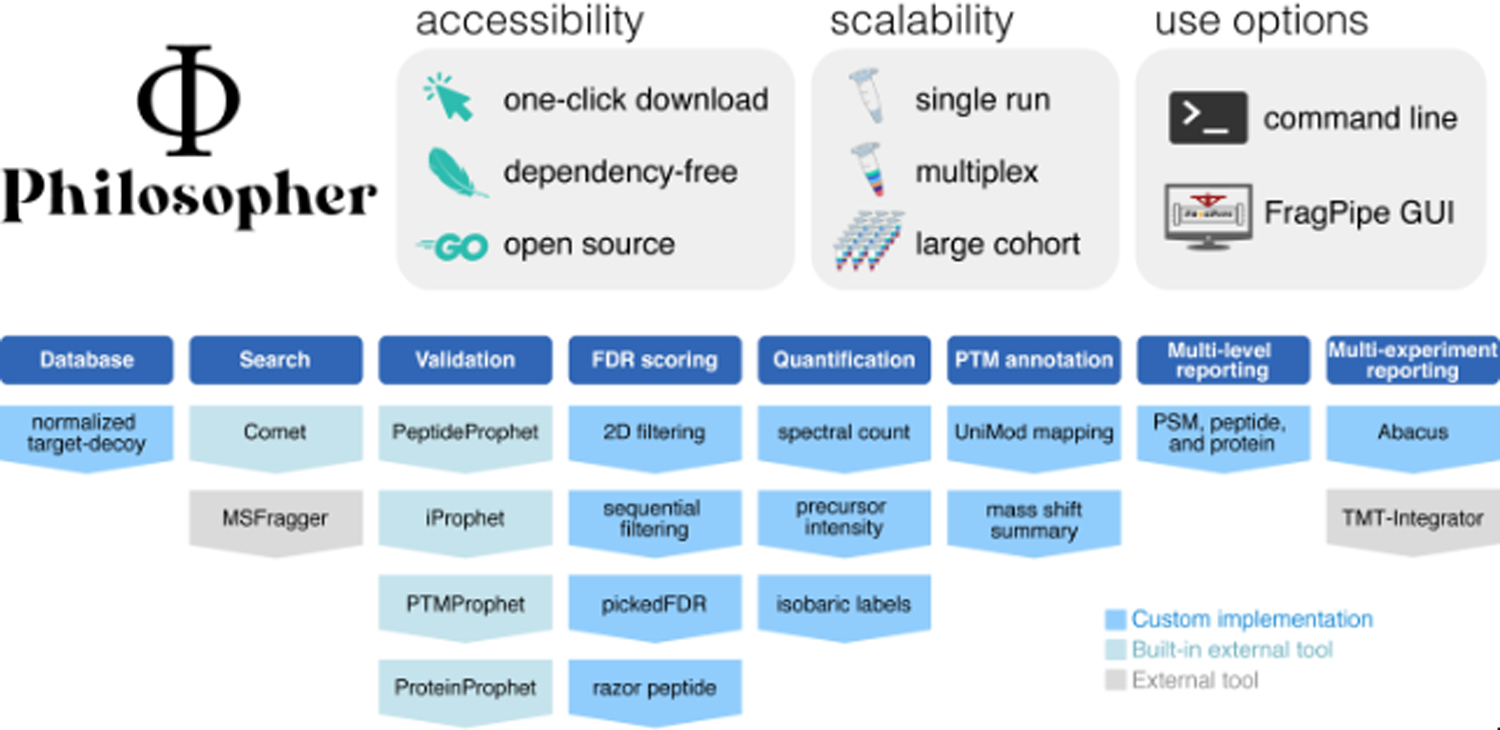
Functions include protein sequence database download and formatting, database searching, peptide-spectrum match validation with PeptideProphet, peptide evidence integration with iProphet, post-translational modification (PTM) localization with PTMProphet, and protein inference with ProteinProphet. Philosopher has multiple options for FDR scoring; performs label-free and labeling-based quantification, PTM annotation, generation of multi-level reports, and quantification comparison between samples. MSFragger and TMT-Integrator must be downloaded separately.
Philosopher includes the database search engine Comet and can use the high-performance search engine MSFragger5 as a separately-downloaded tool. For downstream processing of peptide-spectrum matches (PSMs), Philosopher includes key components of TPP. In addition, it implements best practices for False Discovery Rate (FDR) filtering and data summarization that are not readily available within the TPP, such as picked FDR, two-dimensional or sequential (at PSM and protein levels) filters, and additional options for dealing with peptides whose sequence is present in multiple proteins (e.g. razor peptide approach). As quantification is frequently the goal of modern proteomics experiments, Philosopher includes algorithms for both label-free quantification and isobaric label-based quantification (TMT or iTRAQ). Precursor spectral intensities are retrieved following a method described previously6. Protein-level quantification is estimated using the sum of the three most intense supporting ions. Alternatively, Philosopher can use TMT-Integrator (http://tmt-integrator.nesvilab.org/) as an external tool or output files can be used with downstream quantification and statistical tools such as MSstats7. The rich reports generated by Philosopher are also compatible with other software such as PDV for visualization of peptide assignments to MS/MS spectra8 and CRAPome/REPRINT (https://reprint-apms.org/) for interactome scoring and network visualization using affinity purification mass spectrometry data.
Philosopher is scalable from laptops and desktops to high-performance servers, and can be incorporated in workflow managers such as Galaxy-P9. The power of Philosopher is well illustrated in the recent CPTAC3 consortium publication, where over 800 spectral files from a clear cell Renal Cell Carcinoma cohort, whole proteome and phosphoproteome data, were analyzed in less than 24 hours each with Philosopher and MSFragger on a single Linux server10. While Philosopher is most flexible as a command line tool, its core functions can easily be accessed through our widely used graphical user interface FragPipe (https://fragpipe.nesvilab.org/). In summary, installation and management of computational proteomics tools can be a daunting task, particularly when workflows need to be repeated on different operating systems or scaled from desktop to servers or cloud systems. Philosopher is a versatile and easy-to-use cross-platform toolkit for streamlined proteomics analysis that does not require installation or configuration and can perform full analyses (from spectral files in open mzML/mzXML format to peptide and protein reports) in a single command.
Acknowledgements
We thank the developers of PDV and the TPP tools for useful discussions and technical assistance, and the growing community of Philosopher users for their feedback and suggestions. This work was supported in part by NIH Grants R01-GM-094231 (A.I.N) and U24-CA210967 (A.I.N.).
Footnotes
Code Availability
The source code for the project can be found at https://philosopher.nesvilab.org under the GNU General Public License version 3, along with documentation and tutorials covering various use cases. The DOI for Philosopher (version 3.2.9) is 10.5281/zenodo.3909842.
Competing Interests
The authors declare no competing interests.
References
- 1.Deutsch EW et al. A guided tour of the Trans‐Proteomic Pipeline. Proteomics 10, 1150–1159 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox J & Mann M MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nature biotechnology 26, 1367 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Vaudel M et al. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nature biotechnology 33, 22–24 (2015). [DOI] [PubMed] [Google Scholar]
- 4.da Veiga Leprevost F et al. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics 33, 2580–2582 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D & Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nature methods 14, 513 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Argentini A et al. moFF: a robust and automated approach to extract peptide ion intensities. Nature methods 13, 964–966 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Choi M et al. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Li K, Vaudel M, Zhang B, Ren Y & Wen B PDV: an integrative proteomics data viewer. Bioinformatics 35, 1249–1251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blank C et al. Disseminating metaproteomic informatics capabilities and knowledge using the Galaxy-P framework. Proteomes 6, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark DJ et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 179, 964–983. e931 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]