

Key Points
Teclistamab is a BCMAxCD3 bispecific antibody that can induce T cell–mediated killing of BCMA+ cells.
Teclistamab is currently in a phase 1 clinical trial in patients with relapsed/refractory MM.
Abstract
B-cell maturation antigen (BCMA), a member of the tumor necrosis factor family of receptors, is predominantly expressed on the surface of terminally differentiated B cells. BCMA is highly expressed on plasmablasts and plasma cells from multiple myeloma (MM) patient samples. We developed a BCMAxCD3 bispecific antibody (teclistamab [JNJ-64007957]) to recruit and activate T cells to kill BCMA-expressing MM cells. Teclistamab induced cytotoxicity of BCMA+ MM cell lines in vitro (H929 cells, 50% effective concentration [EC50] = 0.15 nM; MM.1R cells, EC50 = 0.06 nM; RPMI 8226 cells, EC50 = 0.45 nM) with concomitant T-cell activation (H929 cells, EC50 = 0.21 nM; MM.1R cells, EC50 = 0.1 nM; RPMI 8226 cells, EC50 = 0.28 nM) and cytokine release. This activity was further increased in the presence of a γ-secretase inhibitor (LY-411575). Teclistamab also depleted BCMA+ cells in bone marrow samples from MM patients in an ex vivo assay with an average EC50 value of 1.7 nM. Under more physiological conditions using healthy human whole blood, teclistamab mediated dose-dependent lysis of H929 cells and activation of T cells. Antitumor activity of teclistamab was also observed in 2 BCMA+ MM murine xenograft models inoculated with human T cells (tumor inhibition with H929 model and tumor regression with the RPMI 8226 model) compared with vehicle and antibody controls. The specific and potent activity of teclistamab against BCMA-expressing cells from MM cell lines, patient samples, and MM xenograft models warrant further evaluation of this bispecific antibody for the treatment of MM. Phase 1 clinical trials (monotherapy, #NCT03145181; combination therapy, #NCT04108195) are ongoing for patients with relapsed/refractory MM.
Visual Abstract

Introduction
Multiple myeloma (MM) is a malignant plasma cell disorder that leads to clonal proliferation of terminally differentiated plasma cells in the bone marrow (BM) and accounts for ∼10% of all hematologic cancers.1 MM is characterized by overproduction of M protein, which can lead to bone lesions, increased susceptibility to infections, anemia, hypercalcemia, and renal insufficiency.2 Within the past decade, the introduction of proteasome inhibitors, immunomodulatory drugs, and monoclonal antibodies has changed the landscape of myeloma management, leading to improved disease control and prolonged survival.3-12 Despite these therapeutic advances, nearly all patients will eventually relapse and become refractory to available therapies.4,13 Given the poor prognosis and limited treatment options in the relapsed/refractory disease setting, novel therapeutic approaches for MM are needed.
B-cell maturation antigen (BCMA, CD269, TNFRSF17) is a 20 kDa receptor that is selectively expressed in the B-cell lineage and is also widely expressed on MM cells (in addition to smoldering MM and monoclonal gammopathy of undetermined significance).14-16 Upon binding to its ligands, a proliferation-inducing ligand (APRIL; CD256) and BAFF (CD257), BCMA activates p38/NF-κB and induces upregulation of antiapoptotic proteins to regulate B-cell maturation, proliferation, and survival.16-20 Increased levels of a soluble form of BCMA (sBCMA), produced through cleavage at the transmembrane domain by γ-secretase, have been correlated with disease progression and shorter overall survival in patients with MM.21 Altogether, these findings support targeting BCMA for novel treatment approaches for MM.
Key factors for a successful T cell–redirecting therapeutic include selective target expression on the tumor cells with minimal to no expression in other tissues and a potent molecule that can eliminate malignant cells to achieve long-term benefit. Therapeutic approaches such as chimeric antigen receptor T-cell therapies and bispecific T-cell engagers that use T cell–mediated cytotoxicity to target BCMA on plasma cells have shown deep responses in patients with relapsed or refractory disease.21-25 Another class of T-cell redirecting therapy in development for MM is bispecific antibodies. Teclistamab is a humanized immunoglobulin G4-proline, alanine, alanine (IgG-4 PAA) bispecific DuoBody antibody (Genmab). It is hypothesized that teclistamab will induce T cell–mediated cytotoxicity through recruitment of CD3-expressing T cells to BCMA-expressing cells, which will lead to the activation of T cells and subsequent target cell lysis mediated by secreted perforin and various granzymes stored in the secretory vesicles of cytotoxic T cells. The current study evaluated the potential efficacy of teclistamab by using in vitro, ex vivo, and in vivo models of MM.
Materials and methods
Cell lines and cell culture
All cell lines used were of human origin and obtained from either ATCC or DSMZ. Cell lines were cultured in RPMI 1640 medium with 10% fetal bovine serum without antibiotics at 37°C in a 5% carbon dioxide incubator.
Teclistamab (JNJ-64007957) generation
OmniRats (Open Monoclonal Technology) were immunized with BCMA-Fc recombinant protein (R&D Systems) to generate anti-BCMA antibodies, and hits were re-cloned on a relatively silent IgG4-PAA scaffold. The DuoBody antibody (JNJ-64007957 or teclistamab) was generated by controlled Fab-arm exchange of a BCMA antibody and a CD3 parental antibody derived from SP34 clone26 following the method developed by Genmab.27 Null arm controls were generated by controlled Fab-arm exchange of mouse anti-human respiratory syncytial virus neutralizing antibody (Null) with anti-CD3 antibody (NullxCD3) or anti-BCMA antibody (BCMAxNull).28
Flow cytometry analysis of BCMA expression
Human BM mononuclear cells (BM MNCs; ProteoGenex) and MM cell lines (1 × 106) were stained in Live/Dead staining solution (Life Technologies) followed by 5 µg/mL of anti-BCMA antibody (clone 19F2), or corresponding isotype (clone MOPC-173) or anti-CD138 (clones MI15 and DL-101; BioLegend) antibodies in fluorescence-activated cell sorting (FACS) stain buffer. Cells were incubated at 4°C for 60 minutes, washed twice, and analyzed on a BD FACSCanto II cytometer (Becton Dickinson). The BD QuantiBRITE PE Beads kit (BD Biosciences) was used to quantify receptor density by measuring the geometric mean fluorescence intensity of BCMA staining on cells.
Healthy donor whole blood flow cytometry analysis
Fresh peripheral blood from 3 healthy human donors (Janssen donor program) was pipetted into 96-well U-bottom plates in 100 µL aliquots. Cell lineage antibodies were added directly to blood, along with teclistamab, BCMAxNull, NullxCD3, or isotype control antibodies. After 30 minutes of incubation at room temperature and 4 rounds of red blood cell lysis, samples were stained with Live/Dead Near-IR Stain and 1:50 anti-human IgG4-phycoerythrin for 15 minutes at room temperature and analyzed. Cell types were gated by using specific markers (B-cell markers, CD3– and CD19+; natural killer [NK] cell markers, side scatter [SSC]lo, CD3–, CD19–, CD16+, and CD56+; NK T-cell markers, SSClo, CD3+, CD56+, and CD16–; CD4 T-cell markers, CD3+ and CD4+; CD8 T-cell markers, CD3+ and CD8+; monocyte markers, SSChi and CD14+; and neutrophil markers, SSChi, CD45+, CD16+, and CD11b+).
T cell–mediated cytotoxicity and T-cell activation using MM cell lines and healthy donor T-cells
Tumor cell lines (H929, MM.1R, RPMI 8226, and MV4-11) were incubated in 0.5 µM CellTrace CFSE (Life Technologies) for 8 minutes, washed twice, and resuspended at 2.22 × 105 cells/mL in complete medium containing 2 mg/mL of Fc block.29 Assay was established by using 90 µL (2 × 104) of target cells and 90 µL (1 × 105) of freshly thawed normal donor pan-T cells along with 20 µL of 10× concentration of antibody dilutions, and they were incubated at 37°C with 5% carbon dioxide for 48 hours in a 96-well U-bottom plate. After the incubation, plates were spun at 1500 rpm for 3 minutes, and the supernatants were collected for cytokine analysis. The cell pellets were stained with Live/Dead solution and phenotyping antibodies (CD25; BioLegend) for 20 minutes. Samples were analyzed by using BD FACSCanto II. For cytotoxicity, the percentage of dead cells in CFSE+ cells was measured; for T-cell activation, the percentage of CD25+ cells in live CFSE– cells was measured.
T cell–mediated cytotoxicity and T-cell activation using H929 target cells and human healthy whole blood
Healthy human whole blood was collected into heparinized tubes, and the frequency of CD3+ T cells for each donor was calibrated by using labeled H929 cells. The experiment was established by using 90 µL of fresh whole blood, 90 µL of CellTrace carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled and diluted H929 cells to achieve a 5:1 effector to target (E:T) ratio, and 20 µL of teclistamab antibody dilutions per well. Plates were incubated at 37°C for 48 hours, and cells were stained with anti-CD3 and anti-CD25 antibodies for 20 minutes. After the red blood cell lysis, the cells were labeled with Live/Dead stain for 15 minutes, and percent CD25+ events in live CD3+ cells and percent live events in CFSE-labeled target cells were analyzed by using flow cytometry.
T cell–mediated cytotoxicity and T-cell activation using CD138+ BM cells from patients and healthy donor T cells
MM patient BM MNCs (ProteoGenex) and T cells were incubated with 100 ng/mL of interleukin (IL)-6 and teclistamab or control antibody for 48 hours. Samples were spun; washed; labeled with Live/Dead stain, anti-CD3, anti-CD25, and anti-CD138 antibodies; and analyzed for frequency of live CD138+ cells using a BD FACS Canto II cytometer.
Cytokine measurement
Production of interferon-γ, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, and tumor necrosis factor (TNF)-α was assayed by using the Meso Scale Discovery human pro-inflammatory panel 1 kit as per the instructions.30
sBCMA measurement
Plasma samples were from normal subjects (n = 39) and patients with MM (n = 40); donors were acquired from Conversant Biologics and subjected to proteolysis and subsequent mass spectrometry using a reference multiple reaction monitoring sequence (YCNASVTNSVK, MLQMAGQCSQNE). Plasma samples were subjected to depletion of medium and high abundance plasma proteins with IgY14/Supermix resin followed by trypsin digestion and reduction/alkylation before mass spectrometry analysis. sBCMA levels in cell supernatants were measured by using an enzyme-linked immunosorbent assay kit (R&D Systems).
γ-secretase inhibition and BCMA expression
BCMA+ (H929) and BCMA– (MV4-11) cells were treated with a γ-secretase inhibitor (LY-411575; MilliporeSigma) or TAPI-1 (zinc metalloprotease inhibitor; MilliporeSigma) at 0 to 100 nM for 0 to 72 hours. For primary BM MNC, BCMA surface expression was determined after treating the samples with LY-411575 for 48 hours. Teclistamab-mediated cytotoxicity and T-cell activation was tested at a 5:1 E:T ratio as described earlier.30
Ligand-mediated p38 activation and teclistamab agonistic effect
H929 and MM.1R cells were treated with human recombinant APRIL, BAFF, teclistamab, BCMAxNull, or NullxCD3 at 0 to 10 000 ng/mL for 15 minutes after 24 hours of serum starvation. p38 phosphorylation was assessed by using a phospho-p38 antibody (Thermo Fischer Scientific) and normalized to β-actin (Cell Signaling Technology) levels. Total protein was extracted and p38 phosphorylation was quantified by using Wes-Rabbit Master kit (ProteinSimple).
Xenograft studies
For all studies, female NSG (NOD/SCID/γc−/−) mice (The Jackson Laboratory) were used at 6 to 8 weeks of age. All experiments were conducted in accordance with The Guide for the Care and Use of Laboratory Animals31 and were approved by the Institutional Animal Care and Use Committee of Janssen Research & Development.
H929 prophylactic model
Mice were injected IV with 1 × 107 human peripheral blood mononuclear cells in 0.2 mL phosphate-buffered saline (PBS). Seven days after peripheral blood mononuclear cells were engrafted, each mouse received 5 × 106 H929 cells in 0.2 mL of PBS. Cells were implanted subcutaneously in the right flank using a 1-cc syringe and a 26-gauge needle; this was followed by IV administration of 0.2 mL of PBS or teclistamab antibody at 0.1 µg (0.005 mg/kg), 0.5 µg (0.025 mg/kg), or 1 µg (0.05 mg/kg) per animal on days 0, 3, 5, 7, and 10.
RPMI 8226 regression model
Animals were implanted subcutaneously with 1 × 107 RPMI 8226 cells in 0.2 mL of PBS. The day of tumor cell implantation was designated as day 0. On day 25 following tumor cell implantation, animals received an IV injection of 2 × 107 human-purified and expanded T cells in 0.2 mL of PBS. On day 25 following tumor cell implantation, mice were randomized such that all groups had a similar starting tumor volume of ∼115 mm3 (range, 108-126 mm3; n = 10 per group). Treatments were initiated on day 26, with each mouse receiving intraperitoneal administration of 0.2 mL of PBS or teclistamab at 1 µg (0.05 mg/kg), 10 µg (0.5 mg/kg), or 50 µg (2.5 mg/kg) per mouse. NullxCD3 control antibody was dosed at 10 µg per dose. Treatments were administered for a total of 8 doses on days 26, 29, 33, 36, 40, 43, 47, and 50.
For each individual animal, body weight and tumor size were measured at least twice weekly throughout the study. Animals were monitored daily for clinical signs. Percent tumor growth inhibition (TGI) was defined as the difference between mean tumor volumes of the treated and control group, calculated as %TGI = ([TVc – TVt]/TVc) × 100, where TVc is the mean tumor volume of a given control group, and TVt is the mean tumor volume of the treated group. As defined by National Cancer Institute criteria, ≥60% TGI is considered biologically significant.32 Animals were removed from the studies when a maximum tumor volume of ≥1500 mm3 was reached or when adverse clinical signs of pain and distress were noted.
Data analysis
Analysis was performed by using Microsoft Excel (2010) and GraphPad Prism software version 6. Statistical analysis was performed by using the unpaired Student t test with Welch’s correction. Best-fit linear equations, 1-way analysis of variance values, and significance values were calculated in Prism. P < .05 was considered significant. Histograms were created by using FlowJo version 10.1.
Results
Expression of BCMA in MM
Surface expression of BCMA was detected in 3 BCMA+ cell lines (H929, MM.1R, and RPMI 8226) but not in the BCMA– cell line (MV4-11) (Figure 1). BCMA surface expression was also observed in several other MM cell lines (supplemental Figure 1A). Primary BM MNC from patients with MM had a significantly higher CD138+ plasma cell count, and BCMA mean receptor density per cell was 2390 ± 524 compared with 1875 ± 168 for healthy samples (supplemental Figure 1D-E).
Figure 1.

Teclistamab-induced cytotoxicity and activation of T cells in vitro. (A) Cytotoxicity of various BCMA+ MM cell lines (H929, MM.1R, and RPMI 8226) or BCMA– cells (MV4-11) in the presence of teclistamab and healthy T cells for 48 hours. (B) Activation of T cells as evidenced by the CD25 cell surface marker. No lysis or T-cell activation was observed in the BCMA– cell line MV4-11 or with the control antibodies BCMAxNull or NullxCD3. The data points closely aligned with the generated fit curve with minimal donor-to-donor (n = 6 donors) variability. (C) BCMA detection on the surface of various BCMA+ cells (H929, MM.1R, and RPMI 8226) and BCMA– cells (MV4-11) by FACS. Solid red line, BCMA; dotted/gray shading, isotype control. (D) Ligand (APRIL and BAFF) mediated P38 phosphorylation and teclistamab failed to induce any phosphorylation signal as measured by western blot.
Teclistamab can induce T cell–mediated cytotoxicity of BCMA+ cells in vitro
Teclistamab is an IgG4 bispecific antibody that recognizes BCMA on target cells and CD3ε on T cells. It was generated by using the Genmab DuoBody technology and contains the S228P/L234A/L235A mutations in the Fc region to stabilize and minimize its immunological effector functions.29 To assess the activity of this antibody, BCMA+ MM cell lines with varying levels of BCMA expression (H929, MM.1R, and RPMI 8226 were 13 173, 6670, and 3705 BCMA receptors/cell, respectively) were incubated with purified healthy human T cells at a 5:1 E:T ratio in the presence of teclistamab for 48 hours. Teclistamab induced T cell–mediated cytotoxicity of all BCMA+ cell lines (50% effective concentration [EC50] values, 0.15 nM in H929, 0.06 nM in MM.1R, and 0.45 nM in RPMI 8226) without eliciting a cytotoxic response to the BCMA– cell line MV4-11 (Figure 1A). The BCMA receptor number did not correlate with the cytotoxic activity of teclistamab (supplemental Figure 1). As expected, the negative control bispecific antibodies (BCMAxNull and NullxCD3) did not exhibit any cytotoxicity. Teclistamab induced robust T-cell activation in the same assays, as evidenced by the increase in CD25 expression on T cells (EC50 values, 0.21 nM in H929, 0.1 nM in MM.1R, and 0.28 nM in RPMI 8226); no activation was observed when T cells were cultured with BCMA– cells or Null arm control antibodies, except at exceeding concentrations, where low levels of activation were observed (Figure 1B).
When testing T cells for additional markers such as CD107a (T-cell activation marker), programmed cell death-1 (PD-1), and T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) (check point inhibitors) in a killing assay using H929 cells, we found that they showed a pattern similar to that of CD25 and cytotoxicity, as expected (supplemental Figure 2). To further characterize the observed T-cell activation, the levels of secreted cytokines were evaluated from the in vitro H929 and RPMI 8226 functional assays described earlier. Teclistamab treatment led to the secretion of interferon-γ, TNF-α, IL-2, IL-6, IL-8, and IL-10, consistent with T-cell activation (supplemental Table 1). In addition, cytokines such as IL-1β, IL-4, IL-13, and IL-12p70 exhibited detectable but minimal induction upon T-cell activation (data not shown). To determine whether teclistamab can agonize the BCMA receptor, as has been shown with its natural ligands APRIL and BAFF, we measured phospho-P38 levels in H929 cells in the presence of antibody and found no evidence of pathway activation (Figure 1D).
Teclistamab induces cytotoxicity and T-cell activation in cells from patients with MM
The activity of teclistamab was assessed in patient BM MNCs incubated with increasing concentrations of teclistamab and healthy human T cells. In a dose-dependent manner, teclistamab bound to CD138+ BM MNCs, whereas no binding was observed with the control antibody (Figure 2). Teclistamab induced cytotoxicity of BM MNCs with a comparable EC50 (range, 1.5-4.2 nM) across all 3 patient samples. Teclistamab-mediated T-cell activation was also similar in all BM MNC samples, with EC50 values ranging from 0.93 to 1.75 nM. BCMAxNull and NullxCD3 control antibodies did not induce a cytotoxic response or activate T cells, indicating that concomitant binding to CD3 on T cells and BCMA on target cells is necessary to drive activity.
Figure 2.
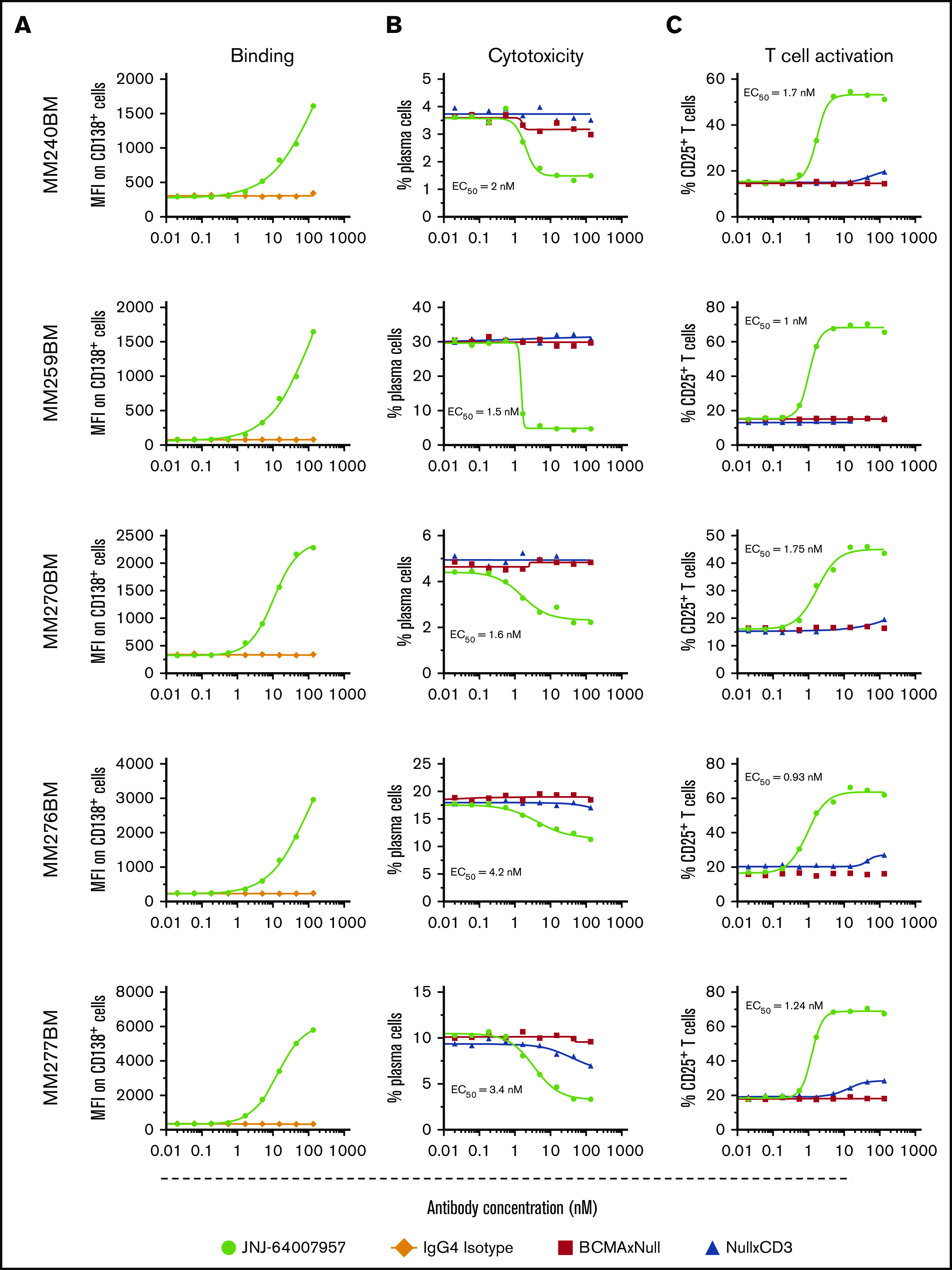
Teclistamab-induced cytotoxicity and activation of MM BM MNC cells ex vivo. Each row represents a different patient sample. Frozen BM MNCs were incubated with various concentrations of teclistamab (0-532 nM) with or without exogenous healthy T cells to measure target binding and killing. BCMA protein density was 2451 receptors per plasma cell in one of the patient samples (ID BM 240 MM). (A) Dose-dependent binding of teclistamab to target cells. (B) Dose-dependent plasma cell depletion. BM MNCs were incubated for 48 hours with exogenous healthy T cells at a 1:1 E:T ratio in the presence of teclistamab and depletion measured as remaining CD138+ cells. (C) Teclistamab-mediated T-cell activation was measured via flow cytometry by gating T cells using CD3 surface marker and CD25 activation marker. Percent CD25+ T cell values were plotted on the y-axis. Teclistamab was able to activate T cells efficiently when incubated with BM MNCs, whereas the control antibodies had no effect.
Teclistamab mediates MM cell lysis when mixed with healthy whole blood
The activity of teclistamab was evaluated in a more clinically relevant setting by using an in vitro whole blood model system. First, we evaluated the binding of teclistamab to various heme cells. The results show that teclistamab specifically bound to T cells (NK, CD4+, and CD8+) but not to other leukocytes from healthy human whole blood (Figure 3A). Second, H929 cells were spiked into the blood of healthy donors, at an E:T ratio of 5:1, along with increasing concentrations of teclistamab for 48 hours to test target cell cytotoxicity and T-cell activation. As expected, teclistamab induced a dose-dependent lysis of H929 cells with concomitant activation of T cells (Figure 3B).
Figure 3.
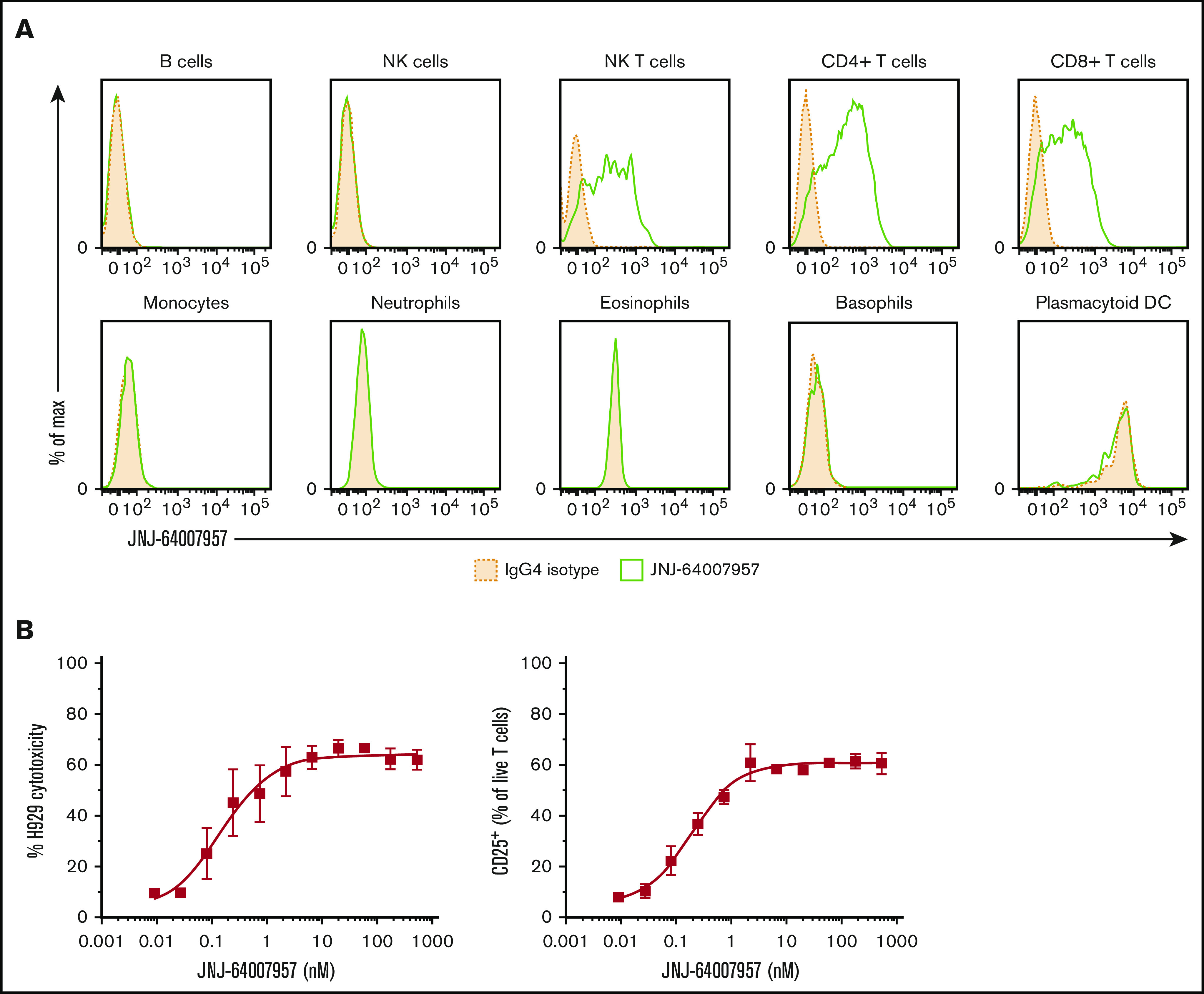
Teclistamab activity in a whole blood assay. (A) Whole blood from 3 healthy human donors was stained with teclistamab. Staining intensity for 1 representative donor is shown in the panels, where solid green lines represent teclistamab, and filled dotted lines are the corresponding isotype. No BCMA expression was observed on lymphocytes (B cells were gated by using CD19 marker), monocytes, granulocytes, NK cells, or plasmacytoid dendritic cells (DCs) in 3 healthy donors. In contrast, the antibody bound to T cells (CD4+ and CD8+) and NK T cells as expected because they express CD3. (B) Dose–response curve of H929 cytotoxicity and T-cell activation as measured by percent CD25+ T-cell population in whole blood after 48-hour incubation with teclistamab. The graphs depict the mean (± standard error of the mean [SEM]) of 6 individual donors.
Teclistamab cytotoxicity is not affected by sBCMA
BCMA can be cleaved by γ-secretase at the transmembrane domain to shed a ∼6 kDa soluble form of BCMA that can exist as free circulating sBCMA in blood.33 To examine whether sBCMA can affect the activity of teclistamab, we first measured the level of sBCMA in healthy donors and myeloma patients by using mass spectrometry. sBCMA levels were significantly higher in myeloma patient samples compared with samples from healthy donors (P ≤ .0001) (Figure 4A).
Figure 4.

Impact of sBCMA on teclistamab activity. (A) Plasma from patients with MM had significantly higher levels of sBCMA than plasma from healthy donors (average, 89.91 vs 8.04 ng/mL, respectively). (B) sBCMA levels were higher in patients with refractory MM compared with patients who were treatment (Tx) naive or had active MM. (C) Cytotoxicity. (D) T-cell activation potential of teclistamab was measured in the presence of various concentrations of sBCMA, APRIL, and BAFF.
Mean sBCMA values were 8.04 ± 1.08 ng/mL and 89.91 ± 26.96 ng/mL in healthy and myeloma serum, respectively. The levels of sBCMA also correlated with disease stage, revealing a significant difference between the plasma samples from treatment-naive patients and patients with active disease compared with refractory patients (Figure 4B). Soluble BCMA was also secreted by only B-cell lineage cell lines and not by other tissue-specific cell lines, as detected by enzyme-linked immunosorbent assay (supplemental Figure 3).
The potency of teclistamab in the cytotoxicity assay was not altered significantly in the presence of sBCMA, APRIL, or BAFF except at the high concentration of sBCMA above 55.5 nM and APRIL above 15.4 nM, at which point a significant decrease in potency was observed (Figure 4C); these concentrations are higher than that observed in myeloma patients.34 In contrast, exogenous BAFF had no impact. The effect of sBCMA, APRIL, and BAFF on teclistamab-induced T-cell activation correlated with the cytotoxicity data, as expected (Figure 4D).
γ-secretase inhibition potentiates teclistamab killing capacity by elevating BCMA surface expression
γ-secretase–mediated cleavage of BCMA was examined in H929 cells treated with the γ-secretase inhibitor LY-411575 or with a negative control zinc metalloprotease inhibitor (TAPI-1). Treatment with LY-411575 increased BCMA surface expression in H929 cells in a dose- and time-dependent manner (Figure 5A). Treatment of H929 cells with TAPI-1 and treatment with either inhibitor in the BCMA– cell line MV4-11 did not lead to any changes in BCMA surface expression. Similar results were observed with BM MNCs from patients with MM treated with LY-411575 for 48 hours (Figure 5B), although the magnitude of BCMA surface expression varied widely across donor samples. To determine if the increased cell surface expression of BCMA in the presence of a γ-secretase inhibitor influences activity, H929 cells were incubated with isolated healthy human pan-T cells at a 5:1 E:T ratio and teclistamab in the presence or absence of LY-411575. Treatment with the γ-secretase inhibitor increased the potency of teclistamab cytotoxicity, shifting the EC50 to 0.004 nM (20-fold) and 0.002 nM (40-fold) with 0.1 nM and 1 nM LY-411575, respectively, compared with an EC50 of 0.09 nM with vehicle control and TAPI-1 treatment. More potent activation of T cells was also observed with teclistamab in the presence of LY-411575 (EC50 of 0.001 and 0.0003 nM) compared with TAPI-1 treatment or vehicle control (EC50 of 0.04 nM). No activity was observed with the BCMAxNull and NullxCD3 control antibodies, as expected (Figure 5C).
Figure 5.
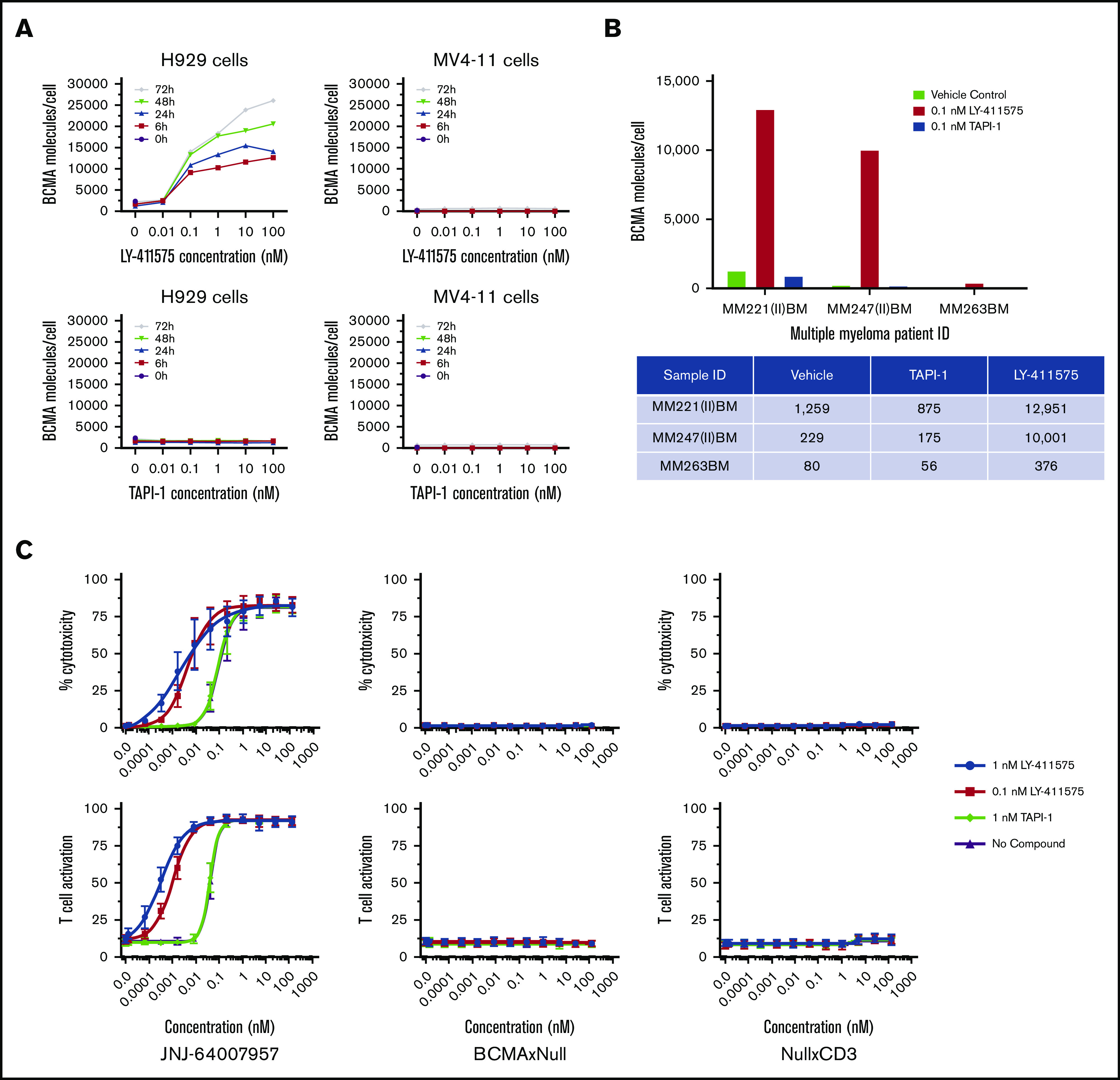
Effects of a γ-secretase inhibitor on BCMA surface expression and teclistamab activity. (A) H929 and MV4-11 cells were incubated with a γ-secretase and TAPI inhibitors at various concentrations for 0 to 72 hours, and BCMA surface expression was measured by using FACS. (B) MM patient BM MNCs were incubated with a γ-secretase inhibitor, and BCMA surface expression was measured. (C) Teclistamab was incubated with H929 cells in the presence of healthy T cells with or without a γ-secretase inhibitor, and cytotoxicity and T-cell activation were measured after 48 hours.
Teclistamab exhibits antitumor activity in xenograft models
To assess the in vivo activity of teclistamab, two MM models were used. In the first H929 prophylactic model, 0.5 and 1 µg doses of teclistamab completely inhibited tumor growth throughout the study period (up to day 22) compared with PBS control. At the teclistamab 0.1 µg dose, no inhibition of tumor growth was observed compared with PBS control (Figure 6A). Although the BCMAxNull control antibody did not exhibit antitumor activity, the NullxCD3 antibody had slight antitumor activity on day 22 (Figure 6A). The antitumor effects of teclistamab were not associated with increased toxicity or graft-versus-host disease, as suggested by the similar change in mean body weight across all treatments (data not shown). In the RPMI 8226 regression model, teclistamab inhibited tumor growth up to day 60, with both the 10 µg and 50 µg doses exhibiting significant tumor reductions compared with PBS control.
Figure 6.
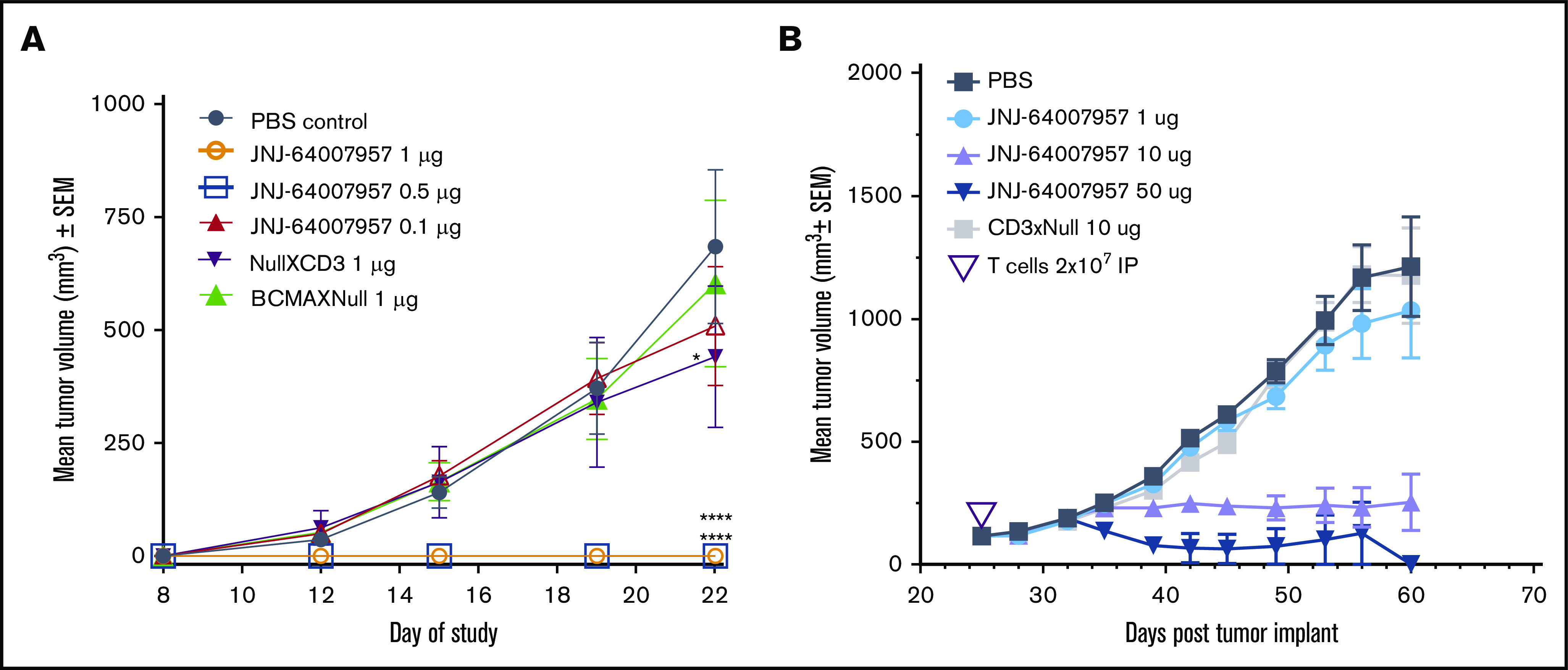
Antitumor activity of teclistamab in xenograft models. (A) In the H929 prophylactic model, teclistamab had antitumor efficacy with significant reduction of tumor formation and growth compared with PBS-treated control mice, at dose levels of either 0.5 or 1 µg per animal; BCMAxNull and NullxCD3 bispecific antibodies, however, failed to suppress tumorigenesis in the model. (B) In the established RPMI 8226 model, teclistamab displayed antitumor effects at both the 10 and 50 µg per animal dose compared with PBS-treated control mice. Human pan-T cells were activated and expanded in vitro by using a T-cell activation and expansion kit (Miltenyi Biotec) and grown in medium containing IL-2 (0.1 µg/µL). Results are presented as average tumor volume, expressed in mm3 ± SEM of each group. Tumor volume was calculated by using the following formula: tumor volume (mm3) = (a × b2/2), where a represents the length and b the width of the tumor as determined by caliper measurements.
However, the lowest dose of 1 μg/kg teclistamab had no antitumor effect (Figure 6B). Control antibodies had no significant antitumor activity, as expected. Mean body weight changes were similar between teclistamab-treated mice and control mice (data not shown). The pharmacokinetic profile for teclistamab was favorable, with a half-life of ∼10 days in NSG mice (supplemental Figure 4), which could potentiate the cytotoxic activity of this antibody in patients with MM.
Discussion
The success of T cell–mediated therapeutics depends, in part, on the selective expression of cell surface target epitopes to minimize the off-tissue toxicity. BCMA is selectively expressed on late-stage B cells and plasma cells, and its expression levels correlate with myeloma tumor burden.35 Data from a BCMA CAR-T clinical study in patients with MM has shown a high overall response with manageable safety, highlighting the potential for targeting BCMA in the clinical setting.36 Here we describe a bispecific antibody (teclistamab) that can recruit T cells to BCMA+ cells. This antibody induces elimination of BCMA+ MM cells both in vitro and in vivo. In vitro, teclistamab induced potent and specific T cell–mediated cytotoxicity of cells expressing various levels of surface BCMA receptors. As expected, the killing of BCMA+ cells corresponded with T-cell activation and cytokine secretion. Cytotoxic activity was also observed ex vivo in BM MNCs from patients with MM, in whom teclistamab induced elimination of plasma cells.
sBCMA is present in the serum and could potentially neutralize BCMA-directed therapy such as T-cell redirection. sBCMA is found at significantly higher levels in plasma from patients with MM than from healthy donors.20 We also observed stratification of sBCMA levels based on the disease stage, with significantly higher levels of sBCMA in plasma from patients with refractory MM, compared with treatment-naive patients or MM patients with active disease. These findings are consistent with previous reports correlating increased sBCMA with poor prognosis in MM.21 Because teclistamab recognizes the same epitope present in both the soluble and transmembrane forms of BCMA, sBCMA could serve as a potential sink for teclistamab. Interestingly, teclistamab cytotoxic activity was only moderately affected in the presence of sBCMA, with cytotoxicity inhibited twofold at 166.5 nM of sBCMA. Because the average concentration of sBCMA in plasma from patients with MM was ∼15 nM in our studies (highest level observed was 125 nM), it is unlikely that sBCMA would inhibit teclistamab efficacy in patients. These results are also consistent with data reported elsewhere with another BCMAxCD3 T-cell engager.37 Moreover, in a physiologically relevant whole blood assay, which contains soluble factors such as APRIL, BAFF, and sBCMA, teclistamab was able to effectively eliminate H929 cells, indicating that shed sBCMA in the blood is unlikely to affect the efficacy of teclistamab.
γ-secretase has been shown to contribute to the shedding of BCMA in the serum. Shedding can lead to reduced surface BCMA levels and affect BCMA-directed therapy. This has led to the hypothesis that γ-secretase inhibitors may be useful in the clinic for maintaining BCMA levels on the cell surface. In vitro, our antibody has been shown to kill BCMA+ cells more potently with the γ-secretase inhibitor LY-411575, consistent with these observations. Resistance to BCMAxCD3 or BCMA CART therapy could also potentially occur through the expansion of BCMA– cells in the original cancer population. To address this, it may become relevant to combine such therapeutics with other T-cell therapies targeting MM targets such as CD38. Another possibility could be to create multispecific antibodies or bispecific CARTs.
In vivo, teclistamab significantly inhibited tumor growth in 2 independent xenograft models: the H929 prophylactic model and the RPMI 8226 regression model. In the aggressive, H929 prophylactic model, teclistamab significantly blocked tumor growth at the 0.5 and 1 µg dose levels tested compared with control-treated mice, indicating that teclistamab has the potential to control the fast-growing myeloma plasma cells in patients. The antibody also significantly regressed tumors in the RPMI 8226 model at 10 and 50 µg of teclistamab per animal. Both mouse models are humanized and are missing components of the immune system (accessory immune cells) and BM interactions that are present in patients. To better understand potential resistance to T-cell redirection drugs in patients, it would be relevant to profile T cells in patients with MM at various stages of treatment and to study interactions with BM stromal components to address T-cell exhaustion, decreased persistence, and clearance.
In conclusion, our study shows the potent and specific elimination of BCMA-expressing cells by teclistamab in MM cell lines, patient samples, and xenograft models of MM. These findings support targeting BCMA for MM therapies and validate the potential use of BCMAxCD3 bispecific antibodies as a treatment approach for MM with a potentially better pharmacokinetic profile than smaller molecular weight biologics. Clinical evaluation of teclistamab in patients with relapsed or refractory MM is currently ongoing (ClinicalTrials.gov #NCT03145181 and #NCT04108195).38
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Brendan Weiss, Blake J. Bartlett, and Jennifer Smit for critical reading of the manuscript.
This study was funded by Janssen Research & Development, LLC. Medical writing support was provided by Tracy T. Cao (Janssen Global Services, LLC) and funded by Janssen Global Services, LLC.
Footnotes
Requests for original data may be submitted to François Gaudet, Janssen Research & Development LLC, 1400 McKean Rd, Spring House, PA 19477; e-mail: fgaudet@its.jnj.com.
Authorship
Contribution: K. Pillarisetti conceived, designed and performed experiments, analyzed data, wrote the manuscript, and coled antibody discovery for JNJ-64007957 (teclistamab); G.P. and E.B. conceived, led antibody generation, and coled antibody discovery for JNJ-64007957 (teclistamab); L.L. and D.C. designed and performed the in vivo rodent experiments; A.B., Y.L., M.M., X.Z., R.N., and N.M. performed experiments and contributed to data; K. Packman oversaw design and execution of in vivo experiments; R.A. and Y.E. oversaw the project; F.G. conceived, contributed to project design, oversaw the project, and cowrote the manuscript; and all authors reviewed and revised the manuscript, approved the final version, and agreed to submit the manuscript for publication.
Conflict-of-interest disclosure: K. Pillarisetti, A.B., X.Z., L.L., R.N., G.P., K. Packman, Y.E., R.A., and F.G. are employees of Janssen Research & Development, LLC; E.B., Y.L., M.M., N.M., and D.C. are former employees of Janssen Research & Development, LLC; and all are shareholders of Johnson & Johnson, the parent company of the Janssen Research & Development, LLC. E.B. is an employee of IBM Watson Health. Y.L. is an employee of Legend Biotech. M.M. and D.C. are employees of Century Therapeutics.
Correspondence: François Gaudet, Janssen Research & Development, LLC, 1400 McKean Rd, Spring House, PA 19477; e-mail: fgaudet@its.jnj.com.
References
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046-1060. [DOI] [PubMed] [Google Scholar]
- 2.Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. [DOI] [PubMed] [Google Scholar]
- 3.Harousseau JL, Attal M, Avet-Loiseau H, et al. Bortezomib plus dexamethasone is superior to vincristine plus doxorubicin plus dexamethasone as induction treatment prior to autologous stem-cell transplantation in newly diagnosed multiple myeloma: results of the IFM 2005-01 phase III trial. J Clin Oncol. 2010;28(30):4621-4629. [DOI] [PubMed] [Google Scholar]
- 4.Kumar SK, Lee JH, Lahuerta JJ, et al. ; International Myeloma Working Group . Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study [published correction appears in Leukemia. 2012;26(5):1153]. Leukemia. 2012;26(1):149-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373(13):1207-1219. [DOI] [PubMed] [Google Scholar]
- 6.Lonial S, Dimopoulos M, Palumbo A, et al. ; ELOQUENT-2 Investigators . Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621-631. [DOI] [PubMed] [Google Scholar]
- 7.Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387(10027):1551-1560. [DOI] [PubMed] [Google Scholar]
- 8.Palumbo A, Dimopoulos M, San Miguel J, et al. Lenalidomide in combination with dexamethasone for the treatment of relapsed or refractory multiple myeloma. Blood Rev. 2009;23(2):87-93. [DOI] [PubMed] [Google Scholar]
- 9.Rajkumar SV, Rosiñol L, Hussein M, et al. Multicenter, randomized, double-blind, placebo-controlled study of thalidomide plus dexamethasone compared with dexamethasone as initial therapy for newly diagnosed multiple myeloma. J Clin Oncol. 2008;26(13):2171-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010;116(5):679-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.San Miguel JF, Schlag R, Khuageva NK, et al. ; VISTA Trial Investigators . Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906-917. [DOI] [PubMed] [Google Scholar]
- 12.Mikhael J, Ismaila N, Cheung MC, et al. Treatment of multiple myeloma: ASCO and CCO Joint Clinical Practice Guideline. J Clin Oncol. 2019;37(14):1228-1263. [DOI] [PubMed] [Google Scholar]
- 13.Kumar SK, Dimopoulos MA, Kastritis E, et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia. 2017;31(11):2443-2448. [DOI] [PubMed] [Google Scholar]
- 14.Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048-2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novak AJ, Darce JR, Arendt BK, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood. 2004;103(2):689-694. [DOI] [PubMed] [Google Scholar]
- 16.Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404(6781):995-999. [DOI] [PubMed] [Google Scholar]
- 17.Marsters SA, Yan M, Pitti RM, Haas PE, Dixit VM, Ashkenazi A. Interaction of the TNF homologues BLyS and APRIL with the TNF receptor homologues BCMA and TACI. Curr Biol. 2000;10(13):785-788. [DOI] [PubMed] [Google Scholar]
- 18.Thompson JS, Schneider P, Kalled SL, et al. BAFF binds to the tumor necrosis factor receptor-like molecule B cell maturation antigen and is important for maintaining the peripheral B cell population. J Exp Med. 2000;192(1):129-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatzoglou A, Roussel J, Bourgeade MF, et al. TNF receptor family member BCMA (B cell maturation) associates with TNF receptor-associated factor (TRAF) 1, TRAF2, and TRAF3 and activates NF-kappa B, elk-1, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. J Immunol. 2000;165(3):1322-1330. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez E, Li M, Kitto A, et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol. 2012;158(6):727-738. [DOI] [PubMed] [Google Scholar]
- 21.Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726-1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao WH, Liu J, Wang BY, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Chen LJ, Yang SS, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019;116(19):9543-9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Topp MS, Duell J, Zugmaier G, et al. Treatment with AMG 420, an anti-B-cell maturation antigen (BCMA) bispecific T-cell engager (BiTE®) antibody construct, induces minimal residual disease (MRD) negative complete responses in relapsed and/or refractory (R/R) multiple myeloma (MM) patients: results of a first-in-human (FIH) phase I dose escalation study. Blood. 2018;132(suppl 1):Abstract 1010. [Google Scholar]
- 26.Pessano S, Oettgen H, Bhan AK, Terhorst C. The T3/T cell receptor complex: antigenic distinction between the two 20-kd T3 (T3-delta and T3-epsilon) subunits. EMBO J. 1985;4(2):337-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Labrijn AF, Meesters JI, de Goeij BE, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A. 2013;110(13):5145-5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canziani GA, Melero JA, Lacy ER. Characterization of neutralizing affinity-matured human respiratory syncytial virus F binding antibodies in the sub-picomolar affinity range. J Mol Recognit. 2012;25(3):136-146. [DOI] [PubMed] [Google Scholar]
- 29.Vafa O, Gilliland GL, Brezski RJ, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65(1):114-126. [DOI] [PubMed] [Google Scholar]
- 30.Pillarisetti K, Edavettal S, Mendonça M, et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood. 2020;135(15):1232-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.National Research Council of the National Academies Guide for the Care and Use of Laboratory Animals. 8th ed. Washington, DC: The National Academies Press; 2011. [Google Scholar]
- 32.Johnson JI, Decker S, Zaharevitz D, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84(10):1424-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laurent SA, Hoffmann FS, Kuhn PH, et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun. 2015;6(1):7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moreaux J, Legouffe E, Jourdan E, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103(8):3148-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seckinger A, Delgado JA, Moser S, et al. Target expression, generation, preclinical activity and pharmacokinetics of the BCMA-T-cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell. 2017;31(3):396-410. [DOI] [PubMed] [Google Scholar]
- 36.Madduri D, Usmani SZ, Jagannath S, et al. Results from CARTITUDE-1: a phase 1b/2 study of JNJ-68284528, a CAR-T cell therapy directed against B-cell maturation antigen (BCMA), in patients with relapsed and/or refractory multiple myeloma (R/R MM) [abstract]. Blood. 2019;134(suppl 1). Abstract 577.
- 37.Hipp S, Tai YT, Blanset D, et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo [published correction appears in Leukemia. 2017;31(10):2278]. Leukemia. 2017;31(8):1743-1751. [DOI] [PubMed] [Google Scholar]
- 38.Usmani SZ, Mateos MV, Nahi H, et al. Phase I study of teclistamab, a humanized B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in relapsed/refractory multiple myeloma (R/R MM). J Clin Oncol. 2020;38(suppl 15):100.31664879 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.