

SUMMARY
Classic osteogenesis imperfecta, an autosomal dominant disorder associated with osteoporosis and bone fragility, is caused by mutations in the genes for type I collagen. A recessive form of the disorder has long been suspected. Since the loss of cartilage-associated protein (CRTAP), which is required for post-translational prolyl 3-hydroxylation of collagen, causes severe osteoporosis in mice, we investigated whether CRTAP deficiency is associated with recessive osteogenesis imperfecta. Three of 10 children with lethal or severe osteogenesis imperfecta, who did not have a primary collagen defect yet had excess post-translational modification of collagen, were found to have a recessive condition resulting in CRTAP deficiency, suggesting that prolyl 3-hydroxylation of type I collagen is important for bone formation.
OSTEOGENESIS IMPERFECTA, OR “BRITTLE BONE DISEASE,” IS AN AUTOsomal dominant genetic disorder characterized by bone fragility with susceptibility to fracture.1 The clinical range of osteogenesis imperfecta (types I through IV)2 extends from mild symptoms to a perinatal lethal condition (Table 1 of the Supplementary Appendix, available with the full text of this article at www.nejm.org). Mutations in type I collagen, the major structural protein of bone and skin, cause most cases of osteogenesis imperfecta.3
Type I collagen is a heterotrimer, composed of two α1(I) and one α2(I) chains. Both α chains have a primary structure of uninterrupted repeats of the tripeptide composed of glycine (Gly) and two other amino acids, often proline (Pro) and hydroxyproline (Hyp), in that order. This primary structure allows the procollagen chains to form a triple helix, with the chains aligning at their carboxyl ends and folding toward the amino termini.4 Heterozygous mutations affecting the primary collagen structure cause moderate and severe osteogenesis imperfecta, whereas decreased collagen production causes mild osteogenesis imperfecta.3 A recessive form of osteogenesis imperfecta has long been suspected in some cases, in which unaffected parents have more than one child with severe bone dysplasia.5–8 Proposals of additional dominant and recessive types of osteogenesis imperfecta (types V through VII) are based on histologic characteristics of bone; these nonlethal forms do not involve primary collagen defects.9–11
Type I procollagen undergoes intracellular post-translational modifications.12 Specific proline and lysine residues throughout the helical region of the collagen chains are hydroxylated to 4-hydroxyproline, 3-hydrox yproline, and hydroxylysine by prolyl 4-hydroxylase, prolyl 3-hydroxylase 1, and lysyl hydroxylase, respectively, with subsequent glycosylation of some hydroxylysine residues.13 4-Hydroxyproline residues are crucial for collagen folding and stability.
Prolyl 3-hydroxylase 1, located in the endoplasmic reticulum, modifies Pro-4Hyp-Gly,14,15 specifically the proline residue in the 986 position (Pro986) in the α1(I) chain.15,16 Prolyl 3-hydroxylase 1 forms a complex with cartilage-associated protein (CRTAP) and cyclophilin B, although CRTAP is not required for prolyl 3-hydroxylase 1 activity in vitro.17,18 CRTAP messenger RNA (mRNA) is detectable in many types of cells, including osteoblasts,18–21 and is secreted into the extracellular matrix of cartilage and long bones.19 In Crtap-null mice, kyphosis and severe osteoporosis develop, with defective osteoid formation18; the α1(I) chains completely lack Pro986 hydroxylation. We screened skin samples from 10 children with lethal or severe osteogenesis imperfecta who had type I collagen with a normal primary structure and identified mutations in the CRTAP gene in infants with decreased levels of CRTAP mRNA.
METHODS
STUDY POPULATION
We screened dermal fibroblasts from 10 children with lethal (type II) or very severe (type III) osteogenesis imperfecta who had type I collagen with a normal primary structure but with excess post-translational modification of the α chain helical region. A well-studied fibroblast cell line from newborn foreskin (American Type Culture Collection 2127) was used as a control. We also examined 1 control patient with classic (type III) osteogenesis imperfecta, 6 normal control subjects, and 11 patients who had severe osteogenesis imperfecta without excess modification and without a collagen defect; none had decreased hydroxylation of Pro986 (data not shown). Written informed consent was obtained from all patients and control subjects or their parents.
DETECTION OF CRTAP MUTATIONS, mRNA LEVELS, AND SPLICING ISOFORMS
The 7 CRTAP exons and surrounding intronic sequences were amplified from the subjects’ genomic DNA with the use of a polymerase-chain-reaction (PCR) assay and were sequenced on a Beckman CEQ2000 assay sequencer. CRTAP mRNA levels in affected infants, their parents, and control fibroblasts were determined with the use of real-time reverse-transcriptase (RT) PCR (Assays-on-Demand, Applied Biosystems). To determine the splicing isoforms of Infant 1, total RNA from cycloheximidetreated fibroblasts was amplified by RT-PCR. Amplified products were subcloned and sequenced.
IN VITRO BIOCHEMICAL ANALYSES
For Western blots of CRTAP protein, total fibroblast lysates were separated by means of sodium dodecyl sulfate–polyacrylamide-gel electrophoresis (SDS-PAGE) and were electroblotted.18 The blots were probed with CRTAP polyclonal antibody. Re-active bands were visualized with the use of a secondary antibody, conjugated with horseradish peroxidase, and chemiluminescence (Pierce Bio-technology). The loading control was γ tubulin. To assess the degree of collagen modification, steady-state procollagen was digested with pepsin and electrophoresed, under nonreducing conditions, with the use of SDS-urea-PAGE. Differential scanning calorimetry and amino acid chromatography were performed according to standard methods. For evaluation of prolyl 3-hydroxylation, electrospray mass spectrometry was performed on tryptic peptides of the α1(I) chains (for details, see the Supplementary Appendix).
RESULTS
CLINICAL FINDINGS OF CRTAP DEFICIENCY
Infant 1, the first child of a couple of Pakistani origin who are full second cousins, was a boy weighing 2370 g who was born at 35 weeks’ gestation after an induced vaginal delivery. Prenatal ultrasonography revealed severe micromelia of the arms and legs. The birth length was 43 cm and the head circumference was 29.5 cm — values at the 50th percentile for an infant at 32 weeks gestation. There was a wide-open anterior fontanel. The eyes showed proptosis and white sclerae. He had pectus excavatum and rhizomelic limb shortening, and held his legs in an externally rotated and abducted position. A radiographic survey revealed over 20 fractures of the long bones and ribs (Fig. 1A through 1D). Ultrasonography revealed moderate hydronephrosis on the left side and a small right kidney. He required 10 days of mechanical ventilation and was discharged at 1 month, receiving nasal oxygen. At 6 months of age, his weight, length, and head circumferences were at the 50th percentile for newborn boys and 1- and 4-month-old boys, respectively. He died at 10 months of age during hospitalization for respiratory syncytial virus bronchiolitis.
Figure 1. Clinical Characteristics of Infants with CRTAP Deficiency.
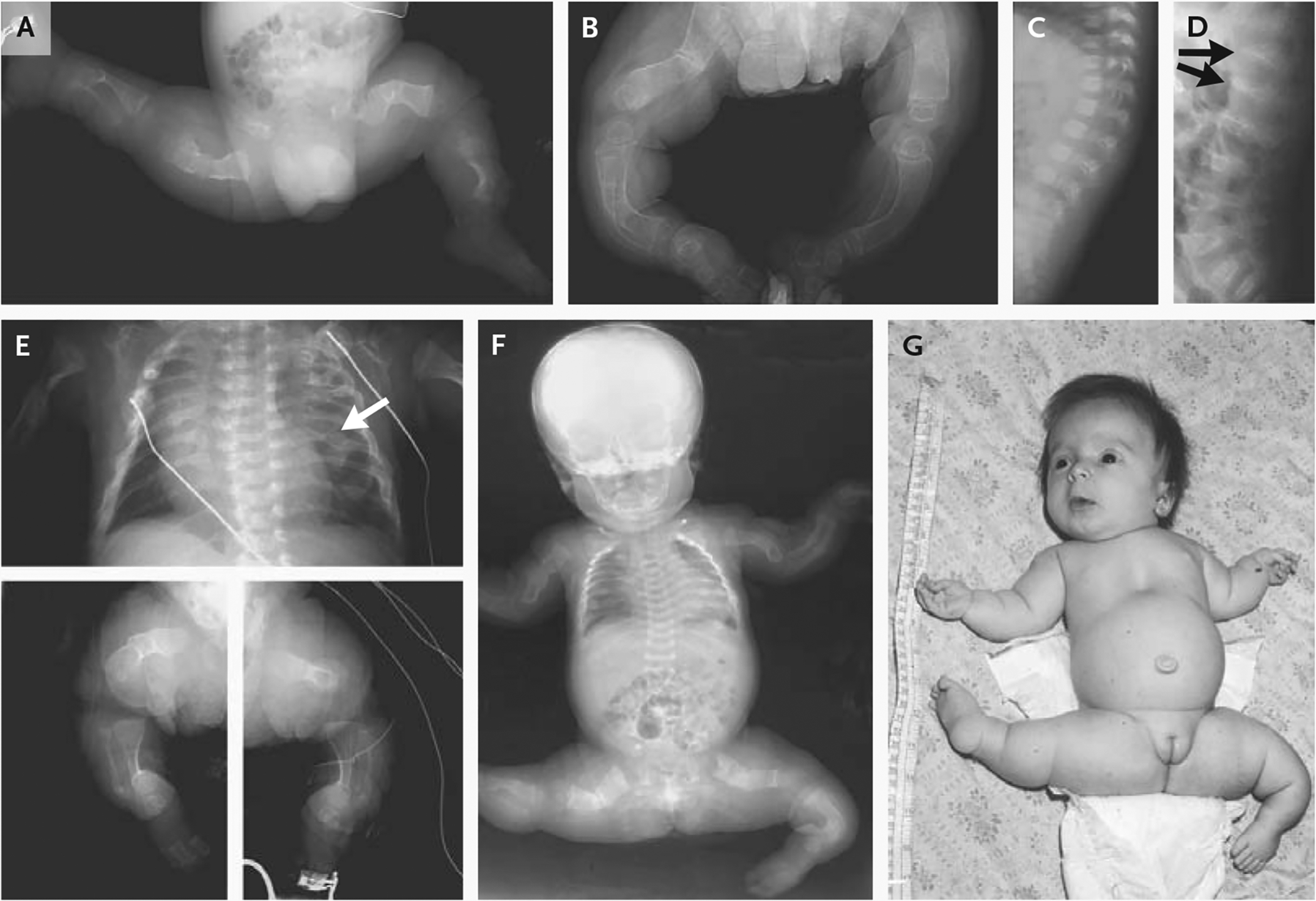
Panels A and B show the legs of Infant 1 at birth and at 8 months of age, respectively. His long bones were severely osteopenic and crumpled at birth; at 8 months, most fractures had healed but the lack of modeling (undertubulation) was indicated by the cylindrical configuration of diaphyses. Panels C and D show lateral views of the spine of Infant 1 at birth and 8 months of age, respectively. The vertebrae were severely osteoporotic at birth but had a normal geometry; by 8 months, there were multiple vertebral compressions (arrows), especially at vertebrae T12 and L1. Panel E shows a composite of radiographs of Infant 2 at birth. The chest was narrow, and the cardiac silhouette was enlarged. Multiple beads of callus (arrow) represent sites of healing fractures. Long bones in the legs were extremely osteopenic and crumpled, with poor modeling and a cylindrical configuration. Panel F shows a radiograph and Panel G a photograph of Infant 3 at 2 months of age. The thorax was narrow, with multiple healing rib fractures and extremely osteoporotic long bones showing crumpled, poorly modeled geometry. Infant 3 held her legs in an abducted position, which is typical of osteogenesis imperfecta; in contrast to infants with osteogenesis imperfecta type II, she had a round face (rather than triangular), a normal profile (rather than a flat midface), and a long philtrum.
Infant 2 was a girl weighing 2435 g who was born at 36 weeks’ gestation to nonconsanguineous black parents after a spontaneous vaginal delivery. The infant’s parents and two half-siblings were healthy. The birth length was 38 cm (50th percentile for infants at 29 weeks’ gestation). The head circumference was 31.7 cm (50th percentile for infants at 33.5 weeks’ gestation); the skull was soft, and the anterior fontanel and the other sutures were wide open. The eyes showed proptosis, and the sclerae were white. The limbs were deformed, and the skeletal survey revealed fractures of the femurs, tibiae, humeri, and multiple ribs (Fig. 1E). The infant’s chest was small, and she required ventilation for respiratory distress. Cardiac catheterization on day 18 revealed an absent right pulmonary artery with a collateral vessel from the proximal descending aorta supplying the trilobed right lung, hypoplastic pulmonary veins, and systemic pulmonary hypertension. The aorta was normal. Echocardiography on day 75 showed right ventricular hypertrophy and dilatation. She died on day 80 from respiratory insufficiency.
Infant 3 was a girl weighing 2600 g (50th percentile for infants at 36 weeks’ gestation) delivered by elective cesarean section at term to nonconsanguineous parents of German descent. The couple had had four children previously, including one with severe skeletal dysplasia who died from pneumonia at 5 months of age. Infant 3 did not require respiratory support at birth. The skull was poorly ossified, and the anterior and posterior fontanels were wide open. The sclerae were light blue. There was micromelia of the arms and legs, and the legs were held in an abducted position. Skeletal surveys showed crumpling of both femurs, multiple rib fractures, and wormian bones of the skull (Fig. 1F). The infant’s cardiorespiratory status allowed her to gain weight as a result of breast-feeding. At 2 months of age, her weight was that of an average newborn, her length was 45 cm, and her head circumference was 36 cm (50th percentile for 34 weeks’ gestation and 5th percentile for age, respectively). There was a pectus excavatum (Fig. 1G). Findings on cardiac and pulmonary examinations were normal. The spine was straight. She had begun to smile and coo. She died at 10 months from respiratory insufficiency and pneumonia.
EXPRESSION OF CRTAP IN FIBROBLASTS FROM INFANTS AND PARENTS
We sampled mRNA from the fibroblasts of the 10 children with lethal or severe osteogenesis imperfecta with excess post-translational modification but without a collagen mutation and screened it for decreased levels of CRTAP mRNA. Infants 1, 2, and 3 had CRTAP mRNA levels that were 0 to 25% of the normal value (Fig. 2A). Fibroblasts from the parents of Infants 1 and 3 had normal CRTAP mRNA levels, suggesting that the infants had more than one defective allele.
Figure 2. Results of Analyses of CRTAP mRNA and Biochemical Analyses of CRTAP and Type I Collagen.
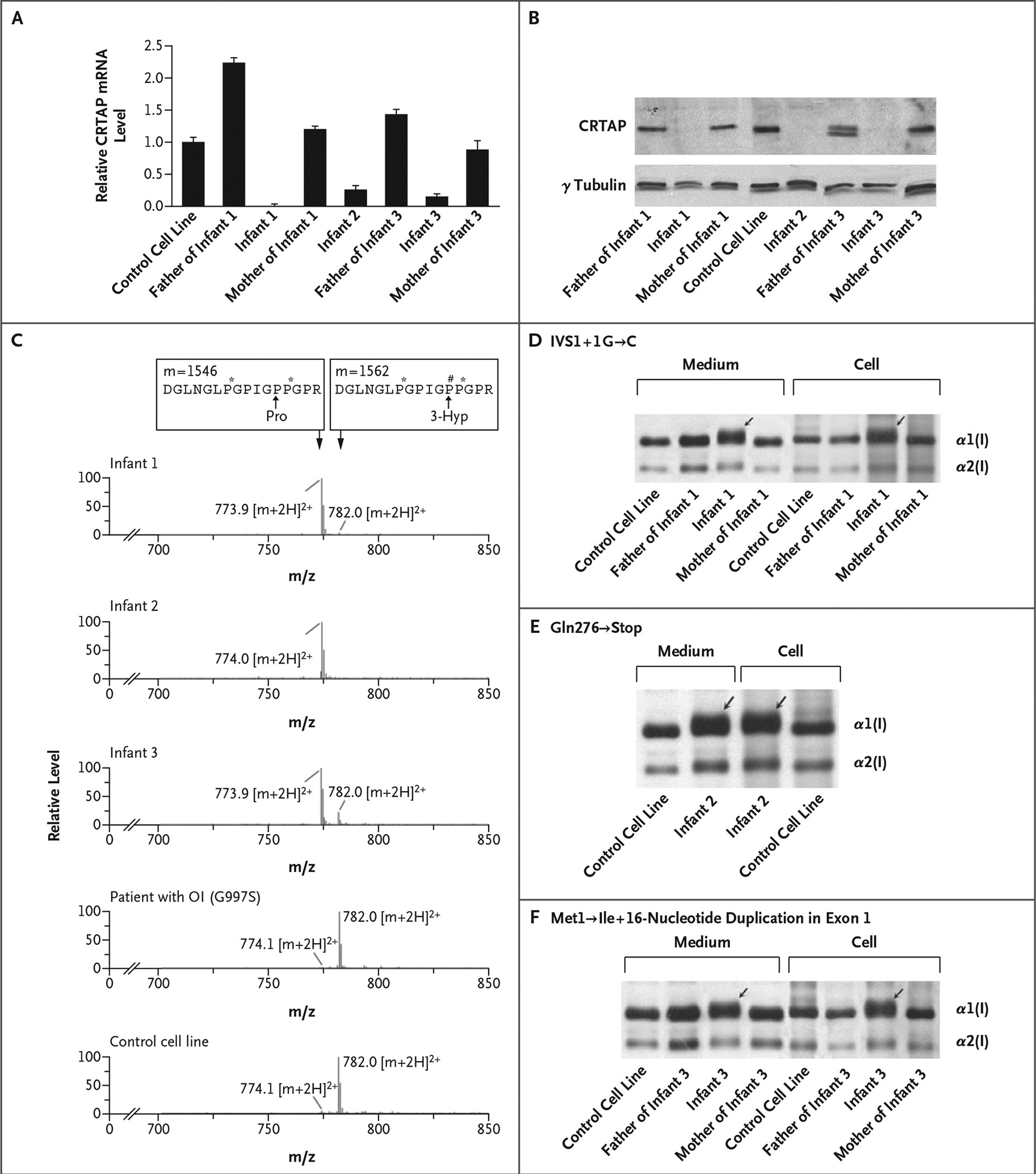
Panel A shows the relative expression of CRTAP in fibroblasts, measured using real-time RT-PCR as the CRTAP mRNA level normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA level and then to the CRTAP mRNA level in the control fibroblast line (which was arbitrarily set at 1). T bars represent standard deviations. Panel B shows a Western blot of fibroblast CRTAP, which was lacking in the affected infants and present in their parents. The father of Infant 3 also had a truncated form of CRTAP. Panel C shows the results of tandem mass spectrometry, revealing a low level of Pro986 3-hydroxylation or none in Infants 1, 2, and 3, in contrast to the normal Pro986 3-hydroxylation in both a patient with classic dominant osteogenesis imperfecta (OI), caused by a glycine substitution (Gly997→Ser) in collagen, and in a control cell line. The tracing shows the cumulative, full scan across the liquid chromatography peak. The top boxes represent the masses (m, in atomic mass units) and the amino acid sequences (capital letters; P# and P* denote 3- and 4-hydroxyproline [Hyp], respectively) of collagen peptides that have either undergone 3-hydroxylation at the proline at position 986 (Pro986) (right-hand box, in which the proline is converted to 3-Hyp) or have not (left-hand box). The levels of the ions relative to one another are shown, as are their mass:charge (m/z) ratios; shown are the double-charged peptide ions ([m+2H]2+) that differ by the 16 mass units of an additional oxygen atom. Peptide ions that have undergone normal Pro986 hydroxylation have an m/z ratio of 782.0, whereas peptides that have not have an m/z ratio of approximately 774. The results of SDS-urea-PAGE of type I collagen from the three infants, their parents, and the control cell line are shown; Infant 1 had a IVS1+1G→C substitution (Panel D); Infant 2, a Gln276→ Stop substitution (Panel E); and Infant 3, a Met1→Ile substitution and a 16-nucleotide duplication in exon 1 (Panel F). The broad bands representing the α1(I) chains indicate the presence of a form with delayed migration in Infants 1, 2, and 3 that was also secreted into the culture medium (arrows).
IDENTIFICATION OF CRTAP MUTATIONS
We sequenced the exons and surrounding intronic sequences of CRTAP from Infants 1, 2, and 3, who had reduced levels of CRTAP expression. Mutations were identified in all three infants (see Fig. 1 in the Supplementary Appendix). Infant 1 was homozygous for a mutation in the splice donor site of exon 1 at the invariant first intronic nucleotide (IVS1+1G→C). Both parents were heterozygous for this mutation (see Fig. 1A1 in the Supplementary Appendix). Infant 2 was homozygous for a point mutation that directly causes a Gln276→Stop substitution in exon 4, which results in the termination of translation at amino acid 275 of 401 CRTAP residues (see Fig. 1B in the Supplementary Appendix). Infant 3 was a compound heterozygote. From her father, she had inherited a point mutation in the CRTAP start codon (AUG→AUA), causing a Met1→Ile substitution, which is expected to eliminate the initiation of CRTAP translation (see Fig. 1C1 in the Supplementary Appendix). From her mother, she had inherited a 16-nucleotide duplication in exon 1, which shifts the reading frame and results in a premature termination codon in exon 2 (see Fig. 1C2 in the Supplementary Appendix). The presence of the parental mutations was confirmed by restriction-enzyme digestion.
The splice donor site mutation in Infant 1 results in four alternatively spliced forms of the rapid degradation of the abnormal mRNA and mRNA transcript (see Fig. 1A2 in the Supple-undetectability of the splicing isoforms with mentary Appendix), each containing a premathe use of an RNA protection assay; the normally ture termination codon in the retained portion spliced mRNA in the parents and the control of intron 1, a finding that is consistent with cell line was detectable (data not shown).
CRTAP DEFICIENCY IN FIBROBLASTS
Intracellular CRTAP was detectable in fibroblasts from the control cell line and from the parents but was undetectable in fibroblasts from Infants 1, 2, and 3 (Fig. 2B), as measured by Western blot. The father of Infant 3, whose mutation alters the start codon, produced both normal CRTAP and CRTAP of smaller size, suggesting that his cells contain a truncated form of CRTAP. Without the normal start codon, protein synthesis may be initiated at the next methionine codon, Met42. We did not detect the truncated form in several independent samples from Infant 3; stabilization of the truncated protein may require the presence of normal CRTAP.
EFFECT OF CRTAP DEFICIENCY ON TYPE I COLLAGEN
Tandem mass spectrometry revealed a lack or reduction of prolyl 3-hydroxylation at Pro986 (Fig. 2A in the Supplementary Appendix) in the α1(I) chain in the infants; Infant 3 had 3-hydroxylation of 21% of α1(I)Pro986 residues, suggesting that she had residual enzyme activity from her paternal allele (Fig. 2C and Table 1). The lack of prolyl 3-hydroxylation, CRTAP function, or both delayed the folding of the collagen helix and allowed for excess modification by lysyl hydroxylase and prolyl 4-hydroxylase along the full length of the helix. Collagen chains from Infants 1, 2, and 3 showed delayed electrophoretic migration on SDS-urea-PAGE (Fig. 2D through 2F), with a 28 to 43% increase in the hydroxylation of lysine residues as compared with normal collagen (Table 1). These findings resemble those from collagen with a structural mutation in the carboxyl end of the helical region, and they are consistent with the increase in the melting temperature of collagen of approximately 1°C on calorimetry (Fig. 2B in the Supplementary Appendix).
Table 1.
Hydroxylation of Type I Collagen.*
| Subject | CRTAP Genotype | Lysine Residues in Type I Collagen Modified by Lysyl Hydroxylase | α1(I) Pro986 Residues with 3-Hydroxylation† | |
|---|---|---|---|---|
| Total‡ | Increase in Patient vs. Control Cell Line | |||
| percent | ||||
| Control cell line | Normal/normal | 24.5 | NA | 94 |
| Infant 1 | IVS1+1G→C/IVS1+1G→C | 35.1 | 43 | 4 |
| Father of Infant 1 | IVS1+1G→C/normal | NS | NS | 91 |
| Mother of Infant 1 | IVS1+1G→C/normal | NS | NS | 88 |
| Infant 2 | Gln276→Stop/Gln276→Stop | 34.7 | 42 | 0 |
| Infant 3 | Met1→Ile/16-nucleotide duplication in exon 1 | 31.3 | 28 | 21 |
| Father of Infant 3 | Met1→Ile/normal | NS | NS | 89 |
| Mother of Infant 3 | 16-nucleotide duplication in exon 1/normal | NS | NS | 89 |
| Patient with classic osteogenesis imperfecta (type III)§ | Gly997→Ser/normal | 34.5 | 41 | 98 |
NA denotes not applicable, and NS not studied.
The percentage was calculated as the number of Pro986 residues with 3-hydroxylation divided by the total number of Pro986 residues. These data were determined by mass spectrometry of tryptic peptides of α1(I) collagen.
The percentage was calculated as the number of hydroxylated lysine residues divided by the total number of lysine residues. These data were determined by high-performance liquid chromatography.
This patient had a structural mutation near the carboxyl end of the α1(I) chain.
DISCUSSION
We identified 10 children who had lethal or severe osteogenesis imperfecta without primary collagen mutations but with excess post-translational modification of type I collagen, indicative of delayed folding of the collagen helix. Most patients with osteogenesis imperfecta and overmodified collagen have a structural mutation in one of the two chains of type I collagen. However, in 10 to 15% of cases of osteogenesis imperfecta, no collagen mutation is found, suggesting that alterations are present in other genes.
In 3 of the 10 children, we identified a recessive form of lethal osteogenesis imperfecta, caused by null mutations in CRTAP, a gene that encodes a protein that is essential for one of the post-translational modifications of types I and II collagen.18 The affected infants had defects in both CRTAP alleles, low levels of CRTAP mRNA, a lack of CRTAP protein, and minimal prolyl 3-hydroxylation of type I collagen. The CRTAP mutations led to premature termination of translation, triggering intracellular destruction of the mutant mRNA transcripts. The parents of Infants 1 and 3, who were available for mutation analysis, were heterozygous carriers; they had minimally reduced levels of CRTAP mRNA, CRTAP protein, and Pro986 hydroxylation in the α1(I) chain. The parents of Infant 2 did not participate in the analysis, but the infant had a homozygous point mutation that directly causes a Gln276→Stop substitution in exon 4 of CRTAP. CRTAP mutations did not account for our other seven cases of severe osteogenesis imperfecta without a primary collagen defect. It is also difficult to know what proportion of all cases of lethal osteogenesis imperfecta is caused by CRTAP mutations, because the genes for type I collagen (COL1A1 and COL1A2) are not sequenced in many cases for which there is a positive biochemical test. A reasonable preliminary estimation is that CRTAP defects cause 2 to 3% of cases of lethal osteogenesis imperfecta.
Although infants with CRTAP deficiency have a clinical presentation similar to that of infants with perinatal lethal type II osteogenesis imperfecta,2 there are distinctive features. In patients with CRTAP deficiency owing to a recessive mutation, head circumference is relatively small, the eyes show proptosis because of shallow orbits, and the sclerae are white or light blue; patients with type II osteogenesis imperfecta and a structural collagen defect have relative macrocephaly and dark blue sclerae. In recessive and dominant lethal osteogenesis imperfecta, the bones are severely undermineralized, have multiple fractures prenatally, and result in an abducted positioning of the legs. On radiography, the long bones in infants with CRTAP deficiency are characterized by a lack of diaphyseal modeling (undertubulation). In both the dominant and recessive forms, respiratory insufficiency causes early death.
Both recessive and dominant lethal osteogenesis imperfecta result in increased modification of the type I collagen helix. Classic dominantly inherited osteogenesis imperfecta involves defects in the primary sequence of the collagen chains22 that delay triple helix formation at the site of the defect,23 exposing the constituent chains to increased collagen modification by lysyl and prolyl hydroxylases at multiple sites along the length of the helix12,13 and causing delayed electrophoretic migration of collagen chains.24 Recessive CRTAP deficiency results in no or minimal Pro986 hydroxyla tion near the carboxyl end of collagen, where triple helix formation begins.4 CRTAP function, 3-hydroxylation, or both must be crucial for triple helix folding, since the lack of these processes also results in excess modification of the helical regions of the chains and the same delayed electrophoretic migration as seen in dominant osteogenesis imperfecta.
Our study shows that CRTAP acts as part of a collagen 3-hydroxylation complex17 and may also directly mediate collagen helical assembly. The human CRTAP gene has been mapped to chromosome 3p22,21 as has been type VII osteogenesis imperfecta, a recessive, severe, nonlethal form of osteogenesis imperfecta with distinctive histologic characteristics of the bone.11,25 A partially functional CRTAP mutant allele was recently identified in patients with type VII osteogenesis imperfecta.18 In contrast, the infants described here had null CRTAP mutations, excess modification of type I collagen, and a condition that was lethal in the first year of life. The spectrum of murine Crtap and human CRTAP mutations suggests that prolyl 3-hydroxylation of type I collagen is crucial for normal bone formation. A lack of prolyl 3-hydroxylation in other collagen types may affect the development of multiple organs (including the heart, kidney, and lung), which may contribute to the lethal outcome.
Supplementary Material
Acknowledgments
Supported by intramural funding from the National Institute of Child Health and Human Development (to Drs. Marini and Leikin) and by grants from the National Institutes of Health (DE016990 to Dr. Lee, HD22657 to Drs. Lee and Eyre, and AR37318 to Dr. Eyre) and the Kimberly V. Talley Chair of Genetics at the University of Oklahoma Health Sciences Center (to Dr. Mulvihill).
We thank the parents of the affected infants for their support, especially the family of Infant 3 for their dedication through years of various investigations; the nurses of the former 9 West Pediatric Unit, National Institutes of Health Clinical Center, for their care of Infants 1 and 3; Melissa A. Scott for her skillful assistance in the preparation of collagen for mass spectrometry analysis; and Anna Galicka, Ph.D., for the sequencing of collagen complementary DNA.
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Marini JC. Osteogenesis imperfecta In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson textbook of pediatrics. 17th ed. Philadelphia: W.B. Saunders, 2004: 2336–8. [Google Scholar]
- 2.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979;16:101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byers P, Cole W. Osteogenesis imperfecta In: Royce PM, Steinmann B, eds. Connective tissue and its heritable disorders: molecular, genetic, and medical aspects. 2nd ed. New York: Wiley-Liss, 2002: 385–430. [Google Scholar]
- 4.Bruckner P, Eikenberry EF. Formation of the triple helix of type I procollagen in cellulo: temperature-dependent kinetics support a model based on cis in equilibrium trans isomerization of peptide bonds. Eur J Biochem 1984;140:391–5. [DOI] [PubMed] [Google Scholar]
- 5.Aitchison K, Ogilvie D, Honeyman M, Thompson E, Sykes B. Homozygous osteogenesis imperfecta unlinked to collagen I genes. Hum Genet 1988;78:233–6. [DOI] [PubMed] [Google Scholar]
- 6.Byers PH, Tsipouras P, Bonadio JF, Starman BJ, Schwartz RC. Perinatal lethal osteogenesis imperfecta (OI type II): a biochemically heterogeneous disorder usually due to new mutations in the genes for type I collagen. Am J Hum Genet 1988;42: 237–48. [PMC free article] [PubMed] [Google Scholar]
- 7.Wallis GA, Sykes B, Byers PH, Mathew CG, Viljoen D, Beighton P. Osteogenesis imperfecta type III: mutations in the type I collagen structural genes, COL1A1 and COL1A2, are not necessarily responsible. J Med Genet 1993;30:492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams EM, Nicholls AC, Daw SC, et al. Phenotypical features of an unique Irish family with severe autosomal recessive osteogenesis imperfecta. Clin Genet 1989;35:181–90. [DOI] [PubMed] [Google Scholar]
- 9.Glorieux FH, Rauch F, Plotkin H, et al. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res 2000;15:1650–8. [DOI] [PubMed] [Google Scholar]
- 10.Glorieux FH, Ward LM, Rauch F, Lalic L, Roughley PJ, Travers R. Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J Bone Miner Res 2002;17:30–8. [DOI] [PubMed] [Google Scholar]
- 11.Ward LM, Rauch F, Travers R, et al. Osteogenesis imperfecta type VII: an autosomal recessive form of brittle bone disease. Bone 2002;31:12–8. [DOI] [PubMed] [Google Scholar]
- 12.Kielty C, Grant M. The collagen family: structure, assembly, and organization in the extracellular matrix In: Royce PM, Steinmann B, eds. Connective tissue and its heritable disorders: molecular, genetic, and medical aspects. 2nd ed. New York: Wiley-Liss, 2002:159–221. [Google Scholar]
- 13.Kivirikko K, Myllyla R. The hydroxylation of prolyl and lysyl residues In: Freedman RB, Hawkins HC, eds. The enzymology of post-translational modification of proteins. London: Academic Press, 1980: 53–104. [Google Scholar]
- 14.Risteli J, Tryggvason K, Kivirikko KI. Prolyl 3-hydroxylase: partial characterization of the enzyme from rat kidney cortex. Eur J Biochem 1977;73:485–92. [DOI] [PubMed] [Google Scholar]
- 15.Fietzek PP, Rexrodt FW, Wendt P, Stark M, Kuhn K. The covalent structure of collagen: amino-acid sequence of peptide 1-CB6-C2. Eur J Biochem 1972;30:163–8. [DOI] [PubMed] [Google Scholar]
- 16.Fietzek PP, Kuhn K. The primary structure of collagen. Int Rev Connect Tissue Res 1976;7:1–60. [DOI] [PubMed] [Google Scholar]
- 17.Vranka JA, Sakai LY, Bachinger HP. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J Biol Chem 2004;279: 23615–21. [DOI] [PubMed] [Google Scholar]
- 18.Morello R, Bertin TK, Chen Y, et al. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006;127:291–304. [DOI] [PubMed] [Google Scholar]
- 19.Castagnola P, Gennari M, Morello R, et al. Cartilage associated protein (CASP) is a novel developmentally regulated chick embryo protein. J Cell Sci 1997;110:1351–9. [DOI] [PubMed] [Google Scholar]
- 20.Morello R, Tonachini L, Monticone M, et al. cDNA cloning, characterization and chromosome mapping of Crtap encoding the mouse cartilage associated protein. Matrix Biol 1999;18:319–24. [DOI] [PubMed] [Google Scholar]
- 21.Tonachini L, Morello R, Monticone M, et al. cDNA cloning, characterization and chromosome mapping of the gene encoding human cartilage associated protein (CRTAP). Cytogenet Cell Genet 1999;87: 191–4. [DOI] [PubMed] [Google Scholar]
- 22.Marini JC, Forlino A, Cabral WA, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raghunath M, Bruckner P, Steinmann B. Delayed triple helix formation of mutant collagen from patients with osteogenesis imperfecta. J Mol Biol 1994;236: 940–9. [DOI] [PubMed] [Google Scholar]
- 24.Bonadio J, Holbrook KA, Gelinas RE, Jacob J, Byers PH. Altered triple helical structure of type I procollagen in lethal perinatal osteogenesis imperfecta. J Biol Chem 1985;260:1734–42. [PubMed] [Google Scholar]
- 25.Labuda M, Morissette J, Ward LM, et al. Osteogenesis imperfecta type VII maps to the short arm of chromosome 3. Bone 2002;31:19–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.