

Abstract
Selenocysteine (Sec) is the 21st proteogenic amino acid in the genetic code. Incorporation of Sec into proteins is a complex and bioenergetically costly process that evokes the question: “Why did nature choose selenium?” An answer that has emerged over the past decade is that Sec confers resistance to irreversible oxidative inactivation by reactive oxygen species (ROS). Here, we explore the question of whether this concept can be broadened to include resistance to reactive electrophilic species (RES) since oxygen and related compounds are merely a subset of RES. To test this hypothesis we inactivated mammalian thioredoxin reductase (Sec-TrxR), a mutant containing alpha-methylselenocysteine ((αMe)Sec-TrxR), and a cysteine-ortholog TrxR (Cys-TrxR) with various electrophiles including acrolein, 4-hydroxynonenal, and curcumin. Our results show that the acrolein-inactivated Sec-TrxR and the (αMe)Sec-TrxR mutant could regain 25% and 30% activity respectively, when incubated with 2 mM H2O2 and 5 mM imidazole. In contrast, the Cys-TrxR did not regain activity under the same conditions. We posit that Sec-enzymes can undergo a repair process via β-syn selenoxide elimination that ejects the electrophile, leaving the enzyme in the oxidized selenosulfide state. (αMe)Sec-TrxR was created by incorporating the non-natural amino acid ((αMe)Sec into TrxR by semisynthesis and allowed for rigorous testing of our hypothesis. This Sec-derivative enables higher resistance to both oxidative and electrophilic inactivation because it lacks a backbone Cα-H, which prevents loss of selenium through formation of dehydroalanine. This is the first time this unique amino acid has been incorporated into an enzyme and is example of state-of-the-art protein engineering.
Graphical Abstract
INTRODUCTION
Thioredoxin reductase (TrxR) is a NADPH dependent pyridine nucleotide-disulfide oxidoreductase whose canonical function is to reduce the small protein, thioredoxin (Trx).1 TrxR, Trx, and NADPH comprise the vital antioxidant thioredoxin system which is responsible for maintaining redox homeostasis in the cell by providing essential reducing equivalents to a number of important protein/enzyme targets.2 Mammalian thioredoxin reductase (mTrxR) contains the rare amino acid selenocysteine (Sec) at the penultimate position of its C-terminus that is involved in the catalytic function of the enzyme.3
The prevailing hypothesis for many years to explain the use of Sec in enzymes relative to cysteine (Cys) has been enhanced catalytic efficiency.4, 5 However, Cys-containing orthologs of TrxR exist in prokaryotes and lower eukaryotes that can catalyze the same reduction of their cognate Trx substrates with comparable catalytic efficiency.6 Thus, nature likely chose to use Sec in enzymes due to other factors related to the chemical difference between selenium and sulfur.7
One hypothesis, first proposed by Chaudière in 1992, and shortly after by Maroney, is that Sec replaces Cys in enzymes such that the Sec-enzyme is much better able to resist irreversible oxidative inactivation.8, 9 This hypothesis has been advocated by us and has been given strong support recently by the work of Conrad.10-12 Here, we broaden that hypothesis as follows: Sec evolved in enzymes to not only resist oxidative stress, but electrophilic stress in general. Oxygen, and related reactive oxygen species (ROS) are just a specific subset of related biological reactive electrophilic species (RES) that selenoenzymes such as TrxR encounter. Many RES contain an α, β-unsaturated carbonyl moiety that makes them Michael-type acceptors for 1,−4 addition reactions.13, 14 Given its low pKa, Sec is an excellent biological nucleophile at neutral pH making it a strong target for adduction by RES as illustrated in Figure 1.5
Figure 1: Alkylation of Sec-enzymes by 1,−4 addition reactions.
Due to the low pKa of Sec, it acts as a nucleophile at neutral pH and can undergo Michael-type addition reactions with RES that contain an α, β-unsaturated carbonyl.
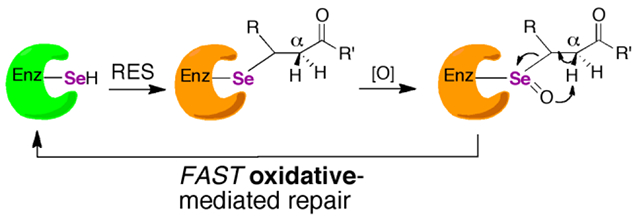
Interactions between Sec-TrxRs and RES, and the biological consequences of such reactions, have been well documented.13-16 Sec-TrxRs adducted by RES are sometimes referred to as “SecTRAPs” (selenium compromised thioredoxin reductase-derived apoptotic proteins).17 SecTRAPs are known to be devoid of thioredoxin reductase activity, however, the reduced flavin coenzyme of mTrxR either by itself, or in combination with the alkylated Sec residue, can donate one electron to molecular oxygen, which then dismutates into hydrogen peroxide (H2O2).17 Thus, while Sec-TrxRs are typically considered antioxidant proteins, when adducted by RES, SecTRAPs are pro-oxidants, increasing cellular oxidant levels.17
The intriguing question we ask here is whether the alkylation of Sec can be reversed such that activity of the Sec-enzyme can be restored. If so, this would provide an additional rationale to justify the large bioenergetic cost of Sec-insertion into a protein. Some evidence of this hypothesis has been previously published by Randall and coworkers who showed that Trx-reductase activity could be restored after alkylation by acrolein in a cellular context, which appeared to involve a reductive mechanism that relied on glutathione (GSH) and Trx.18
Here, we provide direct in vitro evidence for this earlier observation, albeit by a different mechanism. We provide evidence that alkylation of Sec-TrxR by acrolein can be reversed via a β-syn selenoxide elimination. This mechanism makes use of the fact that when the Sec residue in TrxR is alkylated, H2O2 is ultimately produced that could oxidize the selenide to a selenoxide, which could then abstract the α-proton on the α, β-unsaturated carbonyl of the electrophile. This proton would be “beta” relative to the selenium atom. Our proposed β-syn selenoxide elimination reaction would kick off the electrophile, allowing TrxR to return to the oxidized selenosulfide state of the enzyme as illustrated in Figure 2A.
Figure 2: Selenoxide elimination mechanism for reversing alkylation on Sec of TrxR.
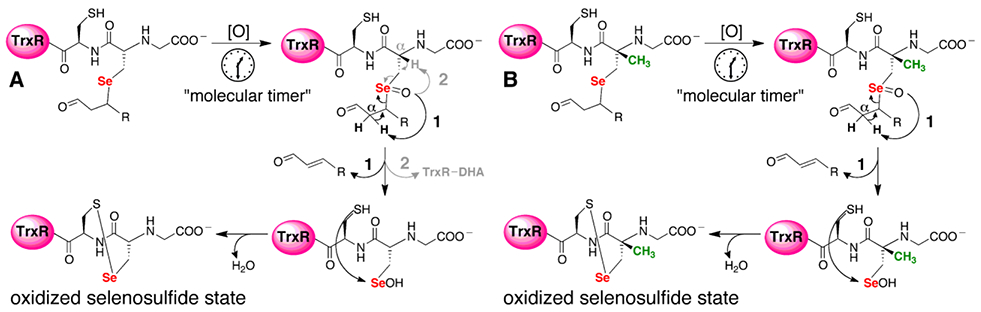
(A) Once Sec is alkylated by an electrophile, mTrx will produce H2O2, which can act as a type of “molecular timer”. Once the concentration of H2O2 is sufficient, the selenide will be converted to a selenoxide which can undergo rapid β-syn elimination via pathway 1 (bold pathway). Abstraction of the Cα-proton of the electrophile leads to elimination of the electrophile and restoration of TrxR back to its oxidized selenosulfide state. However, abstraction of the Cα-H from the enzyme backbone via pathway 2 (gray) results in elimination of selenium from the enzyme and the formation of dehydoalanine (DHA). Without Sec in the active site, enzyme activity cannot be restored. (B) In the mutant mTrxR, the backbone Cα-H is replaced with a Cα-CH3. This replacement means that the mutant can only utilize pathway 1 in the β-syn elimination mechanism. Thus, the mutant should be better able to reverse the electrophilic alkylation and restore Trx-reductase activity.
To test this hypothesis, we replaced the Cα-H of the enzyme backbone of the Sec residue with a Cα-CH3 by synthesizing alpha-methylselenoysteine ((αMe)Sec), then incorporating (αMe)Sec into TrxR via semisynthesis.19-21 This unique mutant can only undergo β-syn selenoxide elimination via pathway 1 and not pathway 2 as illustrated in Figure 2B. Our hypothesis predicts that this mutant should be able to reverse the alkylation and restore activity much better than the wild type (WT) enzyme.
Indeed, here we report that both the WT and mutant TrxR enzymes were able to regain significant activity after acrolein treatment when incubated with 5 mM imidazole (a scavenger for the acrolein eliminated from TrxR) and 2 mM H2O2 at 37 °C for 10 min at pH 7.15. As our hypothesis predicted, the (αMe)Sec-containing mutant regained more activity. The results show that the presence of selenium is key to restoring activity to TrxR, as a Cys-containing ortholog from D. melanogaster (DmTrxR) was not able to regain activity under the same conditions. These results support the broader idea that Sec-enzymes are not only able to resist oxidative stress, but electrophilic stress as well.
MATERIALS AND METHODS
Materials.
Solvents including dichloromethane (DCM), acetonitrile (ACN), dimethylformamide (DMF), methanol, anhydrous diethyl ether, trifluoroacetic acid (TFA), and ethyl acetate (EtOAc), along with Corning™ Costar™ Spin-X™ centrifuge tube filters to remove catalase agarose, and hydrogen peroxide (30%, ACS grade) for enzyme assays were purchased from Fisher Scientific (Waltham, MA). N-Fmoc amino acids and Boc-Cys(Mob)-OH were purchased from RSsynthesis (Louisville, KY). 2-Chlorotrityl chloride resin SS (100-200 mesh), 1-hydroxy-7-azabenzotriazole (HOAt) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) for solid-phase peptide synthesis (SPPS) were purchased from Advanced ChemTech (Louisville, KY). Chitin resin for TrxR semisynthesis was purchased from New England Biolabs (Ipswich, MA). Immobilized catalase beads were obtained from BioVision Incorporated (San Francisco, CA). Acrolein solution (100 μg/mL or 5000 μg/mL) in methanol was purchased from Ultra Scientific (Santa Clara, CA). NADPH for enzyme assays was purchased from AppliChem (Darmstadt, Germany). RapiGest™ SF surfactant for MS analysis of TrxR enzymes was purchased from Waters Corp. (Milford, MA). Sequencing grade modified trypsin for digestion of TrxR enzymes was purchased from Promega (Madison, WI). Imidazole (reagent grade), bovine serum albumin (lyophilized powder) iodoacetamide, 2,2′-dithiobis(5-nitropyridine) (DTNP), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, 99%), selenocysteamine dichloride, pentafluorophenol (>99%), iodoacetaminde (BioUltra), 4-hydroxynonenal (Calbiochem), curcumin (from Curcuma longa, powder), Amicon® ultra centrifugal filter units, and silica gel for column chromatography were purchased from MilliporeSigma (Burlington, MA). Sodium borohydride (98+%, powder), triisopropylsilane (TIS, 98%), piperidine, ninhydrin, formic acid (98+%), N-methylmorpholine (99%), and N, N′-dicyclohexylcarbodiimide (DCC, 99%), were purchased from ACROS Organics (Pittsburgh, PA). All other chemicals were purchased from Sigma-Aldrich (Milwaukee, WI), Fisher Scientific (Waltham, MA), or ACROS Organics (Pittsburgh, PA). Mass spectral analysis was performed via positive ESI on a Thermo Q Exactive mass spectrometer where full MS scans were performed at a resolution of 70,000 in positive ion mode. Collected data files were analyzed using Xcalibur Qual Browser V3 (ThermoScientific, Waltham, MA). Enzyme kinetic assays were performed on a Cary50 UV-Vis spectrophotometer (Walnut Creek, CA).
Synthesis of Boc-Cys(Mob)-OPfp.
Boc-Cys(Mob)-OH (6.0 mmol, 2.05g) was dissolved in 10 mL of dry DCM and cooled to 0 °C via ice bath, under nitrogen gas. To this, DCC (3.0 mmol, 619.0 mg) dissolved in 10 mL of dry DCM was added. The reaction was allowed to proceed ~15 min with constant stirring. A white precipitate was observed to form after ~5 min. Pentafluorophenol (6.0 mmol, 1.1g) dissolved in 20 mL dry ethyl acetate was added under nitrogen gas, and the reaction was immediately removed from ice and allowed to warm to 22 °C, then stirred an additional 1.5 h. The white urea precipitate was removed from solution via filtration, and the filtrate was placed on ice for 15 min to prompt formation of more precipitate and filtered again. The final filtrate was reduced to a yellow oil via roto-evaporation, and column purified using ~25 g silica gel and ~300 mL of 80:20 hexanes:EtOAc as a solvent. Fractions (20 mL) were collected and the desired Boc-Cys(Mob)-OPfp eluted with an Rf = 0.44. The final eluent containing Boc-Cys(Mob)-OPfp was dried under vacuum producing a white, crystalline solid in 89% yield (4.3 g).
Solid phase peptide synthesis of tripeptide H-CUG-OH.
Fmoc-Sec(Mob)-OH, synthesized as described previously,22 was used make the peptide of sequence H-Cys-Sec-Gly- OH (H-CUG-OH). We note that we followed the recommendations of Jones23 for all of the abbreviations of amino acids and reagents for peptide synthesis in this paper. The peptide was synthesized on a 0.1 mmol scale using a glass vessel that was shaken with a model 75 Burrell wrist action shaker. First, 300 mg of 2-chlorotrityl chloride resin SS (100-200 mesh), was swelled in 10 mL of DCM for 30 min. Fmoc-Gly-OH was then coupled directly to the resin using 8 mL of 2% N-methylmorpholine (NMM) in DCM, shaking for 1 h at 22 °C. The resin was then capped using 10 mL of 10% methanol, 10% NMM in DCM. Next, Fmoc-Sec(Mob)-OH was coupled to Gly using a solution of 0.2 mmol amino acid, 1% N,N′-diisopropylcarbodiimide (DIC), and 0.2 mmol 1-hydroxy-7-azabenzotriazole (HOAt) in DMF, shaking for 2 h at 22 °C. The last amino acid, Fmoc-Cys(Trt)-OH, was coupled using a solution of 0.2 mmol amino acid, 1% DIC, and 0.2 mmol HOAt in DMF, shaking for 1 h, at 22 °C. Preactivation of any amino acid was not performed prior to coupling. Between amino acid couplings, the Fmoc protecting group was removed via two 10 min agitations with 10 mL of 20% piperidine in DMF. Success of Fmoc removal steps and amino acid couplings were monitored qualitatively using a ninhydrin test.24 Removal of the final Fmoc protecting group completed the peptide synthesis. H-CUG-OH was next cleaved from the resin as described below.
Solid phase peptide synthesis of H-C(αMe)UG-OH.
The preparation of 2-chlorotrityl chloride resin, coupling and subsequent deprotection of Fmoc-Gly-OH was completed as described above on a 0.1 mmol scale. Next, Fmoc-(αMe)Sec(But)-OH, synthesized as described previously,21 was coupled to N-deprotected Gly using a solution of 0.2 mmol amino acid, 1% DIC, and 0.2 mmol HOAt in DMF, shaking for 2 h, at 22 °C. The Fmoc group was then removed from (αMe)Sec via six, 45 min agitations with 10 mL of 20% piperidine in DMF at 22 °C. Note: ninhydrin test was not used to monitor complete Fmoc removal as we found the (αMe)Sec-amine to be too hindered to react with the ninhydrin dye. To couple Cys to (αMe)Sec, the resin was washed extensively with DCM, then swelled in a minimal volume of DCM and shaken with 2% NMM for 15 min. A solution of 0.22 mmol Boc-Cys(Mob)-OPfp (synthesized as described above) in 5 mL DCM was then added to the resin and shaken for 1.5 h at 22 °C. This was followed by addition of 0.15 mmol HATU in 5 mL of DMF for an additional 1 h. The resin was washed 4x with DMF, then 4x with DCM and the Boc-Cys(Mob)-OPfp coupling step was repeated 10-15x. Note: we were able to scale this tripeptide synthesis up to a 0.3 mmol scale, but for best results typically made the tripeptide on a 0.1 mmol scale. The final N-Boc protecting group was not removed before cleavage from the resin, and the resin cleavage procedure is described below.
Cleavage of peptides from resin.
Peptides (H-CUG-OH and H-C(αMe)UG-OH) were cleaved from the resin via a 4 h reaction in a 10 mL cleavage cocktail consisting of 0.4 mmol of 2,2′-dithiobis(5-nitropyridine) (DTNP),25 2% H2O, 2% TIS, 96% TFA. Following cleavage and side chain deprotection, the resin was washed with TFA and the volume of the cleavage solution was reduced by evaporation with nitrogen gas. The peptide solution was then transferred by pipette into cold, anhydrous diethyl ether to precipitate the peptide and then spun by centrifugation at 3000 rpm on a clinical centrifuge (International Equipment Co., Boston, MA) for 10 min to pellet the peptide. Pelleted peptides were dried, dissolved in a minimal volume of cleavage cocktail, then precipitated in ether and pelleted as described previously. This wash process was repeated 5x for each peptide to remove any unreacted DTNP. After washing, the final peptide pellet was dried, dissolved in a minimal amount of water, lyophilized, and used without further purification.
Semisynthesis of TrxR enzymes.
The production and purification of the recombinant DmTrxR-CCS and the semi-synthetic mTrxR-CUG enzymes used in this study have been previously reported.20, 26 Our semisynthetic method ligates the synthesized tripeptides to a thioester-tagged TrxR polypeptide missing this tripeptide using intein-mediated ligation.19 This procedure has been published multiple times,20 and we used an identical procedure in this work to obtain mTrxR-C(αMe)UG mutant. The concentrations of the final, purified mTrxR-CUG, mTrxR-C(αMe)UG, and DmTrxR-CCG enzymes were determined based on the extinction coefficient of flavin at 460 nm (ε460 = 22.6 mM−1 cm−1 for the dimer).27
Characterization of TrxR, general.
After purification, TrxR enzymes were assayed for activity using Trx, DTNB, selenocysteamine, and hydrogen peroxide substrates, as described previously.28 All activity assays were performed on a Cary 50 UV/VIS spectrophotometer from Varian (Walnut Creek, CA) at 22 °C, in 100 mM potassium phosphate, 1 mM EDTA, pH 7.0. Activity was monitored over 2 min and Vo was determined from the linear slope. Plots of Vo/amount of enzyme (mol) vs. substrate concentration were fit by the Michaelis-Menten equation using KaleidaGraph 4.02 from Synergy Software (Reading, PA) and activities are reported as moles of NADPH consumed per min per mole of TrxR.
Activity Assays with Trx, H2O2, and selenocysteamine substrates.
Activity assays for Trx, H2O2, and selenocysteamine substrates were performed by monitoring NADPH consumption via decrease in absorbance at 340 nm (A340) using an extinction coefficient of 6220 M−1cm−1. A stock solution of 20 mM NADPH in ddI H2O was prepared prior to experiments and frozen in aliquots.
Trx-reductase activity was measured via a 0.5 mL reaction containing 200 μM NADPH and varying concentrations of E. coli Trx that ranged from 0-180 μM. All assays were initiated by addition of the following concentrations of enzyme: 260 nM mTrxR-C(αMe)UG, 2 nM mTrxR-CUG, and 200 nM DmTrxR-CCS.
Stock solutions of H2O2 were freshly prepared in ddI H2O before each experiment and the concentration of the solution was determined spectrophotometrically using an extinction coefficient of 43.6 M−1cm−1 at 240 nm. H2O2-reductase activity was measured via a 0.5 mL reaction containing 200 μM NADPH and varying concentrations of H2O2 that ranged from 0-400 mM. All assays were initiated by addition of the following concentrations of enzyme: 260 nM mTrxR-C(αMe)UG and 6 nM mTrxR-CUG.
Stock solutions of selenocysteamine were freshly prepared in ddI H2O before each experiment. Selenocysteamine-reductase activity was measured in a 0.5 mL reaction containing 200 μM NADPH and varying concentrations of selenocysteamine that ranged from 0-500 μM. All assays were initiated by addition of the following concentrations of enzyme: 104 nM mTrxR-C(αMe)UG and 2 nM mTrxR-CUG. For all substrates, background NADPH consumption was corrected for by subtracting the activity of control assays in which either TrxR enzymes or substrates were omitted from the reaction. All assays were repeated in triplicate for each enzyme.
Activity Assays with DTNB substrate.
Activity assays with DTNB were performed by monitoring formation of TNB− ion via increase in absorbance at 412 nm (A412) using an extinction coefficient of 13600 M−1 cm−1. Stock solutions of DTNB were freshly prepared in ethanol before each experiment. A stock solution of 20 mM NADPH in ddI H2O was prepared prior to experiments and frozen in aliquots. DTNB reductase activity was measured in a 0.5 mL reaction containing 200 μM NADPH and varying concentrations of DTNB that ranged from 0-5 mM. All assays were initiated by addition of the following concentrations of enzyme: 6 nM mTrxRΔ3, 12 nM mTrxR-C(αMe)UG, and 4 nM mTrxR-CUG. Background TNB− formation was corrected for by subtracting the activity of control assays in which enzyme was omitted from the reaction. All assays were repeated in triplicate for each enzyme.
Inactivation of TrxR enzyme activity by H2O2.
Resistance to inactivation by H2O2 for TrxR enzymes was completed by a two-part assay in which TrxR enzymes were first exposed to increasing concentrations of H2O2 (0-100 mM), and second, enzyme activity remaining was assessed relative to a control reaction where H2O2 was omitted. For all such assays, 150 μL reactions were completed in 100 mM potassium phosphate, 1 mM EDTA, pH 7 buffer at 22 °C. To monitor the Trx-reductase activity remaining, mTrxR-CUG (25 nM), mTrxR-C(αMe)UG (1 μM), and DmTrxR-CCS (225 nM) were pre-reduced by 5 min incubation with 200 μM NADPH. Next, TrxR enzymes were incubated with increasing amounts of H2O2 (0 mM – 100 mM), for 25 min followed by addition of 50 μL catalase agarose. Catalase agarose was then removed from the reaction via centrifugation (3200 RPM, 2 min) in Corning™ Costar™ Spin-X™ centrifuge tube filters. The effluent was then combined with an additional 200 μM NADPH, and activity assays were initiated by addition of 110 μM Trx. Trx-reductase activity was measured by monitoring the consumption of NADPH as a decrease in A340 for 3 min. Treatment of mTrxR enzymes with H2O2 and subsequent hydrogen peroxidase activity was monitored exactly as described for Trx substrate, except activity assays were initiated by addition of 250 mM H2O2 substrate. Again, hydrogen peroxidase activity was measured by monitoring the consumption of NADPH as a decrease in A340 for 3 min. To monitor the DTNB-reductase activity remaining, mTrxR-CUG (8 nM), mTrxR-C(αMe)UG (13 nM), and DmTrxR-CCS (75 nM) were pre-reduced by 5 min incubation with 200 μM NADPH. Next, mTrxR enzymes were incubated with increasing amounts of H2O2 for 25 min followed by centrifugation in Amicon filters (30 kDa MWCO) at 13,000 RPM for 2 min. To ensure complete removal of H2O2, a washing step was performed where 300 μL of 100 mM potassium phosphate, 1 mM EDTA, pH 7 buffer was added, and the reaction was spun again (30 kDa MWCO Amicon, 13,000 RPM). The washing step was repeated twice. After spins, 45 μL of retained enzyme solution was combined with 200 μM NADPH and buffer (final volume 150 μL assay), and DTNB-reductase activity was measured by monitoring the formation of TNB− as an increase in A412 for 3 min. For each assay, a no DTNB or no enzyme background was performed. In each of these control experiments, no activity was detected ensuring complete removal of H2O2 by catalase agarose. For all substrates, the change in absorbance was converted to percentage of activity remaining relative to the oxidant-untreated control. All assays were repeated in triplicate for each enzyme.
Determination of the half maximal inhibitory concentration (IC50) of electrophiles.
Inhibition of TrxR enzymes by an electrophile was completed by a two-part assay in which TrxR enzymes were first exposed to increasing concentrations of either curcumin (0-100 μM), acrolein (0-18 nM), or 4-hydroxynonenal (0-800 μM), and second, enzyme activity remaining was assessed relative to a control reaction where the electrophile was omitted. Prior to experimentation, fresh stock solutions of acrolein, 4-hydroxynonenal, or curcumin were prepared in ethanol. For all electrophile inhibition reactions, assays were completed in 100 mM potassium phosphate, 1 mM EDTA, pH 7 buffer at 22 °C. To determine the IC50 of curcumin, mTrxR-CUG (12 nM) or mTrxR-C(αMe)UG (650 nM) were pre-reduced by 5 min incubation with 200 μM NADPH in a 0.5 mL assay. Next, mTrxR enzymes were incubated with increasing amounts of curcumin for 20 min, followed by addition of 200 μM NADPH for the assay. To determine the IC50 of 4-hydroxynonenal, mTrxR-CUG (12 nM) or mTrxR-C(αMe)UG (650 nM) were pre-reduced by 5 min incubation with 200 μM NADPH in a 150 μL assay. Next, mTrxR enzymes were incubated with increasing amounts of 4-hydroxynonenal for 1 h, followed by addition of 200 μM NADPH for the assay. To determine the IC50 of acrolein, mTrxR-CUG (10 nM), mTrxR-C(αMe)UG (780 nM), or DmTrxR-CCS (150 nM) were pre-reduced by 5 min incubation with 200 μM NADPH in a 150 μL assay. Next, mTrxR enzymes were incubated with increasing amounts of acrolein for 10 min, followed by addition of 200 μM NADPH for the assay. For all inhibition experiments, activity assays were initiated by addition of 150 μM Trx. Trx-reductase activity was measured by monitoring the consumption of NADPH as a decrease in A340 for 3 min. For each assay, a no substrate background was performed. For all substrates, the change in absorbance was converted to percentage of activity remaining relative to the electrophile-untreated (ethanol only) control. All assays were repeated in triplicate for each electrophile tested.
Reversal of acrolein inhibition using imidazole and hydrogen peroxide.
Inhibition and subsequent reversal of mTrxR enzymes by acrolein was completed by a three-part assay in which the enzymes were first inhibited by acrolein, then acrolein inhibition was reversed via imidazole/H2O2 combinations, and third, enzyme activity remaining was assessed relative to a control reaction where acrolein was omitted. Prior to experimentation, fresh stock solutions of acrolein (in ethanol) and H2O2 (in ddI H2O) were prepared. The concentration of H2O2 was determined spectrophotometrically using an extinction coefficient of 43.6 M−1cm−1 at 240 nm. Reversal assays utilized a combination of three buffers to facilitate pH changes. For all enzymes, “Buffer A” was composed of 10 mM potassium phosphate, 10 mM sodium acetate (NaOAc), 5 mM KCl, pH 7.0; “Buffer C + imidazole” was composed of 200 mM potassium phosphate, 1 mM EDTA, 210 uM BSA, 21 mM imidazole, pH 7.25; and “Buffer C - imidazole” was composed of 200 mM potassium phosphate, 1 mM EDTA, 210 uM BSA, pH 7.25. For reactions with mTrxR-CUG, “Buffer B” was composed of 50 mM NaOAc, pH 4.5. For reactions with mTrxR-C(αMe)UG, “Buffer B” was composed of 50 mM NaOAc, pH 5.3. For reactions with DmTrxR-CCS, “Buffer B” was composed of 100 mM potassium phosphate, pH 7.
To begin, either mTrxR-CUG (30 nM), mTrxR-C(αMe)UG (780 nM), or DmTrxR-CCS (240 nM) were incubated with 400 μM NADPH in 55 μL of Buffer A for 5 min at 22 °C. To this, 35 μL of Buffer B was added (adjusting the reaction pH to 4.6 for mTrxR-CUG; to pH 5.5 for mTrxR-C(αMe)UG; and to pH 7 for DmTrxR-CCS), followed immediately by addition of either acrolein, or ethanol for control experiments. To achieve 75-85% inhibition: for mTrxR-CUG, 2.5 μM acrolein was used; for mTrxR-C(αMe)UG, 800 nM acrolein was used; and for DmTrxR-CCS, 7 μM acrolein was used. Enzymes were treated with acrolein or ethanol for 10 min at 22 °C.
Next, to reverse acrolein adduction, 40 μL of Buffer C (+/−) imidazole was added to the reaction (i.e. for reversal conditions with imidazole, Buffer C + imidazole was used and for reversal conditions with no imidazole, Buffer C – imidazole was used). For reversal conditions involving H2O2, 2 mM H2O2 was added immediately after addition of Buffer C (+/−) imidazole. Addition of Buffer C (+/−) imidazole adjusted the reaction pH to ~7.15 for all enzymes. The final concentration of imidazole in the reversal experiments was 5 mM. Reversal reactions were incubated for 10 min in a 37 °C water bath. After incubation, to all reactions, 50 μL catalase agarose was added and incubated for 15 min at 22 °C. To remove catalase agarose, reactions were transferred to a Corning™ Costar™ Spin-X™ centrifuge tube filter and centrifuged for 2 min at 3000 RPM. Flow through was collected and an additional 200 μM NADPH was added, then enzymes were assayed with Trx in a final volume of 150 μL (150 μM Trx for assays with mTrxR-CUG and DmTrxR-CCS; and 220 μM Trx for assays with mTrxR-C(αMe)UG). Trx-reductase activity was measured by monitoring the consumption of NADPH as a decrease in A340 for 3 min. For each assay, a “no substrate” background was performed. For all substrates, the change in absorbance was converted to percentage of activity remaining relative to the acrolein-untreated (ethanol only) control.
For the experiments described above, eight replicates were performed for the control (no inhibitor), acrolein inhibited enzymes, and the reversal condition containing both hydrogen peroxide and imidazole. Four replicates were performed for the assays that contained imidazole only and hydrogen peroxide only. Statistical analysis of the data was performed using GraphPad Prism™ (version 8) software, which calculated the standard deviation, the Student’s-test t-value, and corresponding p-value at the 95% confidence limit.
Digestion of TrxR enzymes for liquid chromatography mass spectrometry (LC-MS).
Samples of 15 μg TrxR enzymes were isolated and dried down using a speed-vac. 75 μL of 0.1% RapiGest™ surfactant in 50 mM ammonium bicarbonate pH 7.8 was then added to each sample and incubated at 37 °C for 1 h. Samples where then reduced with 5 μL of 100 mM DTT and heated at 37 °C for 10 min. Next, samples were alkylated with 10.4 μL of 10 mM iodoacetamide and incubated in the dark at RT for 30 min. To each sample, 25 μL of 0.1 μg/μL sequence-grade trypsin was added and then incubated at 37 °C overnight. The following day, samples were dried down via speed-vac. Next, 100 μL of 7% formic acid (FA) in 50 mM ammonium bicarbonate pH 7.8 was added to each sample prior to a 1 h incubation at 37 °C. Samples were dried down again prior to an addition of 100 μL of 0.1% trifluoroacetic acid and incubated for 1 h at 37 °C. Samples were dried down a third time using a speed-vac and then resuspended in 100 μL of 0.1% trifluoroacetic acid. Each sample was then centrifuged at 14,000 rpm at 4 °C for 5 min and the top 95 μL was collected for LC-MS.
LC-MS.
20 μL of each sample was injected onto a Waters X-select HSST3 (3.5 μm, 1.0 × 150 mm) liquid chromatography column at a flow rate of 0.1 mL/min. A 78-minute LC method was used with mobile phase A containing 0.1% formic acid (FA) in H2O and mobile phase B containing 0.1% FA in acetonitrile (ACN). From 0.0 to 4.0 min, B was held at 3%. From 4.0 to 44.0 min, B was raised to 40%. From 44.0 to 48.0 min, B was raised to 60%. From 48.0 to 52.0 min, B was held at 60%. From 52.0 to 58.0 min B was dropped to 3%. Finally, from 58.0 to 78.0 min, B was held at 3%. The LC effluent was directly infused into a Thermo Q Exactive mass spectrometer by ESI, and full MS scans were performed at a resolution of 70,000 in positive ion mode. Collected data files were analyzed using Xcalibur Qual Browser V3 (ThermoScientific, Waltham, MA).
MS Analysis of Full-length and Modified Thioredoxin Reductase Enzymes.
Theoretical trypsin digests of wild type Drosophila melanogaster thioredoxin reductase (UniProtKB-P91938), wild type mitochondrial mouse thioredoxin reductase 2 (UniProtKB- Q9JLT4), and mutant mitochondrial mouse thioredoxin reductase 2 containing a single substitution of Sec489 to (αMe)Sec489 were performed using the online ExPASy PeptideMass tool. Manual adjustments were made for the mutant containing (αMe)Sec489. To determine the presence of full-length enzyme, singly-charged [M+H]+ monoisotopic masses corresponding to the elemental composition of each complete C-terminal peptide were calculated using NIST mass values. However, for selenocysteine-containing peptides, the more abundant 80Se isotope mass was used rather than the monoisotopic 74Se isotope mass. Total peptide elemental composition also factored in the treatment of each cysteine and selenocysteine residue with iodoacetamide to form carbamidomethyl-cysteine (CAM-Cys), carbamidomethyl-selenocysteine (CAM-Sec), and carbamidomethyl-(αMethyl)selenocysteine (CAM-(αMe)Sec). The resulting mass/charge values for singly-charged C-terminal peptides SGLDPTPACCS (C48H79N14O21S2, 1251.50 m/z), SGLEPTVTGCUG (C46H78N13O19SSe, 1285.46 m/z) and SGLEPTVTGC(αMe)UG (C47H80N13O19SSe, 1299.48 m/z) were determined for wild type fruit fly, wild type mouse and mutant mouse thioredoxin reductase enzymes, respectively. Next, data files were opened with Thermo Xcalibur Qual Browser V3 and the LC peaks corresponding to the singly-charged C-terminal peptides were extracted. Spectra were visually evaluated and those containing singly-charged isotopic envelopes differing by no more than 0.05 m/z from the theoretical monoisotopic mass/charge values were used to indicate successful detection of full-length protein.
RESULTS AND DISCUSSION
Synthesis of the tripeptide containing (αMe)Sec.
We previously reported the synthesis of (αMe)Sec, incorporated it into a peptide corresponding to the active site of glutathione peroxidase (GPX),21 and subsequently demonstrated the ability of the (αMe)Sec-containing peptide to resist β-syn elimination from overoxidation. In our previous study, (αMe)Sec was the N-terminal amino acid in the peptide sequence and we did not attempt to couple another amino acid onto the amine of (αMe)Sec during SPPS. Importantly for this study, we sought out to make the peptide of sequence H-Cys-(αMe)Sec-Gly-OH for the semisynthesis of a mutant mTrxR, and therefore had to couple Fmoc-Cys-OH onto the amine of (αMe)Sec, which proved to be a difficult task.
Previous reports have highlighted the synthetic challenges of making peptides that contain sterically hindered and bulky (αMe)AAs.29-33 Specifically, it has been noted that forming an amide bond between an (αMe)AA and the carboxylic acid of the next amino acid in the peptide sequence is difficult due to the steric hinderance of the amine group, which is adjacent to the disubstituted α-carbon, of an (αMe)AA.29, 32
Traditional activation methods employed for amide bond formation, such as carbodiimides, are usually inefficient for (αMe)AA coupling, suffering from low yields, slow reaction rates, and necessitate the use of a large excess of reagents.29-33 Other coupling reagents, such as HATU or HOAt, have also proven to be ineffective in the synthesis of peptides containing (αMe)AAs.30 Albericio and coworkers previously reported synthesis of peptides that consist of difficult sequences with 7-azobenzotriazolyoxytris(pyrrolidino)phosphonium hexafluorophosphate (PyAOP).34 PyAOP has been successfully applied for the preparation of a range of peptides containing hindered amino acids, such as 2-aminoisobutyric acid (Aib).30, 35 PyAOP and the related PyBOP were also used for the synthesis of peptides containing the disubstituted Cα,α-dipropylglycine and Cα,α-diisobutylglycine,32 and α-methyl cysteine.36 However, in our hands, the best yield achieved when attempting to couple Fmoc-Cys(Mob)-OH onto the deprotected (αMe)Sec(But) on 2-chlorotrityl chloride resin using PyBOP was less than 30% on a 0.05 mmol scale despite 5 or more coupling reactions (data not shown). Increasing the scale of the reaction resulted in a further decrease in yield.
Our solution to this problem was to make use of amino acid pentaflurophenol (PfP) esters, which are known to be highly reactive and free from undesired side reactions.37 We synthesized Boc-Cys(Mob)-OPfP to facilitate the acylation between (αMe)Sec and Cys. We synthesized the Boc-protected amino acid instead of the Fmoc-protected amino acid because we found that the multiple couplings that were required to drive the reaction to completion resulted in overcoupling of Cys to the peptide when the Fmoc-derivative was used. This problem was avoided by using the Boc-derivative because Boc is stable to base, whereas Fmoc is base labile. This problem is discussed more extensively in the Supporting Information (see Figure S1).
We began with the synthesis of Boc-Cys(Mob)-OPfP by first forming the symmetric anhydride of Boc-Cys(Mob)-OH via a DCC coupling at 0 °C in dry DCM, then opening up the anhydride by adding pentafluorophenol in dry EtOAc. After silica purification, white, crystalline Boc-Cys(Mob)-OPfP was obtained in 84% yield (SI, Figure S2 and S3). Next, Boc-Cys(Mob)-OPfP was utilized to facilitate the acylation reaction between itself and the bound N-deprotected (αMe)Sec(But) in DCM, with no additional coupling reagent required. To ensure complete formation of the tripeptide, this coupling step was repeated at least 10 times, as depicted in Scheme 1. The tripeptide was then cleaved from the resin in the presence of DTNP to remove the Mob and But protecting groups from Cys and Sec respectively. This effectively replaced each side chain protecting group with a 2-thio(5-nitropyridyl) (5-Npys) adduct, or with the subsequent S-Se bond that forms spontaneously when adjacent Cys/Sec amino acids are treated with DTNP.25, 38, 39 The deprotected tripeptide was washed via ether precipitations to remove any remaining small organic compounds, including excess DTNP. The peptide was not purified before use in the semisynthesis of TrxR enzymes. MS analysis of peptides is given in the Supporting Information as Figures S4 and S5.
Scheme 1: Synthetic route to obtain Cys-(αMe)Sec-Gly tripeptide.
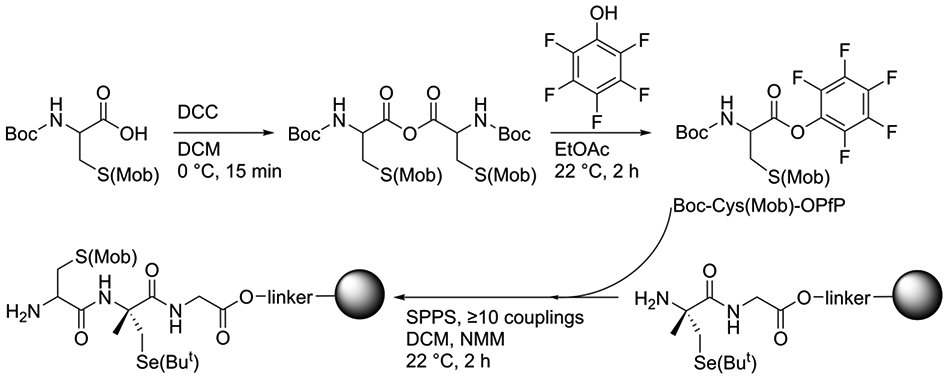
(Top:) Starting with Boc-Cys(Mob)-OH, N,N′-dicyclohexylcarbodiimide (DCC) is used to make a symmetric anhydride intermediate in dichloromethane (DCM), which is then opened up using pentafluorophenol in ethyl acetate (EtOAc). The resulting Boc-Cys(Mob)-OPfP is added directly to the resin (grey sphere) with H-αMeSec(But)-Gly-OH dipeptide attached. SPPS procedures are used to perform ≥10 Boc-Cys(Mob)-OPfP couplings with N-methylmorpholine and no coupling reagents.
Semisynthesis of mTrxR enzymes.
With tripeptides in hand, we went on to obtain mTrxR enzymes: mTrxR-CUG (mouse WT), mTrxR-C(αMe)UG (mouse mutant), and mTrxRΔ3 (truncated mouse). To circumvent the challenges of heterologous production of selenoproteins in E. coli,40, 41 we utilized intein-mediated peptide ligation to join residues 1-487 of mTrxRΔ3 with the tripeptides H-Cys-Sec-Gly-OH or H-Cys-(αMe)Sec-Gly-OH, to produce full length enzyme.20, 22 The final yield of each enzyme from 6 L of E. coli culture was 44 mg for the WT enzyme and 64 mg for the mutant TrxR enzyme.
MS analysis of mTrxR enzymes confirmed the presence of the full-length WT and mutant proteins, as shown in Tables S1 and S2 of the Supporting Information. The mass spectra corresponding to the C-terminal peptide of the WT and of the (αMe)Sec-containing mutant are shown in Figure 3. Mass spectra corresponding to the N-terminal redox center of each enzyme are given in the Supporting Information as Figures S6 and Figure S7.
Figure 3: Mass spectra of the C-terminus redox center peptide for the WT and (αMe)Sec-containing mutant.
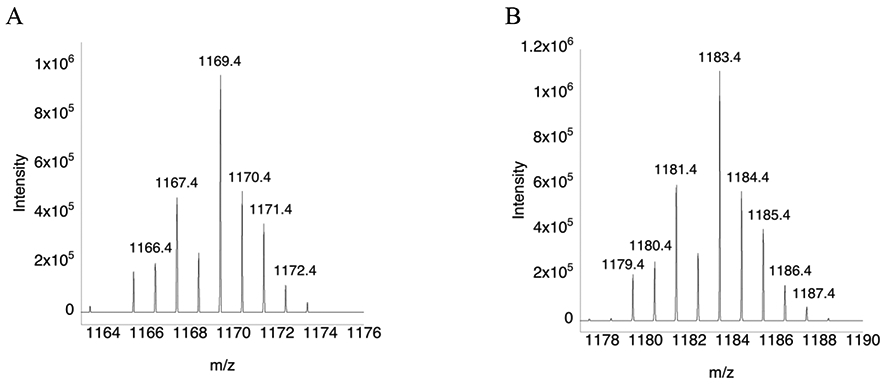
(A) Mass spectrum of peptide SGLEPTVTGCUG that results from a trypsin digest. (B) Mass spectrum of peptide SGLEPTVTGC(αMe)UG that results from a trypsin digest. The mass peaks differ by 14 daltons due to the presence of the αMe group. The sample were not reduced and alkylated prior to MS analysis, so Cys/Sec residues exist in the oxidized form as a selenosulfide bridge.
Kinetic assays of WT- and mutant-TrxR enzymes with various substrates.
We assayed our semisynthetic TrxR enzymes with several known substrates including Trx, H2O2, selenocysteamine, and DTNB. We note that while previously we have used the diselenide selenocystine as a substrate, here we have substituted the more water soluble selenocysteamine as a diselenide substrate instead. The kinetic parameters kcat and Km for the WT, the (αMe)Sec-containing mutant, and truncated enzymes are summarized in Tables 1-4, and the Michaelis-Menten curves are given as Figures S8-S11 of the SI.
Table 1:
Kinetic parameters of mTrxR enzymes with Trx as substrate.
| Enzyme | kcat (min−1) | KM (μM) | kcat/KM (min−1 M−1) |
kcat/KM(mutant) /kcat/KM(WT) |
|---|---|---|---|---|
| mTrxR-CUG | 1770 ± 60 | 42 ± 4.4 | 4.2 x107 | – |
| mTrxR-C(αMe)UG | 15 ± 0.76 | 26 ± 4.8 | 5.8 x105 | 0.0138 |
| mTrxRΔ3 a | NA | NA | NA | – |
The truncated enzyme ends at glycine 487 and is missing the C-terminal tripeptide.
Table 4:
Kinetic parameters of mTrxR enzymes with selenocysteamine as substrate.
| Enzyme | kcat (min−1) | KM (μM) | kcat/KM (min−1 M−1) |
kcat/KM(mutant) /kcat/KM(WT) |
|---|---|---|---|---|
| mTrxR-CUG | 3740 ± 450 | 79 ± 0.7 | 4.7 x107 | – |
| mTrxR-C(αMe)UG | 50 ± 145 | 92 ± 0.9 | 5.4 x105 | 0.011 |
| mTrxRΔ3 a | 76 ± 4 | 1033 ± 114 | 7.3 x104 | 0.0016 |
The truncated enzyme ends at glycine 487 and is missing the C-terminal tripeptide.
For each substrate, our (αMe)Sec mutant is ~100 fold less active than the WT mTrxR with the exception of DTNB. This is likely due to a combination of two factors: (i) the ligation of the tripeptide to the C-terminus of the recombinantly expressed TrxR was not completed in 100% yield (low Sec incorporation) and (ii) the added methyl group to Sec constrains the enzyme backbone such that the C-terminus tail is not as flexible in the mutant than the WT, or perhaps not in the correct conformation for efficient catalysis.
It is well known that for Aib and other (αMe)AAs that rotation about the N-Cα and Cα-CO bond is restricted, resulting in a much more rigid peptide backbone.42, 43 Aib is the most studied (αMe)AA and it is well documented that Aib often induces α-helical structures.32, 42, 44, 45 Therefore, it is not unreasonable to conclude that the lower enzyme activity observed for our mutant mTrxR compared to the WT could be due to an unfavorable C-terminal conformation induced by the incorporation of the (αMe)Sec residue.
Resistance to oxidative inactivation:
Comparison of WT and (αMe)Sec-mutant. We previously showed that (αMe)Sec conferred resistance to oxidative inactivation to a GPX-mimic peptide21 as it is unable to undergo β-syn elimination. We hypothesized that (αMe)Sec would also confer resistance to mTrxR if incorporated into the C-terminal redox center of the enzyme. A study by Snider and coworkers showed that Sec-containing mTrxR was able to resist inactivation by 50 mM H2O2, maintaining ~70% Trx reductase activity,10 while a Cys-containing ortholog lost most of its activity. We hypothesized that replacing Sec with (αMe)Sec would give mTrxR enhanced protection against oxidants such as H2O2, especially at high concentrations. One way in which the WT enzyme can lose activity in the presence of H2O2 is through β-syn elimination, which results in the conversion of Sec to DHA (see Figure 2 of citation 21).
To test if the (αMe)Sec-mutant is more resistant to oxidative inactivation, we reduced mTrxR-CUG, mTrxR-C(αMe)UG, and the Cys-ortholog TrxR from D. melanogaster (abbreviated as DmTrxR-CCS) enzymes with NADPH, and then exposed these enzymes to various concentrations of H2O2 for 25 min in pH 7 buffer at 22 °C. After incubation with H2O2, catalase agarose was added to the reaction to quench any excess H2O2. Catalase agarose was then filtered away from the reaction using Corning™ Costar™ Spin-X™ centrifuge tube filters. Next, the enzymes were assayed for hydrogen peroxidase activity, Trx-reductase activity, and DTNB-reductase activity. The loss of activity of the WT and the (αMe)Sec-mutant enzymes is similar when E. coli Trx is the substrate (Figure 4A), but more dissimilar when either H2O2 (Figure 4B) or DTNB (Figure 4C) are substrates. These points are discussed below.
Figure 4: Resistance to oxidative inactivation of mTrxR-CUG (red), mTrxR-C(αMe)UG (green), and DmTrxR-CCS (blue).
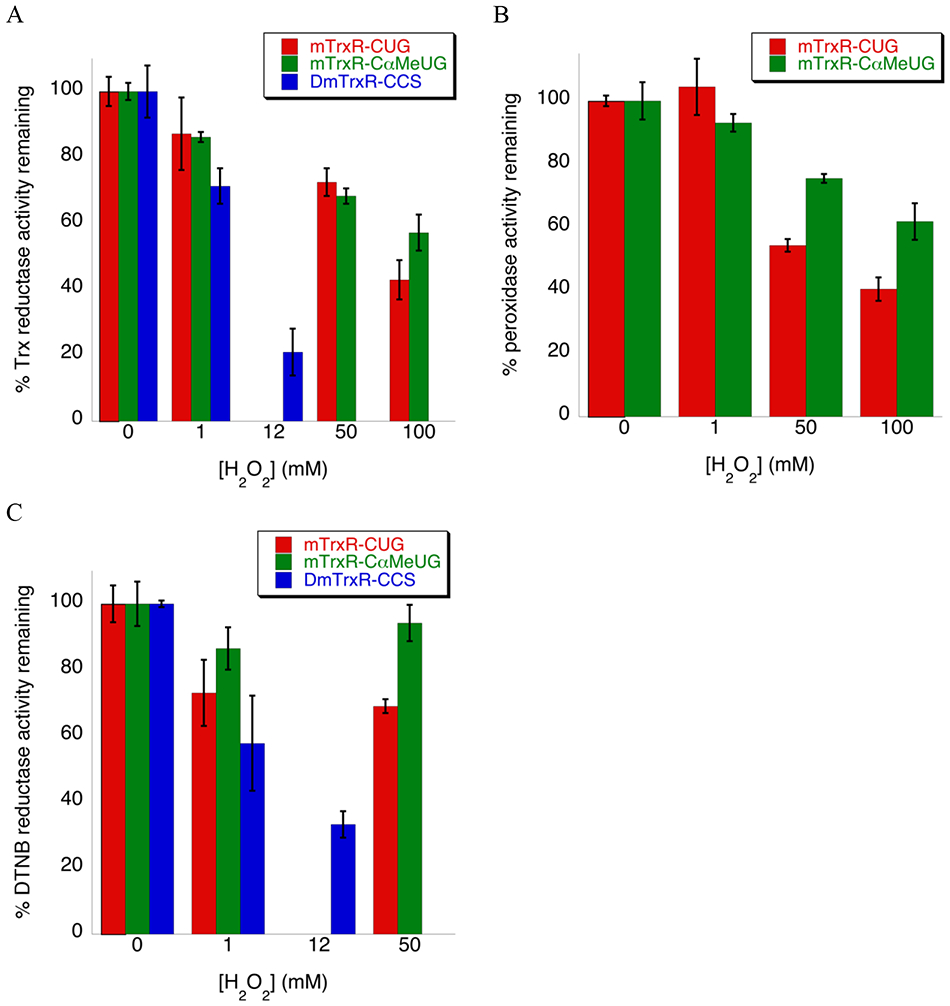
Enzymes were incubated with varying [H2O2] for 25 min at 22 °C, in potassium phosphate buffer pH 7. The percent enzyme activity remaining was monitored spectrophotometrically for substrates: (A) Trx substrate (130 μM Trx), (B) H2O2 substrate (250 mM), and (C) DTNB substrate (2mM DTNB). The activities represent the amount of activity remaining relative to the oxidant-untreated control. For (B), DmTrxR-CCS is not included as it lacks the selenium atom necessary to reduce peroxides. For plots (A-C), the error bars represent the standard deviation from three trials and are normalized to the percentage of remaining activity.
We have previously provided evidence that the reason that mTrxR strongly retains Trx-reductase activity after exposure to oxidant is because the role of Cys497 is to prevent Sec498 from overoxidation to the seleninic acid by rapidly attacking the selenenic acid that forms on Sec498 in the presence of H2O2.46 Since Sec498 resists overoxidation to the seleninic acid in the WT enzyme, the addition of (αMe)Sec does not make the mutant much more resistant to oxidative inactivation. The loss in Trx-reductase activity in both the WT and (αMe)Sec-mutant enzyme could possibly be due to oxidation of Cys497, or oxidation of Cys59 and Cys64 of the N-terminal redox center.
When we use H2O2 as the substrate, the (αMe)Sec-mutant enzyme retains more activity compared to the WT enzyme. This is because the enzymatic mechanism by which the oxidized selenium is reduced is different than the enzymatic mechanism of Trx reduction. The pathway for the reduction of Trx depends upon a reduced Cys497 residue, whereas the reduction of oxidized forms of selenium can be achieved by using either Cys497 or Cys59, or a combination of both of these residues.47 Therefore the ability of the (αMe)Sec-mutant to resist β-syn elimination potentially becomes more important. If the (αMe)Sec residue of the mutant becomes overoxidized to seleninic acid, it can still be reduced by the active site Cys residues. In contrast, if the Sec residue of the WT enzyme is overoxidized, it can undergo β-syn elimination and be inactivated.
Similarly, the mechanism of reduction of DTNB, a low molecular weight disulfide, is different in comparison to the mechanism of the reduction of the disulfide bond of Trx, a macromolecular substrate.47, 48 The (αMe)Sec-mutant is better able to resist overoxidation than the WT enzyme and retains more activity in comparison in this case as well.
Inactivation of TrxR by reactive biological electrophiles.
The adduction of TrxR enzymes by α, β-unsaturated carbonyl-containing compounds (RES) has been well documented.13-16 Adduction and inhibition occurs because the nucleophilic selenolate of TrxR attacks these compounds in a 1,−4 addition reaction, sometimes referred to in the biochemical literature as a “Michael addition”. We note that the formal definition of a Michael addition is the 1,−4 addition of an enolate to an α, β-unsaturated carbonyl.49 Some examples of these types of molecules that have been shown to inhibit TrxR are displayed in Figure 5.
Figure 5: Biological RES.
The α, β-unsaturated carbonyl functional groups (in red) makes these compounds biological Michael acceptors.
A question that we wished to investigate is whether adduction of the Sec residue by an enal or enone is reversible. Evidence that electrophilic modification to TrxR can be reversed was first presented by Randall and coworkers who stated that: “Rather than representing irreversible protein damage, protein alkylation by acrolein and other electrophiles may be an important reversible event in adaptive responses to electrophilic stress or function as an electrophile-specific signaling mechanism analogous to protein S-nitrosylation or S-glutathionylation.”18 They found that cell exposure to acrolein resulted in rapid inactivation of TrxR, which was associated with its alkylation, but that TrxR activity could be restored over several hours in a reductive mechanism that appeared to be mediated by Trx or glutathione (GSH).18
In thinking about a mechanism of reversal depending on thiols, we could only envision a substitution mechanism by which the Se–Cβ bond (Cβ of the electrophile) is broken by attack of a thiolate onto Cβ, restoring a selenolate. Such a mechanism was suggested by Cassidy and coworkers in studying adduction of TrxR by 4-hydroxynonenal.50 They posited that the adduct formed on Cys497 was the thermodynamically stable product, while the adduct formed on Sec498 was the kinetic product.50 In our in vitro experiments, we could not restore activity by addition of exogenous GSH. This mechanism may be more viable when the effective concentration of thiol is very high, such as occurs in an intracellular context or an enzyme active site.
Instead, we envisioned a mechanism of reversal that depended upon the well-studied selenoxide elimination reaction. The β-syn selenoxide elimination is a common synthetic tool used to install alkenes and convert ketones to enones,51, 52 is very rapid under physiological conditions, and is 105 faster in comparison to the corresponding sulfoxide elimination.7 This large difference in elimination rates illustrates another enormous advantage for Sec-enzymes in resisting electrophilic stress in comparison to a Cys-enzyme as a Cys-adduct is largely irreversible. The selenenic acid on TrxR that would result from β-syn selenoxide elimination could be rapidly reduced by the adjacent C497 residue with the elimination of water as we depict in Figure 2. This mechanism is also attractive because it has been shown that adduction of the Sec residue of TrxR by electrophiles results in a modified enzyme whose function changes from an antioxidant to a pro-oxidant as the adducted enzyme is now capable of producing H2O2. We hypothesize that production of H2O2 by the modified TrxR might serve as a type of “molecular timer” that would ultimately result in a reversal of the electrophilic modification via the β-syn selenoxide elimination.
To investigate our hypothesis that electrophilic modification of the Sec residue of TrxR could be reversed through a β-syn selenoxide elimination, we needed to choose between multiple α, β-unsaturated carbonyl-containing compounds. In order to select an appropriate model compound we began by determining the IC50 for various RES. Pre-reduced TrxR enzymes were incubated with various concentrations of RES including acrolein, 4-hydroxynonenal, and curcumin, in 100 mM potassium phosphate, 1 mM EDTA, pH 7 buffer, at 22 °C for various times depending on the RES. The percent enzyme activity remaining was measured spectrophotometrically and used to determine the IC50 value for each RES/enzyme combination. We show the IC50 curve for acrolein in Figure 6. Similar curves for 4-hydroxynonenal and curcumin are shown in the Supporting Information as Figure S12. This data is summarized in Table 5.
Figure 6: Inhibition of TrxR enzymes by acrolein.
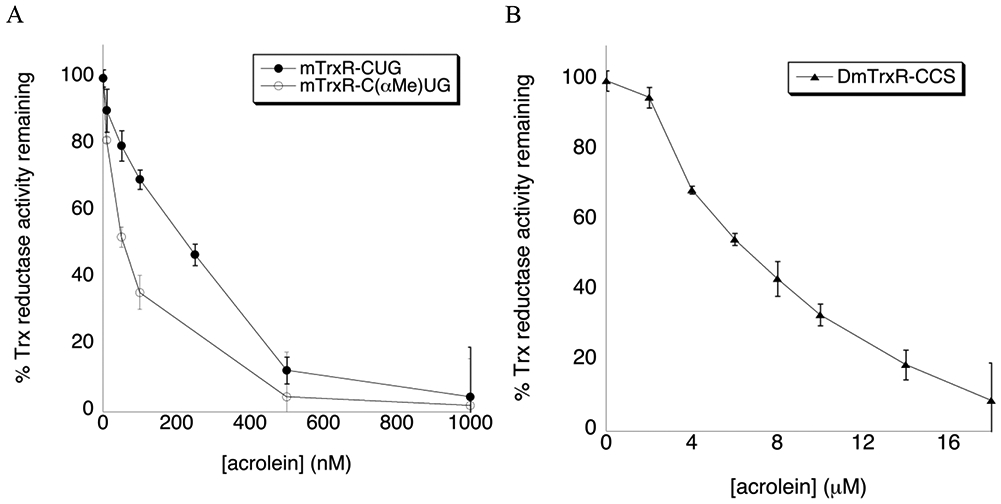
(A) Inhibition of mTrxR-CUG (WT) and mTrxR-C(αMe)UG (mutant) by acrolein after 10 min incubation in 100 mM potassium phosphate, 1 mM EDTA, pH 7 buffer at 22 °C. (B) Inhibition of DmTrxR-CCS by acrolein using the same conditions as mTrxR inhibition described in (A). The error bars represent the standard deviation of three trials.
Table 5:
Half maximal inhibitory concentration (IC50) of RES for TrxR enzymes.
| RES | mTrxR-CUG | mTrxR-C(αMe)UG | DmTrxR-CCS |
|---|---|---|---|
| Curcumin | 34 μM | > 200 μM | - |
| Cinnamaldehyde | 225 μM | 230 μM | - |
| 4-Hydroxynonenal | 235 μM | 118 μM | - |
| Acrolein | 231 nM | 57 nM | 7 μM |
Curcumin is the least potent RES and interestingly, there is a large difference between the IC50 values of curcumin when comparing mTrxR-CUG and mTrxR-C(αMe)UG. In fact, an IC50 value was never reached for the mutant mTrxR as curcumin is only soluble in aqueous solutions up to 100 uM. At 100 uM, ~65% Trx-reductase activity remains for the mutant while an IC50 value of 34 uM curcumin was determined for the WT mTrxR. This large difference can be attributed to the bulky methyl group at the Sec residue of the mutant; curcumin is a relatively large RES and steric hindrance must prevent it from binding to the hindered (αMe)Sec-residue at the active site of the mutant mTrxR.
Acrolein is the simplest and most reactive of the α, β-unsaturated aldehydes tested, and was therefore chosen as the electrophile for our next experiments in which we attempted to reverse the alkylation and restore activity. Acrolein rapidly binds to and depletes cellular nucleophiles such as glutathione.53 It can also adduct protein nucleophiles such as Cys, Lys, and His residues of proteins, and nucleophilic sites in DNA. Acrolein can be released in vivo via biochemical reactions such as lipid peroxidation of polyunsaturated fatty acids. However, the main source of endogenous acrolein is through the degradation of spermine and spermidine in situations of oxidative stress and inflammation.54 Smoking of tobacco products equals or exceeds the total human exposure to acrolein from all other sources.55, 56
Conditions for reversibility of the Sec-acrolein adduct and restoration of activity.
To test whether we could reverse acrolein alkylation of Sec-TrxR and restore enzymatic activity in vitro, we first reduced mTrxR-CUG and mTrxR-C(αMe)UG with NADPH and added acrolein at low pH of (pH 4.6 for WT and pH 5.5 for mutant) for 10 min at 22 °C to maximize chemoselective alkylation of the selenium atom over other enzymic nucleophiles. We attempted to adduct the (αMe)Sec-mutant at pH 4.6 (as we did the WT), however the enzyme precipitated so we chose pH 5.5, which we found to be the lowest pH the mutant enzyme could tolerate for the 10 min acrolein incubation step. We also adducted DmTrxR-CCS with acrolein under similar conditions to test whether alkylation of a Cys-TrxR could be reversed in comparison. However, in this case we conducted the adduction reaction at pH 7 instead of pH 4.5 or 5.5.
For all acrolein adduction experiments, we chose an acrolein concentration that provided 70-80% inhibition of Trx-reductase activity. Using a larger excess of acrolein would undoubtedly inhibit all enzyme activity, but we aimed to use minimal concentrations of acrolein to avoid the unwanted adduction of other amino acids. In addition to using low pH for alkylation, further chemoselectivity comes from the fact that RES are considered “soft” electrophiles and have been shown to preferentially target “soft” nucleophiles, such as Cys.18 Since Sec is even larger, and hence, “softer” than Cys, acrolein should react preferably with the Sec residue over other nucleophilic amino acids.
We next explored conditions by which we could restore Trx-reductase activity in the acrolein adducted enzymes. Under the conditions of our in vitro experiment, we empirically determined that 2 mM H2O2 restored the most activity. We also anticipated needing a scavenger to adduct the acrolein ejected from the enzyme to prevent acrolein from reattaching to the Sec or other enzymic nucleophiles once removed. Imidazole is a good candidate for use as a scavenger since it should react readily with acrolein yet have no side reactions with TrxR or Trx in the subsequent enzyme assay.57 Like hydralazine, which has previously been used as a protective reagent for acrolein, imidazole contains an amidine functionality that reacts strongly with acrolein.58 Our full reversibility pathway is displayed in Figure 7.
Figure 7: Alkylation of mTrxR-CUG with acrolein and subsequent reversibility of the acrolein adduct through a selenoxide elimination pathway.
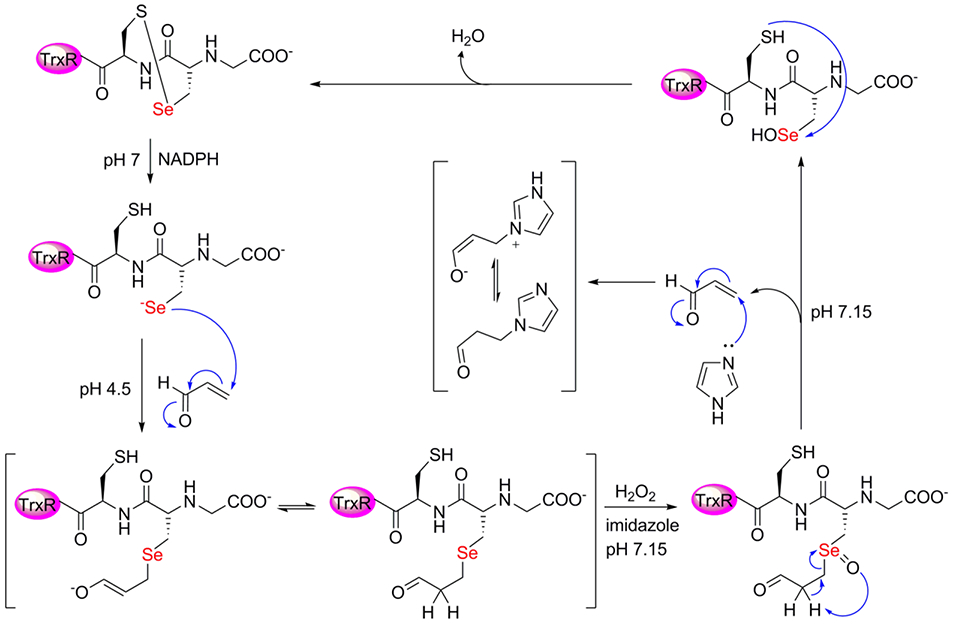
Here, H2O2 converts the acrolein adducted Sec-enzyme to a selenoxide, which can undergo β-elimination with a proton from the acrolein adduct. Imidazole is included to scavenge acrolein once eliminated from the enzyme.
Using the above rationale, we incubated acrolein adducted enzymes with 2 mM H2O2 and 5 mM imidazole in 200 mM potassium phosphate buffer, pH 7.15, at 37 °C for 10 min (Figure 8, condition 4) and were able to restore 30% Trx-reductase activity for the mTrxR-C(αMe)UG mutant, and 25% Trx-reductase activity for the mTrxR-CUG WT. The ability for our (αMe)Sec-mutant to restore more activity than the WT is likely due to the inability of the mutant to undergo β-elimination with the peptide backbone. As shown in Figure 2 it is possible that when acrolein-adducted mTrxR-CUG was oxidized, it underwent β-syn elimination with both the Cα-H of the protein backbone (forming inactive DHA), and the Cα-H of the acrolein adduct. Since both pathways are possible for the WT enzyme, less activity is expected to be recovered, which is what we observed experimentally.
Figure 8: Inhibition of TrxR by acrolein and subsequent reversal of acrolein inhibition using H2O2 and imidazole.
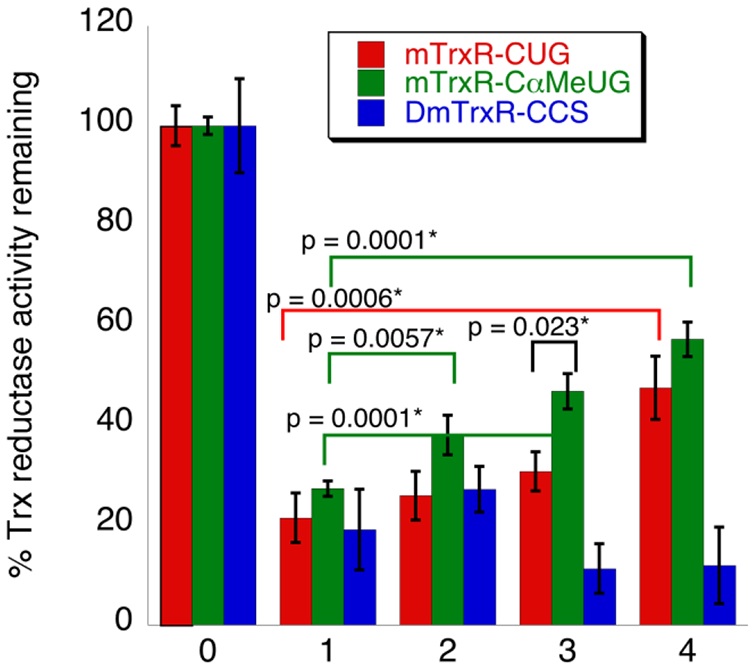
For all, Trx reductase activity was monitored by depletion of NADPH spectrophotometrically at A340. mTrxR-CUG (red), mTrxR-C(αMe)UG (green), and DmTrxR-CCS (blue) were inhibited by acrolein (condition 1), and no reversal reaction was attempted for this condition. Next, acrolein adduction was reversed via one of the following: (condition 2) 5 mM imidazole; (condition 3) 2 mM H2O2; or (condition 4) 2 mM H2O2 and 5 mM imidazole. All “reversal” reactions were conducted in 200 mM potassium phosphate, 1 mM EDTA, pH 7, at 37 °C for 10 min. Condition 0 (control) represents the normalized Trx reductase activity of all enzymes before acrolein inhibition or reversal, and all other bars represent the amount of activity remaining relative to the acrolein-untreated control. The calculated p-values (see Methods) between relevant sets of data are indicated in the figure and the * indicates significance at the 95% confidence level.
Despite efforts to optimize the reversal reactions, we were not able to achieve 100% reversal of Trx-reductase activity for any TrxR tested. This may be due to unwanted acrolein adduction of Cys or His residues (which would not be reversible), despite our rigorous efforts to avoid such occurrence.
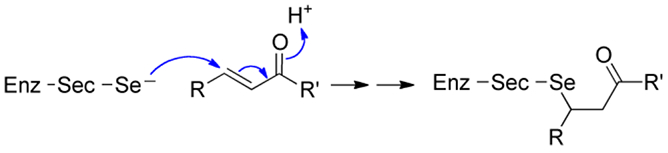
To further explore the mechanism of the reversal reaction, we attempted to reverse acrolein inhibition using either imidazole alone (Figure 8, condition 2) or H2O2 alone (Figure 8, condition 3). Notably, some Trx reductase-activity is restored for both the mTrxR WT and mutant with H2O2 alone. This strongly supports a β-syn selenoxide mechanism as the way in which activity is regained. A smaller percentage of Trx-reductase activity is recovered using imidazole alone for the mTrxR-C(αMe)UG mutant, but the Trx-reductase activity for the WT is within error of the acrolein inhibited enzyme (i.e. the WT did not appear to recover much activity with imidazole alone). The ability of mTrxR-C(αMe)UG to restore Trx-reductase activity with imidazole alone is most likely due to a base-mediated “reverse Michael addition” as depicted in Figure 9. The reason that the (αMe)Sec-mutant could regain more activity compared to the WT enzyme may be explained by the additional rigidity of the C-terminal tail caused by Cα-CH3 substitution. The enhanced rigidity of the C-terminus may prevent the tail from folding back onto the body of the enzyme, making it more accessible to base.
Figure 9: Removal of acrolein from Sec through a base mediated mechanism.
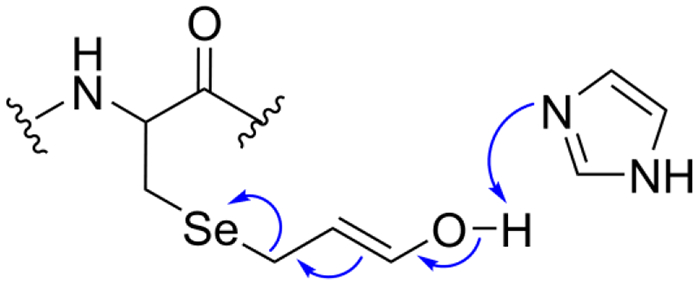
Once adducted, the ketone/enol forms of acrolein are in equilibrium. When in the enol form, imidazole acts as a base to abstract the proton from the acrolein alcohol, which causes removal of the acrolein adduct.
In contrast to the above results, we were not able to restore any activity under the above discussed conditions for the Cys-TrxR, DmTrxR-CCS. In fact, when H2O2 was included in the reversal experiment, DmTrxR-CCS lost more Trx-reductase activity than the non-reversed condition. This is most likely due to oxidative inactivation as DmTrxR-CCS lost activity readily in the presence of oxidants as we showed in Figure 4 and previously.
It has been previously reported by Cai and coworkers that peptide Cys-acrolein adducts can be reversed through a Schiff base mechanism, if the formed Cys-acrolein adduct is in close proximity to an α-amino group on a peptide.59 Notably, ε-amino groups on Lys side chains could not catalyze this reaction, and this chemistry was never tested in proteins. The authors also state ≤ 3% acrolein is reversed from Cys residues in the absence of an α-amino group, highlighting the inherent stability of a Cys-acrolein adduct.59 As recently reviewed by Aye, RES-alkylation of Cys is generally irreversible.60 One notable exception to this is alkylation of Cys by nitro olefin fatty acids. This is due to the much lower pKa of the proton that is beta to the sulfur atom of the adduct (pKa ~17) in comparison to enals and enones that lack such an electron withdrawing group (pKa ~30).60, 61
Our observation that TrxR activity can be recovered after alkylation with an enal like acrolein or other “Michael acceptor” can be explained in terms of the lability of certain types of C–Se bonds that are in close proximity to a carbonyl carbon. As depicted in Figure 10, especially labile C–Se bonds that can be easily cleaved are ones in which the selenium atom is bonded: (i) directly to the carbonyl carbon, (ii) to the carbon alpha to the carbonyl carbon or, (iii) to the carbon beta to the carbonyl carbon. It is well known that selenoesters, like thioesters, are highly labile due to the lack of π-bonding between the chalcogen and the carbonyl carbon.62 It is much less recognized that both α-thio and α-selenocarbonyl compounds can be reduced to the corresponding ketone by soft nucleophiles (middle panel of Figure 10).63, 64 As shown here, and is documented in the literature, oxidation of a β-selenocarbonyl (β-syn selenoxide elimination - last panel of Figure 10) results in formation of the alkene with cleavage of the Cβ–Se bond.51, 52 This last type of bond cleavage is the one needed to impart resistance to RES to a selenoenzyme.
Figure 10: Lability of carbon-selenium bonds in relationship to a carbonyl carbon.
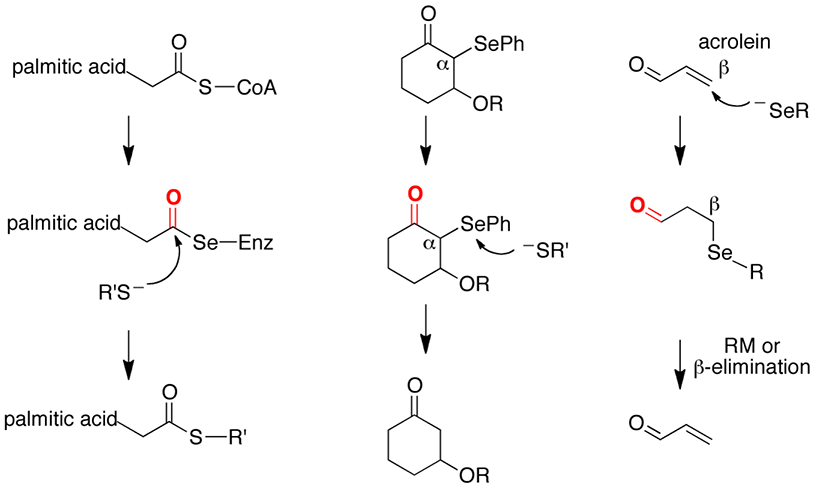
(left panel) Cleavage of a selenoester by a thiolate. (middle panel) reduction of an α-selenoketone by a thiolate. (right panel) β-syn selenoxide elimination or reverse Michael reaction (RM) results in cleavage of the Cβ–Se bond.
Though previous publications have found connections between a “protective function of selenoproteins” in the presence of RES,65 none have explicitly shown that this protective function is due to the reversibility of the RES adduct on Sec. In fact, many studies state that the adduction of Sec in proteins by RES is an irreversible modification.13, 66-70 For example, Park and coworkers observed recovery of TrxR activity after acrolein exposure both in vitro and in vivo, but this was attributed to compensative TrxR gene induction and not repair of the alkylated enzyme .71 While their conclusions may be partially correct, the authors did not discuss the possibility that the formed acrolein adduct on Sec could be reversible in vivo through a selenoxide elimination mechanism. A selenoxide elimination mechanism (as we propose herein) would undoubtedly be feasible in their system since the authors show a significant increase in intracellular peroxide levels.
Support for our idea that electrophilic adduction on the Sec residue of TrxR can be reversed may possibly come from a misinterpreted result from the study of the inhibition of the selenoenzyme GPX with 4-hydroxynonenal by Romero and coworkers.70 This study found that GPX could not be completely inhibited with a great excess of 4-hydroxynonenal (1 mM), and GPX could regain 41% of its activity after inhibition with 0.12 mM 4-hydroxynonenal by addition of 1 mM glutathione.70 The authors attribute the restoration in activity to reversing the alkylation of Lys92 catalyzed by glutathione. This conclusion was reached because it is known that a Schiff base that forms between a Lys side chain and an α, β-unsaturated carbonyl can be reversed by hydrolysis and they considered the alkylation of Sec to be irreversible.70 The authors did not consider the possibility of a β-syn selenoxide elimination despite the fact that H2O2 was used as the substrate in their assays. An alternative explanation is that the additional glutathione added in their rescue experiment was needed to scavenge the 4-hydroxynonenal that was ejected from the β-syn selenoxide elimination.
In this report we have extended the original concept that selenoenzymes resist inactivation by ROS to a broader idea that selenoenzymes can resist electrophilic stress by RES as well. As we previously noted, oxygen and related ROS are just a specific subset of RES, though the mechanism through which the selenoenzyme recovers from each type of stress is different. In the case of recovery from ROS, a selenoenzyme can undergo fast reductive mediated repair by intracellular reducing agents such as glutathione or ascorbate. In the case of recovery from RES, a selenoenzyme can undergo fast oxidative mediated repair via a β-syn selenoxide elimination. These concepts are illustrated in Figure 11 and highlight the gain of function that selenium adds to an enzyme relative to sulfur.
Figure 11: Repair pathways for a selenoenzyme after exposure to RES or ROS.
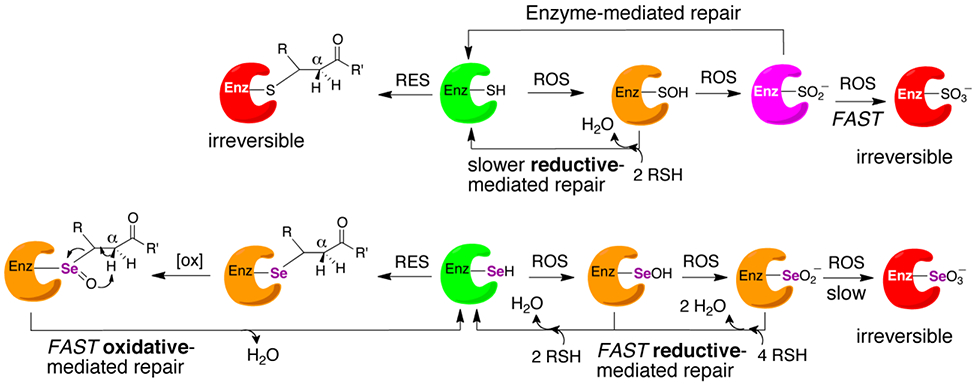
Active enzyme is represented by the color green. Enzyme that is inactivated but repairable by a non-enzymatic reaction is represented by the color orange, red represents irreversible inactivation to the enzyme. The enzyme colored in magenta represents an enzyme that is repairable by an enzyme-mediated repair pathway. In this case, sulfiredoxin is capable of repairing Cys-enzymes that have been overoxidized to sulfinic acid in an ATP-dependent manner.72 The corresponding seleninic acid form of a selenoenzyme can be chemically reduced by GSH or ascorbate and does not need an enzyme catalyzed reaction for repair, highlighting a large advantage for Sec-enzymes compared to the Cys-ortholog. Likewise, Sec-enzymes that are alkylated by an enal or enone can undergo a non-enzymatic repair pathway, but this pathway involves oxidation instead of reduction.
CONCLUSION
Over the past several decades in vitro evidence has emerged the selenoenzymes can resist permanent inactivation by oxidation. Strong in vivo evidence for this hypothesis has recently emerged by the work of Conrad and coworkers studying GPX-4.12 As shown in Figure 11, Se-oxides can be rapidly reduced back to the selenol due to the lability of the Se-O bond. Here, we have provided the first in vitro evidence that selenoenzymes can resist permanent inactivation by alkylation. As we have provided evidence for and discussed, the alkylated Sec-residue can undergo a very rapid β-syn selenoxide elimination that can lead to repair of the selenoenzyme and restore enzymatic activity due to the lability of the C–Se bond. We anticipate that our work will provide a mechanistic foundation for numerous in vivo findings in the future.
Supplementary Material
Table 2:
Kinetic parameters of mTrxR enzymes with H2O2 as substrate.
| Enzyme | kcat (min−1) | KM (μM) | kcat/KM (min−1 M−1) |
kcat/KM(mutant) /kcat/KM(WT) |
|---|---|---|---|---|
| mTrxR-CUG | 1110 ± 62 | 134 ± 16 | 8.2 x103 | – |
| mTrxR-C(αMe)UG | 10.5 ± 0.5 | 35 ± 8.0 | 3.0 x102 | 0.037 |
| mTrxRΔ3 a | NA | NA | NA | – |
The truncated enzyme ends at glycine 487 and is missing the C-terminal tripeptide.
Table 3:
Kinetic parameters of mTrxR enzymes with DTNB as substrate.
| Enzyme | kcat (min−1) | KM (μM) | kcat/KM (min−1 M−1) |
kcat/KM(mutant) /kcat/KM(WT) |
|---|---|---|---|---|
| mTrxR-CUG | 3170 ± 450 | 3 ± 0.7 | 1.1 x109 | – |
| mTrxR-C(αMe)UG | 1125 ± 145 | 5 ± 0.9 | 2.3 x108 | 0.209 |
| mTrxRΔ3 a | 2900 ± 500 | 12 ± 2.7 | 2.4 x108 | 0.218 |
The truncated enzyme ends at glycine 487 and is missing the C-terminal tripeptide.
ACKNOWLEDGMENT
These studies were supported by the National Science Foundation CAREER award MCB-0844478 to Douglas S. Masterson and the National Heart, Lung, and Blood Institute grant HL141146 to Robert J. Hondal. Robert J. Wehrle was supported by the National Science Foundation NRT grant 1449999. Emma J. Ste.Marie was supported by National Heart, Lung, and Blood Institute Grant 5T32HL007594-30 administered by Dr. Kenneth G. Mann and Dr. Robert J. Kelm
Footnotes
SUPPORTING INFORMATION AVAILABLE
There are twelve figures and two tables in the Supporting Information. The supporting information contains data about the synthesis of peptides in this study including 1H-NMR and mass spectral data. In addition, Michaelis-Menten plots are shown with the various substrates tested. The tables summarize peptide mass mapping of the Sec-containing enzymes in this study. This material is available free of charge via the Internet at http://pubs.acs.org
ACCESSION IDENTIFICATION FOR PROTEINS IN THIS STUDY
Drosophila melanogaster thioredoxin reductase (UniProtKB:P91938), wild type mitochondrial mouse thioredoxin reductase 2 (UniProtKB:Q9JLT4).
DEDICATION
This manuscript is dedicated to honor the life and legacy of Professor Hans J. Reich of the University of Wisconsin-Madison in recognition of his many contributions to advancing the field of selenium chemistry. His incredible insight and counsel will be greatly missed.
REFERENCES
- 1.Arnér ES (2009) Focus on mammalian thioredoxin reductases—important selenoproteins with versatile functions, Biochim. Biophys. Acta 1790, 495–526. [DOI] [PubMed] [Google Scholar]
- 2.Arnér ES, and Holmgren A (2000) Physiological functions of thioredoxin and thioredoxin reductase, Eur. J. Biochem 267, 6102–6109. [DOI] [PubMed] [Google Scholar]
- 3.Gladyshev VN, Jeang KT, and Stadtman TC (1996) Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc. Natl. Acad. Sci. U. S. A 93, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gromer S, Johansson L, Bauer H, Arscott LD, Rauch S, Ballou DP, Williams CH Jr., Schirmer RH, Arnér ES (2003) Active sites of thioredoxin reductases: why selenoproteins? Proc. Natl. Acad. Sci. U. S. A 100, 12618–12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnér ES (2010) Selenoproteins: What unique properties can arise with selenocysteine in place of cysteine? Exp. Cell Res. 316, 1296–1303. [DOI] [PubMed] [Google Scholar]
- 6.Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Müller HM, Botella-Munoz J, Schneuwly S, Schirmer R, and Becker K (2001) Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster, Science 291, 643–646. [DOI] [PubMed] [Google Scholar]
- 7.Reich HJ, and Hondal RJ (2016) Why nature chose selenium, ACS Chem. Biol 11, 821–841. [DOI] [PubMed] [Google Scholar]
- 8.Rocher C, Lalanne JL, and Chaudière J (1992) Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase, Eur. J. Biochem 205, 955–960. [DOI] [PubMed] [Google Scholar]
- 9.Choudhury SB, Pressler MA, Mirza SA, Day RO, and Maroney MJ (1994) Structure and redox chemistry of analogous nickel thiolato and selenolato complexes: Implications for the nickel sites in hydrogenases, Inorg. Chem 33, 4831–4839. [Google Scholar]
- 10.Snider GW, Ruggles EL, Khan N, and Hondal RJ (2013) Selenocysteine confers resistance to inactivation by oxidation in thioredoxin reductase: Comparison of selenium and sulfur enzymes, Biochemistry 52, 5472–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maroney MJ, and Hondal RJ (2018) Selenium versus sulfur: reversibility of chemical reactions and resistance to permanent oxidation in proteins and nucleic acids, Free Radic. Biol. Med 127, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, Mehr L, Aichler M, Walch A, Lamp D, Jastroch M, Miyamoto S, Wurst W, Ursini F, Arnér ESJ, Fradejas-Villar N, Schweizer U, Zischka H, Angeli JPF, and Conrad M (2018) Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis, Cell 172, 1–14. [DOI] [PubMed] [Google Scholar]
- 13.Cai W, Zhang L, Song Y, Wang B, Zhang B, Cui X, Hu G, Liu Y, Wu J, and Fang J (2012) Small molecule inhibitors of mammalian thioredoxin reductase, Free Radic. Biol. Med 52, 257–265. [DOI] [PubMed] [Google Scholar]
- 14.Gan F-F, Kaminska KK, Yang H, Liew C-Y, Leow P-C, So C-L, Tu LN, Roy A, Yap C-W, and Kang T-S (2013) Identification of Michael acceptor-centric pharmacophores with substituents that yield strong thioredoxin reductase inhibitory character correlated to antiproliferative activity, Antioxid. Redox Signal 19, 1149–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saccoccia F, Angelucci F, Boumis G, Carotti D, Desiato G, E Miele A, and Bellelli A (2014) Thioredoxin reductase and its inhibitors, Curr. Protein Pept. Sci 15, 621–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Zhang B, Li X, Han X, Liu R, and Fang J (2019) Small molecule inhibitors of mammalian thioredoxin reductase as potential anticancer agents: an update, Med. Res. Rev. 39, 5–39. [DOI] [PubMed] [Google Scholar]
- 17.Anestål K, Prast-Nielsen S, Cenas N, and Arnér ES (2008) Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells, PloS one 3, e1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Randall MJ, Hristova M, and van der Vliet A (2013) Protein alkylation by the α, β-unsaturated aldehyde acrolein. A reversible mechanism of electrophile signaling?, FEBS Lett. 587, 3808–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans TC, Benner J, and Xu MQ (1998) Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci. 7, 2256–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eckenroth BE, Harris K, Turanov AA, Gladyshev VN, Raines RT, and Hondal RJ (2006) Semisynthesis and characterization of mammalian thioredoxin reductase, Biochemistry 45, 5158–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wehrle RJ, Ste. Marie EJ, Hondal RJ, and Masterson DS (2019) Synthesis of alpha‐methyl selenocysteine and its utilization as a glutathione peroxidase mimic, J. Pept. Sci 25, e3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hondal RJ, and Raines RT (2002) Semisynthesis of proteins containing selenocysteine, Methods Enzymol. 347, 70–83. [DOI] [PubMed] [Google Scholar]
- 23.Jones JH (2006) Abbreviations and symbols in peptide science: a revised guide and commentary, J. Pept. Sci 12, 1–12. [DOI] [PubMed] [Google Scholar]
- 24.Kaiser E, Colescott R, Bossinger C, and Cook P (1970) Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides, Analyt. Biochem 34, 595–598. [DOI] [PubMed] [Google Scholar]
- 25.Harris KM, Flemer S, and Hondal RJ (2007) Studies on deprotection of cysteine and selenocysteine side‐chain protecting groups, J. Pept. Sci 13, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eckenroth BE, Lacey BM, Lothrop AP, Harris KM, and Hondal RJ (2007) Investigation of the C-terminal redox center of high Mr thioredoxin reductase by protein engineering and semisynthesis, Biochemistry 46, 9472–9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong L, and Holmgren A (2000) Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations, J. Biol. Chem 275, 18121–18128. [DOI] [PubMed] [Google Scholar]
- 28.Arnér ES, Zhong L, and Holmgren A (1999) Preparation and assay of mammalian thioredoxin and thioredoxin reductase, Methods Enzymol. 300, 226–239. [DOI] [PubMed] [Google Scholar]
- 29.Fu Y, Hammarström LG, Miller TJ, Fronczek FR, McLaughlin ML, and Hammer RP (2001) Sterically hindered C α, α-disubstituted α-amino acids: Synthesis from α-nitroacetate and incorporation into peptides, J. Org. Chem 66, 7118–7124. [DOI] [PubMed] [Google Scholar]
- 30.Katritzky AR, Todadze E, Angrish P, and Draghici B (2007) Efficient peptide coupling involving sterically hindered amino acids, J. Org. Chem 72, 5794–5801. [DOI] [PubMed] [Google Scholar]
- 31.Humphrey JM, and Chamberlin AR (1997) Chemical synthesis of natural product peptides: coupling methods for the incorporation of noncoded amino acids into peptides, Chem. Rev 97, 2243–2266. [DOI] [PubMed] [Google Scholar]
- 32.Fu Y (2002) Artificial peptides containing Cα,α-disubstituted amino acids: synthesis, conformational studies, and application as β-strand mimics. Ph. D. thesis, Louisiana State University and Agricultural and Mechanical College. [Google Scholar]
- 33.Fu Y, and Hammer RP (2002) Efficient acylation of the N-terminus of highly hindered Cα,α-disubstituted amino acids via amino acid symmetrical anhydrides, Org. Lett 4, 237–240. [DOI] [PubMed] [Google Scholar]
- 34.Albericio F, Cases M, Alsina J, Triolo SA, Carpino LA, and Kates SA (1997) On the use of PyAOP, a phosphonium salt derived from HOAt, in solid-phase peptide synthesis, Tetrahedron Lett. 38, 4853–4856. [Google Scholar]
- 35.Frérot E, Coste J, Pantaloni A, Dufour M-N, and Jouin P (1991) PyBOP and PyBroP: Two reagents for the difficult coupling of the α, α-dialkyl amino acid, Aib, Tetrahedron 47, 259–270. [Google Scholar]
- 36.Masterson DS, Kedrowski BL, and Blair A (2010) An improved method for the preparation of protected (R)-2-methylcysteine: solution-phase synthesis of a glutathione analogue, Synlett 19, 2941–2943. [Google Scholar]
- 37.Atherton E, Cameron LR, and Sheppard RC (1988) Peptide synthesis: Part 10. Use of pentafluorophenyl esters of fluorenylmethoxycarbonylamino acids in solid phase peptide synthesis, Tetrahedron 44, 843–857. [Google Scholar]
- 38.Schroll AL, Hondal RJ, and Flemer S Jr (2012) The use of 2, 2′‐dithiobis (5‐nitropyridine)(DTNP) for deprotection and diselenide formation in protected selenocysteine‐containing peptides, J. Pept. Sci 18, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schroll AL, Hondal RJ, and Flemer S Jr. (2012) 2,2’-Dithiobis(5-nitropyridine) (DTNP) as an effective and gentle deprotectant for common cysteine protecting groups, J. Pept. Sci 18, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tormay P, and Bock A (1997) Barriers to heterologous expression of a selenoprotein gene in bacteria, J. Bacteriol 179, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu X, Söll D, and Sevostyanova A (2018) Challenges of site-specific selenocysteine incorporation into proteins by Escherichia coli, RNA biology 15, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karle IL, and Balaram P (1990) Structural characteristics of. alpha.-helical peptide molecules containing Aib residues, Biochemistry 29, 6747–6756. [DOI] [PubMed] [Google Scholar]
- 43.Yokum TS, Gauthier TJ, Hammer RP, and McLaughlin ML (1997) Solvent effects on the 310-/α-helix equilibrium in short amphipathic peptides rich in α, α-disubstituted amino acids, J. Am. Chem. Soc 119, 1167–1168. [Google Scholar]
- 44.Venkataram BVV, Balaram P, and Benedetti E (1984) The stereochemistry of peptides containing α-aminoisobutyric acid, Crit. Rev. Biochem 16, 307–348. [DOI] [PubMed] [Google Scholar]
- 45.Marshall GR, Hodgkin EE, Langs DA, Smith GD, Zabrocki J, and Leplawy MT (1990) Factors governing helical preference of peptides containing multiple alpha, alpha-dialkyl amino acids, Proc. Natl. Acad. Sci. U. S. A, 87, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Payne NC, Barber DR, Ruggles EL, and Hondal RJ (2019) Can dimedone be used to study selenoproteins? An investigation into the reactivity of dimedone toward oxidized forms of selenocysteine, Protein Sci. 28, 41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lothrop AP, Ruggles EL, and Hondal RJ (2009) No selenium required: reactions catalyzed by mammalian thioredoxin reductase that are independent of a selenocysteine residue, Biochemistry 48, 6213–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lacey BM, Eckenroth BE, Flemer S Jr, and Hondal RJ (2008) Selenium in thioredoxin reductase: a mechanistic perspective, Biochemistry 47, 12810–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michael A (1887) On the addition of sodium acetoacetate and sodium malonic acid esters to the esters of unsaturated acids, Journal fur Praktische Chemi 35, 3490356. [Google Scholar]
- 50.Cassidy PB, Edes K, Nelson CC, Parsawar K, Fitzpatrick F, and Moos PJ (2006) Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles, Carcinogenesis 27, 2538–2549. [DOI] [PubMed] [Google Scholar]
- 51.Reich HJ, Renga JM, and Reich IL (1975) Organoselenium chemistry. Conversion of ketones to enones by selenoxide syn elimination, J. Am. Chem. Soc 97, 5434–5447. [Google Scholar]
- 52.Reich HJ, and Wollowitz S (1993) Preparation of α, β‐unsaturated carbonyl compounds and nitriles by selenoxide elimination, Organic reactions 44, 4–281. [Google Scholar]
- 53.Kehrer JP, and Biswal SS (2000) The molecular effects of acrolein, Toxicol. Sci 57, 6–15. [DOI] [PubMed] [Google Scholar]
- 54.Stevens JF, and Maier CS (2008) Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease, Mol. Nutr. Food Res 52, 7–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carmella SG, Chen M, Zhang Y, Zhang S, Hatsukami DK, and Hecht SS (2007) Quantitation of acrolein-derived (3-hydroxypropyl) mercapturic acid in human urine by liquid chromatography− atmospheric pressure chemical ionization tandem mass spectrometry: effects of cigarette smoking, Chem. Res. Toxicol 20, 986–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carmines E, and Gaworski C (2005) Toxicological evaluation of glycerin as a cigarette ingredient, Food Chem. Toxicol 43, 1521–1539. [DOI] [PubMed] [Google Scholar]
- 57.Davies SS, and Zhang LS (2017) Reactive carbonyl species scavengers – novel therapeutic approaches for chronic diseases. Curr. Pharmacol. Rep 3, 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moghe A, Ghare S, Lamoreau B, Mohammad M, Barve S, McClain C, and Joshi-Barve S (2015) Molecular mechanisms of acrolein toxicity: relevance to human disease, Toxicol. Sci 143, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai J, Bhatnagar A, and Pierce WM Jr (2009) Protein modification by acrolein: formation and stability of cysteine adducts, Chem. Res. Toxicol 22, 708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parvez S, Long MJ, Poganik JR, and Aye Y (2018) Redox signaling by reactive electrophiles and oxidants, Chem. Rev 118, 8798–8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Batthyany C, Schopfer FJ, Baker PR, Durán R, Baker LM, Huang Y, Cerveñansky C, Branchaud BP, and Freeman BA (2006) Reversible post-translational modification of proteins by nitrated fatty acids in vivo, J. Biol. Chem 281, 20450–20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Makriyannis A, Guenther WH, and Mautner HG (1973) Selenol esters as specific reagents of the acylation of thiol groups, J. Am. Chem. Soc 95, 8403–8406. [Google Scholar]
- 63.Oki M, Funakoshi W, and Nakamura A (1971) The reaction of α-carbonyl sulfides with bases. I. The reaction between α-carbonyl sulfides with thiolates, Bull. Chem. Soc. Jpn 44, 828–832. [Google Scholar]
- 64.Shimizu M, Takeda R, and Kuwajima I (1981) Oxidation of olefins into α-phenylseleno carbonyl compounds. Highly regioselective anti-Markownikoff type oxidation of allylic alcohol derivatives, Bull. Chem. Soc. Jpn 54, 3510–3517. [Google Scholar]
- 65.Moos PJ, Edes K, Cassidy P, Massuda E, and Fitzpatrick F (2003) Electrophilic prostaglandins and lipid aldehydes repress redox-sensitive transcription factors p53 and hypoxia-inducible factor by impairing the selenoprotein thioredoxin reductase, J. Biol. Chem 278, 745–750. [DOI] [PubMed] [Google Scholar]
- 66.Myers CR, and Myers JM (2009) The effects of acrolein on peroxiredoxins, thioredoxins, and thioredoxin reductase in human bronchial epithelial cells, Toxicology 257, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fang J, and Holmgren A (2006) Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo, J. Am. Chem. Soc 128, 1879–1885. [DOI] [PubMed] [Google Scholar]
- 68.Fang J, Lu J, and Holmgren A (2005) Thioredoxin reductase is irreversibly modified by curcumin a novel molecular mechanism for its anticancer activity, J. Biol. Chem 280, 25284–25290. [DOI] [PubMed] [Google Scholar]
- 69.Nordberg J, Zhong L, Holmgren A, and Arnér ES (1998) Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue, J. Biol. Chem 273, 10835–10842. [DOI] [PubMed] [Google Scholar]
- 70.Bosch-Morell F, Flohé L, Marín N, and Romero FJ (1999) 4-Hydroxynonenal inhibits glutathione peroxidase: protection by glutathione, Free Radic. Biol. Med 26, 1383–1387. [DOI] [PubMed] [Google Scholar]
- 71.Park YS, Misonou Y, Fujiwara N, Takahashi M, Miyamoto Y, Koh YH, Suzuki K, and Taniguchi N (2005) Induction of thioredoxin reductase as an adaptive response to acrolein in human umbilical vein endothelial cells, Biochem. Biophys. Res. Comm 327, 1058–1065. [DOI] [PubMed] [Google Scholar]
- 72.Akter S, Fu L, Jung Y, Conte ML, Lawson JR, Lowther WT, Sun R, Liu K, Yang J, and Carroll KS (2018) Chemical proteomics reveals new targets of cysteine sulfinic acid reductase, Nat. Chem. Biol 14, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.