

Abstract
Emergence of resistance is a major factor limiting the efficacy of molecularly targeted anticancer drugs. Understanding the specific mutations, or other genetic or cellular changes, that confer drug resistance can help in the development of therapeutic strategies with improved efficacies. Here, we outline recent progress in understanding chemotype-specific mechanisms of resistance and present chemical strategies, such as designing drugs with distinct binding modes or using proteolysis targeting chimeras, to overcome resistance. We also discuss how targeting multiple binding sites with bifunctional inhibitors or identifying collateral sensitivity profiles can be exploited to limit the emergence of resistance. Finally, we highlight how incorporating analyses of resistance early in drug development can help with the design and evaluation of therapeutics that can have long-term benefits for patients.
Graphical Abstract
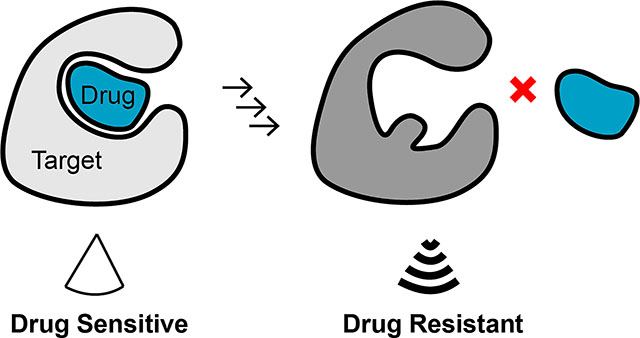
Chemical inhibitors that selectively block their target’s functions can be valuable as probes for dynamic cellular processes, for testing therapeutic hypotheses and as useful starting points for developing drugs. When these inhibitors are active in vivo, they can lead to new molecularly targeted therapeutics, many of which have provided new paradigms for treating diseases such as cancer. For example, aberrant signaling of the BCR-ABL fusion in leukemia or the upregulated activity of epidermal growth factor receptor (EGFR) kinase mutants in lung cancer can be blocked using potent chemical inhibitors and result in improved clinical outcomes1,2. However, the long-term efficacy of such targeted therapeutics can be limited as resistance against them inevitably arises3,4.
The emergence of resistance is driven by evolutionary pressures exerted by drugs on growing cells and can involve multiple mechanisms. Extensive studies of antiviral, antimicrobial and anticancer agents have established paradigms for understanding mechanisms of drug resistance (for reviews see refs. 5,6,7). For example, resistance to antiviral drugs commonly arises due to mutations in the target proteins that can prevent drug binding8. Selection of the resistant virus can occur rapidly, as viral populations consist of ensembles of related genotypes (also termed viral quasispecies or swarms9) that may arise due to high mutation rates during replication10. Emergence of single-point mutations often leads to acquired drug resistance in cells (e.g., bacteria or cancer cells), but unique constraints in different cellular, multicellular and organismal contexts can also lead to a wide range of resistance mechanisms. For example, horizontal gene transfer in bacteria can give rise to acquired resistance by selection of genetic elements that facilitate modifications of drugs and render them ineffective (e.g., hydrolysis of β-lactam antibiotics by β-lactamase)11. In cancer cells, mechanisms contributing to resistance can also include reduction of cellular drug abundance by upregulating xenobiotic pathways that promote drug metabolism, as well as increased expression of genes leading to nonspecific multidrug resistance (MDR; for a recent review see ref. 12). Consistent with these studies, drug-resistance mechanisms in patients can be complex, and new chemical strategies are needed to address the emergence of drug resistance and to develop therapeutics with long-term benefits.
Here, we focus on chemotype-specific resistance to chemical inhibitors in cancer, as these mechanisms are now being addressed by innovations in chemistry and chemical biology. In the following sections, we highlight recent examples of drug-resistance analyses and chemical approaches that can help address resistance (Fig. 1).
Fig. 1 |. Strategies to overcome resistance against molecularly targeted therapeutics.
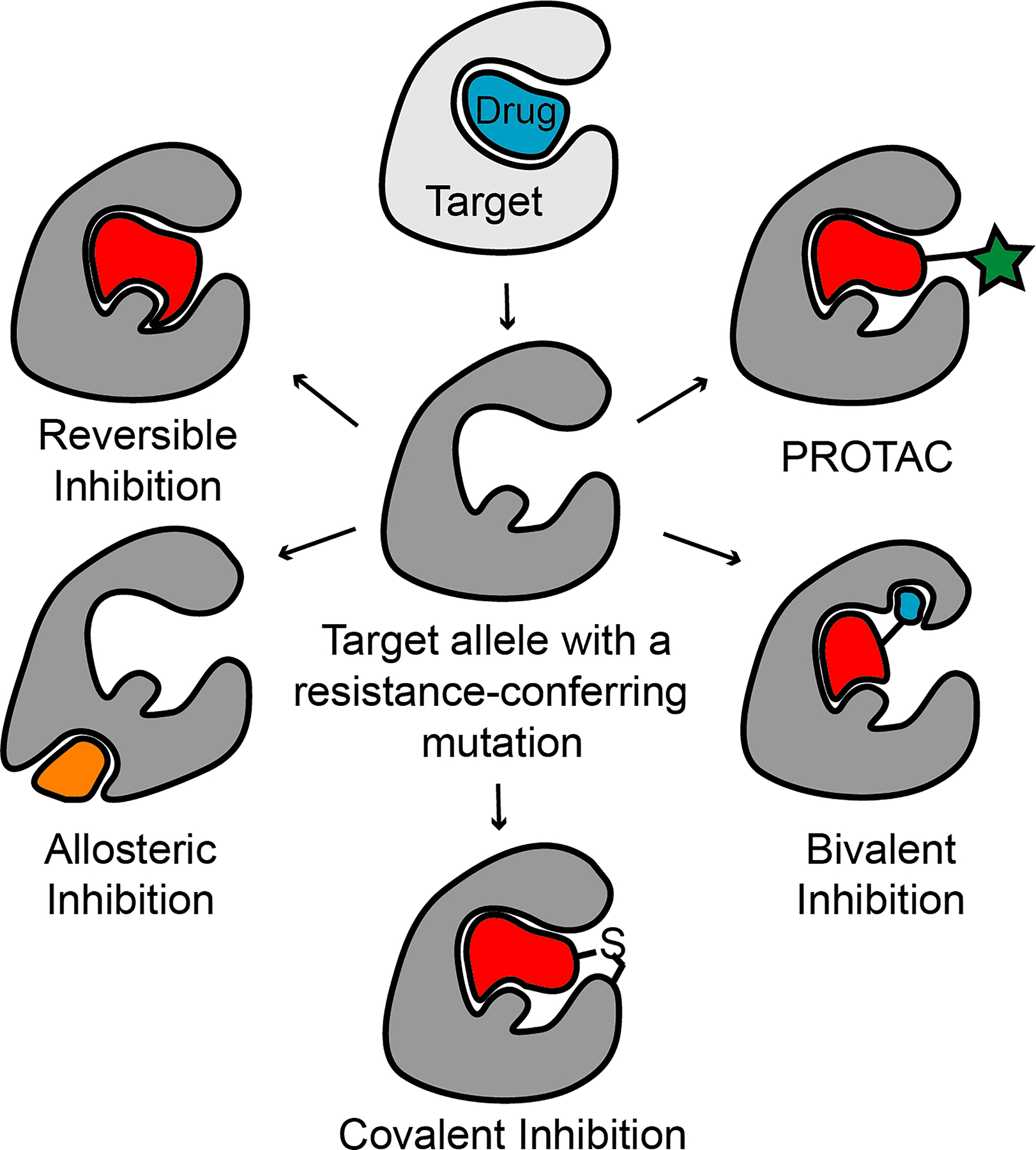
Schematic shows strategies, which are highlighted in this Review, to overcome chemotype-specific resistance to inhibitors. The activity of resistance-conferring alleles (dark gray, center) can be blocked by inhibitors with distinct binding modes, allosteric inhibitors, covalent inhibitors, or bivalent compounds. Resistance-conferring alleles can also be targeted for degradation by the proteasome using PROTACs (red ligand with a green star, see text for details).
Designing inhibitors with distinct binding modes
Resistance to small-molecule anticancer agents can result from mutations in genes encoding the target proteins (e.g., BCR-ABL, EGFR or ALK, Table 1) that prevent or reduce drug binding3,13,14. An important example of this type of resistance in cancer cells is the mutation of the gatekeeper residue that can prevent binding of drugs targeting the nucleotide-binding site of oncogenic kinases15. For instance, the T315I gatekeeper mutation often arises in BCR-ABL-driven leukemias and prevents the binding of different inhibitors targeting the active site of Abl1 kinase such as imatinib or dasatinib16 (Table 1). Similarly, sustained treatment of anaplastic lymphoma kinase (ALK)-rearranged lung cancers with ATP-competitive inhibitors such as crizotinib invariably leads to emergence of resistance-conferring mutations, including the gatekeeper mutation (ALK-L1196M, Table 1)14,17. In these cases, for which the drug resistance mechanisms are known, new drugs and chemical strategies have been designed to address resistance18,19.
Table 1.
Selected drugs discussed in the manuscript
| Chemical structure | Name(s) (Brand name) | Target(s) | Examples of resistance- conferring mutationsRef | Mode of Action |
|---|---|---|---|---|
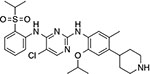
| Crizotinib (Xalkori™) | ALK | ALK-L1196M, C1156Y14,17,19 | ATP-competitive | |
 |
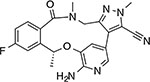
Ceritinib (Zykadia™) | ALK | ALK-L1198F19,20 | ATP-competitive |
 |
Lorlatinib (Lorbrena™) | ALK | ALK-C1156-L1198F19,20 | ATP-competitive |
| Imatinib (Gleevec™) | BCR-ABL | BCR-ABL-T315I3,16 | ATP-competitive | |
| Dasatinib (Sprycel™) | BCR-ABL | BCR-ABL-T315I3,16 | ATP-competitive | |
 |
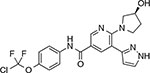
Vandetanib (Caprelsa™) | BCR-ABL-V299L | BCR-ABL-V299L-E255K, V299L-F317L78 | ATP-competitive |
| EGFR | ||||
 |
ABL001 Asciminib | BCR-ABL-WT | BCR-ABL-A337C, P465S, V468F40 | allosteric |
| BCR-ABL-T315I | ||||
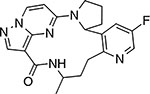 |
LOXO-195 | TrkA, B, C | N/A | ATP-competitive |
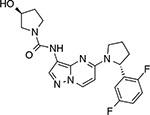 |
Larotrectinib LOXO-101 (Vitrakvi™) | TrkA, B, C | TrkA-F589L, G595R, G667C24,25 | ATP-competitive |
| TrkC-G623R, G696A24,25 | ||||
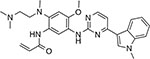 |
Osimertinib (Tagrisso™) | EGFR-T790M | EGFR-C797S, L718Q35 | covalent |
| WZ4020 | EGFR-T790M | EGFR-C797S35 | covalent | |
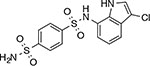 |
Indisulam | RBM39 | RBM39-G268V,W,R,E67 | “molecular glue” |
| DCAF15 | ||||
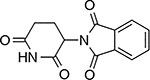 |
Thalidomide | CRBN | IKFZ1-Q146H64,65 | “molecular glue” |
| IKFZ1 | IKFZ3-Q147H64,65 | |||
| IKFZ3 | CRBN-W386A64,65 | |||
| CRBN-W400A64,65 | ||||
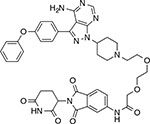 |
MT-802 | CRBN | N/A | PROTAC |
| BTK(C481S) |
A successful approach to overcome resistance to crizotinib, which is often a result of mutations of the gatekeeper residue L1196M, centers on targeting the active site of ALK with inhibitors that adopt a distinct binding mode compared to that of crizotinib (Fig. 2a and Table 1). For example, ceritinib binds ALK in the active site and can inhibit a number of crizotinib-resistant alleles including the gatekeeper mutant ALK-L1196M19. However, as the binding modes of crizotinib and ceritinib partially overlap (Fig. 2a,b), ceritinib can only block a subset of resistance-conferring mutant alleles in ALK kinase19. To overcome resistance to crizotinib and ceritinib (e.g., due to a mutation outside of the active site, C1156Y), a third generation macrocyclic drug called lorlatinib was developed20 (Fig. 2c,d). Even though all three inhibitors bind in the ALK active site, they can overcome resistance by leveraging distinct inhibitor–residue contacts to achieve selectivity and potency. However, treatment with lorlatinib can also lead to emergence of mutations such as ALK-C1156Y-L1198F that confer resistance to this drug20. Interestingly, the ALK-C1156Y-L1198F double mutant can bind crizotinib (Fig. 2e) and re-sensitizes the resistant cancer cells to crizotinib treatment20. Taken together, these studies establish a paradigm for how designing drugs with distinct binding modes can help overcome resistance to targeted therapy.
Fig. 2 |. overcoming resistance by designing inhibitors with distinct binding modes.
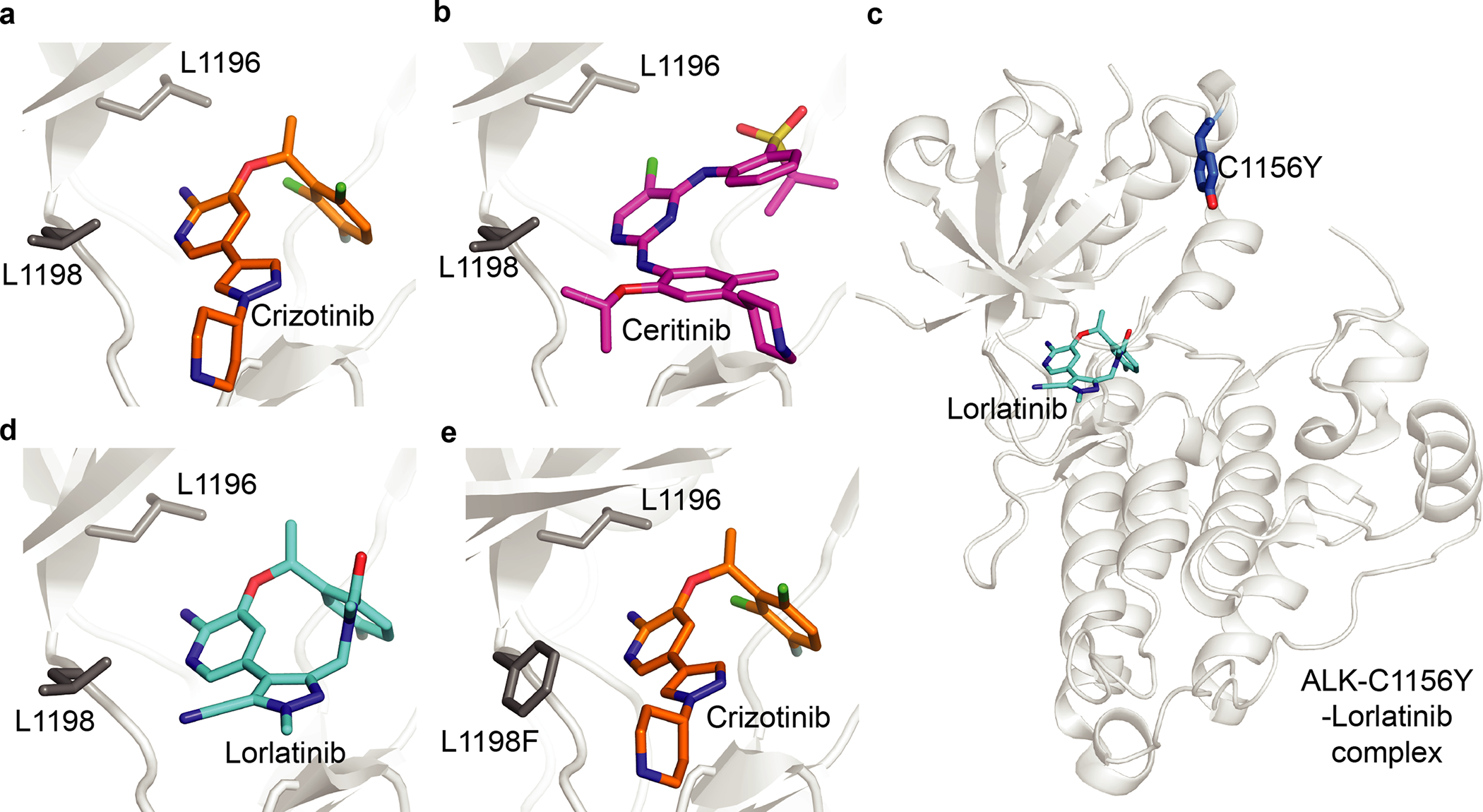
a–e, Structural models of ALK kinase alleles in complex with crizotinib (a, ALK-WT, PDB: 2XP2; e, ALK-C116Y-L1198F, PDB: 5AAB), ceritinib (b, ALK-WT, PDB: 4MKC) and lorlatinib (c,d, ALK-C1156Y, PDB: 5A9U). Selected residues are indicated (stick representation).
Combining preclinical analyses of resistance and compound testing is becoming a successful approach to anticipate and overcome resistance21. Entrectinib and larotrectinib are first-generation, ATP-competitive inhibitors for treating cancers driven by lesions in NTRK1–3 genes that encode tropomyosin receptor kinases (TrkA, TrkB and TrkC)22,23. As is the case with other molecularly targeted therapeutics, acquired resistance to these compounds was found to arise upon treatment with these inhibitors24,25. Analyses of resistance in tumor samples from patients and in cell culture models of NTRK-driven cancers revealed a number of mutations that confer resistance to larotrectinib and entrectinib21. In particular, binding of these drugs was suppressed by mutations in the solvent-exposed loop at the front of the kinase nucleotide-binding pocket (e.g., G595R and G667C in TrkA and G623R in TrkC) and in the activation loop of the DFG motif (e.g., G667C in TrkA). Testing compound libraries against these mutant alleles revealed a series of 13-member macrocyclic scaffolds, based on the larotrectinib core scaffold, that potently block the activity of Trk alleles resistant to larotrectinib and entrectinib21. One of the identified compounds, LOXO-195 (Table 1), was rapidly advanced to clinical trials. In these trials, patients that developed resistance to larotrectinib, possibly due to rapid selection of pre-existing resistant clones, were treated with LOXO-195. Remarkably, treatment with LOXO-195 resulted in tumor regression in most patients, likely because of on-target inhibition21. These encouraging results illustrate how analyses of resistance can be valuable for designing therapeutics with improved efficacies. Therefore, for the promise of molecularly targeted drugs for long-term cancer treatment to be realized26, integrating analyses of resistance early in drug development, by approaches such as resistance analysis during design (RADD, Box 1), is needed.
Box 1. Resistance analysis during design (RADD).
Binding of inhibitors to their targets is driven by the their complementary steric and electrostatic interactions92. Mutations that are functionally silent and lead to changes in the shape and electrostatics of the drug-binding sites can reveal such protein–inhibitor interactions. Therefore, identifying key residues and inhibitor–protein contacts can be useful not only for anticipating resistance and validating targets of inhibitors in cells93, but also for optimizing inhibitor potency and specificity. An approach termed resistance analysis during design (RADD)94 has recently been used for the design of spastazoline, a cell-permeable, potent and selective chemical probe of spastin95. In particular, RADD involves engineering biochemically silent mutations in the target protein and testing compounds against them. Mutations altering compound potency identify residues that form key inhibitor–target interactions and predict inhibitor–target binding poses. Briefly, to identify target–inhibitor interactions needed for the design of the pyrazolylpyrrolopyrimidine-based spastazoline, selected inhibitor scaffolds were tested against spastin mutant alleles that retain enzymatic activity (Box 1 figure). These analyses helped develop a compound-binding model that guided optimization of the inhibitor. Furthermore, testing diaminotriazole-based scaffolds, which are chemically unrelated to spastazoline, against spastin mutant alleles revealed how even minor modifications of the inhibitors can lead to distinct binding modes in the active site94. Together, these studies indicate that analyses of resistance can be valuable for the inhibitor design process94,95.
RADD can be especially useful for evaluating unoptimized compounds (e.g., screening hits) as models of inhibitor–target interactions for these compounds are often not readily available. In addition, identifying starting scaffolds with distinct binding modes could facilitate the design of new inhibitors to overcome resistance. Recent advances in genome editing (e.g., CRISPR–Cas9 technology) can help introduce resistance-conferring mutations in cells, such as those identified by RADD or using unbiased target-focused approaches, and analyze those that alter compound activity96,97,98,99,100. Finally, analyzing compound–target interactions using RADD could help identify combinations of drugs with non-overlapping resistance signatures that may delay and even prevent emerging drug resistance (see text).
Box1 figure:
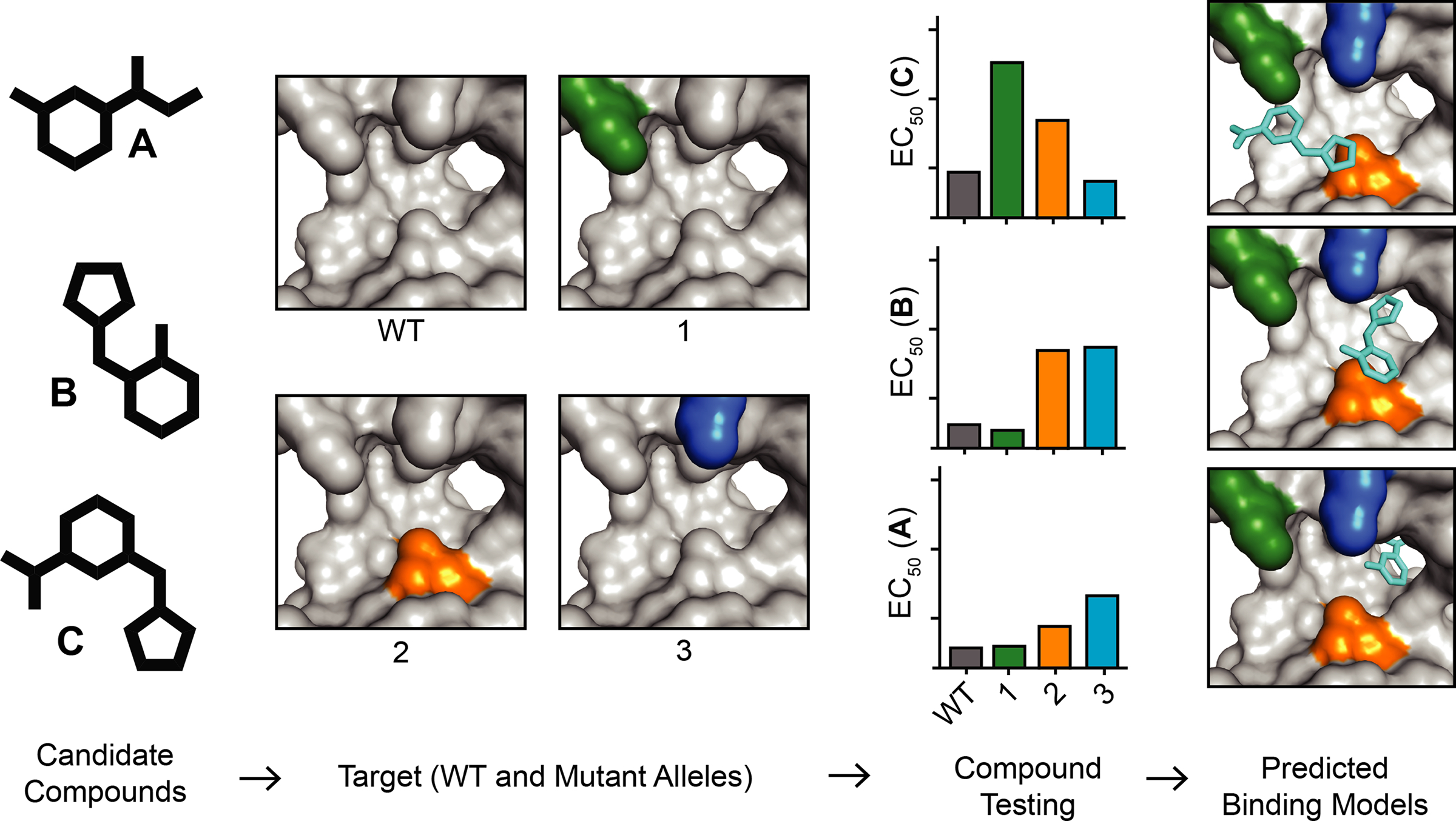
RADD approach for developing inhibitor-target binding models. Schematic shows the RADD approach. Selected candidate compounds are tested against wild-type (WT) and mutant alleles of a chosen target to identify key inhibitor–target interactions that can guide the development of robust binding models.
Designing cysteine-targeting covalent inhibitors
Another chemical strategy to overcome resistance involves the design of covalent inhibitors27,28. This approach hinges on the presence of an electrophilic moiety (e.g., acrylamide, epoxide or α-halocarbonyl group) in the inhibitor that can form a covalent bond with a nucleophilic residue in the binding site (e.g., cysteine or lysine)27. A number of drug targets contain a native cysteine that can be leveraged for inhibitor design. For example, the EGFR-T790M mutant allele, which is the most common resistance-conferring mutation that arises upon prolonged treatment of EGFR-driven cancers with ATP-competitive inhibitors such as gefitinib or erlotinib2,3,29, does not prevent binding of these compounds in the active site. Instead, this mutant allele binds the ATP substrate with increased affinity, which has been proposed to contribute to drug resistance30. EGFR contains a cysteine residue at the entrance of the ATP-binding site, and electrophilic compounds that form a covalent bond with this residue (C797) can potently block the activity of EGFR-T790M and substantially increase the inhibitor’s occupancy in comparison to that of reversible inhibitors31,32. Optimizing interactions between the covalent inhibitors such as osimertinib33 or WZ4002 (ref. 31) and the methionine side chain of the mutant gatekeeper residue (T790M) can also help reduce the undesired side effects due to the inhibition of wild-type EGFR in noncancerous cells31. These studies illustrate how targeting native cysteines in the active sites of proteins can be leveraged for designing compounds that can overcome resistance to targeted anticancer drugs.
Designing inhibitors targeting allosteric binding sites
The efficacy of ATP-competitive or covalent inhibitors can be compromised by active site mutations that can arise upon prolonged drug treatment16,34,35 (Fig. 3a). A complementary strategy to target the ATP-binding site is designing inhibitors that can bind distinct pockets on the protein, such as regulatory sites or allosteric pockets, that become accessible when the protein adopts alternative conformations (e.g., the inactive state in kinases36,37). These pockets can be less conserved than ATP-binding sites, and inhibitors targeting them can potentially achieve high selectivity and have additional favorable properties (e.g., longer on-target residence times)38,39. For example, inhibitors targeting the myristate pocket of Abl1 kinase can potently block kinase activity40,41. In addition, inhibitors targeting the myristate pocket, such as ABL001 (also termed asciminib, Table 1), can also block the activity of BCR-ABL alleles with mutations in the active site that confer resistance to ATP-competitive inhibitors such as imatinib or dasatinib40. Therefore, targeting alternative pockets on proteins can be a useful strategy to overcome resistance.
Fig. 3 |. inhibitors with non-overlapping resistance profiles to overcome drug resistance.
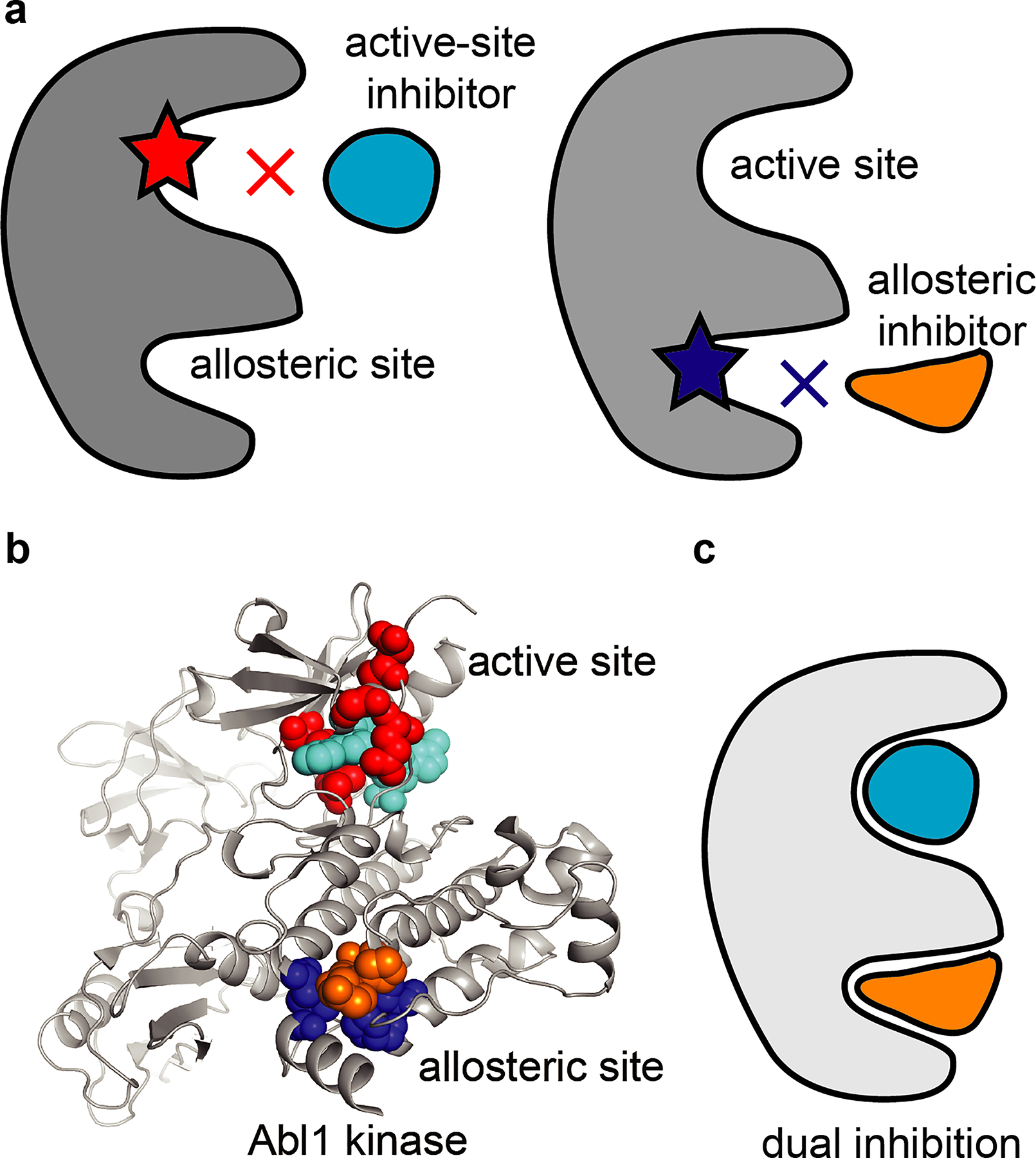
a, Schematics show mutant alleles of a target protein with resistance-conferring mutations in the active site (red star, left) and the allosteric site (blue star, right). Schematics of active-site and allosteric-site inhibitors are also shown (cyan and orange, respectively). b, Structural model of Abl1 kinase with inhibitors (sphere representation) bound in the active site (inhibitor in cyan) and the allosteric site (inhibitor in orange). Residues (sphere representation) that can be mutated without loss of protein activity and that confer resistance to inhibitors in the active site (residues in red) or the allosteric site (residues in blue) are shown. PDB: 5MO4. c, Schematic of a target protein with two inhibitors concomitantly bound in the active and allosteric sites.
However, mutations can also arise in the allosteric pockets and confer resistance to these inhibitors (Fig. 3a). Studies of acquired resistance to asciminib reveal that mutations in the myristate pocket of Abl1 kinase, the proposed binding site of the inhibitor, can confer resistance and prevent binding of the drug40,42 (Fig. 3b). In this case, mutations that block the binding of asciminib are distinct from those that confer resistance to active-site inhibitors40 (Fig. 3b and Table 1). BCR-ABL alleles with mutations in the allosteric pocket do not substantially alter kinase activity and can be blocked by active site inhibitors such as imatinib or dasatinib (Table 1). Interestingly, combining allosteric and active site inhibitors to block BCR-ABL in cells can help reduce and even prevent the acquisition of on-target resistance40 (Fig. 3c). Further studies will be needed to determine whether such drug combinations could also limit the development of resistance to BCR-ABL targeting drugs in patients.
Designing compounds targeting multiple binding pockets
mTOR kinase plays critical roles in the PI3K–Akt–mTOR pathway, which is frequently hyperactivated in human cancers43, and inhibitors that bind mTOR in the rapamycin-binding site (rapalogs) or the ATP-binding site (TOR kinase inhibitors, TORKi’s) have been evaluated in clinical trials44,45 (Fig. 4a,b). However, mutations in these drug-binding sites can arise and confer resistance46. Rapalog binding is prevented by mutations (e.g., A2034V or F2108L) in the FKBP12-rapamycin-binding domain (FRB domain) that have also been reported to confer resistance to everolimus in patients47. Interestingly, the M2327I mutation, which confers resistance to the TOR kinase inhibitor AZD8055, maps to the kinase domain ~15 Å away from the ATP-binding site and does not prevent compound binding46. This mutation leads to an approximately threefold increase in the basal kinase activity, which likely contributes to resistance against inhibitors targeting the active site of mTOR in cells.
Fig. 4 |. Bivalent inhibitors to overcome drug resistance.
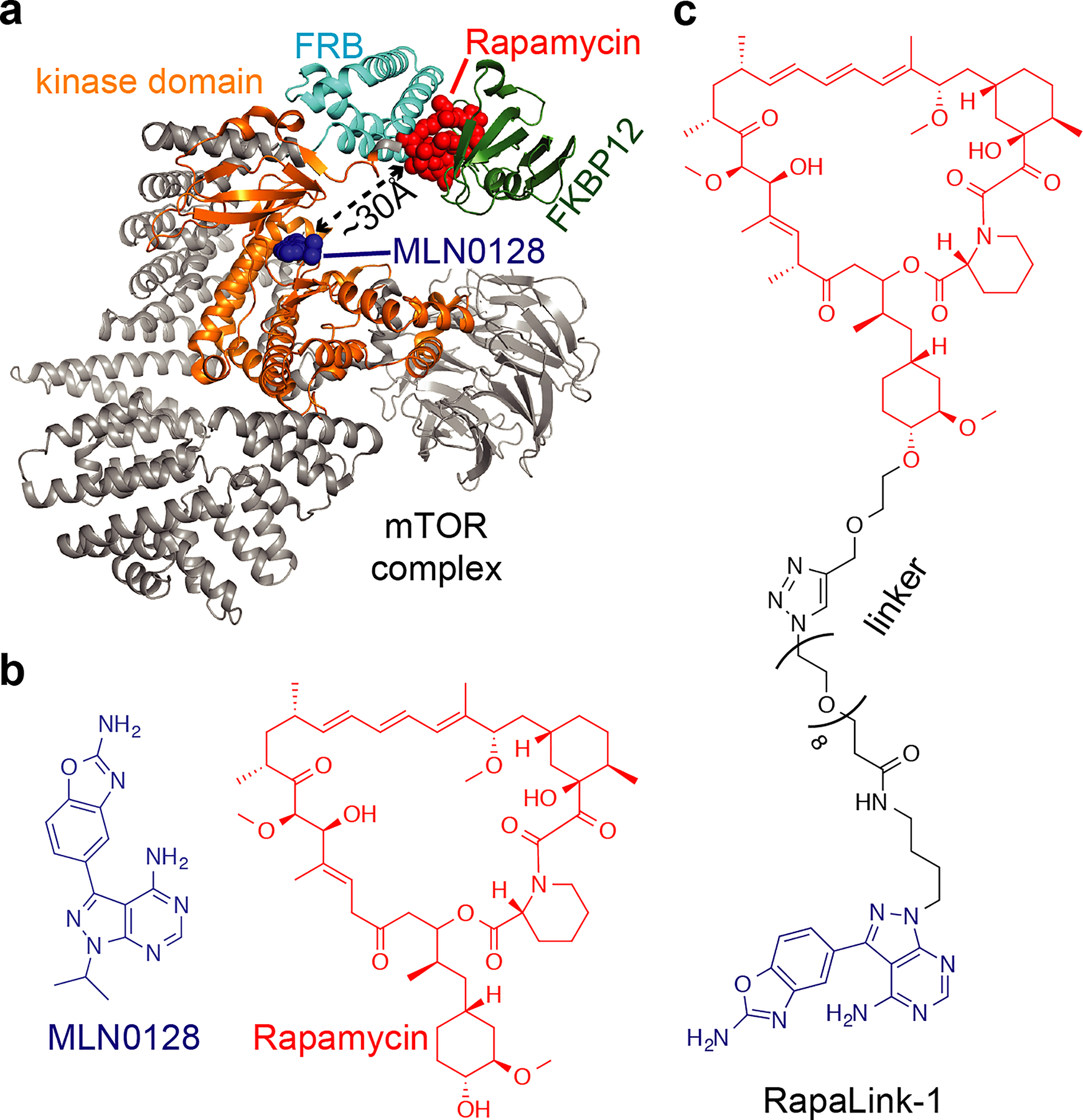
a, Model of mTOR kinase (gray, constructed from structural models of PDB IDs 4JT5 and 1FAP). FKBP12 (green), FRB (cyan) and kinase (orange) domains are indicated. Inhibitors bound in the kinase active site and the FRB site are also shown (blue and red spheres, respectively). Shortest distance between rapamycin and MLN0128 inhibitors is indicated. b, Chemical structures of mTOR inhibitors binding in the active site (MLN0128, blue) and the FRB-site (rapamycin, red). c, Chemical structure of the bivalent mTOR inhibitor RapaLink-1.
Resistance to the two classes of mTOR inhibitors was recently addressed by designing a bivalent compound that can bind the rapamycin-binding site and the ATP-binding site in the mTOR kinase46 (Fig. 4a). This inhibitor, termed RapaLink-1 (Fig. 4c), has a linker between the rapalog and the TORKi moieties that allows the compound to simultaneously interact with the rapamycin- and ATP-binding sites. Studies in cells indicate that RapaLink-1 limits proliferation by blocking mTOR signaling and suggest that the compound binds in both pockets of the target46. Interestingly, RapaLink-1 also inhibits activity of mTOR alleles with mutations in both FRB and the kinase domains even though this double mutant confers resistance to rapalogs, TORKi’s, as well as their combinations46. Bivalent compounds such as RapaLink-1 could also help reduce off-target toxicities associated with each drug, as the linker moiety and the topology of the drug conjugate can be tuned to improve selectivity for the chosen target. It will be important to examine whether similar approaches could be applied to other drugs and cancer pathways for which we also have extensive structural and biochemical data.
Overcoming resistance by targeted degradation
PROteolysis Targeting Chimeras (PROTACs48) belong to a class of heterobifunctional compounds that can induce targeted degradation of proteins (Fig. 5a). The use of PROTACs has recently emerged as a promising strategy to modulate protein function and also to target drug-resistant alleles in cancer49. PROTACs are composed of a ligand that recruits E3 ubiquitin ligase enzymes such as von Hippel-Lindau (VHL) or cereblon (CRBN) ligases50,51. This ligand is covalently attached via a linker to a second moiety that interacts with the protein of interest52. The concomitant binding of PROTACs to the target protein and the E3 ligase can lead to the formation of a ternary complex that promotes ubiquitination of the target by the E3 ligase and degradation of the ubiquitinated target by the proteasome53 (Fig. 5a). However, to engage two distinct targets, the PROTAC concentration must be precisely adjusted to avoid formation of ‘unproductive’ dimers with the target protein or the E3 ligase at high concentrations that can preclude ternary complex formation (so called ‘hook effect’54). In addition to complex pharmacology, developing PROTACs can be challenging, as designing optimal linkers can also be difficult. Interestingly, PROTACs can act sub-stoichiometrically as degradation of the ubiquitylated proteins releases the PROTAC molecule, which can then bind another molecule of the target protein and repeat the degradation cycle. This mode of action differs from the common ‘occupancy-driven’ pharmacology of inhibitor–target binding that may require high doses of the inhibitor to effectively block activity of the target. An additional advantage of targeted degradation compared to reversible inhibition is the longer sustained blockade of the target, as its function and activity can only be restored by protein re-synthesis55.
Fig. 5 |. Schematic of PROTAC and collateral sensitivity strategies.
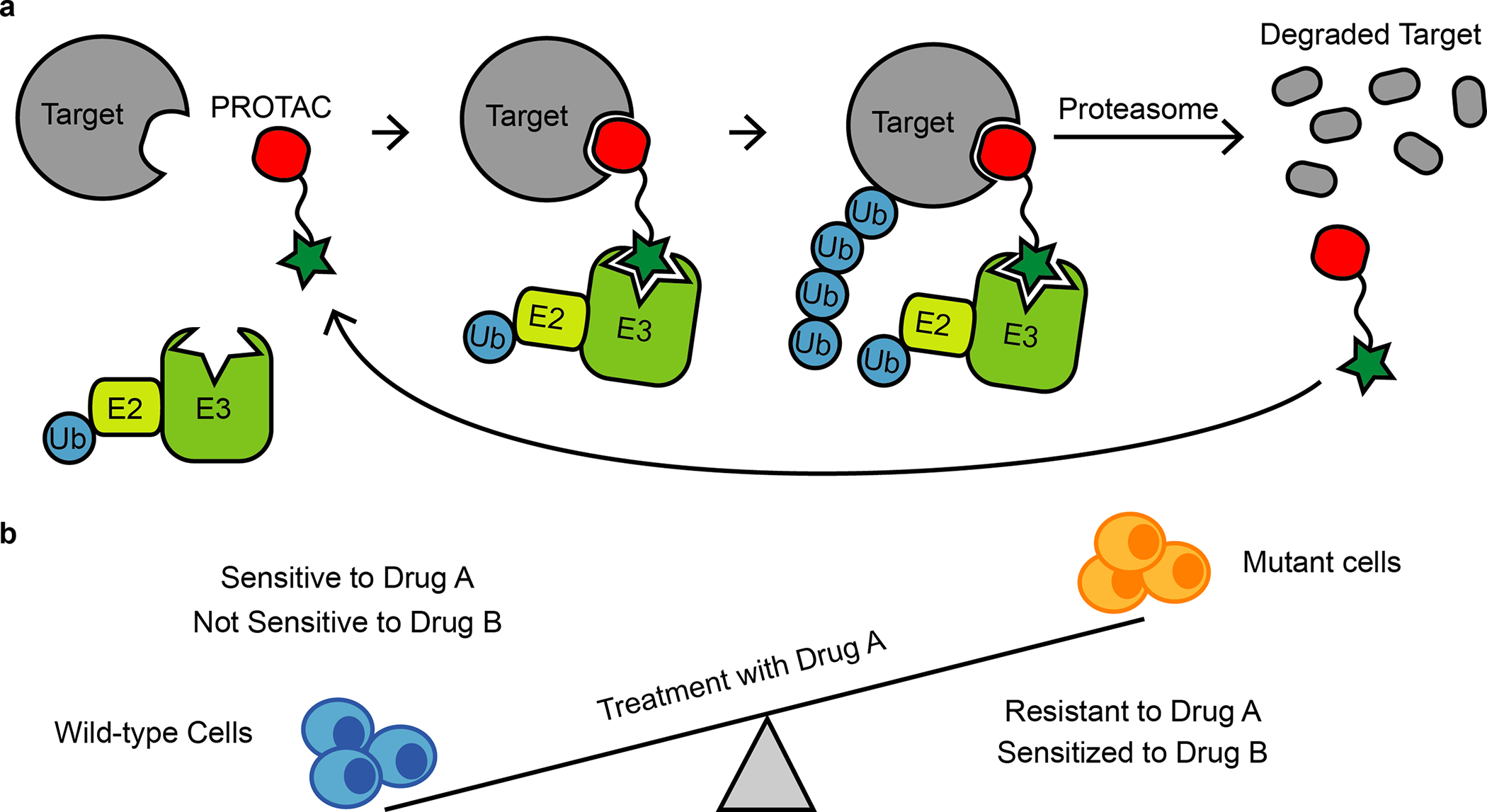
a, Schematic of the PROTAC strategy. A ternary complex formed upon binding of PROTAC, a target protein (e.g., drug-resistant mutant allele of a selected protein) and an E3 ligase complex can promote target protein ubiquitination (Ub) and degradation by the proteasome.
b, Schematic showing wild-type and drug A-resistant cells (blue and orange, respectively). Wild-type cells are sensitive to inhibition with drug A but not drug B. Cells that acquire resistance to drug A can become collaterally sensitive to drug B.
Targeting proteins for proteasomal degradation using the PROTAC approach has now been demonstrated for a number of proteins in cancer (e.g., BCR-ABL56, EGFR55 or Bruton’s kinase (BTK)57). Recent advances also suggest that PROTACs could help overcome resistance associated with mutations in these targets. For example, resistance-conferring alleles in Bruton’s kinase often arise by a substitution of the active-site cysteine residue to a serine (C481S), which reduces binding of the covalent inhibitor ibrutinib58. Interestingly, PROTACs based on the ibrutinib scaffold (e.g, MT-802, Table 1), which can still bind to the mutant protein, albeit with lower potency, can induce robust degradation of the BTK alleles that confer resistance to the parental compound57,59. This indicates that alleles conferring resistance to anticancer drugs may be targeted using the PROTAC approach.
In cases in which resistant mutations completely preclude binding, PROTACs targeting distinct binding pockets on the protein may be needed to provide an alternative mode for degradation (e.g., the myristate pocket of BCR-ABL60). Combining PROTACs targeting different sites on a protein (e.g., an active site and allosteric sites) could also be a strategy to limit resistance similar to what has been observed for ‘occupancy-driven’ drug combinations (see below).
Recent data show that resistance to PROTACs can arise via mutations not only in the target protein but also within the ubiquitination machinery61,62. Interestingly, these studies indicate a limited overlap of mutations that confer resistance to cereblon- and VHL-targeted degraders. This difference could be leveraged for designing robust treatment regimens that could potentially limit the emergence of resistance against PROTAC-based therapeutics.
Analogous ‘molecular glue’ mechanism of actions have been demonstrated for thalidomide-related compounds (also referred to as immunomodulatory drugs, or IMiDs) that promote formation of a tertiary complex with E3 ligase cereblon (CRBN) and an endogenous target protein (e.g., IKAROS family zinc finger proteins 1 and 3 (IKFZ1/3))51,63,64,65,66. Recent studies have also identified aryl sulfonamide-based inhibitors67, such as indisulam and tasisulam, as ‘molecular glues’ that induce protein–protein interactions. Studies applying an approach similar to DrugTargetSeqR (see below) in HCT116 cells revealed mutations that map to the RBM39 gene (RNA binding motif splicing factor 39) and confer resistance to a number of aryl sulfonamide compounds67,68. Biochemical and in vivo studies indicate that these compounds facilitate formation of a ternary complex between the RBM39 protein and DCAF15-CUL4-RBX1-DDB1 E3 ubiquitin ligase that can promote RBM39 ubiquitination and its degradation by the proteasome. Resistance-conferring mutations in RBM39 block aryl sulfonamide binding and RBM39 association with the E3 ubiquitin ligase. Disrupting this complex protects the RBM39 splicing factor from degradation and can promote cancer growth. Recent structural characterizations of E3 ligases in complex with aryl sulfonamides match the predicted binding sites of these compounds and suggest the molecular basis of their selectivity69,70. It is possible that these different molecular glues can be modified to induce the degradation of additional cellular targets, which could help address resistance mechanisms. These studies also raise the possibility that endogenous molecular glues that regulate protein–protein interactions may exist and could be discovered.
Overcoming resistance by using combinations of drugs
Combination therapy is a well-established clinical approach for cancer treatment and can also help address emerging resistance. Using cocktails of multiple chemotherapeutics has been a successful approach to cancer therapy, as drug combinations can have synergistic effects and provide more potent anticancer outcomes than single drugs alone (for reviews see refs. 5,71,72,73). Here, we highlight recent advances of drug combination strategies that leverage principles of non-overlapping resistance profiles and synthetic lethality to help overcome drug resistance.
Resistance to drugs often arises due to genetic changes in the drug’s target (e.g., point mutations, deletions or misregulation of expression), and multiple changes would be needed to confer resistance to drug combinations that target distinct proteins or binding sites. However, multiple genetic changes (e.g., single-point mutations) are less likely to occur simultaneously in a single cell. Therefore, using combinations of drugs with non-overlapping profiles of resistance-conferring mutations (e.g., due to targeting distinct sites on a protein or by targeting distinct protein targets) could be a powerful strategy to limit and prevent the emergence of resistance.
A nice example of drug combinations that have distinct patterns of resistance include drugs targeting BCR-ABL kinase. For instance, the active site inhibitor dasatinib in combination with an allosteric site-targeting inhibitor asciminib can substantially limit the emergence of resistant cells in preclinical models of the disease and may even lead to tumor eradication40. Additional studies will be needed to identify combinations of drugs with distinct resistance profiles.
Identifying combinations of drugs can not only enhance efficacy of targeted therapies but also uncover synthetic lethal strategies to suppress the evolution of drug resistance. For example, targeting mutant alleles of EGFR in lung cancers can elicit beneficial therapeutic responses, but is often followed by a relapse associated with acquired resistance29. Although the precise evolutionary trajectories leading to acquisition of the resistant phenotype in these cancers are not known, recent evidence suggests that the activity of Aurora A kinase contributes to the development of resistance in response to treatment with anti-EGFR agents74. Preclinical studies indicate that suppression of Aurora A kinase activity can improve outcomes of EGFR inhibition and can even limit the emergence of resistance to EGFR inhibitors in xenograft tumor models74. These studies suggest that drug combination regimens could be designed to help prevent developing resistance to targeted therapeutics. Further studies will be needed to determine whether this approach could also limit emergence of resistance to other classes of targeted therapeutics.
Synergistic drug combinations (i.e., when drugs induce larger effects in combination than predicted by their individual activities) often lead to increased potency of the two drugs in cancer cells. Interestingly, though, in the case of antimicrobial agents, synergistic drug combinations were shown to promote acquisition of resistance75. Interestingly, some antagonistic combinations (i.e., when the effect of drugs in combination is less than the predicted effect of each drug alone) of antimicrobial drugs can reduce and may even invert the selection pressure for resistance75. However, these experiments are currently limited mainly to bacterial systems, and additional studies will be needed to establish whether using antagonistic combinations of inhibitors could also limit the emergence of resistance in cancer cells.
Exploiting collateral sensitivity to limit emerging resistance
A potential strategy to reduce acquisition of a drug-resistant phenotype, as well as to treat disease after relapse, can be to target distinct evolutionary trajectories that promote emergence of resistance76. In principle, cells can acquire genetic changes during evolution of resistance to one drug that may sensitize these cells toward another drug (Fig. 5b).
This approach, first described in bacteria77 and termed collateral sensitivity, exploits drug-specific vulnerabilities and trade-offs that can emerge during the evolution of resistant phenotypes78,79,80. For example, BCR-ABL leukemia cells treated with the active-site-targeting drugs dasatinib or bosutinib often acquire multiple resistance-conferring mutations in the active site (e.g., V299L-E255K or V299L-F317L double mutants78). In these cases, the intermediate state of clonal evolution (i.e., cells that acquired the BCR-ABL-V299L mutant allele) can represent a particularly stable and robust state that is susceptible to treatment with other drugs. This ‘sensitization window’, also named ‘temporal collateral sensitivity’, is lost when cells acquire the double mutant genotype78. In particular, chemical screens revealed that proliferation of cell subpopulations expressing the single-point mutant allele BCR-ABL-V299L were sensitized to inhibitors such as vandetanib or foretinib that are chemically dissimilar to known BCR-ABL inhibitors (e.g., dasatinib or bosutinib)78. Interestingly, although vandetanib and foretinib can bind a number of distinct kinases, studies of isogenic cell lines suggest that the collateral sensitivity in these cells likely arises due to on-target inhibition of the BCR-ABL-V299L allele78. Molecular modeling and biochemical studies also indicate that these drugs can exploit different binding modes in the active site of BCR-ABL and potently inhibit the ‘sensitized’ mutant allele (V299L). Most notably, treating animals with BCR-ABL-V299L-driven tumor models with vandetanib or foretinib can lead to significant increase in survival in comparison to animals with tumors expressing wild-type BCR-ABL.
Together, these studies suggest that identifying and predicting evolutionary trajectories of cancer cells in response to drug treatment could be exploited for chemotherapy, although further studies will be needed to establish whether targeting temporal collateral sensitivity could be achieved in the clinic. Further studies will also be required to establish rational approaches and methods to systematically identify evolutionary trajectories and collaterally sensitive stages of cancer cells that could be targeted to overcome resistance.
‘Crash-testing drugs’
It is becoming clear that understanding resistance mechanisms can help design therapeutic strategies. Therefore, unbiased analyses of resistance-conferring mechanisms in different cell lines (e.g., established cancer cell lines or patient-derived cell lines) need to be developed and should be incorporated at early stages of drug development.
Chemical biology approaches, such as the DrugTargetSeqR81,82,83 that can be used for drug target identification in cells, can also help uncover high-frequency mechanisms of resistance. The first step of DrugTargetSeqR is to isolate cells resistant to the inhibitor. In the case of toxic compounds, such resistant clones can be selected from compound-treated genetically diverse cell populations. Genomic analyses, such as RNA-seq or exome sequencing, of the resistant clones can then be used to identify lesions that likely confer resistance to the compound. As passenger mutations will likely be unique to each clone, the lesions mapping in multiple independent clones to the same gene or a set of genes in a common pathway (e.g., kinase signaling cascade) likely indicate the targets of the inhibitor. In many cases, these high-frequency mutations are sufficient to confer resistance67,81,82,83. Importantly, these mutations can also match those that confer drug resistance in the clinic. For example, single-point substitutions in the PSMB5 gene (e.g., M104V) often arise and confer resistance in cultured cell lines upon prolonged treatment with the proteasome inhibitor bortezomib82 (Velcade). Analogous mutations have been identified in patient samples following relapse following bortezomib therapy84. Therefore, ‘crash testing’ drugs in different cancer cell lines can help reveal mechanisms of chemotype-specific resistance.
Variations in cancer cell genomes and karyotypes impose distinct constraints on the emergence of drug resistance. For example, applying DrugTargetSeqR in near-haploid and diploid cancer cell lines revealed distinct mechanisms of resistance to the mitotic kinesin CENP-E inhibitor GSK923925. This compound was evaluated in clinical trials for cancer, as blocking CENP-E activity can be toxic to cancer cells. However, similar to other targeted therapeutics, resistance can emerge in cells upon prolonged treatment with the inhibitor. In diploid cells, resistance to GSK923295 was found to arise via single-point mutations in the N-terminal motor domain of CENP-E, which can prevent inhibitor binding83. In contrast, analysis of GSK923295-resistant near-haploid cells revealed a deletion of the C-terminal domain of CENP-E. In contrast to chemical inhibition of CENP-E protein, which leads to cell death in diploid and haploid cells, genetic knockout of the CENPE gene does not block cell growth in haploid cells, and thus disrupting the gene in these cells can confer resistance. Though additional studies are needed to examine the emergence of drug resistance in cells with different ploidies, this recent study suggests that different karyotypes in cancer cells can constrain the trajectories of the genetic changes underlying drug resistance.
Cancer cell lines with impaired DNA mismatch repair (MMR) pathways exhibit increased mutation rates85, and identifying resistance-conferring alleles in these cells can be efficient86. However, most cells and cancer cell lines with intact MMR harbor low mutation frequencies, which can hinder selection of resistant clones. This can pose a barrier for incorporating analyses of resistance to established drug development pipelines. New approaches have recently been developed to address this limitation. For example, genetic ablation of MSH2, a gene involved in MMR pathways, in a number of cancer cell lines can induce higher mutation rates and facilitate selection of drug resistance-conferring alleles87. Incorporating these techniques in drug development projects could be particularly valuable for identifying mechanisms of resistance in cancer models with low mutational rates (e.g., childhood retinoblastomas or Ewing sarcomas88,89).
Conclusion and outlook
We have outlined a number of recent innovations in chemical biology for analyzing and addressing chemotype-specific resistance to anticancer therapeutics. As resistance to targeted drugs inevitably arises, new approaches are needed to develop drugs with improved efficacies and to devise strategies to address drug resistance. In particular, we highlighted how analyses of resistance can help the design of new drugs with distinct binding poses. In addition, simultaneous targeting of multiple sites on a protein (e.g., the active site and the allosteric site) can reduce the emergence of resistance, as multiple mutations blocking compound binding are less likely to occur. Identifying compounds with non-overlapping resistance signatures should therefore be prioritized early in the design process to help develop drugs that could reduce and even prevent the emergence of drug resistance. Innovative chemical biology strategies (e.g., the design of bivalent compounds that bind their target simultaneously in two proximal binding sites46) can also help in this process and can provide new leads for the development of therapeutics. Predicting trajectories of resistance and sensitivity to drugs in cancer cells using computational modeling will also help improve efficacies of new therapeutics90,91. Finally, advances in computational drug design and machine learning will likely facilitate the discovery of drugs that have binding poses distinct from those of other drugs and could help overcome resistance. Moving forward, integrating preclinical resistance analyses and new chemical approaches to tackle resistance should help realize the promise of targeted therapeutics for cancer treatment.
Acknowledgements
The authors would like to thank NIH/NIGMS for funding (R35 GM130234-01). R.P. is grateful to the Tri-Institutional Program in Chemical Biology for support. The authors regret any omissions of prior work due to limits on space and the number of citations.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Capdeville R, Buchdunger E, Zimmermann J & Matter A Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov 1, 493–502 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Paez JG et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi S et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 352, 786–792 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Gorre ME et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Baym M, Stone LK & Kishony R Multidrug evolutionary strategies to reverse antibiotic resistance. Science 351, aad3292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richman DD Antiviral drug resistance. Antivir. Res 71, 117–121 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Konieczkowski DJ, Johannessen CM & Garraway LA A convergence-based framework for cancer drug resistance. Cancer Cell 33, 801–815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clavel F & Hance AJ HIV drug resistance. N. Engl. J. Med 350, 1023–1035 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Andino R & Domingo E Viral quasispecies. Virology 479–480, 46–51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Margeridon-Thermet S et al. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J. Infect. Dis 199, 1275–1285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salverda MLM, De Visser JAGM & Barlow M Natural evolution of TEM-1 β-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol. Rev 34, 1015–1036 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Gottesman MM, Lavi O, Hall MD & Gillet J-P Toward a better understanding of the complexity of cancer drug resistance. Annu. Rev. Pharmacol. Toxicol 56, 85–102 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Shah NP et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Choi YL et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med 363, 1734–1739 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Blencke S et al. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem. Biol 11, 691–701 (2004). [DOI] [PubMed] [Google Scholar]
- 16.O’Hare T, Eide CA & Deininger MWN Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 110, 2242–2249 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Katayama R et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med 4, 120ra17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Hare T, Deininger MWN, Eide CA, Clackson T & Druker BJ Targeting the BCR-ABL signaling pathway in therapy-resistant Philadelphia chromosome-positive leukemia. Clin. Cancer Res 17, 212–221 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Friboulet L et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 4, 662–673 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw AT et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N. Engl. J. Med 374, 54–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper (Ref. 20) describes exemplary approaches to overcome acquired resistance to drugs targeting ALK in cancer.
- 21.Drilon A et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 7, 963–972 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drilon A et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med 378, 731–739 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ardini E et al. Entrectinib, a pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer Ther 15, 628–639 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Russo M et al. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discov. 6, 36–44 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Drilon A et al. What hides behind the MASC: clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann. Oncol 27, 920–926 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strebhardt K & Ullrich A Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 8, 473–480 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Dalton SE & Campos S Covalent small molecules as enabling platforms for drug discovery. ChemBioChem 21, 1080–1100 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Singh J, Petter RC, Baillie TA & Whitty A The resurgence of covalent drugs. Nat. Rev. Drug Discov 10, 307–317 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Pao W et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, e73 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yun C-H et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl Acad. Sci. USA 105, 2070–2075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou W et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engelman JA et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 67, 11924–11932 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Cross DAE et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4, 1046–1061 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thress KS et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med 21, 560–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ercan D et al. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin. Cancer Res 21, 3913–3923 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shukla D, Meng Y, Roux B & Pande VS Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat. Commun 5, 3397 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Z et al. Exploration of type II binding mode: a privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol 9, 1230–1241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang Z, Grütter C & Rauh D Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol 8, 58–70 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Müller S, Chaikuad A, Gray NS & Knapp S The ins and outs of selective kinase inhibitor development. Nat. Chem. Biol 11, 818–821 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Wylie AA et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Schoepfer J et al. Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J. Med. Chem 61, 8120–8135 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Qiang W et al. Mechanisms of resistance to the BCR-ABL1 allosteric inhibitor asciminib. Leukemia 31, 2844–2847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vivanco I & Sawyers CL The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Basu B et al. First-in-human pharmacokinetic and pharmacodynamic study of the dual m-TORC 1/2 inhibitor AZD2014. Clin. Cancer Res 21, 3412–3419 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagle N et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 4, 546–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodrik-Outmezguine VS et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 534, 272–276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper (Ref. 46) reports the elegant design of a bifunctional inhibitor to overcome resistance to drugs targeting the ATP-binding and FRB sites of mTOR kinase.
- 47.Wagle N et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med 371, 1426–1433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burslem GM & Crews CM Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 181, 102–114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai AC & Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov 16, 101–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Min J-H et al. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 296, 1886–1889 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Fischer ES et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paiva S-L & Crews CM Targeted protein degradation: elements of PROTAC design. Curr. Opin. Chem. Biol 50, 111–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakamoto KM et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work (Ref. 53) describes a proof-of-concept strategy for targeted degradation of proteins using proteolysis targeting chimeras (PROTACS).
- 54.Douglass EF Jr., Miller CJ, Sparer G, Shapiro H & Spiegel DA A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc 135, 6092–6099 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burslem GM et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem. Biol 25, 67–77. e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai AC et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl 55, 807–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobrovolsky D et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 133, 952–961 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Furman RR et al. Ibrutinib resistance in chronic lymphocytic leukemia. N. Engl. J. Med 370, 2352–2354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buhimschi AD et al. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57, 3564–3575 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Burslem GM et al. Targeting BCR-ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation. Cancer Res. 79, 4744–4753 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang L, Riley-Gillis B, Vijay P & Shen Y Acquired resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol. Cancer Ther 18, 1302–1311 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Ottis P et al. Cellular resistance mechanisms to targeted protein degradation converge toward impairment of the engaged ubiquitin transfer pathway. ACS Chem. Biol 14, 2215–2223 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Ito T et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010). [DOI] [PubMed] [Google Scholar]; This paper (Ref. 63) describes the long-awaited identification of cellular targets of thalidomide.
- 64.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krönke J et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chamberlain PP et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol 21, 803–809 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Han T et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, eaal3755 (2017). [DOI] [PubMed] [Google Scholar]; This work (Ref. 67) describes the discovery of cellular targets of anticancer arylsulfonamides using an approach similar to DrugTargetSeqR.
- 68.Uehara T et al. Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat. Chem. Biol 13, 675–680 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Bussiere DE et al. Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat. Chem. Biol 16, 15–23 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Faust TB et al. Structural complementarity facilitates E7820-mediated degradation of RBM39 by DCAF15. Nat. Chem. Biol 16, 7–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palmer AC & Sorger PK Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 171, 1678–1691. e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jia J et al. Mechanisms of drug combinations: interaction and network perspectives. Nat. Rev. Drug Discov 8, 111–128 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Han K et al. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol 35, 463–474 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shah KN et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med 25, 111–118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chait R, Craney A & Kishony R Antibiotic interactions that select against resistance. Nature 446, 668–671 (2007). [DOI] [PubMed] [Google Scholar]
- 76.McGranahan N & Swanton C Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Szybalski W & Bryson V Genetic studies on microbial cross resistance to toxic agents. I. Cross resistance of Escherichia coli to fifteen antibiotics. J. Bacteriol 64, 489–499 (1952). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao B et al. Exploiting temporal collateral sensitivity in tumor clonal evolution. Cell 165, 234–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dhawan A et al. Collateral sensitivity networks reveal evolutionary instability and novel treatment strategies in ALK mutated non-small cell lung cancer. Sci. Rep 7, 1232 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Imamovic L & Sommer MOA Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci. Transl. Med 5, 204ra132 (2013). [DOI] [PubMed] [Google Scholar]
- 81.Kasap C, Elemento O & Kapoor TM DrugTargetSeqR: a genomics- and CRISPR-Cas9-based method to analyze drug targets. Nat. Chem. Biol 10, 626–628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wacker SA, Houghtaling BR, Elemento O & Kapoor TM Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat. Chem. Biol 8, 235–237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pisa R, Phua DYZ, and Kapoor TM Distinct Mechanisms of Resistance to a CENP-E Inhibitor Emerge in Near-Haploid and Diploid Cancer Cells. Cell Chem. Biol 27, 7, 850–857. e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; Refs. 81–83 describe DrugTargetSeqR-like approaches for ‘crash testing’ drugs.
- 84.Barrio S et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 33, 447–456 (2019). [DOI] [PubMed] [Google Scholar]
- 85.Supek F & Lehner B Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521, 81–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Glaab WE & Tindall KR Mutation rate at the hprt locus in human cancer cell lines with specific mismatch repair-gene defects. Carcinogenesis 18, 1–8 (1997). [DOI] [PubMed] [Google Scholar]
- 87.Povedano JM et al. Engineering forward genetics into cultured cancer cells for chemical target identification. Cell Chem. Biol 26, 1315–1321. e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang J et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481, 329–334 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crompton BD et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 4, 1326–1341 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Liu R, Zhang G & Yang Z Towards rapid prediction of drug-resistant cancer cell phenotypes: single cell mass spectrometry combined with machine learning. Chem. Commun. (Camb.) 55, 616–619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Menden MP et al. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One 8, e61318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Whitesides GM & Krishnamurthy VM Designing ligands to bind proteins. Q. Rev. Biophys 38, 385–395 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kapoor TM & Miller RM Leveraging chemotype-specific resistance for drug target identification and chemical biology. Trends Pharmacol. Sci 38, 1100–1109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pisa R, Cupido T, Steinman JB, Jones NH & Kapoor TM Analyzing resistance to design selective chemical inhibitors for AAA proteins. Chem. Biol 26, 1263–1273. e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper (Ref. 94) describes the resistance analysis during design (RADD) approach. RADD can help develop robust inhibitor-target binding models and anticipate resistance-conferring mutations to drugs.
- 95.Cupido T, Pisa R, Kelley ME & Kapoor TM Designing a chemical inhibitor for the AAA protein spastin using active site mutations. Nat. Chem. Biol 15, 444–452 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shi J et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol 33, 661–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ma L et al. CRISPR-Cas9-mediated saturated mutagenesis screen predicts clinical drug resistance with improved accuracy. Proc. Natl Acad. Sci. USA 114, 11751–11756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ipsaro JJ et al. Rapid generation of drug-resistance alleles at endogenous loci using CRISPR-Cas9 indel mutagenesis. PLoS One 12, e0172177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Neggers JE et al. Target identification of small molecules using large-scale CRISPR-Cas mutagenesis scanning of essential genes. Nat. Commun 9, 502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jost M & Weissman JS CRISPR approaches to small molecule target identification. ACS Chem. Biol 13, 366–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]