

Abstract
The main neuropathological hallmarks of Parkinson’s disease (PD) are loss of dopaminergic neurons in the substantia nigra and intraneuronal protein aggregates immunoreactive for α-synuclein phosphorylated at serine 129 (pS129). Most cases of PD are idiopathic; however, genetic mutations have been identified in several genes linked to familial PD. Mutations in the gene encoding α-synuclein are causally linked to dominantly inherited forms of PD and mutations in the PTEN-induced kinase-1 (PINK1) gene are linked to recessively inherited forms of PD. Because abnormal α-synuclein protein aggregates appear spontaneously in PINK1 knockout (KO) rats, we hypothesize that PINK1-deficiency causes endogenous α-synuclein to be more prone to aggregation. α-Synuclein aggregation does not normally occur in mice or rats, however, it can be induced by intracranial injection of α-synuclein pre-formed fibrils (PFFs), which also induces loss of dopaminergic nigral neurons 3-6 months post-injection. Because PINK1-deficiency is linked to early-onset PD, we further hypothesize that PINK1 KO rats will show earlier PFF-induced neurodegeneration compared to wild-type (WT) rats. Herein, we report that intracranial injection of α-synuclein PFFs into the dorsal striatum induced more abundant pS129 α-synuclein in PINK1 KO rat brains compared to WT littermate controls. Moreover, the synuclein extracted from the brains of PFF-injected PINK1 KO rats was more insoluble compared to PFF-injected WT littermates, suggesting greater progression of α-synuclein pathology in PINK1 KO rats. Four weeks post-injection, PFFs caused significant loss of dopaminergic neurons in the substantia nigra of PINK1 KO rats, but not WT controls. Together, our results indicate that PINK1 deficiency increases vulnerability to α-synuclein aggregation and dopaminergic neurodegeneration in vivo.
Keywords: Parkinson’s disease, synuclein, PINK1, Pre-formed fibrils, Lewy bodies, inclusions, aggregation
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder. The two primary neuropathological criteria for PD diagnosis postmortem are loss of pigmented dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of α-synuclein immunoreactive protein aggregates including Lewy bodies and Lewy neurites. These aggregates are composed of abnormal fibril forms of α-synuclein, which is an abundant protein normally loosely associated with presynaptic vesicle membranes (Spillantini et al., 1997;Jensen et al., 1998;Spillantini et al., 1998;Kahle et al., 2000;Rhoades et al., 2006). Antibodies specific for α-synuclein phosphorylated at serine 129 (pS129) are selective for pathological forms of α-synuclein (Fujiwara et al., 2002). The etiology of PD remains uncertain. Most cases occur apparently sporadically while rare inherited forms of PD are causally linked to mutations in genes including Parkin, PINK1, DJ-1, LRRK2 and α-synuclein (Polymeropoulos et al., 1997;Kitada et al., 1998;Bonifati et al., 2003;Paisan-Ruiz et al., 2004;Valente et al., 2004;Zimprich et al., 2004). Loss-of-function PINK1 mutations cause early onset PD with high penetrance and symptoms typical of idiopathic PD, including tremor, rigidity, bradykinesia and postural instability (Valente et al., 2004). The mechanisms by which PINK1-deficiency causes PD remains the topic of intense research. PINK1 is a kinase that phosphorylates ubiquitin and Parkin, which activates the E3 ubiquitin ligase activity of Parkin and targets depolarized mitochondria for degradation by autophagy (Narendra et al., 2008;Kondapalli et al., 2012;Kazlauskaite et al., 2014a;Kazlauskaite et al., 2014b;Koyano et al., 2014;Barodia et al., 2017). Aside from functioning together with Parkin in mitochondrial quality control, several additional functions of PINK1 have recently been identified, including mitochondrial antigen presentation and regulation of innate immune signaling (Matheoud et al., 2016;Sliter et al., 2018;Matheoud et al., 2019). PINK1 has also been shown to promote the removal of α-synuclein aggregates and to protect against α-synuclein-mediated neuronal injury (Liu et al., 2017;Yang et al., 2018).
Although the normal function of α-synuclein remains uncertain, abnormal fibril forms of α-synuclein are implicated in PD pathogenesis on multiple grounds. First, their presence upon postmortem examination is pathognomonic for PD (Michotte, 2003). Second, genetic point mutations in α-synuclein are linked to early onset PD (Polymeropoulos et al., 1997). Third, genetic duplication and triplication of the gene encoding α-synuclein (without any point mutations) causes PD with gene dose-dependent severity (Singleton et al., 2003;Chartier-Harlin et al., 2004). Together, this strongly suggests that PD-associated neurodegeneration can be caused by increasing the concentration of α-synuclein or by increasing the propensity of α-synuclein to undergo conformational changes to fibril forms that could impinge on normal cell functions and form large aggregates such as Lewy bodies and Lewy neurites (Goldberg and Lansbury, 2000). This theory has recently been reinforced by studies showing PD-like pathology in rats and mice induced by intracranial injection of pre-formed fibril (PFF), but not monomer, forms of α-synuclein (Volpicelli-Daley et al., 2011;Luk et al., 2012;Paumier et al., 2015;Chung et al., 2019). Moreover, recent studies show that aggregated α-synuclein can impair the function of Parkin, which could exacerbate the effects of PINK1-deficiency (Jesko et al., 2019;Wilkaniec et al., 2019).
Autopsies have revealed the presence of Lewy bodies and Lewy neurites in familial forms of PD, including some cases of PINK1-linked PD (Gandhi et al., 2006;Samaranch et al., 2010). The relationship between PD-linked PINK1 mutations and α-synuclein aggregation is important to understand yet relatively unexplored. Lewy bodies and other synuclein aggregates are often observed in aged human brains, even without symptoms of disease (Bayer et al., 1999;Braak et al., 1999;Tsuboi et al., 2001). By contrast, spontaneous age-dependent formation of α-synuclein aggregates has generally not been found in mouse or rat animal models of PD without overexpression of α-synuclein or other interventions to induce α-synuclein aggregation. PINK1 KO rats are exceptional because they exhibit spontaneous formation of α-synuclein aggregates as early as age 4 months, with increasing abundance with age (Grant et al., 2015;Creed and Goldberg, 2018a;b). Here, we sought to exploit the unique features of the PFF and the PINK1 KO rat models of PD to test the hypothesis that PINK1 deficiency increases α-synuclein aggregation and neurodegeneration induced by intracranial injection of α-synuclein PFFs.
Experimental Procedures
Animals
PINK1 KO rats on a Long-Evans genetic background were obtained with a breeding license from Horizon Discovery and bred in our colony to obtain homozygous PINK1 knockout and wild-type (WT) littermate controls (Dave et al., 2014). Animals were maintained on a 12-hour light/dark cycle and were allowed food and water ad libitum. All animal experiments were reviewed and approved in advance by the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Preparation and Characterization of ɑ-Synuclein Pre-Formed Fibrils
α-Synuclein fibrils (gift from Andrew West and Laura Volpicelli-Daley) were generated as previously described (Abdelmotilib et al., 2017). Briefly, 100 μl Aliquots of 5mg/ml mouse α-synuclein monomeric protein (in 50 mM Tris, pH 7.5, and 150 mM KCl) were shaken at 700 RPM at 37°C for 7 days, then stored at −80°C until use. Immediately before surgeries, fibrils were thawed at room temperature (RT) and sonicated using a probe tip sonicator (Fisher Model 500 Sonic Dismembrator) at 30% power, 1 second on, 1 second off, for 15 seconds, then placed on ice for 2 minutes to dissipate heat. This was repeated twice more for a total of 75 seconds, with 2 minutes on ice between each 15 second interval. Thioflavin T fluorescence was measured in 96-well plates with 200 μl of 20 μM Thioflavin T in phosphate-buffered saline (PBS) mixed with 2 μl (10 μg) of α-synuclein monomer or PFFs. After incubating 15 minutes at room temperature, plates were read on a fluorescence plate reader with 440 nm excitation and 508 nm emission filters. Dynamic Light Scattering was used to calculate the PFF hydrodynamic radius from the time-dependent fluctuations in scattered light intensity at 25 °C using a Wyatt Technology DynaPro NanoStar instrument with DYNAMICS software.
Surgeries
WT (45 animals) and PINK1 KO (49 animals) rats were administered unilateral intracranial injections of α-synuclein PFFs or monomer control at age 13 weeks (~3 months). Rats were anaesthetized with vaporized isoflurane and fitted to a stereotaxic frame (David Kopf). 4μl of 5 mg/ml α-synuclein PFFs (kept at RT for up to 8 hours) or monomer (kept on ice) was injected at a rate of 0.5μl/minute. The syringe was held in place for an additional 4 minutes to ensure that all the solution dispended into the brain. All injections were targeted to the right dorsal striatum at the following coordinates measured from bregma: A/P + 0.48, M/L −3.0, D/V −5.6. Incisions were closed by suture and rats were returned to their home cages. 2 or 4 weeks later, rats were deeply anesthetized with isoflurane and transcardially perfused with PBS. Brains were removed, post-fixed for 24 hours in 10% formalin at 4°C, and then transferred to 30% sucrose in PBS solution at 4°C for at least 3 days for cryoprotection prior to sectioning and staining.
Immunofluorescence and Immunohistochemistry
Brains were sectioned in the coronal plane at 30μm thickness using a freezing microtome. Free-floating brain sections were thoroughly washed in 1X PBS to remove cryoprotectant and then blocked in 1% normal goat serum (NGS) in PBS for 1 hour, then incubated with anti-tyrosine hydroxylase (TH) primary antibody (Millipore, cat #: AB152, 1:3000) for ~ 48 hours in the same buffer at 4°C. Sections were incubated with biotinylated goat anti-rabbit secondary antibody for 2 hours at RT, followed by avidin-biotin peroxidase complex solution (Vector Laboratories ABC Elite) for 2 hours at RT. Tissue sections were then developed using DAB chromogen (Vector Laboratories) for ~3 minutes.
For immunofluorescence, tissues sections were incubated in blocking buffer (10% normal donkey serum in PBS with 0.3% Triton-X-100) for 1 hour at RT, then incubated for ~48 hours at 4°C with primary antibodies (pS129 α-synuclein, BioLegend #825701, 1:10,000) in blocking buffer containing 1% normal goat serum. Following three washes in PBS, sections were incubated with Alexa-conjugated secondary antibodies (AlexaFlour 555-conjugated goat anti-mouse IgG2a) for 2 hours at RT in blocking buffer containing 1% normal goat serum. Sections were washed in PBS for 5 minutes, then stained for 5 minutes with Hoechst 33342 (Sigma) diluted in 1X PBS, 1:5000. Following washing 3 x 5 minutes in PBS, sections were mounted on microscope slides, air dried overnight and cover slipped using Vectashield H-1400 hardest mounting medium (Vector Laboratories). Images were acquired with a Nikon Ti-S epifluorescence microscope and pS129 α-synuclein immunoreactive cells were counted using Nikon NIS elements software.
TH fiber analysis was performed according to previously described methods (Daher et al., 2015). Briefly, free floating sections were rinsed with ice cold 1X Tris-buffered saline (TBS), then twice washed 5 minutes in TBS, then incubated in antigen retrieval buffer (10mM citrate, 0.05% tween-20, pH 6.0) shaking at 40 rpm for 1 hour at 37°C. Sections were then washed in TBS 3 x 5 minutes, followed by blocking (5% Goat serum in TBS, 0.3% Tx-100) for one hour RT. Sections were incubated in rabbit anti-TH (Millipore, abl52, 1:2,000) primary antibody (in TBS plus 5% Goat serum, 0.1% Triton X-100) for 48 hours at 4°C. Sections were then incubated in LiCOR IRDye 680, Goat anti-rabbit secondary (1:20,000) overnight at 4°C, mounted on slides and scanned using a LiCOR Odyssey scanner and quantified using Image Studio Lite software.
Proteinase-K Immunohistochemistry
Immediately prior to DAB immunohistochemistry, free-floating coronal sections (N= 5 WT, 5 KOs) were washed with PBS and treated with 2 mg/ml proteinase K (Fisher Scientific 50-751-7334) for 10 minutes at RT. Sections were then washed thoroughly with PBS and analyzed by DAB immunohistochemistry with anti-pS129 α-synuclein, as described above. Quantification of proteinase K-resistant α-synuclein immunoreactivity was scored by an investigator blind to treatment and genotype as follows: Score = 0: no aggregates per section; Score = 1: fewer than 10 aggregates; Score = 2: 10-50 aggregates; Score = 3: greater than 50 aggregates. Three sections from each animal were analyzed and averaged.
Stereology
Every 6th section (spanning the SNpc) was selected and DAB stained with anti-TH antibody as above. The optical fractionator probe of Stereoinvestigator software (MicroBrightFeild) was used to obtain an unbiased estimate of TH-positive neurons in the SNpc. Contours were drawn around the SNpc using a 4x objective. Neurons were counted using a 1.42 NA 60x oil objective with a 50 x 50μm counting frame and a grid size set at 100 x 100 μm. Only TH-positive cells with a nucleus that comes into focus inside the counting frame or touching the two green sides of the frame were counted by an investigator blind to genotype and treatment.
Biochemical assay
Immediately upon euthanasia, brains from 6 WT and 7 KO rats were removed, hemisected, frozen on dry ice and stored at −80°C until use. The right and left hemisphere of each animal were individually weighed and dounce homogenized on ice in PBS buffer (20%w/v) supplemented with protease and phosphatase inhibitor cocktails (Sigma Aldrich). Following homogenization, sarkosyl was added to 1% and samples were sonicated at 50% amplitude, 1 second on/off for 10 seconds to shear DNA. Samples were then centrifuged at 200,000xg for 60 minutes at 4°C in a tabletop centrifuge (Beckman coulter). Pellets were washed with PBS-1% sarkosyl, and then resuspended in 500μl urea buffer (8M urea, 3% SDS in 50mM Tris-HCl, pH 8.5). For gel electrophoresis, 50μg protein (measured with BCA assay) from each sample was separated by SDS-PAGE 1 hour in a 4-20% mini protean gel (Bio-Rad). Proteins were then transferred onto a 0.2μM PVDF membrane then blocked in 1:1 LI-COR Odyssey blocking buffer and TBS with 0.05% Tween 20 (TBS-T) for 1 hour at RT, then incubated with primary antibody overnight at 4°C: pS129 α-synuclein (phospho-solutions, #p157-129, 1:1,000), GAPDH (Millipore, #MAB374, 1:5,000), TBK1 (Abcam ab40676, 1,1000), phospho-TBKl (Abcam abl09272, 1:1000), or p62 (Abeam ab56416, 1:1,000). After washing with TBS-T, membranes were incubated with LI-COR Odyssey secondary antibodies for 2 hours at RT and imaged using a LI-COR Odyssey Scanner.
Statistical Analysis
GraphPad Prism 8 software was used for statistical analysis and graphing. All values are shown as mean +/− SEM. Student’s t-test was used for comparison between genotypes. Student’s t-test with Welch’s correction was used where variances were not equal. Data were found to be normally distributed by both D’Angostino & Pearson’s test and the Shapiro-Wilk’s test. For analyses utilizing multiple factors, two-way ANOVA was used and Sidak’s multiple comparison’s test was used where appropriate. A p-value of less than 0.05 was considered statistically significant.
Results
PFF-induced α-synuclein aggregates appear as early as two weeks post-injection.
Previous studies in mice and rats have demonstrated that intrastriatal injection of α-synuclein PFFs induces the formation of α-synuclein protein aggregates in various brain regions that project to the striatum, including the cortex, amygdala and SNpc (Luk et al., 2012;Paumier et al., 2015;Abdelmotilib et al., 2017). Moreover, the PFF-induced aggregates are immunoreactive for α-synuclein phosphorylated at serine 129, which is a selective marker of pathological forms of synuclein in diseased postmortem human brains (Fujiwara et al., 2002). To date, most studies of PFF-injected mice and rats include a 3–6-month interval between intracranial PFF injection and harvesting brains for immunohistochemical analysis, to allow for the PFFs to seed the conversion of endogenous monomeric α-synuclein to oligomeric fibrillar synuclein aggregates and to cause neurodegeneration (Luk et al., 2012;Paumier et al., 2015). However, recent studies have shown that smaller size PFF particles, produced by increased sonication time, induce greater pathology in both rats and mice (Abdelmotilib et al., 2017). Such protocols have produced pSer129-α-synuclein aggregates in the cortex of mice as early as two weeks post-injection of PFFs into the striatum (Froula et al., 2019). Therefore, to determine the extent to which PINK1-deficiency affects PFF-induced α-synuclein aggregation and neurodegeneration, we injected the striatum of PINK1 KO and WT rats unilaterally with monomeric α-synuclein or PFFs generated using increased sonication time, then harvested brains for analysis 2 weeks and 4 weeks post-injection. Later time points were not included so as to avoid confounding effects of the age-dependent spontaneous α-synuclein aggregation and neurodegeneration in PINK1 KO rats reported by multiple independent studies (Dave et al., 2014;Grant et al., 2015;Villeneuve et al., 2016;Creed and Goldberg, 2018a;b). The PFFs generated using increased sonication time—but not the monomeric α-synuclein starting material—showed substantial Thioflavin T fluorescence, indicative of amyloid-like beta-sheet structure (Figure 1A). Further characterization of the PFFs using dynamic light scattering showed light scattering consistent with particle diameters of 5-10 nm (Figure 1B).
Figure 1. Biophysical characterization of α-Synuclein pre-formed fibrils.
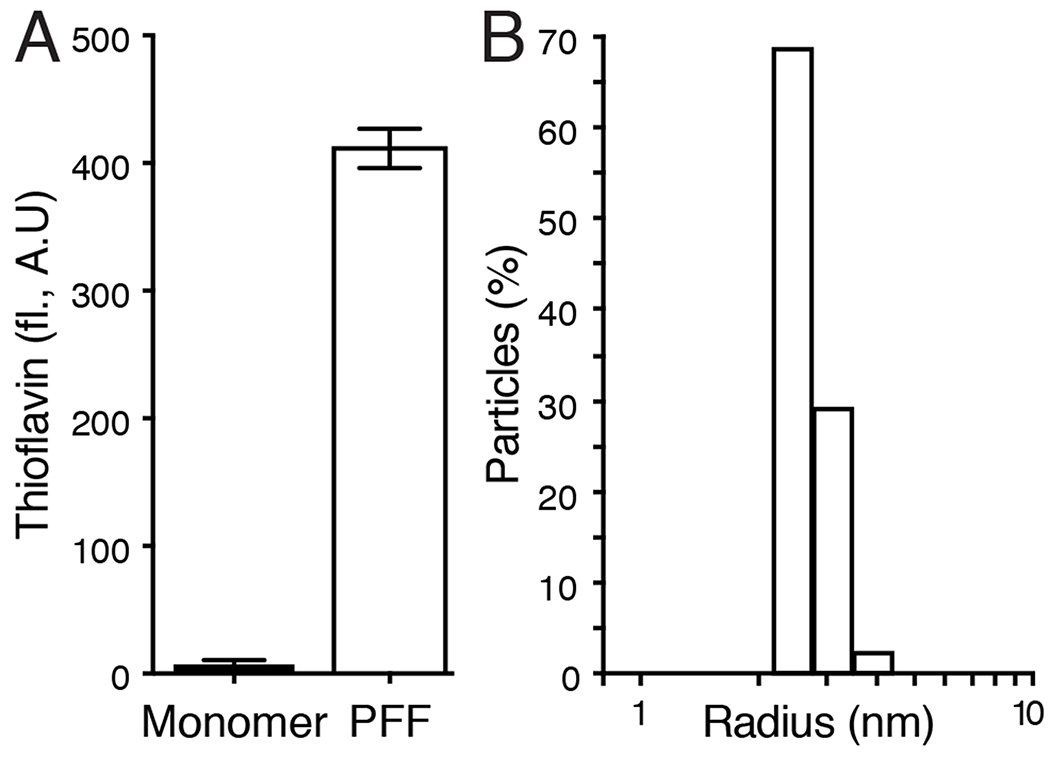
(A) Thioflavin T fluorescence of monomeric α-synuclein and α-synuclein PFFs. Bars show mean +/− SEM fluorescence (arbitrary units) of 3 measurements. (B) Dynamic light scattering of α-synuclein PFFs. Bars show percent of particles at each hydrodynamic radius calculated by the time-dependent fluctuations in scattered light intensity using a Wyatt Technology DynaPro NanoStar instrument with DYNAMICS software.
At two weeks post-injection, both WT and PINK1 KO rat brains had prominent pS129 α-synuclein immunoreactive aggregates in various brain regions including the ipsilateral (but not the contralateral) SNpc (Figure 2A). At this time point, quantification of the pS129 α-synuclein burden (mean number of immunoreactive cells) showed no significant difference between WT and PINK1 KO rats by students t-test (n = 5 rats/group) (Figure 2B). To further characterize the α-synuclein aggregates, we also treated tissue sections with proteinase K to remove non-aggregated α-synuclein prior to immunostaining. This was scored on a scale of 0-3 by an investigator blind to genotype and treatment, as previously described (Froula et al., 2019). Proteinase K-resistant α-synuclein was present to a similar extent in the SNpc of WT and PINK1 KO rats two weeks post-PFF injection (Figure 2C–D). Furthermore, we observed proteinase K-resistant α-synuclein in the subiculum and the amygdala of WT and PINK1 KO rats to a similar extent 2 weeks post-PFF injection (Figure 2E).
Figure 2. α-Synuclein pathology in WT and PINK1 KO rats 2 weeks post PFF injection.
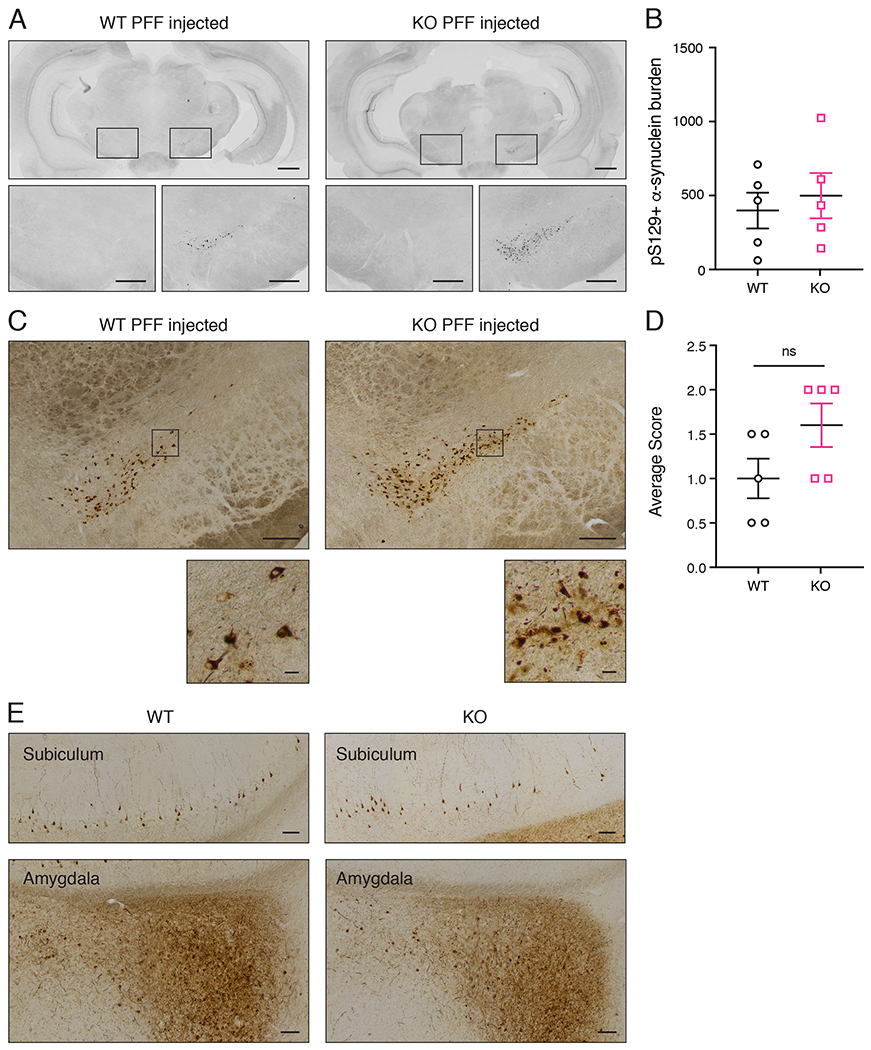
(A) pS129 α-synuclein immunofluorescence in coronal brain sections from PFF injected WT and PINK1 KO rats 2 weeks post injection. Grayscale fluorescence intensity is inverted to reveal neuroanatomy. For both WT and PINK1 KO rats, the injected hemisphere is on the right. Scale bars are 1 mm. Boxed regions around the contralateral and ipsilateral substantia nigra are shown at higher magnification below with 250 micron scale bars. (B) Quantification of α-synuclein burden (mean number of pS129 α-synuclein immunoreactive cells counted by an investigator blind to treatment and genotype) showed no differences between WT and PINK1 KO rats by t-test (n= 5/group, p > 0.05, student’s t-test). (C) pS129 α-synuclein DAB immunohistochemistry of proteinase K-treated sections of WT and PINK1 KO rats 2 weeks post PFF injection. Scale bars are 20 microns. Boxed regions within the ipsilateral substantia nigra are shown at higher magnification below. Scale bars are 20 microns. (D) Quantification of proteinase K-resistant α-synuclein immunoreactivity was scored by an investigator blind to treatment and genotype as follows: Score = 0: no aggregates per section; Score = 1: fewer than 10 aggregates; Score = 2: 10-50 aggregates; Score = 3: greater than 50 aggregates. Three sections from each animal were analyzed and averaged. There is no significant difference between WT and PINK1 KO rats (n= 5/group, p > 0.05, student’s t-test). (E) WT and PINK1 KO rat subiculum and amygdala pS129 α-synuclein DAB immunohistochemistry following proteinase K-treatment of tissue sections. Scale bars are 100 microns. Error bars represent mean +/− SEM.
Greater α-synuclein burden in PINK1 KO rats compared to WT rats 4 weeks post injection.
4 weeks post surgery, the injected hemisphere of PINK1 KO rats showed significantly greater pS129 α-synuclein burden (mean number of immunoreactive cells; *p = 0.0447, unpaired t-test with Welch’s correction: t(27.61)= 2.103) in the midbrain but not the amygdala compared to WT littermate controls (Figure 3, A–C). Other brain regions, such as the motor cortex, showed abundant pS129 α-synuclein pathology but without a significant difference between PINK1 KO and WT rats (Figure 3D–E). We did not observe any pS129 α-synuclein immunoreactivity in the brains of any animals injected with an equivalent amount of monomeric α-synuclein (Figure 3F).
Figure 3. pS129 α-synuclein burden in WT and PINK1 KO rats 4 weeks post PFF injection.
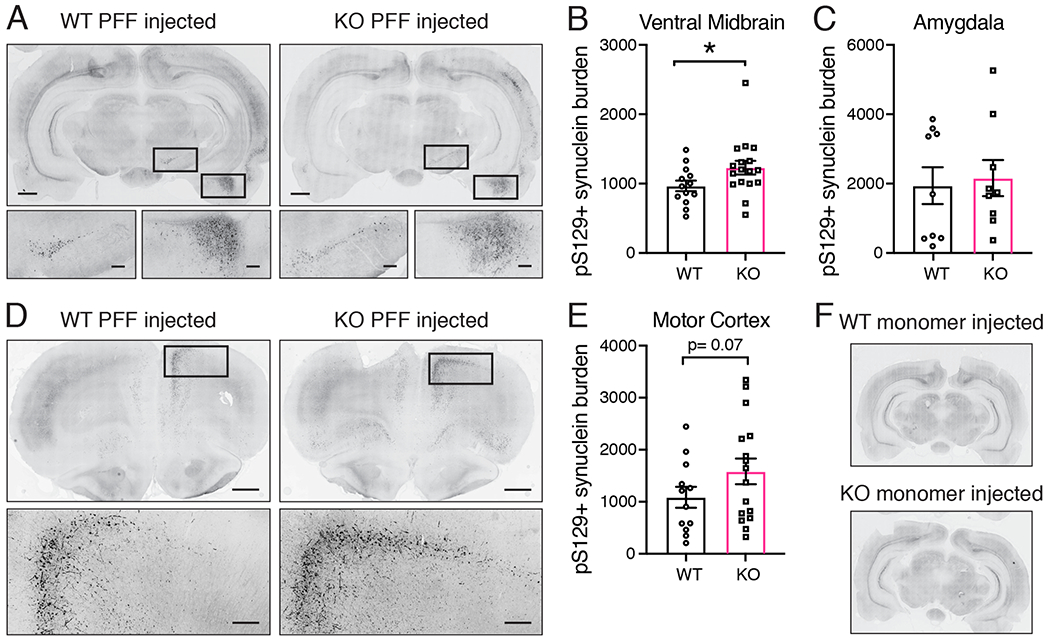
(A) pS129 α-synuclein immunofluorescence in coronal brain sections from PFF injected WT and PINK1 KO rats 4 weeks post injection. Grayscale fluorescence intensity is inverted to reveal neuroanatomy. Scale bar is 1 mm. Boxed regions around the ipsilateral substantia nigra and amygdala are shown at higher magnification below with 250 micron scale bars. (B) Quantification of pS129 α-synuclein burden (mean number of immunoreactive cells) shows an increase in pS129 α-synuclein in the ipsilateral ventral midbrain of PINK1 KO rats compared to WT rats (n=13 WT, 17 KO, *p = 0.0447, unpaired t-test with Welch’s correction: t(27.61)= 2.103; normal data according to the Kolmogorov-Smirnov normality test). (C) Quantification of pS129 α-synuclein burden in the amygdala showed no difference between WT and PINK1 KO rats (p > 0.05, student’s t-test). (D) pS129 α-synuclein immunofluorescence of rostral coronal sections from PFF injected WT and PINK1 KO rats. The injected hemisphere is on the right. Scale bars are 1 mm. Boxes in ipsilateral motor cortex are shown at higher magnification below with 250 micron scale bars. (E) Quantification of pS129 α-synuclein burden in motor cortex shows no difference between WT and PINK1 KO rats (p > 0.05, student’s t-test). (F) No pS129 α-synuclein immunofluorescence above background was detected in coronal sections from WT and PINK1 KO rats injected with α-synuclein monomer. Bars represent mean +/− SEM.
Increased insoluble α-synuclein in PFF-injected PINK1 KO rats.
The α-synuclein aggregates induced by PFF injections share common biophysical properties with human Lewy pathology such as greater insolubility (Paumier et al., 2015). To compare the extent to which α-synuclein aggregates were insoluble in PFF-injected PINK1 KO rats compared to WT littermate controls, we extracted sarkosyl-soluble and sarkosyl-insoluble fractions from both PFF injected and non-injected hemispheres of WT and PINK1 KO rats. As shown in a representative western blot (Figure 4A) and the mean densitometry of 5-7 animals (Figure 4B), we found no difference between genotypes in the level of sarkosyl-soluble pS129 α-synuclein (Figure 4B). By contrast, there was an increase in the level of pS129 α-synuclein in the sarkosyl-insoluble fraction of the injected hemisphere of PINK1 KO rats compared to WT controls (Figure 4C, 2-way ANOVA main effect of genotype p = 0.0032; Sidak’s post hoc comparison p= 0.0332). While not statistically significant, we observed a trend for increased pS129 α-synuclein in the un-injected hemisphere of PINK1 KO rats compared to WT rats, (p= 0.0907, Sidak’s post hoc comparison).
Figure 4. Levels of sarkosyl-soluble and insoluble α-synuclein in WT and PINK1 KO rat brains 4 weeks post PFF injection.
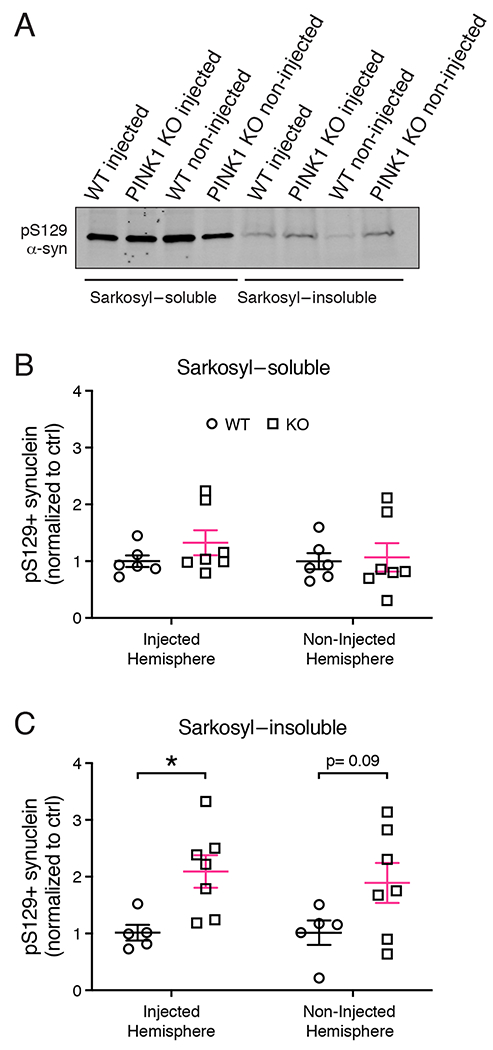
(A) pS129 α-synuclein western analysis of sarkosyl-soluble and insoluble fractions from whole brain lysates of PINK1 KO and WT rats 4 weeks post PFF injections. (B) Densitometric quantification of pS129 α-synuclein levels in the sarkosyl-soluble fraction. (C) Quantification of pS129 α-synuclein levels in the sarkosyl-insoluble fraction. Two-way ANOVA with Sidak’s multiple comparison showed a significant main effect of genotype F(1, 20)=11.21), *p = 0.0032. (n= 5 WT, 7 KO). Sidak’s multiple comparisons shows significantly increased pS129 α-synuclein in PINK1 KO rats in the injected hemisphere (*p = 0.0332), but not in the non-injected hemisphere (p = 0.09). Data were normal according to the Kolmogorov-Smirnov normality test. Error bars represent mean +/− SEM.
PFF-induced dopaminergic neurodegeneration in PINK1 KO rats.
Previous studies have observed neurodegeneration as early as 2 months post PFF injection in WT rats (Paumier et al., 2015). Because PINK1 deficiency is causally linked to early-onset PD, we hypothesized that PINK1 KO rats would show neurodegeneration at an earlier time point post PFF injection compared to WT rats. Therefore, we used unbiased stereology to determine whether PINK1 KO rats were more susceptible to PFF-induced neurodegeneration at 4 weeks post injection. Unlike WT rats, PINK1 KO rats showed a significant decrease in TH+ neurons in the ipsilateral SNpc, as a percentage of TH+ neurons in the non-injected hemisphere (Fig 5 A–B; WT rats (n= 14 WT, 16 KO) *p = 0.0464, unpaired t-test with Welch’s correction, t(19.71)= 1.125). This indicates that PINK1 KO rats are more susceptible to nigral cell loss induced by fibrillar α-synuclein. Analysis of the density of TH+ fibers in the striatum showed no difference between PINK1 KO and WT rats (Fig 5 C–D). This suggests that there is no net loss of dopaminergic innervation of the striatum.
Figure 5. PFF induced neurodegeneration in PINK1 KO rats.
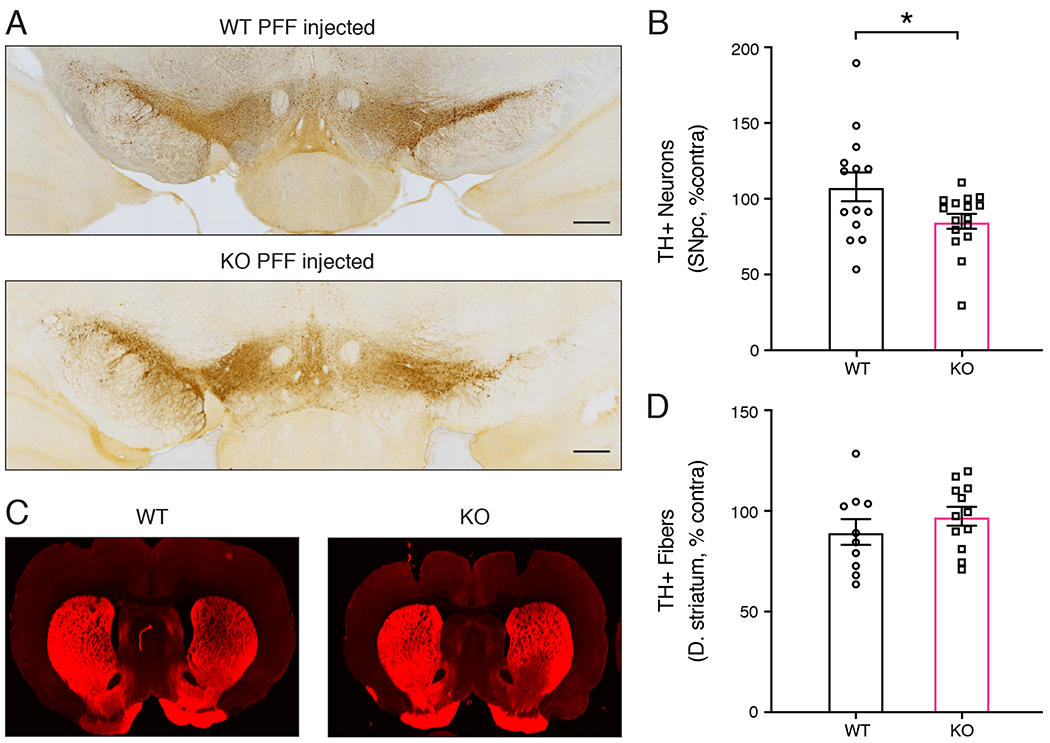
(A) Dopaminergic neurons analyzed by tyrosine hydroxylase (TH) immunohistochemistry of coronal brain sections WT and PINK1 KO rats 4 weeks post PFF injection. The region of the substantia nigra is shown. The injected hemisphere is on the right. Scale bars are 500 microns. (B) Quantification of TH+ neurons in the SNpc of WT and PINK1 KO rats by unbiased stereology. PFF injection caused a significant loss of dopaminergic neurons in PINK1 KO rats compared to WT rats (n= 14 WT, 16 KO) *p = 0.0464, unpaired t-test with Welch’s correction, t(19.71)= 1.125. (C) TH immunofluorescence of the dorsal striatum of WT and PINK1 KO rats 4 weeks post PFF injection. Data were normal according to the Kolmogorov-Smirnov normality test and the Shapiro Wilks test. (D) Quantification of TH immunofluorescence intensity in the dorsal striatum shows no differences between WT and PINK1 KO rats 4 weeks post PFF injection.
Decreased phosphorylation of TBK1 in PFF-injected PINK1 KO rats.
Tank Binding Protein 1 (TBK1) is important for the degradation of misfolded protein aggregates (Korac et al., 2013). Furthermore, PINK1 has been shown to be required for TBKl phosphorylation and activation of its downstream effects (Lazarou et al., 2015). The increased PFF-induced α-synuclein pathology in PINK1 KO rats prompted us to examine whether the TBKl-mediated clearance pathway could be altered in PINK1 KO rat brains. Western analysis of the sarkosyl-soluble fraction showed a significant decrease (*p = 0.0447, unpaired t-test with Welch’s correction, t(8)= 1.932) in the ratio of phosphorylated TBK1 (pTBKl) to total TBK1 in brain homogenates from PFF-injected PINK1 KO rats compared to PFF-injected WT controls (Figure 6A–C). We also examined whether global autophagy was affected in PINK1 KO rats. Western analysis the levels of P62, a widely-used marker of autophagic flux, indicated that global autophagy remained unchanged in PINK1 KO rats compared to WT littermate controls (Figure 6D).
Figure 6. Reduced phospho-TBK1 (pTBK1) in PFF-injected PINK1 KO rats.
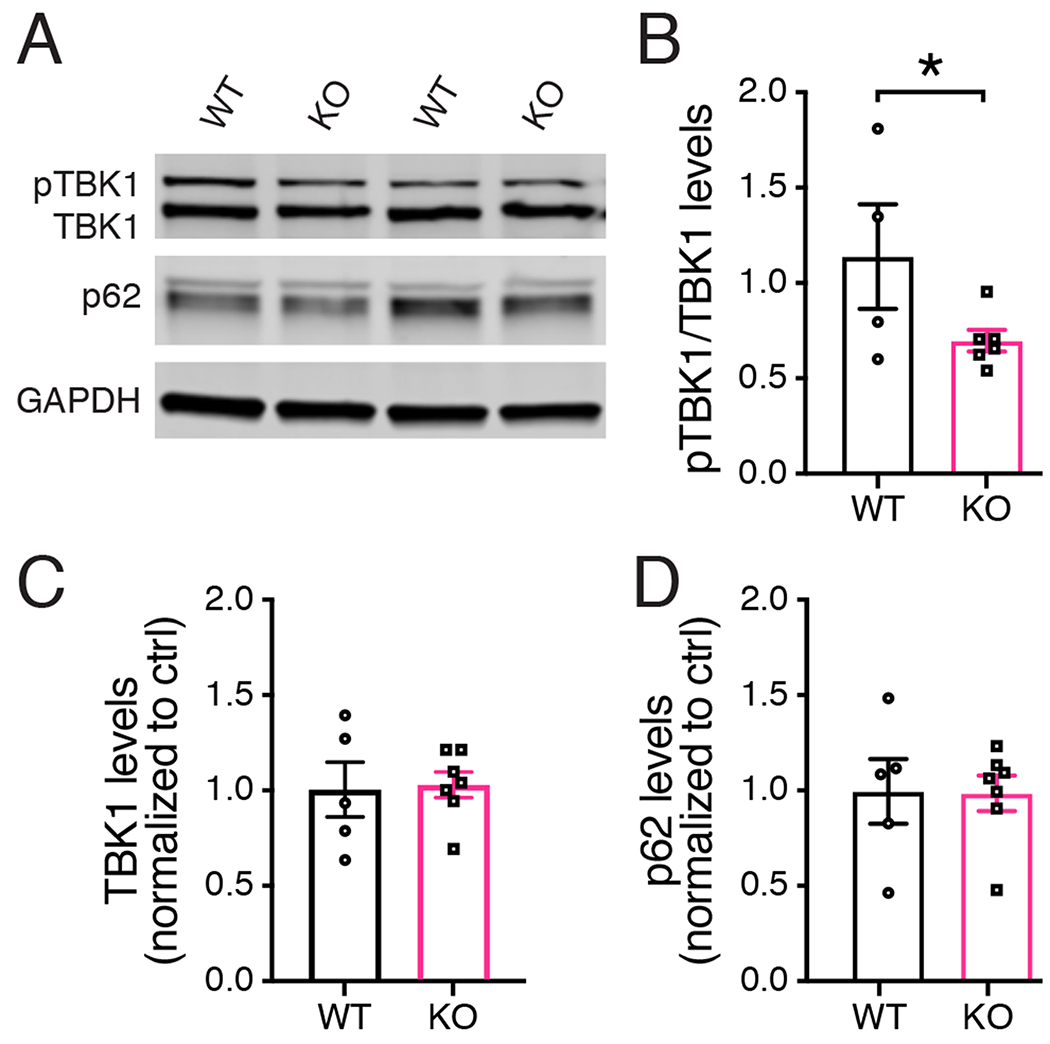
(A) Western analysis of sarkosyl-soluble whole brain homogenates 4-weeks post PFF injection using antibodies specific for pTBKl, total TBK1, the autophagy marker p62 and GAPDFI as loading control. (B) The ratio of pTBKl compared to total TBK1 is significantly lower in PINK1 KO rats compared to WT controls (n = 4 WT, 6 KO) *p = 0.0447, unpaired t-test with Welch’s correction, t(8)= 1.932. Data were normal according to the Shapiro Wilks test. (C) There is no significant difference in the level of total TBK1 in WT and PINK1 KO rats (D). There is no significant difference in the level of p62 in WT and PINK1 KO rats. Bars represent mean +/− SEM.
Discussion
Our results include several important findings in the context of what is currently known about the roles of PINK1 and α-synuclein in PD pathogenesis. Despite the identification of mutations in several genes that cause inherited forms of PD with high penetrance, including those encoding PINK1 and α-synuclein, it has been surprisingly challenging to obtain animal models that recapitulate all the key features of PD. The recent development of the α-synuclein PFF animal model of PD has been a major advance because it reproduces both the conversion of endogenous α-synuclein into pathological pS129 α-synuclein aggregates resembling Lewy bodies and Lewy neurites as well as the subsequent loss of nigral dopaminergic neurons (Luk et al., 2012;Paumier et al., 2015). WT rats have generally not shown loss of nigral neurons prior to 2-3 months post intracranial injection of α-synuclein PFFs (Paumier et al., 2015;Duffy et al., 2018;Patterson et al., 2019). Our finding that PINK1 KO rats, but not WT rats, have significant loss of nigral dopaminergic neurons only one month post striatal PFF injection indicates that PINK1-deficiency renders nigral neurons more susceptible to α-synuclein-induced degeneration. Although statistically significant, the magnitude of nigral cell loss (~15%) in PINK1 KO rats 4 weeks post PFF injection is modest (Figure 5B). This did not coincide with a measurable loss of striatal TH immunofluorescence (Figure 5C–D), suggesting that the striatum remained fully innervated by dopaminergic terminals. This could be due to increased TH expression or collateral sprouting of axon terminals from remaining dopaminergic neurons to compensate for the nigral cell loss. There is evidence that similar compensatory mechanisms occur at the early stages of PD and we observed a similar loss of nigral neurons without loss of striatal TH intensity in our previous studies of Parkin KO mice (Frank-Cannon et al., 2008;Kordower et al., 2013). Such modest neurodegeneration 4 weeks post PFF-injection would not be expected to significantly alter the behavior of PINK1 KO rats, although it has previously been reported that PINK1 KO rats begin to show locomotor behavior deficits at age 4 months and spontaneous degeneration at age 6-9 months even without any injections (Dave et al., 2014;Villeneuve et al., 2016). Notably, PD patients lose the majority of both nigral cells and projections to the striatum prior to the manifestation of motor symptoms (Cheng et al., 2010;Kordower et al., 2013).
Consistent with our results in PINK1 KO rats, previous studies using PINK1 KO mice showed greater nigral cell loss and significantly higher levels of phosphorylated α-synuclein neuropathology compared to WT mice after viral overexpression of α-synuclein (Oliveras-Salva et al., 2014). Moreover, A53T mutant α-synuclein transgenic mice have earlier onset of locomotor deficits and greater neuropathology when crossed with PINK1 KO mice (Gispert et al., 2015).
The mechanism by which lack of PINK1 promotes α-synuclein aggregation remains uncertain. Many studies point to increased production of mitochondrial reactive oxygen species as a potential mechanism by which loss of function of PINK1 leads to α-synuclein pathology (Blesa et al., 2015;Puspita et al., 2017). Indeed, studies from iPSC-derived midbrain dopamine neurons from PINK1 patients showed mitochondrial dysfunction and α-synuclein accumulation (Chung et al., 2016). Alternatively, PINK1 has recently been shown to interact with α-synuclein in the cytosol and to promote the removal of excess α-synuclein by activating autophagy (Liu et al., 2017). Moreover, PINK1 prevented α-synuclein association with mitochondria, an association which has been shown to promote mitochondrial dysfunction in PD (Di Maio et al., 2016).
It is also possible that PINK1 deficiency leads to increased steady state intracellular levels of α-synuclein, which would be predicted to increase the rate of aggregation and fibril formation, consistent with the dose-dependent severity of early onset PD linked to duplication and triplication of the otherwise nonmutant human α-synuclein gene (Singleton et al., 2003;Chartier-Harlin et al., 2004). We previously reported that, although the total levels of α-synuclein protein are unchanged in the brains of PINK1 KO rats compared to WT rats, we found a significant shift of α-synuclein from the cytosolic fraction to the synaptic vesicle-enriched fraction of PINK1 KO brain homogenates compared to WT (Creed and Goldberg, 2018a). Several studies suggest this shift towards greater concentration of membrane-associated α-synuclein could affect the propensity of α-synuclein to aggregate (Dikiy and Eliezer, 2012;Galvagnion et al., 2015;Ysselstein et al., 2015;Galvagnion et al., 2016;Samuel et al., 2016;Nuber et al., 2018).
In this study, we also found evidence that the increased fibrillar α-synuclein and the loss of TH+ neurons in PINK1 KO rats could be due to impairment of TBKl-mediated protein clearance. PINK1-deficiency has been shown to impair the phosphorylation and activation of TBK1, which mediates numerous downstream pathways, including proteophagy and innate immunity (Shu et al., 2013;Lazarou et al., 2015;Richter et al., 2016). Recently, it has been shown that upon exhaustive exercise or other mitochondrial stress, PINK1 KO mice show increased inflammation mediated by STING (stimulator of interferon genes), an activator of innate immunity (Sliter et al., 2018). Moreover, TBK1 activation leads to degradation of STING (Liu et al., 2019). This raises the possibility that the increased aggregation and neurodegeneration we observed in PINK1 KO rats injected with PFFs could be due not only to impairments in TBKl-mediated protein clearance but also due to increased STING-mediated inflammation triggered by fibrillar α-synuclein. These possibilities are not mutually exclusive. Additional studies are needed to further establish the extent to which these mechanisms contribute to the increased pS129 α-synuclein and loss of TH+ neurons in PINK1 KO rats upon intrastriatal injection of α-synuclein PFFs. Moreover, further studies are warranted to investigate functional deficiencies in PINK1 or the activity of proteins downstream from PINK1 kinase activity in idiopathic PD because these could serve as potential therapeutic targets.
Together, our data show that PINK1 deficiency, which is causally linked to early-onset PD, leads to greater PFF-induced α-synuclein aggregation and nigral dopaminergic neurodegeneration in rats. These results further establish the utility of both the PINK1 KO rat model of PD and the intracranial PFF animal model of PD for studies of the role of α-synuclein in PD-related neurodegeneration. Our results suggest potential mechanisms by which PINK1 may promote the degradation of proteinaceous α-synuclein inclusions and prevent PFF-induced neurodegeneration.
Highlights:
α-Synuclein aggregates appear as early as two weeks after injection of α-synuclein pre-formed fibrils into rat striatum
PD-linked PINK1-deficiency causes greater pS129 α-synuclein pathology in rats injected with α-synuclein pre-formed fibrils
PINK1 KO rats are more susceptible to nigral cell loss induced by intracranial injection of α-synuclein pre-formed fibrils
Acknowledgements
The authors thank Andrew West, Laura Volpicelli-Daley, and Valentina Krendelchtchikova for providing recombinant purified α-synuclein and fibrils as well as guidance and advice.
Funding
This research was supported by the National Institute of Neurological Disorders and Stroke under NIH award numbers R01NS082565 to MSG and F99NS108458 to RBC, and by grants from the Michael J. Fox Foundation for Parkinson’s Research. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no competing financial or non-financial interests
References
- Abdelmotilib H, Maltbie T, Delic V, Liu Z, Hu X, Fraser KB, Moehle MS, Stoyka L, Anabtawi N, Krendelchtchikova V, Volpicelli-Daley LA, and West A (2017). alpha-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neuroblol Dis 105, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barodia SK, Creed RB, and Goldberg MS (2017). Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res Bull 133, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer TA, Jakala P, Hartmann T, Havas L, Mclean C, Culvenor JG, Li QX, Masters CL, Falkai P, and Beyreuther K (1999). Alpha-synuclein accumulates in Lewy bodies in Parkinson’s disease and dementia with Lewy bodies but not in Alzheimer’s disease beta-amyloid plaque cores. Neurosci Lett 266, 213–216. [DOI] [PubMed] [Google Scholar]
- Blesa J, Trigo-Damas I, Quiroga-Varela A, and Jackson-Lewis VR (2015). Oxidative stress and Parkinson’s disease. Front Neuroanat 9, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, Van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, Van Dongen JW, Vanacore N, Van Swieten JC, Brice A, Meco G, Van Duijn CM, Oostra BA, and Heutink P (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. [DOI] [PubMed] [Google Scholar]
- Braak H, Sandmann-Keil D, Gai W, and Braak E (1999). Extensive axonal Lewy neurites in Parkinson’s disease: a novel pathological feature revealed by alpha-synuclein immunocytochemistry. Neurosci Lett 265, 67–69. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, and Destee A (2004). Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, and Burke RE (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Ho HA, Perez-Acuna D, and Lee SJ (2019). Modeling alpha-Synuclein Propagation with Preformed Fibril Injections. J Mov Disord 12, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SY, Kishinevsky S, Mazzulli JR, Graziotto J, Mrejeru A, Mosharov EV, Puspita L, Valiulahi P, Sulzer D, Milner TA, Taldone T, Krainc D, Studer L, and Shim JW (2016). Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and alpha-Synuclein Accumulation. Stem Cell Reports 7, 664–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed RB, and Goldberg MS (2018a). Analysis of alpha-Synuclein Pathology in PINK1 Knockout Rat Brains. Front Neurosci 12, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed RB, and Goldberg MS (2018b). New Developments in Genetic rat models of Parkinson’s Disease. Mov Disord 33, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JP, Abdelmotilib HA, Hu X, Volpicelli-Daley LA, Moehle MS, Fraser KB, Needle E, Chen Y, Steyn SJ, Galatsis P, Hirst WD, and West AB (2015). Leucine-rich Repeat Kinase 2 (LRRK2) Pharmacological Inhibition Abates alpha-Synuclein Gene-induced Neurodegeneration. J Biol Chem 290, 19433–19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave KD, De Silva S, Sheth NP, Ramboz S, Beck MJ, Quang C, Switzer RC 3rd, Ahmad SO, Sunkin SM, Walker D, Cui X, Fisher DA, Mccoy AM, Gamber K, Ding X, Goldberg MS, Benkovic SA, Haupt M, Baptista MA, Fiske BK, Sherer TB, and Frasier MA (2014). Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol Dis 70, 190–203. [DOI] [PubMed] [Google Scholar]
- Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, Mccoy J, Chu CT, Burton EA, Hastings TG, and Greenamyre JT (2016). alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med 8, 342ra378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikiy I, and Eliezer D (2012). Folding and misfolding of alpha-synuclein on membranes. Biochim Biophys Acta 1818, 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy MF, Collier TJ, Patterson JR, Kemp CJ, Luk KC, Tansey MG, Paumier KL, Kanaan NM, Fischer DL, Polinski NK, Barth OL, Howe JW, Vaikath NN, Majbour NK, El-Agnaf OMA, and Sortwell CE (2018). Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflammation 15, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Trevino I, O’brien DE, Casey B, Goldberg MS, and Tansey MG (2008). Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci 28, 10825–10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froula JM, Castellana-Cruz M, Anabtawi NM, Camino JD, Chen SW, Thrasher DR, Freire J, Yazdi AA, Fleming S, Dobson CM, Kumita JR, Cremades N, and Volpicelli-Daley LA (2019). Defining alpha-synuclein species responsible for Parkinson’s disease phenotypes in mice. J Biol Chem 294, 10392–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, and Iwatsubo T (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4, 160–164. [DOI] [PubMed] [Google Scholar]
- Galvagnion C, Brown JW, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, Sparr E, and Dobson CM (2016). Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein. Proc Natl Acad Sci U S A 113, 7065–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvagnion C, Buell AK, Meisl G, Michaels TC, Vendruscolo M, Knowles TP, and Dobson CM (2015). Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol 11, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Muqit MM, Stanyer L, Healy DG, Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees AJ, Latchman DS, Holton JL, Wood NW, and Revesz T (2006). PINK1 protein in normal human brain and Parkinson’s disease. Brain 129, 1720–1731. [DOI] [PubMed] [Google Scholar]
- Gispert S, Brehm N, Weil J, Seidel K, Rub U, Kern B, Walter M, Roeper J, and Auburger G (2015). Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum Mol Genet 24, 1061–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, and Lansbury PT Jr. (2000). Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson’s disease? Nat Cell Biol 2, E115–119. [DOI] [PubMed] [Google Scholar]
- Grant LM, Kelm-Nelson CA, Hilby BL, Blue KV, Paul Rajamanickam ES, Pultorak JD, Fleming SM, and Ciucci MR (2015). Evidence for early and progressive ultrasonic vocalization and oromotor deficits in a PINK1 gene knockout rat model of Parkinson’s disease. J Neurosci Res 93, 1713–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PH, Nielsen MS, Jakes R, Dotti CG, and Goedert M (1998). Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem 273, 26292–26294. [DOI] [PubMed] [Google Scholar]
- Jesko H, Lenkiewicz AM, Wilkaniec A, and Adamczyk A (2019). The interplay between parkin and alpha-synuclein; possible implications for the pathogenesis of Parkinson’s disease. Acta Neurobiol Exp (Wars) 79, 276–289. [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, Van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, and Haass C (2000). Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J Neurosci 20, 6365–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kelly V, Johnson C, Baillie C, Hastie CJ, Peggie M, Macartney T, Woodroof HI, Alessi DR, Pedrioli PG, and Muqit MM (2014a). Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol 4, 130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, and Muqit MM (2014b). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, and Shimizu N (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. [DOI] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, and Muqit MM (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2, 120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, and Dikic I (2013). Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126, 580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, and Bartus RT (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136, 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, and Matsuda N (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, Sehgal SA, Wang X, Wang J, Shang Y, Jiang Z, Liu L, and Chen Q (2019). STING directly activates autophagy to tune the innate immune response. Cell Death Differ 26, 1735–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang X, Lu Y, Duan C, Gao G, Lu L, and Yang H (2017). Pink1 interacts with alpha-synuclein and abrogates alpha-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis 8, e3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’brien P, Trojanowski JQ, and Lee VM (2012). Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, Ducrot C, Laplante A, Bourque MJ, Zhu L, Cayrol R, Le Campion A, Mcbride HM, Gruenheid S, Trudeau LE, and Desjardins M (2019). Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(−/−) mice. Nature 571, 565–569. [DOI] [PubMed] [Google Scholar]
- Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau LE, Burelle Y, Gagnon E, Mcbride HM, and Desjardins M (2016). Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell . [DOI] [PubMed] [Google Scholar]
- Michotte A (2003). Recent developments in the neuropathological diagnosis of Parkinson’s disease and parkinsonism. Acta Neurol Belg 103, 155–158. [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, and Youle RJ (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Rajsombath M, Minakaki G, Winkler J, Muller CP, Ericsson M, Caldarone B, Dettmer U, and Selkoe DJ (2018). Abrogating Native alpha-Synuclein Tetramers in Mice Causes a L-DOPA-Responsive Motor Syndrome Closely Resembling Parkinson’s Disease. Neuron 100, 75–90 e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveras-Salva M, Macchi F, Coessens V, Deleersnijder A, Gerard M, Van Der Perren A, Van Den Haute C, and Baekelandt V (2014). Alpha-synuclein-induced neurodegeneration is exacerbated in PINK1 knockout mice. Neurobiol Aging 35, 2625–2636. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, Van Der Brug M, Lopez De Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, De Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, and Singleton AB (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Patterson JR, Duffy MF, Kemp CJ, Howe JW, Collier TJ, Stoll AC, Miller KM, Patel P, Levine N, Moore DJ, Luk KC, Fleming SM, Kanaan NM, Paumier KL, El-Agnaf OMA, and Sortwell CE (2019). Time course and magnitude of alpha-synuclein inclusion formation and nigrostriatal degeneration in the rat model of synucleinopathy triggered by intrastriatal alpha-synuclein preformed fibrils. Neurobiol Dis 130, 104525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM, and Sortwell CE (2015). Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82, 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, and Nussbaum RL (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Puspita L, Chung SY, and Shim JW (2017). Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain 10, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades E, Ramlall TF, Webb WW, and Eliezer D (2006). Quantification of {alpha}-Synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, and Dikic I (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 113, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J, and Pastor P (2010). PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133, 1128–1142. [DOI] [PubMed] [Google Scholar]
- Samuel F, Flavin WP, Iqbal S, Pacelli C, Sri Renganathan SD, Trudeau LE, Campbell EM, Fraser PE, and Tandon A (2016). Effects of Serine 129 Phosphorylation on alpha-Synuclein Aggregation, Membrane Association, and Internalization. J Biol Chem 291, 4374–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu C, Sankaran B, Chaton CT, Herr AB, Mishra A, Peng J, and Li P (2013). Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure 21, 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, and Gwinn-Hardy K (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C, and Youle RJ (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, and Goedert M (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, and Goedert M (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Tsuboi Y, Ahlskog JE, Apaydin H, Parisi JE, and Dickson DW (2001). Lewy bodies are not increased in progressive supranuclear palsy compared with normal controls. Neurology 57, 1675–1678. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, and Wood NW (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Villeneuve LM, Purnell PR, Boska MD, and Fox HS (2016). Early Expression of Parkinson’s Disease-Related Mitochondrial Abnormalities in PINK1 Knockout Rats. Mol Neurobiol 53, 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, and Lee VM (2011). Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkaniec A, Lenkiewicz AM, Czapski GA, Jesko HM, Hilgier W, Brodzik R, Gassowska-Dobrowolska M, Culmsee C, and Adamczyk A (2019). Extracellular Alpha-Synuclein Oligomers Induce Parkin S-Nitrosylation: Relevance to Sporadic Parkinson’s Disease Etiopathology. Mol Neurobiol 56, 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Wang X, Liu J, Duan C, Gao G, Lu L, Yu S, and Yang H (2018). PINK1 suppresses alpha-synuclein-induced neuronal injury: a novel mechanism in protein phosphatase 2A activation. Oncotarget 9, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ysselstein D, Joshi M, Mishra V, Griggs AM, Asiago JM, Mccabe GP, Stanciu LA, Post CB, and Rochet JC (2015). Effects of impaired membrane interactions on alpha-synuclein aggregation and neurotoxicity. Neurobiol DIs 79, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Caine DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, and Gasser T (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]