

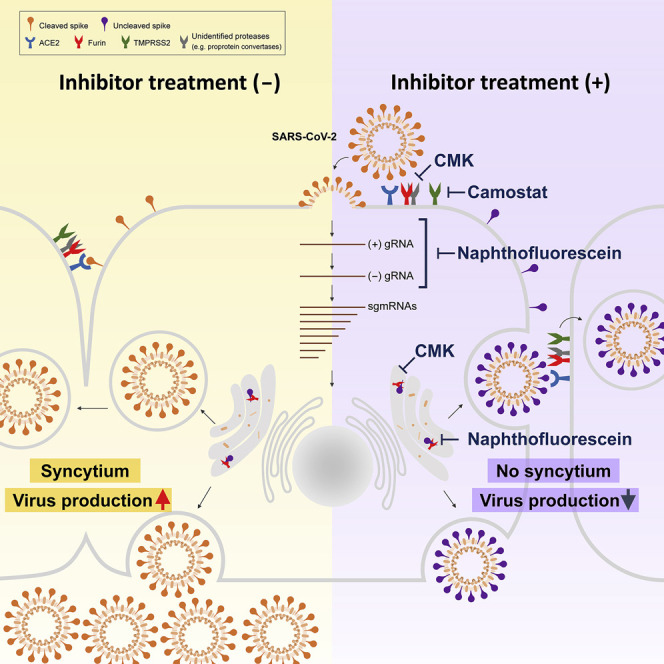
Abstract
Development of specific antiviral agents is an urgent unmet need for SARS-coronavirus 2 (SARS-CoV-2) infection. This study focuses on host proteases that proteolytically activate the SARS-CoV-2 spike protein, critical for its fusion after binding to angiotensin-converting enzyme 2 (ACE2), as antiviral targets. We first validate cleavage at a putative furin substrate motif at SARS-CoV-2 spikes by expressing it in VeroE6 cells and find prominent syncytium formation. Cleavage and the syncytium are abolished by treatment with the furin inhibitors decanoyl-RVKR-chloromethylketone (CMK) and naphthofluorescein, but not by the transmembrane protease serine 2 (TMPRSS2) inhibitor camostat. CMK and naphthofluorescein show antiviral effects on SARS-CoV-2-infected cells by decreasing virus production and cytopathic effects. Further analysis reveals that, similar to camostat, CMK blocks virus entry, but it further suppresses cleavage of spikes and the syncytium. Naphthofluorescein acts primarily by suppressing viral RNA transcription. Therefore, furin inhibitors may be promising antiviral agents for prevention and treatment of SARS-CoV-2 infection.
Keywords: SARS-CoV-2, spike, furin, syncytium, cytopathic effect
Graphical Abstract
Highlights
-
•
The furin cleavage site in the SARS-CoV-2 spike protein mediates syncytium formation
-
•
The SARS-CoV-2 spike-mediated syncytium is suppressed by specific furin inhibitors
-
•
Furin inhibitors block SARS-CoV-2 virus entry and virus replication
-
•
Furin inhibitors are potential antiviral agents for SARS-CoV-2 infection and pathogenesis
Development of effective antiviral agents is an urgent unmet need for SARS-CoV-2 infection. Cheng et al. find that cleavage of the furin substrate site in the viral spike protein is critical for virus production and cytopathic effects. Two inhibitors targeting furin are potential antiviral agents to control SARS-CoV-2 infection and pathogenesis.
Introduction
SARS-coronavirus 2 (SARS-CoV-2) has caused more than 24,000,000 infections and 800,000 deaths after spreading into 184 countries (WHO, 2020). The pandemic is very difficult to contain at present, and SARS-CoV-2 will very likely become a CoV with sustained ability to infect humans, similar to predecessor CoVs causing the common cold, such as NL63, OC43, and HKU-1 (Gaunt et al., 2010; Zeng et al., 2018).
The medical demand for SARS-CoV-2 control is similar to that for seasonal influenza control. In addition to developing vaccines to actively protect naive people, specific antiviral prophylactic or therapeutic agents are needed for people who are already infected, especially those in the high-risk group for serious illness. Unfortunately, no drug or vaccine has so far been approved to treat human CoVs. Currently, active trials repurpose approved or in-development (IND) drugs, including remdesivir, favipiravir, hydroxychloroquine (HCQ) and azithromycin, lopinavir-ritonavir, and convalescent plasma, for treatment of coronavirus disease 2019 (COVID-19) patients (Sanders et al., 2020). Preliminary results from limited cases are promising, and more rigorous randomized clinical studies are warranted. Among the tested drugs, remdesivir was granted emergency use authorization (EUA) by the US Food and Drug Administration (FDA) for patients hospitalized with severe COVID-19, which, however, appeared to be only mildly beneficial for disease recovery in randomized clinical trials (Goldman et al., 2020; Wang et al., 2020b). Chloroquine (CQ) and HCQ were also granted EUA by the FDA for hospitalized COVID-19 patients, but this was revoked by the FDA because of ineffectiveness and potentially serious side effects (Li et al., 2020). Moreover, these regimens may not be specific enough for SARS-CoV-2 because the mechanisms of action of this virus are neither clear nor confirmed.
The molecules targeted by specific antiviral agents should be essential for the viral life cycle or immune clearance. Such molecules can be divided into two categories: viral molecules and host molecules required for completion of virus replication and infection (Tu et al., 2020). Among the numerous structural and nonstructural proteins encoded by SARS-CoV-2, viral enzymes are attractive targets for development of antiviral agents (Li and De Clercq, 2020; Zumla et al., 2016). Approaches to repurpose existing SARS-CoV and Middle East respiratory syndrome (MERS) CoV inhibitors for SARS-CoV-2 were successful and identified molecules potently inhibiting the SARS-CoV-2 MPro protease (Ton et al., 2020; Zhang et al., 2020); numerous studies are ongoing worldwide. In addition, replication of SARS-CoV-2 requires several host molecules that have become candidate targets for drug development. Our previous study identified that the cellular kinase glycogen synthase kinase 3 beta (GSK3β) is important for viral nucleoprotein phosphorylation and nested viral RNA transcription in the mouse hepatitis virus (MHV) model (Wu et al., 2009, 2014). Imatinib also showed an antiviral effect on SARS-CoV (Coleman et al., 2016).
Currently, attention has been focused specifically on the spike (S) protein, the key determinant for virus entry. The S protein contains an N-terminal S1 domain for binding with the host angiotensin-converting enzyme 2 (ACE2) receptor and a C-terminal S2 domain for membrane fusion (Li et al., 2003; Tortorici and Veesler, 2019). Small molecules targeting ACE2 were developed to reduce viral infection, results from clinical trials are pending (Gurwitz, 2020). Although binding to ACE2 is a key step in establishing infection, proteolytic activation of the S protein by host proteases at the putative cleavage site located at the S1/S2 boundary (S1/S2 cleavage site) and within the S2 domain (S2′ cleavage site) has been documented to be critical for its fusion activity in CoVs (Belouzard et al., 2009). Several host proteases, including endosomal cathepsins, cell surface TMPRSS2 proteases, furin, and trypsin, have been identified to be responsible for S protein cleavage during virus entry or viral protein biogenesis in CoVs, depending on their distribution in cells. In addition to regulating virus entry, S protein cleavage might also regulate the host tropism and pathogenesis of CoV infection (Coutard et al., 2020; Hoffmann et al., 2020b; Park et al., 2016; Wang et al., 2020a).
The SARS-CoV-2 genomic sequences indicate that the viral S protein contains conserved putative motifs for several cellular proteases. Via a pseudovirus approach, Hoffmann et al. (2020b) reported that camostat, an inhibitor of the TMPRSS2 protease, can inhibit virus entry. In addition to TMPRSS2, which targets the S2′ cleavage site, furin has been proposed as another host protease mediating cleavage of the SARS-CoV-2 S protein at the S1/S2 cleavage site (Hoffmann et al., 2020a). The S1/S2 boundary features a polybasic stretch of an RRAR motif, matching the consensus sequence of the substrate for furin and related proprotein convertase (PC) family members (Seidah and Prat, 2012). This site was identified in SARS-CoV-2 but not in SARS-CoV or other lineages of β-CoVs, although it is preserved in some other human CoVs, including HCoV-OC43, MERS-CoV, and HKU1 (Coutard et al., 2020). A recent study showed that abolishing this site in pseudotyped SARS-CoV-2 S viral particles did not affect infectivity (Walls et al., 2020). However, the function of this furin cleavage site in viral pathogenesis, especially spreading and cytopathic effects (CPEs), needs to be addressed more thoroughly in a virus infection system.
Cleavage by furin and related PCs has been documented as a key event in activating the envelope glycoprotein for its fusion activity, critical for virus entry and pathogenesis, in numerous pathogenic virus families, including herpesviruses, CoVs, flaviviruses, togaviruses, bornaviruses, bunyaviruses, filoviruses, orthomyxoviruses, paramyxoviruses, pneumoviruses, and retroviruses (Braun and Sauter, 2019; Izaguirre, 2019). Furin/PCs thus are attractive therapeutic targets for various infectious diseases. In recent years, several peptide-based and small-molecule inhibitors targeting furin/PCs have been developed as putative antiviral agents, many of which can block maturation of viral envelope proteins and, thus, their fusion activity in various viruses (Braun and Sauter, 2019; Izaguirre, 2019). We tested four furin/PC inhibitors in SARS-CoV-2-infected VeroE6 cells. In addition to confirming cleavage at the putative furin site at the S1/S2 boundary, we evaluated the inhibitory effects on the virus replication cycle and CPEs, such as syncytium formation. The results supported an essential role of host furin/PC proteases in virus replication and pathogenesis of SARS-CoV-2 infection. Thus, furin/PC inhibitors may be specific antiviral leads warranting further development.
Results
Cleavage of the SARS-CoV-2 S Protein at a Putative Furin Substrate Motif Is Critical for S-Mediated Syncytium Formation in VeroE6 Cells
We first tried to validate the function of the furin cleavage site at R685/S686 in the RRAR↓S motif in the SARS-CoV-2 S protein. To this end, two expression plasmids for S proteins that were codon optimized for human expression—one for the wild-type (WT) protein and the other for the R682A mutant (in which the furin cleavage site was mutated from RRAR↓S to ARARS)—with a hemagglutinin (HA) tag at the C terminus were constructed (Figure 1 A). After transfection into VeroE6 cells, SARS-CoV-2 S protein cleavage was examined in cell lysates by western blotting using anti-S or anti-HA tag antibodies (Abs). The WT full-length S protein was readily processed into smaller ones, indicating efficient cleavage. However, the R682A mutant S protein remained largely intact with minimal cleavage (Figure 1B). These results indicate that the furin cleavage motif at the S1/S2 boundary is functional.
Figure 1.

Cleavage of the SARS-CoV-2 S Protein at the Putative Furin Substrate Site Is Critical for Syncytium Formation in S-Only-Expressing VeroE6 Cells
(A) Top panel: schematic illustration of the SARS-CoV-2 S protein, including the N-terminal domain (NTD), receptor binding domain (RBD), and transmembrane (TM) domain. The putative furin cleavage site at the S1/S2 boundary is indicated by a red arrowhead. Bottom panel: sequence of amino acids (aa) 676–688 in the WT and mutant S-R682A proteins. The SARS-CoV-2-specific RRAR sequence is marked in red, and the mutation site is underlined.
(B) Immunoblot analysis of lysates from WT S- and R682A mutant S-expressing VeroE6 cells. Immunoblots were probed with anti-S or anti-HA tag Abs. The full-length (FL) S protein and cleaved S fragments are marked as indicated; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was included as the loading control.
(C and D) Microscopic observation and (D) immunofluorescence staining with 4′,6-diamidino-2-phenylindole (DAPI) (blue) or an anti-S Ab (green) of VeroE6 cells transfected with vector (control), WT S, or mutant S-R682A plasmids 24 h after transfection. The syncytial phenotype is indicated by the multinucleated giant cells. Scale bars, 100 μm (C) and 200 μm (D).
Furthermore, expression of the WT S protein alone induced an extensive syncytial phenotype, with fused cells containing multiple nuclei visible under light and fluorescence microscopy (Figures 1C and 1D). However, this syncytial phenotype did not occur in cells expressing the R682A mutant S protein (Figures 1C and 1D) despite similar protein expression levels. Therefore, cleavage of the SARS-CoV-2 S protein at the S1/S2 furin substrate site occurs and possibly contributes to syncytium formation.
Furin/PC Inhibitors, but Not the TMPRSS2 Inhibitor, Suppress SARS-CoV-2 S Protein Cleavage and Development of the Syncytial Phenotype in VeroE6 Cells
To further explore this possibility, we evaluated the effects of several furin/PC inhibitors on SARS-CoV-2 S protein cleavage and syncytium formation. Four potent furin/PC inhibitors—two peptide inhibitors (decanoyl-RVKR-chloromethylketone [CMK] and hexa-D-arginine amide [D6R]) and two small-molecule inhibitors (SSM 3 trifluoroacetate [SSM3] and naphthofluorescein)—were evaluated (Coppola et al., 2008; Henrich et al., 2003; Remacle et al., 2010; Sarac et al., 2002). Camostat, an inhibitor targeting TMPRSS2, another S cleavage protease, was used as a control. VeroE6 cells expressing the WT S protein were treated with the inhibitors for 24 h at an effective dose, as suggested in previous studies (Coppola et al., 2008; Croissandeau et al., 2002; Hoffmann et al., 2020b; Sarac et al., 2002).
After treatment with CMK or naphthofluorescein, immunoblot analysis of cell lysates showed a dramatic decrease in the levels of processed S protein fragments in VeroE6 cells. The full-length but not the cleaved S protein was detected after treatment with either of these two inhibitors. In contrast, no suppression of S protein cleavage was detected in cells treated with the other inhibitors (Figure 2 A). Notably, cleavage of cellular integrin αV, a furin substrate, into heavy- and light-chain fragments was decreased in cells treated with CMK and naphthofluorescein but not the other inhibitors (Figure 2A), suggesting that CMK and naphthofluorescein, but not the other inhibitors, suppressed furin/PC activity in VeroE6 cells.
Figure 2.

The Furin/PC Inhibitors, but Not the TMPRSS2 Inhibitor, Suppress SARS-CoV-2 S Protein Cleavage and Development of the Syncytial Phenotype in S-Only-Expressing VeroE6 Cells
(A) Immunoblot of lysates from VeroE6 cells transfected with the SARS-CoV-2 S (WT) expression construct and treated with different inhibitors as indicated (the furin/PC inhibitors CMK [50 μM], D6R [50 μM], SSM3 [25 μM], and naphthofluorescein [20 μM] and the TMPRSS2 inhibitor camostat [500 μM]) and harvested 24 h after treatment. The immunoblot was probed with anti-S and anti-HA Abs, and the FL and cleaved S proteins are marked as indicated. The furin substrate in VeroE6 cells, integrin αV, was included as an indicator of the inhibitory efficacy of the furin inhibitors, with pro-integrin αV and the cleaved integrin αV light-chain fragment as indicated; GAPDH was included as the loading control.
(B and C) Microscopic observation and (C) immunofluorescence staining with DAPI (blue) or an anti-S Ab (green) of VeroE6 cells transfected with plasmids and subjected to inhibitor treatment as described in (A). Cells were harvested 24 h after treatment with the indicated specific inhibitors. The syncytial phenotype is indicated by the multinucleated giant cells. Scale bars, 100 μm (B) and 200 μm (C).
Consistent with these observations, microscopic observation revealed significant inhibition of syncytium formation by CMK and naphthofluorescein but not by the other inhibitors (Figures 2B and 2C). Thus, SARS-CoV-2 S protein cleavage in VeroE6 cells and syncytium formation were confirmed to be dependent on active furin/PC function in host cells.
The Furin/PC Inhibitors CMK and Naphthofluorescein Suppress Virus Production and CPEs in SARS-CoV-2-Infected VeroE6 Cells
Next we evaluated the possible antiviral effects of these furin/PC inhibitors in the virus infection system. VeroE6 cells were pretreated with the inhibitors at the doses indicated in Figure 2 for 1 h before viral infection (MOI = 1), and the inhibitors were maintained in the medium until 24 h after infection.
Microscopic observation showed that prominent CPEs were induced by SARS-CoV-2 infection and that these CPEs were suppressed by treatment with CMK or naphthofluorescein but not by treatment with D6R or SSM3 (Figure 3 A). These results were similar to those obtained in S-expressing VeroE6 cells. The levels of not only the cleaved S1/S2 subunits but also of the full-length S protein and the nucleocapsid (N) protein in cell lysates were diminished by treatment with CMK or naphthofluorescein (Figure 3B). Moreover, the virus titers of progeny viruses harvested from the supernatant were decreased significantly by CMK and naphthofluorescein (Figure 3C). The inhibitory effect of these two inhibitors was validated further by a multi-step virus growth cycle setup, in which VeroE6 cells were pretreated with the inhibitors for 1 h before viral infection (MOI = 0.01) and harvested 24 and 48 h after infection for analysis. The virus titer and viral proteins were decreased significantly 24 and 48 h after infection by CMK and naphthofluorescein (Figure S1). Therefore, these data suggest that CMK and naphthofluorescein effectively inhibit CPEs and virus production in SARS-CoV-2-infected VeroE6 cells. We found that treatment with camostat, the control inhibitor, also inhibited CPEs and virus production in association with decreased viral proteins in VeroE6 cells in this SARS-CoV-2 infection system (Figures 3A–3C).
Figure 3.

The Furin/PC Inhibitors and the TMPRSS2 Inhibitor Block CPE and Virus Production in SARS-CoV-2-Infected VeroE6 Cells
(A) Microscopic observation of CPEs in VeroE6 cells infected with SARS-CoV-2 (MOI = 1) in the absence or presence of the specific furin/PC inhibitors (CMK, 50 μM; D6R, 50 μM; SSM3, 25 μM; naphthofluorescein, 20 μM) or the TMPRSS2 inhibitor (camostat, 500 μM), which were applied 1 h before infection and maintained in the medium until cell assessment 24 h after treatment. Scale bars, 100 μm.
(B) Immunoblot of lysates from VeroE6 cells infected with SARS-CoV-2 (MOI = 1) that were treated with different inhibitors as described in (A). The immunoblot was probed with anti-S and anti-N Abs. FL S proteins, cleaved S proteins, and nucleocapsid (N) proteins are marked as indicated. GAPDH was included as the loading control.
(C) A plaque assay was performed to determine the plaque-forming units (PFUs) of SARS-CoV-2 virus in the supernatant of VeroE6 cells infected and subjected to inhibitor treatment as described in (A). A representative photo of plaque assay is shown in the left panel. A bar graph and statistic results of the triplicate plaque assay results are shown in the right panel (mean ± SD, ∗∗p < 0.01, ∗∗∗p < 0.001).
See also Figures S1 and S2.
In addition to VeroE6 cells, the similar antiviral effects of these five inhibitors of SARS-CoV-2 infection were validated in MK2 cells, which are also susceptible to SARS-CoV-2 infection (Figure S2).
CMK, Naphthofluorescein, and Camostat Dose-Dependently Decrease Virus Production and CPEs in Association with Reduced Viral RNA and Protein Levels
Then we conducted a dose-response experiment with CMK, naphthofluorescein, and camostat to assess suppression of CPEs and virus production. The results confirmed the suppressive effects of the inhibitors on CPEs and virus production (by plaque assay) and showed that the effective dose for CMK was as low as 5 μM (Figures 4A and 4B). Naphthofluorescein was effective at 10–20 μM (Figures 4E and 4F), and the effective dose for camostat was 100 μM (Figures 4I and 4J). To further investigate the efficacy and cytotoxicity of these three inhibitors, we checked their selectivity index (SI) individually. The 50% cytotoxic concentration (CC50) values evaluated by CCK-8 assay were 318.2 μM for CMK, 57.44 μM for naphthofluorescein, and more than 2,000 μM for camostat. The half-maximal inhibitory concentration (IC50) values determined by plaque reduction assay were 0.057 μM for CMK, 9.025 μM for naphthofluorescein, and 0.025 μM for camostat. The SI was calculated to be 5,567 for CMK, 6.36 for naphthofluorescein, and more than 81,004 for camostat (Figures 4C, 4G, and 4K).
Figure 4.

CMK, Naphthofluorescein, and Camostat Treatment Dose-Dependently Decreased Virus Production and CPEs in Association with Reduced Viral RNA and Protein Levels in SARS-CoV-2-Infected VeroE6 Cells
(A, E, and I) Microscopic observation of CPEs in VeroE6 cells infected with SARS-CoV-2 (MOI = 1) in the absence or presence of serial doses of (A) CMK, (E) naphthofluorescein, and (I) camostat, which were applied 1 h before infection and maintained in the medium until cell assessment 24 h after treatment. Scale bars, 100 μm.
(B, F, and J) The virus titers (PFUs per milliliter) in the supernatant of SARS-CoV-2-infected VeroE6 cells treated with serial doses of (B) CMK, (F) naphthofluorescein, and (J) camostat were determined by plaque assay (mean ± SD of triplicates, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(C, G, and K) The selectivity index (SI) of (C) CMK, (G) naphthofluorescein, and (K) camostat was determined by CC50/IC50. The left y axis indicates the inhibition of virus titer (percent) relative to that of the untreated control group. The right y axis indicates the cell viability (percent) relative to that of the untreated control group. The x axis indicates the concentration of inhibitors. The CC50, IC50, and SI values for each inhibitor are shown above the figures.
(D, H, and L) Viral protein and viral RNA in SARS-CoV-2-infected VeroE6 cells treated with serial doses of (D) CMK, (H) naphthofluorescein, or (L) camostat were detected by immunoblot (top panels) and northern blot (bottom panels) analyses. Viral RNA isolated from VeroE6 cells was quantified by qRT-PCR targeting the E gene, as indicated below the northern blot results.
Immunoblot analysis revealed a parallel decrease in the levels of viral S and N proteins in VeroE6 cells treated with different doses of the inhibitors (Figures 4D, 4H, and 4L, top panels). Our study identified a difference between S-only-expressing cells and the SARS-CoV-2 virus infection system regarding the significant reduction in S protein levels (not only the cleaved but also the full-length form) in virus-infected VeroE6 cells treated with CMK, naphthofluorescein, and camostat. To study the mechanisms of these inhibitors in addition to S cleavage, we used northern blotting to further examine viral RNA species in infected VeroE6 cells after treatment. The results showed that the levels of all viral RNA species were reduced significantly by treatment with these three inhibitors: CMK at doses of 5 μM or more, naphthofluorescein at doses of more than 10 μM, and camostat at doses of 100 μM or more (Figures 4D, 4H, and 4L; bottom panels). These results were validated by qRT-PCR with an amplicon at the E gene (Figures 4D, 4H, and 4L, at the bottom of the northern blot results). The concordant decrease in viral RNA and viral protein levels implies that CMK, naphthofluorescein, and camostat may suppress the viral life cycle at a stage upstream of viral RNA transcription.
CMK and Camostat Affect the Early Stage of the Virus Replication Cycle, but Naphthofluorescein Decreases the Viral RNA Level Rather Than Virus Entry
To clarify the stage of the virus replication cycle targeted by CMK, naphthofluorescein, and camostat, we treated VeroE6 cells with individual inhibitors at different time point during virus infection. In addition to co-administration of inhibitors with virus infection (Figure S3A), the cells were treated with the inhibitors before and after virus infection; the protocols are illustrated schematically in Figure 5 A (preinfection treatment) and Figure 5E (postinfection treatment).
Figure 5.

CMK and Camostat Treatment Affects the Early Stage of the Virus Replication Cycle, but Naphthofluorescein Decreases Viral RNA Levels after Virus Entry
(A and E) Schematic illustration of (A) the preinfection treatment experiments and (E) the postinfection treatment experiments.
(B and F) Microscopic observation of CPEs in VeroE6 cells assessed at the end of (B) preinfection treatment and (F) postinfection treatment with specific inhibitors (CMK [50 μM], D6R [50 μM], SSM3 [25 μM], naphthofluorescein [20 μM], or camostat [500 μM]). Scale bars, 100 μm.
(C and G) The virus titers (PFUs per milliliter) in the supernatant of SARS-CoV-2-infected VeroE6 cells at the end of the (C) preinfection treatment experiments and (G) postinfection treatment experiments were determined by plaque assay (mean ± SD of triplicates, ∗∗p < 0.01, ∗∗∗p < 0.001).
(D and H) The viral protein and viral RNA in VeroE6 cells at the end of the (D) preinfection treatment experiments and (H) postinfection treatment experiments were analyzed by immunoblotting (top panels) and northern blotting (bottom panels). Viral RNA isolated from VeroE6 cells was quantified by qRT-PCR targeting the E gene, as indicated below the northern blot results.
(I) Immunoblot of SARS-CoV-2 viral proteins in supernatant harvested from VeroE6 cells at the end of the postinfection treatment experiments. The immunoblot was probed with anti-S and anti-N Abs.
(J) Representative results of a plaque assay to determine the PFUs of SARS-CoV-2 virus in the supernatant of VeroE6 cells at the end of the postinfection treatment experiments. #, the lower number of PFUs in the 10−3 dilution than in the 10−4 dilution of the supernatant could be due to the inhibitory effect of camostat on virus entry in the higher-dose 10−3 dilution.
See also Figures S3–S5.
In the preinfection treatment experiments, CPEs were abolished by treatment with CMK or camostat but only mildly by treatment with naphthofluorescein (Figure 5B). Consistent with this result, the virus titer was decreased strikingly by treatment with CMK or camostat but less significantly by naphthofluorescein (Figure 5C). The northern blot and western blot analysis results further showed a marked reduction in viral RNA and protein only with CMK and camostat treatment but not with naphthofluorescein (Figure 5D). Co-administration of CMK, camostat, and naphthofluorescein with virus infection effectively decreased the virus titer in the supernatant and viral RNA and protein levels in infected cells (Figure S3). The results suggested that CMK and camostat, but not naphthofluorescein, might target the early stage of the viral life cycle before viral RNA synthesis.
In the postinfection treatment experiments, the CPEs and virus titer were reduced by treatment with naphthofluorescein (Figures 5F and 5G). Consistent reductions in the viral RNA and protein levels were identified only in naphthofluorescein-treated VeroE6 cells (Figure 5H). Therefore, CMK and camostat act primarily to inhibit early virus entry, but naphthofluorescein might affect a replication stage downstream of virus entry into cells. To further confirm the effect of naphthofluorescein on viral transcription, we monitored the level of viral RNA transcripts 24 and 48 h after infection. The northern blot results did support this possibility, showing a lower rate of increase in viral RNA transcripts by naphthofluorescein treatment in a dose-dependent manner (Figure S4).
Because CMK and camostat affected the virus entry stage, we examined whether they disrupt binding of the S protein to the ACE2 receptor. Via an in vitro binding assay, we determined that binding of the SARS-CoV-2-S protein (receptor binding domain [RBD])-Fc to human ACE2-expressing 293T cells was not affected by treatment with these inhibitors (Figure S5), eliminating the possibility that binding disruption is a mechanism of these inhibitors.
Blockade of Furin/PC-Mediated S Protein Cleavage after SARS-CoV-2 Infection Decreases CPEs but Not Virus Production and Infectivity
Interestingly, although postinfection treatment with CMK did not inhibit expression of the S protein, it did significantly reduce S protein cleavage into the S1 and S2 subunits (the S protein appears mainly as the uncleaved form; Figure 5H). Consistent with these findings, uncleaved S protein was enriched in virus harvested from the supernatant after treatment with CMK (Figure 5I). Moreover, as revealed by plaque assay, the virus titer and infectivity of these virions from CMK-treated cells were not decreased compared with control virions (Figures 5G and 5J), indicating that cleavage of the S protein by furin/PCs during biogenesis might not be essential for virus assembly and infectivity.
Notably, although postinfection treatment with CMK did not affect virus replication and assembly, it decreased the CPEs induced by SARS-CoV-2 infection. Such a decrease in CPEs did not occur in camostat-treated cells (Figure 5F). Therefore, from a therapeutic viewpoint, blockade of furin/PC-mediated S protein cleavage after viral infection likely ameliorates CPE and, thus, virulence and pathogenicity.
Discussion
Similar to numerous pathogenic viruses, cleavage of the S protein by host proteases has been documented to be important for its fusion activity after its binding to the host receptor in several CoVs (Heald-Sargent and Gallagher, 2012; Hoffmann et al., 2018). The putative furin substrate site located at the S1/S2 boundary and the putative TMPRSS2 substrate site located within the S2 domain of the SARS-CoV-2 S protein attract considerable interest. Our study not only confirmed the critical role of the furin cleavage site in the fusion activity of the S protein and contributing to virus production and syncytium formation but also identified two related antiviral lead compounds targeting the furin cleavage site in SARS-CoV-2, laying the groundwork for further development of antiviral agents against SARS-CoV-2.
Furin/PC-mediated processing of the SARS-CoV-2 S protein may be clinically significant. In autopsies of COVID-19 victims, many multinucleated giant cells resulting from syncytia of pneumocytes have been found in the lungs (Xu et al., 2020). In infection with other CoVs, syncytium formation confers multiple pathogenic advantages over cell-free spread. Direct cell-to-cell spread of CoV is more efficient than cell-free spread, which requires engagement with cell-membrane-specific receptors. In addition, syncytium formation allows the virus to evade innate humoral and cellular defenses (Sattentau, 2008, 2011). Therefore, for many infections with CoVs, syncytium formation has been found to be associated with increased viral pathogenesis (Frana et al., 1985; Nakagaki et al., 2005; Park et al., 2016); for example, greatly enhancing the infectivity of avian coronavirus infectious bronchitis (Yamada et al., 2009). Therefore, suppression of syncytium formation in SARS-CoV-2 infection by furin/PC inhibitors could be a strategy to reduce virus spread and ameliorate virulence and disease progression.
In support of the role of furin/PC-mediated S protein cleavage in formation of syncytia from SARS-CoV-2-infected cells, insertion of the furin cleavage site at the S1/S2 boundary of the SARS-CoV genome, which lacks such a site, greatly increased syncytium formation but not viral infectivity (Watanabe et al., 2008). This result was consistent with our postinfection treatment experiment showing that syncytium formation was suppressed; however, neither the virus titer nor infectivity was affected by CMK. Furin/PC-mediated cleavage of the S protein during biogenesis and assembly, which is accomplished mainly by furin/PC proteases localized at the trans-Golgi network, is required for syncytium formation but not essential for efficient virion assembly or infectivity in VeroE6 cells.
However, preinfection treatment with CMK did block the early stage of the viral life cycle before viral RNA synthesis. As noted, release of the fusion peptide from the S protein via cleavage by host proteases is a prerequisite for virus-cell membrane fusion and, thus, virus entry (Ou et al., 2016). Because CMK treatment did not affect binding of S to the ACE2 receptor, it suggests that preinfection treatment with CMK might suppress cleavage and release of the fusion peptide from the S protein after its binding with the receptor. In addition to furin, the putative target proteases for CMK at the membrane to help release the fusion peptide could be other PC family members or other off-target proteases (for example, TMPRSS2); these possibilities are worthy of investigation.
As shown in our virus infection system, CMK, naphthofluorescein, and camostat showed antiviral activity but targeted different replication stages. CMK and camostat effectively blocked virus entry, but naphthofluorescein acted after virus entry to reduce viral RNA levels. Both furin/PC inhibitors equivalently blocked cleavage of the S protein in S-only-expressing VeroE6 cells, suggesting their ability to inhibit the furin/PC activity mediating cleavage of newly synthesized S protein. Our finding raised the question of why naphthofluorescein and CMK showed distinct antiviral effects by targeting different SARS-CoV-2 replication stages. CMK, a peptidomimetic, is a cell-permeable competitive inhibitor and is considered highly specific for most PCs, including furin and PC1, PC2, PC4, PACE4, PC5/6, and PC7 (Becker et al., 2010; Henrich et al., 2003; Imran et al., 2019; Izaguirre, 2019). However, naphthofluorescein, a non-competitive small-molecule inhibitor of furin, identified by high-throughput screening of a compound library, might have different target specificity than CMK. The reporter established for the screen was based on a hybrid reporter protein containing the cleavage site from the trans-Golgi compartment-specific substrate stromelysin-3 (Coppola et al., 2007). Naphthofluorescein might thus act differently for furin substrates located in different compartments. Alternatively, it might have off-target effects on decreasing viral RNA levels. Therefore, in addition to studying the exact mechanism of naphthofluorescein to decrease viral transcription, it remains to be clarified whether this mechanism depends on furin activity or other new targets.
Although the mechanism underlying the distinct effects of these two furin/PC inhibitors in the virus replication cycle still remains to be investigated, our study proposes that these three inhibitors might be used for different antiviral strategies. The results of the preinfection treatment experiments indicated that CMK and camostat might function to prevent virus entry. Therefore, they could be used as prophylactics to prevent viral spread to uninfected individuals or to naive cells in infected individuals. However, the results of the postinfection treatment experiments showed that these two inhibitors did not block virus production in infected cells; instead, naphthofluorescein markedly decreased viral RNA levels. Thus, naphthofluorescein might be a lead compound for a potential therapeutic agent for COVID-19 patients. CMK, despite its failure to suppress virus replication after infection, could still suppress syncytium formation and might be applied to control viral pathogenesis.
It is noteworthy that inhibitors like CMK, naphthofluorescein, and camostat, which target host proteases, do not specifically inhibit SARS-CoV-2 production and CPEs. Inhibition of these proteases also affects their functions in regulating normal cellular processes. Moreover, as documented by in vitro and ex vivo studies, a cleavage redundancy exists among the constitutive PCs toward numerous substrates, especially among furin, PC5/6, and PACE4 (Seidah et al., 2008); thus, none of the proposed furin/PC inhibitors are highly specific to only one convertase. Because PCs are required for activation of hundreds of cellular substrates to maintain normal physiological functions, long-term blockage of furin/PCs by CMK or naphthofluorescein might cause detrimental side effects and may not be suitable for chronic treatment. The inhibitors could be more valuable for blocking SARS-CoV-2 infection in acute treatment, and local application of the inhibitors to the upper respiratory tract may be more feasible than systemic therapy, which needs further development.
Most successful predecessor antiviral drugs target virus-encoded essential enzymes, such as the proteases or polymerases of HIV, HCV, and influenza viruses (Adalja and Inglesby, 2019; De Clercq, 2002; Izaguirre, 2019; Patick and Potts, 1998). However, because virus replication occurs rapidly and has a higher mutation rate than host cell DNA replication, drug-resistant mutants emerge soon after single-agent antiviral therapy. Therefore, a combination of multiple antiviral agents (a cocktail) is frequently needed. This observation suggests that a regimen including more than one antiviral agent is probably required for successful anti-SARS-CoV-2 activity. On the other hand, host molecules are good targets, and targeting of host molecules minimizes the issue of drug resistance if the toxicities can be tolerated. The current study identified two inhibitors that block steps in virus production—virus entry or virus replication—and diminish pathogenic syncytium formation induced by the furin-cleaved S protein in SARS-CoV-2-infected individuals. These inhibitors might become lead compounds for further development of prophylactic or therapeutic antiviral agents.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-SARS-CoV/SARS-CoV-2 (COVID-19) spike antibody | Laboratory of Shiou-Hwei Yeh | N/A |
| Rabbit anti-SARS-CoV/SARS-CoV-2 (COVID-19) nucleocapsid antibody | Laboratory of Shiou-Hwei Yeh | N/A |
| Mouse anti-SARS-CoV/SARS-CoV-2 (COVID-19) spike antibody [1A9] | Genetex | Cat# GTX632604; RRID:AB_2864418 |
| Rabbit anti-integrin alpha V antibody | Genetex | Cat# GTX54357 |
| Rabbit anti-GAPDH | Genetex | Cat# GTX100118; RRID:AB_1080976 |
| Goat Anti-Mouse IgG antibody (HRP) | Genetex | Cat# GTX213111-01; RRID:AB_10618076 |
| Goat Anti-Rabbit IgG antibody (HRP) | Genetex | Cat# GTX213110-01; RRID:AB_10618573 |
| Mouse anti-HA antibody | LTK BioLaboratories | N/A |
| Bacterial and Virus Strains | ||
| hCoV-19/Taiwan/NTU03/2020 | Laboratory of Sui-Yuan Chang | GISAID (Accession ID:EPI_ISL_413592) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Camostat Mesylate | Medchemexpress | Cat# HY13512 |
| Hexa-D-Arginine-Amide | Tocris Bioscience | Cat# 4711 |
| Decanoyl-RVKR-CMK | Tocris Bioscience | Cat# 3501 |
| Naphthofluorescein | Sigma-Aldrich | Cat# 61419 |
| SSM 3 Trifluoroacetate | Tocris Bioscience | Cat# 5253 |
| Lipofectamine-2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668500 |
| Critical Commercial Assays | ||
| Cell Counting Kit-8 | Dojindo Molecular Technologies | Cat# CK04-05 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | Cat# 18080051 |
| QuikChange II Site-Directed Mutagenesis Kit | Agilent | Cat# 200521 |
| NucleoSpin RNA, Mini kit for RNA purification | Macherey-Nagel | REF 740955.50 |
| Deposited Data | ||
| hCoV-19/Taiwan/NTU03/2020 | GISAID | Accession ID: EPI_ISL_413592 |
| Experimental Models: Cell Lines | ||
| VERO C1008 [Vero 76, clone E6, Vero E6] | ATCC | Cat# CRL-1586; RRID:CVCL_0574 |
| LLC-MK2 Original | ATCC | Cat# CCL-7; RRID:CVCL_3009 |
| 293T/hACE2 stable cell | Laboratory of Mi-Hao Tao | N/A |
| Oligonucleotides | ||
| nCoV19-N cDNA probe F: AAGCTGGACTTCCCTATGGTGC | Mission Biotech | N/A |
| nCoV19-N cDNA probe R: CCTTGGGTTTGTTCTGGACCACG | Mission Biotech | N/A |
| Spike-R682A-F: ACGCTCCGGGCTCTTGCGGGAGAGT TTGTCTG |
Mission Biotech | N/A |
| Spike-R682A-R: CAGACAAACTCTCCCGCAAGAGCCC GGAGCGT |
Mission Biotech | N/A |
| E_Sarbeco_F1: ACAGGTACGTTAATAGTTAATAGCGT | Mission Biotech | N/A |
| E_Sarbeco_R2: ATATTGCAGCAGTACGCACACA | Mission Biotech | N/A |
| PBGD probe-F: GGTGACCAGCACACTTTGGG | Purigo | N/A |
| PBGD probe-R: AGCCGGGTGTTGAGGTTTCC | Purigo | N/A |
| PBGD-F: GCATCGCTGAAAGGGCCTTCC- | Mission Biotech | N/A |
| PBGD-R: TCATCCTCAGGGCCATCTTCATGC | Mission Biotech | N/A |
| Recombinant DNA | ||
| pUC57-2019-nCoV-S (humanized) | Laboratory of Che Ma | N/A |
| pcDNA3.0- SARS-CoV-2 S (Hu)-HA | This paper | N/A |
| pcDNA3.0- SARS-CoV-2 S-R682A (Hu)-HA | This paper | N/A |
| Software and Algorithms | ||
| SigmaPlot | Systat Software | https://systatsoftware.com/products/sigmaplot/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shiou-Hwei Yeh (shyeh@ntu.edu.tw)
Materials Availability
All materials in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The original sequencing datasets for hCoV-19/Taiwan/NTU03/2020 can be found on the GISAID under Accession ID: EPI_ISL_413592.
Experimental Model and Subject Details
Viruses
Sputum specimens obtained from SARS-CoV-2-infected patients were maintained in viral transport medium. Virus in the specimens was propagated in VeroE6 cells in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 μg/mL tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma-Aldrich). Culture supernatant was harvested when CPE were seen in more than 70% of cells, and virus titers were determined by a plaque assay. The virus isolate used in the current study is hCoV-19/Taiwan/NTU03/2020 (GISAID:: EPI_ISL_413592)
Plaque assay
The plaque assay was performed as previously described with minor modifications (Su et al., 2008). In brief, VeroE6 cells (2 × 105 cells/well) were seeded in triplicate in 24-well tissue culture plates in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics one day before infection. SARS-CoV-2 was added to the cell monolayer for 1 hr at 37°C. Subsequently, virus was removed, and the cell monolayer was washed once with PBS before being overlaid with medium containing 1% methylcellulose and incubated for 5-7 days. Cells were fixed with 10% formaldehyde overnight. After removal of the overlay medium, cells were stained with 0.7% crystal violet, and plaques were counted. The vial titer presented in a histogram from the mean of three independent experiments.
Plasmid construction
pUC57-2019-nCoV-S (humanized) (GenScript) was kindly provided by Dr. Che Ma at the Institute of Genomics Research Center, Academia Sinica, Taiwan. The spike sequence was cloned into the pcDNA3.0-HA vector with the addition of an HA tag at the C terminus via NheI and XbaI sites. The spike-R682A mutant construct was generated by site-directed mutagenesis using a QuikChange II Site-Directed Mutagenesis Kit (Agilent) by following manufacturer’s instructions, with the primer set of 5′-ACGCTCCGGGCTCTTGCGGGAGAGTTTGTCTG-3′ and 5′-CAGACAAACTCTCCCGCAAGAGCCCGGAGCGT-3′.
Cell culture experiments
VeroE6 cells were cultured at 37°C in DMEM containing 10% FBS (HyClone, GE Healthcare Life Sciences) in a 5% CO2 incubator. The indicated plasmid was transfected into VeroE6 cells with Lipofectamine™ 2000 (Thermo Fisher Scientific). The inhibitors were added to the medium at the indicated concentrations 2 hr post transfection. Cells were harvested or fixed 24 hr post transfection for subsequent western blot or immunofluorescence analysis. The stock inhibitors tested in this study were all prepared in DMSO solutions, including CMK (10 mM), D6R (5 mM), SSM3 (25 mM) (Tocris Bioscience), naphthofluorescein (10 mM) (Sigma-Aldrich) and camostat (100 mM) (Medchemexpress). The concentration and percentage of DMSO (v/v) in the working solution of these inhibitors after dilution of the stock solutions are as follows: CMK 50 μM (0.5% DMSO); D6R 50 μM (1% DMSO); SSM3 25 μM (0.1% DMSO); naphthofluorescein 20 μM (0.2% DMSO); camostat 500 μM (0.5% DMSO).
Method Details
Western blot analysis
Western blotting was performed as previously described (Wu et al., 2009). In brief, cell lysates were harvested in 1X RIPA buffer (Merck Millipore) containing 1X proteinase inhibitor (Merck Millipore) and 1X phosphatase inhibitor (Calbiochem). Protein samples were added to 4X SDS loading dye and denatured for 10 min at 95°C. Proteins were separated on 10% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% milk in 1X TBST prior to incubation with primary antibodies at 4°C overnight. Membranes were then reacted with a secondary antibody. Antigen-antibody complexes were visualized using Western Lightning Plus-ECL (PerkinElmer). The antibodies used for western blot analysis were as follows: rabbit anti-SCoV/SARS-CoV-2 (COVID-19) spike (generated by our laboratory), rabbit anti-SCoV/SARS-CoV-2 nucleocapsid (generated by our laboratory), mouse anti-SARS-CoV/SARS-CoV-2 (COVID-19) spike [1A9] (Genetex, GTX632604), rabbit anti-integrin alpha V (Genetex, GTX54357), rabbit anti-GAPDH (Genetex, GTX100118), mouse anti-HA (LTK BioLaboratories), horseradish peroxidase-conjugated mouse IgG (Genetex, GTX213111-01) and rabbit IgG (Genetex, GTX213110-01).
Immunofluorescence microscopy
VeroE6 cells were fixed with 4% paraformaldehyde for 10 min at room temperature. After three washes with 1X PBS, cells were permeabilized with 0.1% Triton X-100 at 4°C for 10 min. Next, cells were incubated in blocking buffer (1X PBS containing 5% FBS+3% BSA and 0.1% Triton X-100) at room temperature for 30 min. Cells were then incubated with primary antibodies (mouse anti-SARS-CoV/SARS-CoV-2 (COVID-19) spike antibody [1A9] (Genetex, GTX632604) in 1X PBS containing 5% FBS+3% BSA overnight at 4°C. Alexa Fluor-488 or Alexa Fluor −594 IgG (Thermo Fisher Scientific) and 4’,6-diamidino-2-phenylindole (DAPI) nuclear counterstaining were applied for 1 hr at room temperature. Images were acquired using a Zeiss AXIO Imager A1 microscope.
RNA extraction and Northern blot analysis
RNA was extracted using NucleoSpin RNA Kit (Macherey-Nagel) according to the instruction manual. Northern blotting was performed as previously described (Wu et al., 2014). In brief, 0.5 μg of RNA was denatured and processed for electrophoresis on an 0.8% agarose/formaldehyde gel at 70 V for 2 hr and 100 V for 3 hr. Before capillary transfer, the agarose gel was submerged in 50 mM NaOH for 50 min, washed with 100 mM Tris-HCl (pH 7.5) for 30 min, and incubated in 20X SSC buffer for 20 min. A Hybond-N nylon membrane (Amersham Biosciences) was presoaked in 2X SSC buffer for capillary transfer overnight. The next day, RNA was immobilized by UV crosslinking (1800 × 100 μJ/cm) and hybridized at 50°C overnight with digoxigenin (DIG)-labeled probes generated with a PCR DIG probe synthesis kit (Roche Diagnostics). The primers used to synthesize the DIG-labeled nCoV19-N cDNA probe were 5′-AAGCTGGACTTCCCTATGGTGC-3′ and 5′-CCTTGGGTTTGTTCTGGACCACG-3′. The probes of porphobilinogen deaminase (PBGD) used as the internal control in Northern blot were 5′-GGTGACCAGCACACTTTGGG-3′ and 5′-AGCCGGGTGTTGAGGTTTCC-3′.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
RNA extracted from VeroE6 cells infected with SARS-CoV-2 was reverse transcribed using SuperScript III Reverse Transcriptase System (Thermo Fisher Scientific). Quantitative PCR of E gene was performed using FastStart DNA SYBR Green on LightCycler 1.5 (Roche Diagnostics). The primers at E gene were E_Sarbeco_F1: 5′- ACAGGTACGTTAATAGTTAATAGCGT-3′ and E_Sarbeco_R2: 5′- ATATTGCAGCAGTACGCACACA-3′. The expression level of E gene in the SARS-CoV-2 infected VeroE6 cells was determined in relative to the internal control of cellular PBGD gene, with primer set of 5′- GCATCGCTGAAAGGGCCTTCC-3′ and 5′- TCATCCTCAGGGCCATCTTCATGC-3′
Cytotoxicity assay
The cytotoxicity effect of specific inhibitors was evaluated by a CCK-8 assay (Dojindo Molecular Technologies). In brief, 5x103 VeroE6 cells were seeded in 96-well plates for 24 hr before treatment with serial doses of the inhibitors. Ten microliters of CCK-8 reagent was added to each 96-well plate after 24 hr of inhibitor treatment and further incubated in a 37°C incubator for 2 hr before measurement of the optical density at 450 nm. The data from three independent experiments was used to calculate the CC50 by nonlinear regression using GraphPad Prism V5.0 software.
Spike-ACE2 in vitro binding assay
Flow cytometry analysis was performed to detect the blockage of receptor binding domain (RBD)-hACE2 receptor binding. DMSO diluents, 3 μg of soluble hACE2 (generated by laboratory of Mi-Hua Tao), or inhibitors were incubated with 0.2 μg of SARS-CoV-2 RBD containing a C-terminal mFc (Sino Biological, 40592-V05H) for 1 hr at room temperature. Then, 2x105 293T/hACE2 cells resuspend in 100 μL of 1%FBS-DPBS were incubated with 0.2 μg of RBD-ACE2 binding reactant for 1 hr on ice, followed by incubation with 100 μL of phycoerythrin (PE)-labeled goat anti-mouse IgG-Fc antibody (1:200; Jackson 115-116-146) for 30 min. After staining, cells were resuspended in 300 μL of 1% FBS-DPBS containing 7-amino-actinomycin D (7-AAD; 1:100) to exclude non-viable cells and analyzed by flow cytometry.
IC50 determination by plaque reduction assay
In brief, VeroE6 cells (2 × 105 cells/well) were seeded in triplicate in 24-well tissue culture plates in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics one day before infection. The VeroE6 cells were treated by CMK, naphthofluorescein, or camostat for 1 hr before 100 PFU/well SARS-CoV-2 was added to the cell monolayer for 1 hr at 37°C. Subsequently, virus was removed, and the cell monolayer was washed once with PBS before being overlaid with medium containing 1% methylcellulose and incubated for 5-7 days. Cells were fixed with 10% formaldehyde overnight. After removal of the overlay medium, cells were stained with 0.7% crystal violet, and plaques were counted. The results from three independent experiments were used to calculate the IC50 by nonlinear regression using GraphPad Prism V5.0 software.
Quantification and Statistical Analysis
The virus titers quantified by plaque assay in triplicate were shown as the means ± standard deviations. The difference between the control cells (without inhibitor treatment) and the cells treated with specific inhibitors were evaluated by Student’s t test. The P values of 0.05 or lower were considered statistically significant (∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001).
Acknowledgments
This study was supported by grants from the Ministry of Science and Technology, Taiwan (MOST109-2327-B-002-009, MOST109-2634-F-002-043, and MOST109-3114-Y-001-001) and the Center of Precision Medicine from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) of Taiwan. We would like to acknowledge the service provided by the biosafety level 3 laboratory of the First Core Laboratory, National Taiwan University College of Medicine. We also thank the staff of the Biomedical Resource Core at the First Core Labs, National Taiwan University College of Medicine, for technical assistance.
Author Contributions
Y.-W.C., C.-L.L., S.-H.Y., and S.-Y.C. designed the research. Y.-W.C. performed the SARS-CoV-2 spike transfection experiments. T.-L.C. performed viral infection experiments. Y.-H.P., C.-H.L., Y.-M.T., and W.-H.L. contributed to the viral infection experiments, plaque assay, and virus titer determination. L.-T.J. cloned pcDNA3.0-SARS-CoV-2 spike-HA. Y.-W.C. cloned SARS-CoV-2 spike-R682A-HA. Y.-W.C. performed western and northern blotting. S.-H.W. contributed to northern blotting. T.-C.H. contributed to western blotting. M.-F.C. performed immunofluorescence microscopy. M.-H.T. and P.-Y.W. contributed to the ACE2 neutralization assay. Y.-W.C. and C.-L.L. performed the CCK-8 assay. C.-L.L. performed qRT-PCR. Y.-W.C., C.-L.L., S.-H.Y., and S.-Y.C. analyzed data. Y.-W.C., C.-L.L., S.-Y.C., and S.-H.Y. drafted the manuscript. P.-J.C. critically revised the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108254.
Supplemental Information
References
- Adalja A., Inglesby T. Broad-spectrum antiviral agents: a crucial pandemic tool. Expert Rev. Anti Infect. Ther. 2019;17:467–470. doi: 10.1080/14787210.2019.1635009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker G.L., Sielaff F., Than M.E., Lindberg I., Routhier S., Day R., Lu Y., Garten W., Steinmetzer T. Potent inhibitors of furin and furin-like proprotein convertases containing decarboxylated P1 arginine mimetics. J. Med. Chem. 2010;53:1067–1075. doi: 10.1021/jm9012455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard S., Chu V.C., Whittaker G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA. 2009;106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun E., Sauter D. Furin-mediated protein processing in infectious diseases and cancer. Clin. Transl. Immunology. 2019;8:e1073. doi: 10.1002/cti2.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman C.M., Sisk J.M., Mingo R.M., Nelson E.A., White J.M., Frieman M.B. Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus fusion. J. Virol. 2016;90:8924–8933. doi: 10.1128/JVI.01429-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola J.M., Hamilton C.A., Bhojani M.S., Larsen M.J., Ross B.D., Rehemtulla A. Identification of inhibitors using a cell-based assay for monitoring Golgi-resident protease activity. Anal. Biochem. 2007;364:19–29. doi: 10.1016/j.ab.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola J.M., Bhojani M.S., Ross B.D., Rehemtulla A. A small-molecule furin inhibitor inhibits cancer cell motility and invasiveness. Neoplasia. 2008;10:363–370. doi: 10.1593/neo.08166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N.G., Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croissandeau G., Basak A., Seidah N.G., Chrétien M., Mbikay M. Proprotein convertases are important mediators of the adipocyte differentiation of mouse 3T3-L1 cells. J. Cell Sci. 2002;115:1203–1211. doi: 10.1242/jcs.115.6.1203. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- Frana M.F., Behnke J.N., Sturman L.S., Holmes K.V. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: host-dependent differences in proteolytic cleavage and cell fusion. J. Virol. 1985;56:912–920. doi: 10.1128/jvi.56.3.912-920.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunt E.R., Hardie A., Claas E.C.J., Simmonds P., Templeton K.E. Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J. Clin. Microbiol. 2010;48:2940–2947. doi: 10.1128/JCM.00636-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman J.D., Lye D.C.B., Hui D.S., Marks K.M., Bruno R., Montejano R., Spinner C.D., Galli M., Ahn M.Y., Nahass R.G., GS-US-540-5773 Investigators Remdesivir for 5 or 10 days in patients with severe COVID-19. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2015301. Published online May 27, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurwitz D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020;81:537–540. doi: 10.1002/ddr.21656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald-Sargent T., Gallagher T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses. 2012;4:557–580. doi: 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrich S., Cameron A., Bourenkov G.P., Kiefersauer R., Huber R., Lindberg I., Bode W., Than M.E. The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat. Struct. Biol. 2003;10:520–526. doi: 10.1038/nsb941. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Hofmann-Winkler H., Pöhlmann S. Priming time: How cellular proteases arm coronavirus spike proteins. In: Böttcher-Friebertshäuser E., Garten W., Klenk H.D., editors. Activation of Viruses by Host Proteases. Springer; 2018. pp. 71–98. [Google Scholar]
- Hoffmann M., Kleine-Weber H., Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell. 2020;78:779–784.e5. doi: 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imran M., Saleemi M.K., Chen Z., Wang X., Zhou D., Li Y., Zhao Z., Zheng B., Li Q., Cao S., Ye J. Decanoyl-Arg-Val-Lys-Arg-chloromethylketone: an antiviral compound that acts against flaviviruses through the inhibition of furin-mediated prM cleavage. Viruses. 2019;11:1011. doi: 10.3390/v11111011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaguirre G. The proteolytic regulation of virus cell entry by furin and other proprotein convertases. Viruses. 2019;11:837. doi: 10.3390/v11090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV) Nat. Rev. Drug Discov. 2020;19:149–150. doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wang Y., Agostinis P., Rabson A., Melino G., Carafoli E., Shi Y., Sun E. Is hydroxychloroquine beneficial for COVID-19 patients? Cell Death Dis. 2020;11:512. doi: 10.1038/s41419-020-2721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagaki K., Nakagaki K., Taguchi F. Receptor-independent spread of a highly neurotropic murine coronavirus JHMV strain from initially infected microglial cells in mixed neural cultures. J. Virol. 2005;79:6102–6110. doi: 10.1128/JVI.79.10.6102-6110.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X., Zheng W., Shan Y., Mu Z., Dominguez S.R., Holmes K.V., Qian Z. Identification of the fusion peptide-containing region in betacoronavirus spike glycoproteins. J. Virol. 2016;90:5586–5600. doi: 10.1128/JVI.00015-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.E., Li K., Barlan A., Fehr A.R., Perlman S., McCray P.B., Jr., Gallagher T. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl. Acad. Sci. USA. 2016;113:12262–12267. doi: 10.1073/pnas.1608147113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patick A.K., Potts K.E. Protease inhibitors as antiviral agents. Clin. Microbiol. Rev. 1998;11:614–627. doi: 10.1128/cmr.11.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle A.G., Gawlik K., Golubkov V.S., Cadwell G.W., Liddington R.C., Cieplak P., Millis S.Z., Desjardins R., Routhier S., Yuan X.W. Selective and potent furin inhibitors protect cells from anthrax without significant toxicity. Int. J. Biochem. Cell Biol. 2010;42:987–995. doi: 10.1016/j.biocel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders J.M., Monogue M.L., Jodlowski T.Z., Cutrell J.B. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;323:1824–1836. doi: 10.1001/jama.2020.6019. [DOI] [PubMed] [Google Scholar]
- Sarac M.S., Cameron A., Lindberg I. The furin inhibitor hexa-D-arginine blocks the activation of Pseudomonas aeruginosa exotoxin A in vivo. Infect. Immun. 2002;70:7136–7139. doi: 10.1128/IAI.70.12.7136-7139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattentau Q. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008;6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]
- Sattentau Q.J. The direct passage of animal viruses between cells. Curr. Opin. Virol. 2011;1:396–402. doi: 10.1016/j.coviro.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Seidah N.G., Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012;11:367–383. doi: 10.1038/nrd3699. [DOI] [PubMed] [Google Scholar]
- Seidah N.G., Mayer G., Zaid A., Rousselet E., Nassoury N., Poirier S., Essalmani R., Prat A. The activation and physiological functions of the proprotein convertases. Int. J. Biochem. Cell Biol. 2008;40:1111–1125. doi: 10.1016/j.biocel.2008.01.030. [DOI] [PubMed] [Google Scholar]
- Su C.T., Hsu J.T., Hsieh H.P., Lin P.H., Chen T.C., Kao C.L., Lee C.N., Chang S.Y. Anti-HSV activity of digitoxin and its possible mechanisms. Antiviral Res. 2008;79:62–70. doi: 10.1016/j.antiviral.2008.01.156. [DOI] [PubMed] [Google Scholar]
- Ton A.T., Gentile F., Hsing M., Ban F., Cherkasov A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inform. 2020;39:e2000028. doi: 10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M.A., Veesler D. Structural insights into coronavirus entry. Adv. Virus Res. 2019;105:93–116. doi: 10.1016/bs.aivir.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y.F., Chien C.S., Yarmishyn A.A., Lin Y.Y., Luo Y.H., Lin Y.T., Lai W.Y., Yang D.M., Chou S.J., Yang Y.P. A review of SARS-CoV-2 and the ongoing clinical trials. Int. J. Mol. Sci. 2020;21:2657–2675. doi: 10.3390/ijms21072657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Qiu Y., Li J.Y., Zhou Z.J., Liao C.H., Ge X.Y. A unique protease cleavage site predicted in the spike protein of the novel pneumonia coronavirus (2019-nCoV) potentially related to viral transmissibility. Virol. Sin. 2020;35:337–339. doi: 10.1007/s12250-020-00212-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhang D., Du G., Du R., Zhao J., Jin Y., Fu S., Gao L., Cheng Z., Lu Q. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–1578. doi: 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R., Matsuyama S., Shirato K., Maejima M., Fukushi S., Morikawa S., Taguchi F. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved S protein as revealed by pseudotype virus bearing cleaved S protein. J. Virol. 2008;82:11985–11991. doi: 10.1128/JVI.01412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . 2020. Coronavirus disease 2019 (COVID-19) weekly epidemiological updates.https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports [Google Scholar]
- Wu C.H., Yeh S.H., Tsay Y.G., Shieh Y.H., Kao C.L., Chen Y.S., Wang S.H., Kuo T.J., Chen D.S., Chen P.J. Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J. Biol. Chem. 2009;284:5229–5239. doi: 10.1074/jbc.M805747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.H., Chen P.J., Yeh S.H. Nucleocapsid phosphorylation and RNA helicase DDX1 recruitment enables coronavirus transition from discontinuous to continuous transcription. Cell Host Microbe. 2014;16:462–472. doi: 10.1016/j.chom.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y., Liu X.B., Fang S.G., Tay F.P., Liu D.X. Acquisition of cell-cell fusion activity by amino acid substitutions in spike protein determines the infectivity of a coronavirus in cultured cells. PLoS ONE. 2009;4:e6130. doi: 10.1371/journal.pone.0006130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z.Q., Chen D.H., Tan W.P., Qiu S.Y., Xu D., Liang H.X., Chen M.X., Li X., Lin Z.S., Liu W.K., Zhou R. Epidemiology and clinical characteristics of human coronaviruses OC43, 229E, NL63, and HKU1: a study of hospitalized children with acute respiratory tract infection in Guangzhou, China. Eur. J. Clin. Microbiol. Infect. Dis. 2018;37:363–369. doi: 10.1007/s10096-017-3144-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A., Chan J.F., Azhar E.I., Hui D.S., Yuen K.Y. Coronaviruses - drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016;15:327–347. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original sequencing datasets for hCoV-19/Taiwan/NTU03/2020 can be found on the GISAID under Accession ID: EPI_ISL_413592.