

Abstract
STAT proteins can regulate both pro- and anti-inflammatory cytokine signaling. Therefore, identifying consequences of modulating expression of a given STAT is ultimately critical for determining its potential as a therapeutic target and for defining the mechanisms through which immune-mediated disease variants in STAT genes contribute to disease pathogenesis. Genetic variants in the STAT1/STAT4 region are associated with multiple immune-mediated diseases, including inflammatory bowel disease (IBD). These diseases are characterized by dysregulated cytokine secretion in response to pattern-recognition receptor (PRR) stimulation. We found that the common IBD-associated rs1517352 C risk allele increased both STAT1 and STAT4 expression in human monocyte-derived macrophages (MDMs). We therefore hypothesized that the STAT1/STAT4 variant might regulate PRR-initiated responses in a complementary and cooperative manner because of the important role of autocrine/paracrine cytokines in modulating PRR-initiated signaling. STAT1 and STAT4 were required for PRR- and live bacterial-induced secretion of multiple cytokines. These outcomes were particularly dependent on PRR-initiated autocrine/paracrine IL-12–induced STAT4 activation to generate IFN-γ, with autocrine IFN-γ then signaling through STAT1. STAT1 and STAT4 also promoted bacterial-induced cytokines in intestinal myeloid cells and PRR-enhanced antimicrobial pathways in MDMs. Importantly, MDMs from rs1517352 C IBD risk allele carriers demonstrated increased TLR4-, IFN-γ– and IL-12–induced STAT1 and STAT4 phosphorylation and cytokine secretion and increased TLR4-enhanced antimicrobial pathways. Taken together, STAT1 and STAT4 expression is coregulated by a shared genetic region, and STAT1/STAT4-immune disease–associated variants modulate IFN-γ– and IL-12–associated outcomes, and in turn, PRR-induced outcomes, highlighting that these genes cooperate to regulate pathways relevant to disease pathogenesis.
Proper responses to microbial products detected by pattern-recognition receptors (PRRs) on myeloid cells are critical for immune homeostasis at mucosal surfaces such as the intestine. A key PRR-induced response is cytokine secretion, and this cytokine secretion can, in turn, dramatically amplify the initial response to microbial products (1). Consistently, one of the hallmarks of immune-mediated diseases, including inflammatory bowel disease (IBD), is dysregulated responses to and/or production of cytokines (1). STAT proteins are critical for mediating responses to cytokines, such that the JAK-STAT pathway is being actively investigated as a therapeutic target in immune-mediated diseases. However, an important challenge in targeting these pathways is that these proteins can mediate responses to both proinflammatory and anti-inflammatory cytokines. As such, it is critical to clearly define the distinct contributions of each of these family members to this cytokine balance. Furthermore, mouse and human cells can show significant differences in inflammatory pathways (2), such it is important to understand how these STAT proteins regulate outcomes in human myeloid-derived cells. Genetic variants in these genes can help elucidate consequences of this regulation.
Common genetic variants in the STAT1/STAT4 region confer altered susceptibility to IBD (3) and systemic lupus erythematosus (SLE) (4). STAT1 and STAT4 can complement each other as regulators of cytokine-induced outcomes. STAT1 mediates type I IFN– and IFN-γ–initiated signaling, and STAT4 can mediate signaling by type I IFNs and IL-12 and is required for IL-12–induced IFN-γ (5). STAT1 has been reported to promote TLR-induced outcomes in mouse macrophages (6); the role of STAT4 in PRR-initiated responses in human macrophages has not been clearly defined. Genetic associations in each STAT1 and STAT4 have focused predominantly on rare coding mutations. As such, two rare coding variants resulting in defective STAT1 function result in decreased IFN-α– and IFN-γ–initiated responses; infant carriers of these mutations can develop severe mycobacterial and viral infections (7). In contrast, rare coding gain-of-function STAT1 mutations associated with STAT1 hyperactivation in T cells lead to excessive IFN-γ signaling, resulting in impaired Th17 immunity and chronic mucocutaneous candidiasis (8). Consistent with its role in type I IFN and IL-12 signaling, a STAT4 gain-of-function intronic polymorphism associated with SLE results in increased IFN-α responses in human PBMCs (9). To our knowledge, how common IBD-associated genetic variants in the STAT1/STAT4 region regulate myeloid cell outcomes has not been defined.
The common rs1517352 polymorphism located in an intronic region of STAT4 and upstream of STAT1 is associated with IBD (3) and SLE (4). The C risk allele has a frequency ranging from 0.33 to 0.40 in European ancestry individuals per the Single Nucleotide Polymorphism Database. How this common polymorphism contributes to risk is incompletely defined. One study found that the C risk allele is part of a haplotype associated with increased STAT4 expression in human osteoblasts and lymphoblasts and increased anti-dsDNA in SLE patients (10). How this polymorphism regulates STAT4 protein expression, STAT4 activation, or STAT4-dependent cytokine secretion, including to PRR stimulation, is not clear. Furthermore, STAT1 is located ~50 kb from this polymorphism, and it is unclear if rs1517352 modulates STAT1 expression or STAT1-dependent outcomes. The coregulation of these two STAT proteins from a common genetic variant would have implications for how these STAT proteins might cooperate with each other in immune responses. We therefore asked how STAT1 and STAT4 regulate PRR-initiated outcomes in human monocyte-derived macrophages (MDMs), given the importance of macrophages in immune-mediated diseases, and how the common immune-mediated disease-associated STAT1/STAT4 region polymorphism modulates these outcomes.
In this study, using primary human MDMs, we found that STAT1 and STAT4 were required for optimal signaling and cytokine secretion following stimulation of a broad range of PRRs and with live bacteria. Mechanisms contributing to STAT1- and STAT4-dependent PRR-initiated cytokine regulation included cooperative autocrine IFNGR and IL-12R signaling, respectively. STAT1 and STAT4 also promoted cytokine secretion by intestinal myeloid cells following Salmonella Typhimurium coculture and induction of antimicrobial pathways following PRR stimulation of myeloid cells. MDMs from disease-associated rs1517352 C risk allele carriers in the STAT1/STAT4 region demonstrated higher expression levels of both STAT1 and STAT4, as well as increased PRR-, IFN-α–, and IL-12–induced STAT1 and STAT4 activation and downstream outcomes. Taken together, we identify that STAT1 and STAT4 regulate PRR-induced outcomes, identify mechanisms leading to these outcomes, and determine consequences for STAT1/STAT4 region immune disease-associated variants in PRR-and cytokine-induced outcomes, highlighting the importance of these pathways in immune homeostasis.
Materials and Methods
Myeloid cell isolation
Human cell studies were conducted as approved by the institutional review board at Yale University. Cells were genotyped for the rs1517352 variant by TaqMan (Life Technologies, Grand Island, NY). Monocytes were purified from human PBMCs by adhesion and cultured with M-CSF (10 ng/ml) (Shenandoah Technology, Warwick, PA) for 7 d for MDM differentiation.
Myeloid cell stimulation
Human MDMs were treated with lipid A (Peptides International, Louisville, KY), Pam3Cys (EMD Millipore, Billerica, MA), polyinosinic-polycytidylic acid [poly(I:C)], CpG DNA (Invivogen, San Diego, CA), IL-12, IFN-γ (R&D Systems, Minneapolis, MN), IFN-α (Biovision, Milpitas, CA), or cocultured with Salmonella enterica serovar Typhimurium at multiplicity of infection (MOI) 10:1. Supernatants were assayed for IL-6, IL-10, TNF, IL-1β, IFN-γ (BD Biosciences), IL-12 (eBioscience, San Diego, CA), or IFN-γ (BioLegend) by ELISA. In some cases, cells were pretreated with 5 μg/ml fludarabine (STAT1 inhibitor), 150 μM lisofylline (STAT4 inhibitor) (Cayman Chemical Company), Upadacitinib (JAK1 inhibitor), or BMS-986165 (TYK2 inhibitor) (MedChemExpress).
Intestinal lamina propria cell isolation
Intestinal lamina propria cells were isolated from colonic resection specimens from uninvolved intestine in non-IBD patients undergoing surgery for diverticular disease or colon cancer as in Ref. 11. Briefly, intestinal resections were washed and then digested (RPMI containing 0.2 mM EDTA, 2-ME) in a 37°C shaker to remove epithelial cells. Epithelial cells and intraepithelial lymphocytes were discarded, and intestines were washed, cut into 1-mm2 pieces, and incubated in a buffer consisting of RPMI, 75 mg/ml collagenase type VIII, DNase (MilliporeSigma), and HEPES for 45 min in a 37°C shaker. The cells were filtered through a 40-μm filter, selected on a Ficoll gradient, washed, and then used in studies.
Transfection of small interfering RNA
One hundred nanomolar scrambled or ON-TARGETplus or siGENOME SMARTpool small interfering RNA (siRNA) against STAT1, STAT4, STAT3, JAK1, TYK2, IL12RB2, IFNGR1, IFNAR, IL10RA, ERK, p38, JNK, or NEMO (Dharmacon, Lafayette, CO) (four pooled siRNAs for each gene) was transfected into myeloid cells using Amaxa nucleofector technology (Lonza).
Phosphoprotein and total protein detection
Cell surface or intracellular proteins (permeabilized cells) were detected by flow cytometry with fluorophore-labeled Abs to phospho-ERK, phospho-p38, phospho-JNK, phospho-IκBα, phospho-STAT1, phospho-STAT4, phospho-JAK1, phospho-JAK2, phospho-TYK2 (Cell Signaling Technology, Danvers, MA), STAT1, STAT4, IL-10RA, LC3β, and NOS2 (Santa Cruz Biotechnology, Santa Cruz, CA).
mRNA expression analysis
Following stimulation, total RNA was isolated, reverse transcribed, and quantitative PCR performed as in Ref. 12 on the ABI Prism 7000 (Applied Biosystems). Each sample was run in duplicates and normalized to GAPDH. Primers sequences as per Supplemental Table I.
Intracellular reactive oxygen species measurement
Intracellular reactive oxygen species (ROS) production was measured by flow cytometry using 10 μM cell-permeant 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA) (Invitrogen).
Bacterial entry
Macrophages were cocultured with 2.5 × 107/ml E. coli-FTIC bioparticles (Molecular Probes) or 5 × 107 CFU/ml live bacteria S. Typhimurium-GFP (kindly provided by Jorge E. Galan) for 20 min. Cell surface fluorescence was quenched with 0.25 mg/ml trypan blue for 1 min, and after 4% paraformaldehyde fixation cells were analyzed by flow cytometry.
Intracellular bacterial clearance
Human MDMs were infected with adherent-invasive Escherichia coli (AIEC) (strain LF82; a generous gift from Dr. E. Mizoguchi) or Salmonella enterica serovar Typhimurium at 10:1 MOI for 20 min, washed with PBS, and incubated in HBSS medium with 20 μg/ml gentamicin for a total of 2 h. Cells were washed, lysed with 1% Triton X-100, and plated on MacConkey or Luria-Bertani agar.
Statistical analysis
Significance was assessed using two-tailed Student t test. A one-way ANOVA with Tukey posttest or t test with Bonferroni–Holm correction was applied as appropriate for multiple comparisons. To keep cytokines on same axis, a multiplier was applied for the lower cytokine levels of IFN-α and IFN-γ as shown in figure keys. A p value <0.05 was considered significant. Lines over adjacent bars indicate identical p values for these bars.
Results
MDMs from rs1517352 C risk carriers in the STAT1/STAT4 region demonstrate increased PRR-induced cytokine secretion and increased STAT1 and STAT4 expression and activation
PRRs on myeloid-derived cells are critical for recognition and responses to microbes by the host, and these responses are essential for intestinal immune homeostasis (1); PRR-induced signaling and cytokine secretion are dramatically regulated by autocrine/ paracrine cytokines. We therefore assessed if the rs1517352 polymorphism in the STAT1/STAT4 region associated with IBD (3) regulates PRR-induced cytokine secretion in MDMs. We examined the PRR TLR4 using the ligand lipid A and measured IL-12 secretion. MDMs from disease-associated rs1517352 CC carriers secreted higher levels of IL-12 upon lipid A treatment (Fig. 1A). We observed similar results when examining IFN-γ secretion (Supplemental Fig. 1A). We also examined the anti-inflammatory cytokine IL-10 and found it underwent similar regulation (Fig. 1A), indicating that the rs1517352 polymorphism similarly regulates pro- and anti-inflammatory cytokines. We observed similar genotype-dependent cytokine regulation at more than one dose of lipid A treatment (Fig. 1A, Supplemental Fig. 1A). Given the broad range of PRRs to which cells are exposed to in intestinal tissues, we also examined TLR2-, TLR3- and TLR9-induced cytokine secretion and found a similar rs1517352 genotype–dependent regulation (Fig. 1B, Supplemental Fig. 1B).
FIGURE 1.

MDMs from rs1517352 CC disease risk carriers secrete increased levels of PRR-induced cytokines relative to AA carrier MDMs. Human MDMs from rs1517352 CC, CA, and AA carriers (n = 10 per genotype) were treated for 24 h with the indicated doses of (A) lipid A (TLR4 ligand), or (B) Pam3Cys (TLR2), poly(I:C) (TLR3), or CpG DNA (TLR9). Cytokine secretion + SEM. One-way ANOVA with Tukey posttest. *p < 0.05, **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5.
The rs1517352 polymorphism is located in an intronic region of STAT4, and a previous study found rs1517352 to be part of a haplotype that regulates STAT4 mRNA expression (10). However, given that the STAT1 gene is in close proximity to STAT4, and that both these proteins can regulate cytokine signaling (5), we asked if this polymorphism modulates PRR-induced cytokines through regulating the expression of these two genes as well as possibly other genes in the region. The coregulation of STAT1 and STAT4 might allow for these genes to cooperate through complementary functions. We first assessed if either STAT1 or STAT4 expression was regulated upon TLR4 stimulation. Neither STAT1 nor STAT4 mRNA (Supplemental Fig. 1C) nor protein (Supplemental Fig. 1D) expression was altered upon TLR4 stimulation. Consistent with the prior report (10), MDMs from CC carriers showed increased STAT4 mRNA expression compared with A carriers (Fig. 2B). Importantly, CC carrier MDMs also showed increased STAT1 mRNA expression compared with A carriers (Fig. 2A). Expression of additional detected genes located ~500 kb on each side of the polymorphism was not regulated by rs1517352 genotype (Supplemental Fig. 1E, Fig. 1F). We confirmed rs1517352 genotype–dependent regulation of STAT1 (Fig. 2C) and STAT4 (Fig. 2D) protein expression. Consistent with upregulated protein expression and PRR-induced cytokines, rs1517352 C carrier MDMs demonstrated increased TLR4-induced STAT1 (Fig. 2E) and STAT4 (Fig. 2F) activation relative to A carriers. Taken together, MDMs from immune-mediated disease-associated rs1517352 C carriers show increased STAT1 and STAT4 mRNA and protein expression and increased PRR-induced STAT1 and STAT4 activation and cytokine secretion relative to A carriers.
FIGURE 2.

MDMs from rs1517352 C disease risk carriers express higher levels of STAT1 and STAT4 and show increased TLR4-induced STAT1 and STAT4 activation relative to A carriers. (A–D) Human MDMs from rs1517352 CC, CA, and AA carriers were assessed for the following: (A) STAT1 and (B) STAT4 mRNA expression (n = 10 per genotype) or (C) STAT1 and (D) STAT4 protein expression with (left) representative flow cytometry and mean fluorescence intensity (MFI) values shown and (right) summarized data (n = 8 per genotype). (E and F) MDMs from rs1517352 CC, CA, and AA carriers (n = 10 per genotype) were treated for 30 min with 0.1 μg/ml lipid A. Fold (E) STAT1 and (F) STAT4 phosphorylation was assessed with representative flow cytometry and summarized data. Mean + SEM. One-way ANOVA with Tukey posttest. *p < 0.05, **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5.
STAT1 and STAT4 are required for optimal PRR-induced cytokine secretion from MDMs
Given the rs1517352 genotype–dependent modulation of STAT1 and STAT4 expression (Fig. 2) and of PRR-induced cytokines (Fig. 1), we directly assessed if STAT1 and STAT4 regulate PRR-induced cytokine secretion in human MDMs. Upon effective knockdown of STAT1 and STAT4 (Supplemental Fig. 2A) and ensuring cell viability was intact under these knockdown conditions (Supplemental Fig. 2B), we examined cytokine secretion following TLR4 stimulation. As we surmised that the ability of STAT1 and STAT4 to regulate PRR-induced outcomes would be through autocrine/paracrine cytokine loops, we examined select cytokines that signal in response to or are regulated by STAT1 and STAT4, including IL-6, IL-12, IFN-γ, and IFN-γ (5). As pro- and anti-inflammatory cytokines can be differentially regulated, we also examined the anti-inflammatory cytokine IL-10. Both STAT1 and STAT4 knockdown led to a reduction in TLR4-induced secretion of these cytokines (Fig. 3A). Relative to individual knockdown of these STAT proteins, combined STAT1 and STAT4 knockdown showed a stronger decrease in the cytokines examined (Fig. 3A). In contrast, knockdown of multiple other genes in the STAT1/STAT4 region (Supplemental Fig. 2C) did not affect TLR4-induced cytokine secretion (Supplemental Fig. 2D). To establish the contribution of STAT1 and STAT4 to TLR4-induced cytokines through an independent approach, we used a STAT1 inhibitor (fludarabine) (13) and a STAT4 inhibitor (lisofylline) (14–16), alone and in combination. Regulation was similar to that observed with the knockdown approach (Fig. 3B). We verified specificity of each inhibitor relative to other STAT family members (Supplemental Fig. 2E). Cell viability was intact with inhibitor usage (Supplemental Fig. 2F). The regulation of cytokine secretion by STAT1 and STAT4 was also observed upon stimulation of additional PRRs, including TLR2, TLR3, and TLR9 (Fig. 3C). Moreover, with knockdown of STAT1 and STAT4, secretion of both pro- and anti-inflammatory cytokines was reduced upon coculture of MDMs with the live bacteria S. Typhimurium (Fig. 3D). Taken together, these data demonstrate that STAT1 and STAT4 are required for secretion of cytokines upon stimulation of a broad range of PRRs and with live bacteria.
FIGURE 3.

STAT1 and STAT4 are required for optimal secretion of cytokines in MDMs upon stimulation through a broad range of PRRs. (A, C, and D) Human MDMs were transfected with scrambled siRNA or with STAT1 or STAT4 siRNA, alone or in combination, and then treated for 24 h with the following: (A) 0.1 μg/ml lipid A (TLR4) (n = 6, similar results seen in an independent n = 8), (C) 10 μg/ml Pam3Cys (TLR2), 100 μg/ml poly(I:C) (TLR3), or 10 μg/ml CpG DNA (TLR9) (n = 6, similar results seen in an independent n = 8), or (D) S. Typhimurium (n = 6). (B) MDMs were pretreated for 1 h with a STAT1 inhibitor (fludarabine) or a STAT4 inhibitor (lisofylline), alone or in combination, or with vehicle (DMSO) and then with 0.1 μg/ml lipid A for 24 h (n = 4). (A–D) Mean cytokine secretion + SEM. Significance is shown compared with stimulated, scrambled siRNA–transfected cells or stimulated, vehicle control-treated cells. t test with Bonferroni–Holm correction. **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. scr, scrambled; Tx, treatment.
Autocrine IFN-γ and IL-12 are required for optimal PRR-induced cytokine secretion from MDMs
Given the regulation of PRR-induced cytokines by STAT1 and STAT4, we hypothesized that the autocrine/paracrine IFN-γ, IFN-α, and IL-12 produced during PRR stimulation (Fig. 3), and which signal through STAT1 and STAT4 (5), respectively, would cooperate as mechanisms amplifying secretion of additional cytokines through these STAT family members upon PRR stimulation. We therefore knocked down receptors required for IFN-γ, IFN-α, and IL-12 signaling (Supplemental Fig. 3A), ensured cell survival was intact (Supplemental Fig. 3B), and measured cytokine secretion. A decrease in the expression of either IFN-γR1 or IL-12Rβ2 dramatically reduced TLR4-induced cytokine secretion, whereas knockdown of IFNAR did not affect this cytokine secretion (Fig. 4). We ensured functional efficacy of the IFNAR knockdown as it prevented IFN-α–induced cytokines (Supplemental Fig. 3C). These data suggest that although IFN-α induces cytokines from human MDMs and is produced upon TLR4 stimulation, autocrine IFN-α is not necessary for TLR4-induced cytokines. We also examined if various other cytokines reported to signal through either STAT1 or STAT4 might be contributing in an autocrine/paracrine manner to TLR4-induced cytokines by knocking down either the cytokines or their respective receptors (Supplemental Fig. 3D). These autocrine/ paracrine cytokines either did not contribute (Supplemental Fig. 3E, left) or did not contribute to the same degree (Supplemental Fig. 3E, right) as did IFN-γ and IL-12 to TLR4-induced cytokines. Taken together, both IFN-γ and IL-12 feedback in an autocrine/ paracrine manner to promote PRR-induced cytokines. We will, therefore, focus on IFN-γ and IL-12 autocrine/paracrine signaling in the studies that follow.
FIGURE 4.

Autocrine/paracrine IL-12 and IFN-γ secretion promotes TLR4-induced cytokine secretion. Human MDMs (n = 6, similar results in an additional six donors) were transfected with scrambled, IFNGR1, IFNAR, or IL-12Rβ2 siRNA and then treated with 0.1 μg/ml lipid A for 24 h. Mean cytokine secretion + SEM. Significance is shown compared with scrambled siRNA–transfected, lipid A–treated cells. ***p < 0.001, †p < 1 × 10−4. scr, scrambled; Tx, treatment.
IFN-γ and IL-12 signal through STAT1 and STAT4, respectively, and this activation is required for secretion of additional cytokines in human MDMs
Given the important role for autocrine IFN-γ and IL-12 in TLR4-induced cytokines (Fig 4), we sought to more clearly define if these cytokines functioned in an autocrine/paracrine manner to regulate TLR4-induced STAT1 and STAT4 activation. We therefore first assessed the relative activation of these STAT family members by IFN-γ and IL-12 treatment in human MDMs. IFN-γ preferentially activated STAT1, and IL-12 preferentially activated STAT4 (Fig. 5A). Consistent with this, upon knockdown of IFNGR1 in MDMs to prevent autocrine/paracrine IFN-γ signaling, lipid A-induced STAT1 activation was significantly reduced, whereas lipid A-induced STAT4 activation was less affected (Fig. 5B). With IL-12Rβ2 knockdown to prevent IL-12 autocrine/ paracrine signaling, lipid A-induced STAT4 activation was reduced to a greater degree than was STAT1 activation (Fig. 5B). Given the preferential STAT1 and STAT4 activation by IFN-γ and IL-12, respectively, we next sought to determine how each of these STAT members, in turn, regulated IFN-γ– and IL-12–induced cytokine secretion. Optimal IFN-γ–induced cytokine secretion required STAT1 in MDMs, whereas STAT4 was minimally required (Fig. 5C). To determine if a similar selective STAT1 dependency was observed with type I IFNs, we examined IFN-α–induced cytokines and found a similar dependency on STAT1, but not STAT4 (Fig. 5C). In contrast, both STAT4 and STAT1 knockdown decreased IL-12–induced cytokine secretion, although the reduction was generally greater with STAT4 knockdown (Fig. 5C). These data suggest that the STAT4-dependent IL-12–induced IFN-γ then acts in an autocrine/paracrine manner to signal through STAT1, such that both STAT1 and STAT4 cooperate in a complementary manner to mediate IL-12–induced cytokine secretion in MDMs. Finally, given that with PRR stimulation, early STAT1 and STAT4 signaling depended on autocrine IFN-γ and IL-12, respectively, we assessed if IFN-γ and IL-12 were secreted early after lipid A treatment. As autocrine/paracrine cytokines are consumed, which can make the low levels of cytokines present at early time points difficult to detect, we used neutralizing Abs to the respective receptors to minimize this consumption. Low levels of both IFN-γ and IL-12 were observed in the supernatant 15 min after lipid A treatment (Supplemental Fig. 3F). Consistent with this early protein secretion, blocking transcription with actinomycin did not affect the early, low levels of these cytokines in supernatants (Supplemental Fig. 3F); the actinomycin was effective as accumulation of cytokines at 24 h was impaired (data not shown). Taken together, autocrine IFN-γ and IL-12 signaling promote TLR4-induced STAT1 and STAT4 activation, respectively, and this activation in turn is required for optimal secretion of additional TLR4-induced cytokines.
FIGURE 5.

IFN-γ and IL-12 activate STAT1 and STAT4, respectively, and this activation contributes to subsequent cytokine secretion. (A) Human MDMs (n = 6; similar results in an additional six donors) were treated with 10 ng/ml IFN-γ or 10 ng/ml IL-12 for 60 min. Fold STAT1 and STAT4 phosphorylation. 0.1 μg/ml lipid A is shown as a control. (B) MDMs (n = 4) were transfected with scrambled, IFNGR1, or IL-12Rγ2 siRNA, and then treated with 0.1 μg/ml lipid A for 30 min. Fold STAT1 and STAT4 phosphorylation. (C) MDMs were transfected with scrambled, STAT1, or STAT4 siRNA. Cells were then treated with 10 ng/ml IFN-γ, IFN-α, or IL-12 for 24 h. Cytokine secretion (n = 8; similar results in an additional eight donors). Mean + SEM. Significance with t test in (A) and with Bonferroni-Holm correction in (B) and (C). *p < 0.05, **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. scr, scrambled; Tx, treatment.
IFN-γ and IL-12 treatment results in a low level of MAPK and NF-κB signaling in MDMs
Autocrine/paracrine IFN-γ and IL-12 were required for optimal levels of TLR4-induced cytokine secretion (Fig. 4), but STAT1 or STAT4 knockdown only partially reduced secretion of additional cytokines upon IFN-γ and IL-12 treatment (Fig. 5) or upon PRR stimulation (Fig. 3). We therefore hypothesized that IFN-γ and IL-12 signal through other pathways in addition to STAT1 and STAT4, which then contribute to PRR-induced cytokine secretion. Given the important role of MAPK and NF-κB pathways in PRR-induced cytokines (17), we examined these pathways. Both IFN-γ and IL-12 induced ERK, p38, JNK, and IκBα activation in MDMs (Fig. 6A). Consistently, knockdown of each of these signaling pathways (Supplemental Fig. 3G) resulted in a partial decrease IFN-γ− and IL-12−induced cytokine secretion (Fig. 6B). Cell viability was intact under these knockdown conditions (Supplemental Fig. 3H). Taken together, in addition to STAT1 and STAT4, MAPK and NF-κB pathways are also required for optimal IFN-γ− and IL-12−induced cytokine secretion.
FIGURE 6.

IL-12 and IFN-γ activate MAPK and NF-κB pathways. (A) MDMs (n = 6, similar results in an additional six donors) were treated with 10 ng/ml IFN-γ or 10 ng/ml IL-12 for 30 min. (Left) Representative flow cytometry of phosphoproteins with mean fluorescence intensity (MFI) values shown. (Right) Summary of fold phosphorylation of MAPKs and IκBα. (B) MDMs (n = 6, similar results in an additional eight donors) were transfected with scrambled or the indicated siRNA. Cells were treated with 10 ng/ml IFN-γ or 10 ng/ml IL-12 for 24 h. Cytokine secretion. Mean + SEM. Significance is shown compared with cytokine-treated, scrambled siRNA-transfected cells. *p < 0.05, **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. scr, scrambled; Tx, treatment.
STAT1 and STAT4 are required for S. Typhimurium–induced cytokines in human intestinal myeloid cells
The rs1517352 variant in the STAT1/STAT4 region is associated with IBD (3), a disease characterized by dysregulated cytokines in intestinal tissues (18). We therefore next assessed if STAT1 and STAT4 regulate cytokine secretion by human intestinal myeloid cells upon microbial exposure. Intestinal myeloid-derived cells secrete very low levels of cytokines upon microbial ligand exposure compared with peripheral myeloid cells (19, 20), but live S. Typhimurium coculture induces cytokines in intestinal myeloid-derived cells (21, 22). We used the STAT1 and STAT4 inhibitors described in Fig. 3B and Supplemental Fig. 2E, 2F. Intestinal myeloid cells cocultured with S. Typhimurium secreted reduced levels of IL-β and IL-8 following both STAT1 and STAT4 inhibition (Fig. 7). Reduction was more pronounced with combined inhibition of these pathways (Fig. 7). We confirmed that S. Typhimurium–induced IL-1β and IL-8 secretion was reduced in peripheral MDMs upon STAT1 and STAT4 inhibition (Fig. 7). Taken together, STAT1 and STAT4 are required for cytokine secretion in human intestinal myeloid cells following coculture with live S. Typhimurium.
FIGURE 7.

STAT1 and STAT4 promote S. Typhimurium–induced cytokine secretion in human intestinal myeloid cells. Human intestinal myeloid cells (n = 6 donors) or peripheral MDMs (n = 6 donors) were preincubated for 1 h with either fludarabine (STAT1 inhibitor) or lisofylline (STAT4 inhibitor), alone or in combination, and then cocultured with S. Typhimurium (S. Typhim) at MOI 10:1 for 24 h. Cytokine secretion + SEM. Significance with t test with Bonferroni–Holm correction. ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. Inh, inhibitor.
The rs1517352 variant in the STAT1/STAT4 region modulates IFN-γ– and IL-12–induced signaling and cytokines
Given the increased TLR4-induced STAT1 and STAT4 activation in rs1517352 CC risk carrier MDMs (Fig. 2E, 2F), and the role of autocrine/paracrine IFN-γ and IL-12 in TLR4-induced cytokines (Fig. 4), we assessed if MDMs from rs1517352 CC risk carriers also show increased IFN-γ–induced STAT1 activation and IL-12–induced STAT4 activation relative to AA carriers. We found this to be the case (Fig. 8A). Consistently, rs1517352 CC risk carrier MDMs demonstrated increased IFN-γ– and IL-12–induced cytokines relative to AA carriers (Fig. 8B). MDMs from heterozygote carriers generally showed intermediate results (Fig. 8). Therefore, MDMs from immune-mediated disease-associated rs1517352 CC risk carriers show increased IFN-γ– and IL-12–induced STAT1 and STAT4 activation and cytokine secretion relative to AA carriers.
FIGURE 8.

MDMs from rs1517352 CC disease risk carriers demonstrate increased IFN-γ– and IL-12–induced signaling and cytokines relative to AA carrier MDMs. (A) Human MDMs from rs1517352 CC, CA, and AA carriers were treated with 10 ng/ml IFN-γ or 10 ng/ml IL-12. (A) Fold STAT1 and STAT4 phosphorylation at 60 min (n = 8 per genotype). (B) Cytokine secretion at 24 h (n = 12 per genotype). Mean + SEM. Significance with one-way ANOVA with Tukey posttest. *p < 0.05, **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5.
STAT1 and STAT4 regulate TLR4-induced cytokines in a manner distinct to JAK members or STAT3
STATs can mediate signaling to both proinflammatory and anti-inflammatory cytokines. STAT3 is a key protein for IL-10 signaling, and we and others have found that with STAT3 deletion in myeloid cells PRR-induced proinflammatory cytokines can in fact increase in the context of reduced negative feedback from anti-inflammatory molecules (23, 24). In contrast, with STAT1 and STAT4 knockdown, we have found that TLR4-induced proinflammatory cytokines are decreased despite the reduction in IL-10 secretion, suggesting that STAT1 and STAT4 play a more important role in regulating inflammatory cytokines. Consistently, preventing autocrine IL-10 negative feedback through effectively knocking down IL-10RA (Supplemental Fig. 4A) in STAT1-/STAT4-deficient MDMs resulted in a dramatic increase in proinflammatory cytokines (Fig. 9A).
FIGURE 9.

JAK1 and TYK2 regulate TLR4-induced cytokines in a manner distinct to STAT1 and STAT4. (A) MDMs were transfected with scrambled, STAT1 and STAT4, or IL10RA (to block autocrine IL-10), alone or in combination, and then treated with 0.1 μg/ml lipid A for 24 h. Cytokine secretion (n = 6). (B–E) MDMs were pretreated with vehicle (DMSO) or the indicated doses of either (B and C) Upadacitinib (Upa; JAK1 inhibitor) or (D and E) BMS-986165 (BMS; TYK2 inhibitor) for 1 h. (B and D) Fold induction of the indicated phosphoproteins with 0.1 μg/ml lipid A for 15 min (n = 5; similar results in an additional n = 4) (earlier time point assessed to minimize signaling through autocrine/paracrine cytokine loops). (C and E) Cells were treated with 0.1 μg/ml lipid A or 10 ng/ml IFN-γ or IL-12. Cytokines at 24 h (n = 6; similar results in an additional n = 4 for lipid A). (F) MDMs were transfected with scrambled, JAK1, TYK2, or STAT3 siRNA, and then treated with 0.1 μg/ml lipid A for 24 h. Cytokine secretion (n = 6). Mean + SEM. Significance is to scrambled siRNA–transfected, lipid A–treated cells for (F), to vehicle and cytokine- or lipid A–treated cells in (C) and (E), or as indicated. t test for (A) combined with Bonferroni–Holm correction for (B)–(F). *p < 0.05, **p < 0.01, ***p < 0.001, †p < 1 × 10−4. scr, scrambled; Tx, treatment.
We had found that TLR4-induced autocrine IFN-γ and IL-12 signal through STAT1 and STAT4, respectively. With respect to upstream JAKs, IFN-γ preferentially signals through JAK1 and IL-12 preferentially signals through TYK2; note that these JAK family members mediate signaling to a wide range of cytokines. We and others have previously reported that with deletion of the upstream JAK members in myeloid cells, PRR-induced proinflammatory cytokines, in fact, increase in the context of reduced negative feedback from anti-inflammatory molecules (25–27). We had found that this outcome depends on the threshold of JAK signaling (27). Therefore, JAK1 and TYK2 do not phenocopy STAT1 and STAT4 regulation, despite the overlap in mediating IFN-γ and IL-12 responses. We confirmed the differential regulation by JAK1 and TYK2 through an alternative approach using inhibitors. Upadacitinib is a JAK1 inhibitor that is an effective treatment for rheumatoid arthritis (28, 29) and is being investigated as a therapy for various other immune-mediated diseases, including ulcerative colitis (30) and Crohn disease (31) which recently showed efficacy in Phase 2 trials. We confirmed progressive reduction in JAK1 phosphorylation with increasing Upadacitinib doses (Fig. 9B). At higher doses, Upadacitinib can also inhibit JAK2; we observed slight reduction in TLR4-induced JAK2 activation at the highest selected dose of Upadacitinib (Fig. 9B). BMS-986165 is a TYK2 inhibitor that is similarly being investigated in various immune-mediated diseases. We confirmed progressive reduction in TYK2 phosphorylation with increasing BMS-986165 doses (Fig. 9D). At higher doses, BMS-986165 can also inhibit JAK1; we observed slight reduction in TLR4-induced JAK1 activation at the highest selected dose of BMS-986165 (Fig. 9D). Cell viability was intact with both these inhibitors (Supplemental Fig. 4B). With lower doses of each of these inhibitors, both TLR4-induced proinflammatory and anti-inflammatory cytokines decreased (Fig. 9C, 9E). However, at higher levels of JAK1 and TYK2 inhibition, proinflammatory cytokines increased, whereas IL-10 progressively decreased (Fig. 9C, 9E). Consistent with these results, upon knockdown of JAK1 and TYK2 using siRNA (Supplemental Fig. 4C, 4D), we observed increased proinflammatory cytokines in the context of reduced IL-10 secretion (Fig. 9F). We also confirmed a similar reciprocal pattern of regulation with STAT3 knockdown (Fig. 9F, Supplemental Fig. 4C, 4D). We further confirmed relative selectively for IFN-γ–induced signaling through JAK1 (Fig. 9C) and IL-12–induced signaling through TYK2 (Fig. 9E). Interestingly, in contrast to PRR stimulation, when stimulating with either IFN-γ or IL-12, higher levels of inhibition of these JAK family members did not lead to reciprocal regulation of proinflammatory and anti-inflammatory cytokines (Fig. 9C, 9E). Therefore, JAK family members and STAT3 demonstrate a pattern of TLR-induced cytokine regulation in human macrophages wherein once expression/activity falls below a threshold, proinflammatory cytokines increase in the context of a progressive reduction in anti-inflammatory cytokines; this is distinct to the pattern of regulation observed with STAT1 and STAT4.
STAT1 and STAT4 promote TLR4-induced bacterial uptake
Macrophages play a key role in bacterial clearance. As IFN-γ and IL-12 can promote antimicrobial pathways in macrophages, we next assessed the role of STAT1 and STAT4 in mediating antimicrobial outcomes. The initial step in bacterial clearance requires uptake of bacteria. STAT1 or STAT4 knockdown, either alone or in combination, did not alter the low level of uptake of S. Typhimurium–GFP in MDMs under baseline conditions (Supplemental Fig. 4E). Similar results were observed with uptake of E. coli bioparticles (Supplemental Fig. 4E). Cells in the intestinal environment are continuously exposed to microbial products, and we (32) and others (33, 34) have found that prolonged PRR stimulation can enhance antimicrobial pathways. We therefore examined MDMs after chronic PRR stimulation. We confirmed increased bacterial uptake after treatment of MDMs with lipid A for 48 h (Fig. 10). STAT1 and STAT4 were required for this TLR4-enhanced bacterial uptake (Fig. 10), and they cooperated in this process (Fig. 10). Prolonged IFN-γ and IL-12 treatment also increased bacterial uptake (Fig. 10). IFN-γ–enhanced bacterial uptake required STAT1, but not STAT4 (Fig. 10). In contrast, IL-12–enhanced bacterial uptake required both STAT1 and STAT4 (Fig. 10), consistent with the STAT4-dependent, IL-12–induced IFN-γ secretion, which then feeds back to activate STAT1 (Fig. 5), in particular under these prolonged treatment conditions. Taken together, STAT1 and STAT4 promote TLR4-, IFN-γ−, and IL-12–induced bacterial uptake.
FIGURE 10.

STAT1 and STAT4 are required for optimal TLR4-induced bacterial uptake. MDMs were transfected with scrambled siRNA or with STAT1 or STAT4 siRNA, alone or in combination (comb). Cells were then treated for 48 h with 0.1 μg/ml lipid A, 10 ng/ml IFN-γ, or 10 ng/ml IL-12. Uptake of S. Typhimurium–GFP or E. coli–FITC was assessed (n = 10 from two independent experiments) as per Materials and Methods. Representative flow cytometry and summary graphs. Mean fluorescence intensity (MFI) + SEM. t test with Bonferroni–Holm correction. ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. NT, no treatment; scr, scrambled; Tx, treatment.
STAT1 and STAT4 are required for TLR4-enhanced bacterial clearance
We next assessed if STAT1 and STAT4 regulate clearance of intracellular bacteria in macrophages. In untreated MDMs, knockdown of STAT1 and STAT4 led to reduced clearance of S. Typhimurium (Fig. 11A). Similar results were observed with AIEC (Fig. 11A), which are increased in the ilea of Crohn disease patients (35). Furthermore, upon TLR4 stimulation, despite the reduced uptake of bacteria in STAT1- and STAT4-deficient MDMs (Fig. 10), both STAT1- and STAT4-deficient MDMs were less able to clear the lower levels of bacteria that had been taken up (Fig. 11B). STAT1 and STAT4 cooperated in mediating this intracellular bacterial clearance (Fig. 11B). Prolonged IFN-γ treatment similarly increased bacterial clearance, and this required STAT1 (Fig. 11B), whereas IL-12–enhanced intracellular bacterial clearance required both STAT1 and STAT4 (Fig. 11B).
FIGURE 11.

STAT1 and STAT4 are required for optimal intracellular bacterial clearance. MDMs were transfected with scrambled siRNA or with STAT1 or STAT4 siRNA, alone or in combination (comb). Cells were then (A) left untreated (n = 6) or (B) treated with 0.1 μg/ml lipid A, 10 ng/ml IFN-γ, or 10 ng/ml IL-12 for 48 h (n = 6; similar results in an additional n = 4). Intracellular bacterial clearance. Mean CFU + SEM. Significance with t test with Bonferroni–Holm correction. **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. scr, scrambled; Tx, treatment.
To assess mechanisms mediating STAT1- and STAT4-dependent intracellular bacterial clearance, we considered antimicrobial pathways that these signaling proteins might be promoting. We first assessed ROS given the important role ROS production plays in mediating bacterial clearance (1). The low level of ROS in untreated macrophages was not regulated by STAT1 or STAT4 (data not shown). However, upon TLR4 stimulation, both STAT1 and STAT4, and particularly in combination, were required for ROS production in MDMs (Fig. 12A). Reactive nitrogen species (RNS) can also contribute to bacterial clearance, and a combination of ROS and RNS pathways is central in maintaining homeostasis in the intestinal mucosa (36). Both STAT1 and STAT4 were required for TLR4-induced NOS2 expression in MDMs (Fig. 12B). Autophagy is another key bacterial clearance mechanism induced with PRR stimulation (37), and polymorphisms in the autophagy-associated gene ATG16L1 confer altered susceptibility to Crohn disease (3). STAT1 and STAT4, particularly in combination, were required for TLR4-induced expression of the autophagy marker LC3II (Fig. 12C). Moreover, STAT1 was required for IFN-γ induction, and both STAT1 and STAT4 were required for IL-12 induction of each of these antimicrobial pathways. Taken together, STAT1 and STAT4 are required for TLR4-induced ROS, RNS, autophagy, and intracellular bacterial clearance in human MDMs.
FIGURE 12.

STAT1 and STAT4 promote TLR4-induced antimicrobial pathways. MDMs were transfected with scrambled siRNA or with STAT1 or STAT4 siRNA, alone or in combination (comb). Cells were then left untreated or treated with 0.1 μg/ml lipid A, 10 ng/ml IFN-γ or 10 ng/ml IL-12 for 48 h (n = 6) and assessed by flow cytometry for the following: (A) ROS production, (B) NOS2 expression, or (C) LC3II expression. Representative flow cytometry and summary graphs (mean fluorescence intensity [MFI]). Mean + SEM. Significance with t test with Bonferroni–Holm correction. ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. NT, no treatment; scr, scrambled; Tx, treatment.
MDMs from rs1517352 C risk carriers in the STAT1/STAT4 region demonstrate increased microbial-induced cytokines and TLR4-induced antimicrobial pathways
We next assessed if human MDMs from rs1517352 C IBD risk carriers in the STAT1/STAT4 region that show increased STAT1 and STAT4 expression and TLR-induced cytokines might be at an advantage with respect to induction of antibacterial mechanisms. We first assessed S. Typhimurium-induced proinflammatory (TNF, IL-1β) and anti-inflammatory (IL-10) cytokines and found that similar to TLR stimulation, MDMs from rs1517352 C IBD risk carriers showed increased S. Typhimurium-induced cytokine secretion compared AA carriers (Fig. 13A). In addition to the previously examined cytokines (Fig. 1), TLR4-induced TNF and IL-1β were also increased in MDMs from rs1517352 C IBD risk carriers (Fig. 13B). We further found that TLR4-enhanced uptake of S. Typhimurium and E. coli bioparticles was increased in MDMs from rs1517352 C IBD risk carriers compared with AA carriers (Fig. 14). Finally, rs1517352 C IBD risk MDMs demonstrated increased TLR4-induced ROS (Fig. 15A), RNS (Fig. 15B), and autophagy (Fig. 15C), and in turn, increased intracellular clearance of Enterococcus faecalis, AIEC, and S. Typhimurium (Fig. 15D) compared with AA carriers. Therefore, MDMs from rs1517352 C IBD risk carriers more effectively induce antimicrobial pathway responses.
FIGURE 13.

MDMs from rs1517352 CC disease risk carriers secrete increased levels of S. Typhimurium–induced cytokines relative to AA carrier MDMs. Human MDMs from rs1517352 CC, CA, and AA carriers (n = 10 per genotype) were treated for 24 h with (A) S. Typhimurium or (B) 0.1 μg/ml lipid A. Cytokine secretion + SEM. Significance with one-way ANOVA with Tukey posttest. *p < 0.05, **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. Tx, treatment.
FIGURE 14.

MDMs from rs1517352 CC disease risk carriers demonstrate increased bacterial uptake relative to AA carrier MDMs. Human MDMs from rs1517352 CC, CA, and AA carriers (n = 10 per genotype) were left untreated or treated for 48 h with 0.1 μg/ml lipid A, and then assessed for bacterial uptake. (Left) Representative flow cytometry with mean fluorescence intensity (MFI). (Right) Summary of S. Typhimurium or E. coli uptake. Mean + SEM. Significance with one-way ANOVA with Tukey posttest. *p < 0.05, **p < 0.01, †p < 1 × 10−4, ††p < 1 × 10−5. NT, no treatment; Tx, treatment.
FIGURE 15.
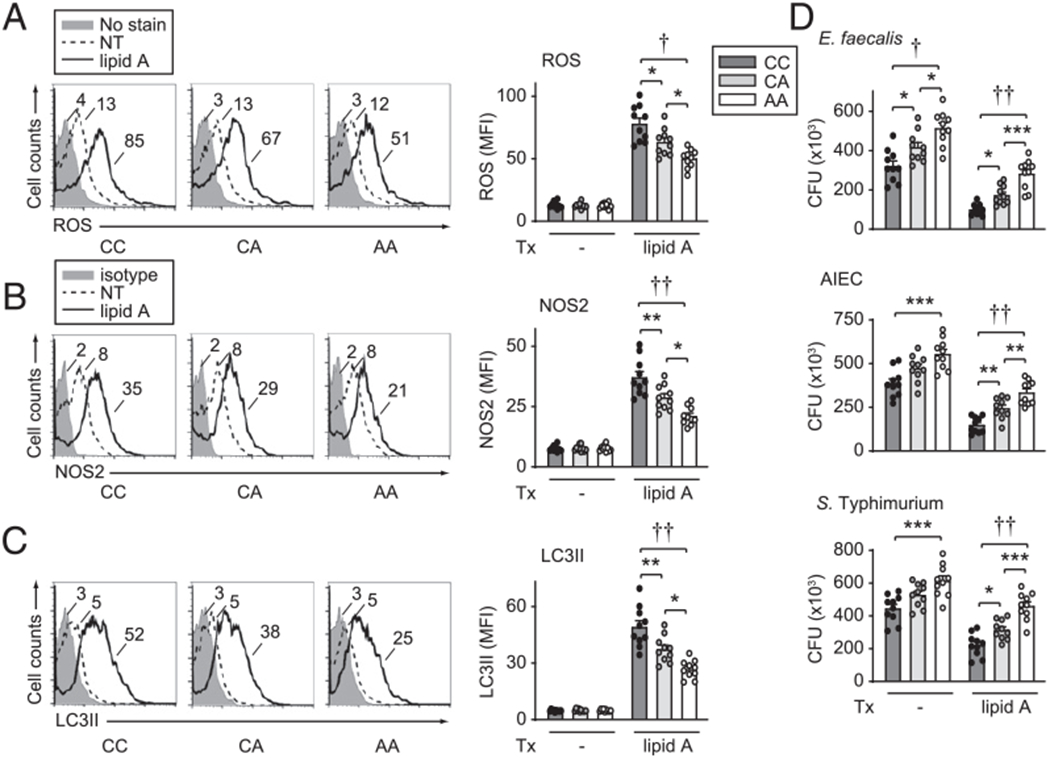
MDMs from rs1517352 CC disease risk carriers demonstrate increased intracellular bacterial clearance relative to AA carrier MDMs. Human MDMs from rs1517352 CC, CA, and AA carriers (n = 10 per genotype) were left untreated or treated for 48 h with 0.1 μg/ml lipid A. (A) ROS production. (B) NOS2 expression. (C) LC3II expression. Representative flow cytometry and summary graphs (mean fluorescence intensity [MFI]). (D) Intracellular bacterial clearance (CFU). Mean + SEM. Significance with one-way ANOVA with Tukey posttest. *p < 0.05, **p < 0.01, ***p < 0.001, †p < 1 × 10−4, ††p < 1 × 10−5. NT, no treatment; Tx, treatment.
Discussion
In this study, we identify that STAT1 and STAT4 are required for optimal secretion of multiple cytokines upon stimulation of a broad range of PRRs and by live bacteria in human MDMs, and we establish that autocrine/paracrine IFN-γ and IL-12 are mechanisms contributing to these outcomes. We further identify that STAT1 and STAT4 are required for optimal TLR-, IFN-γ– and IL-12–enhanced uptake and clearance of bacteria. Importantly, the rs1517352 C variant in the STAT1/STAT4 region conferring increased risk for IBD (3) increases expression and activation of both STAT1 and STAT4, and enhances TLR-, bacterial-, IFN-γ– and IL-12–induced cytokines and TLR-induced antimicrobial pathways in MDMs compared with A carrier MDMs. We find that IL-12 preferentially activates STAT4, and STAT4 is required for optimal levels of IL-12–induced IFN-γ secretion, with IFN-γ preferentially activating STAT1 to mediate secretion of a range of cytokines. Therefore, STAT1 and STAT4 are coregulated from from a common genetic locus and then cooperate functionally in a complementary manner. We further determine that IFN-γ– and IL-12–mediated cytokine secretion in human myeloid cells requires not only STAT1 and STAT4 signaling but is also dependent on MAPK and NF-κB pathways. Moreover, we find that STAT1 and STAT4 are required for cytokine secretion by intestinal myeloid cells following live S. Typhimurium coculture. Taken together, this study elucidates mechanisms for how immune disease-associated polymorphisms in the STAT1/STAT4 region modulate cytokine secretion and antimicrobial pathways in macrophages; STAT1 and STAT4 are coregulated in expression and then cooperate to regulate outcomes, thereby highlighting a previously undefined interaction of STAT1 and STAT4 in the immune response (Supplemental Fig. 4F).
Both the STAT1 and STAT4 pathways are elevated in IBD patients (38–40). Studies examining experimental models of colitis have found that transgenic STAT4 expression can promote inflammatory CD4+ T cell responses (41). Consistent with these animal studies, our findings show that the rs1517352 C variant in the STAT1/STAT4 region–conferring–increased risk for IBD leads to increased expression of both STAT1 and STAT4 and, in turn, increased PRR-induced signaling and cytokines in human macrophages. Conversely, individuals with STAT1/STAT4 variants leading to reduced STAT1 and STAT4 expression and activation and reduced inflammatory innate responses highlight potential benefits for inhibiting this pathway in IBD patients. The specific mechanism through which the rs1517352 variant, or potentially a variant in linkage disequilibrium with rs1517352, modulates gene expression is not known. rs1517352 is within a region that might serve as a binding site for aae-miR-11901, although this would need to be tested experimentally. Of note, these findings highlight that the STAT1 and STAT4 pathways do not demonstrate a threshold of signaling in innate immune cells below which one observes a reciprocal inflammatory outcome wherein proinflammatory cytokines increase as anti-inflammatory cytokines decrease, as has been observed with JAK inhibition (27) or with STAT3 deficiency (23); we confirm the distinct regulation of JAK and STAT3 pathways in the current study. Therefore, despite the ability of STAT1 and STAT4 to regulate both inflammatory and anti-inflammatory mediators in myeloid cells in response to microbial products, the dominant effect is that on inflammatory mediators.
Polymorphisms in the JAK/STAT pathway, including JAK2, TYK2, STAT3, STAT5, STAT1, and STAT4, have been associated with multiple immune-mediated diseases, including IBD (3), indicating that this pathway has a central role in mediating immune homeostasis. Consistent with this, JAK pathway inhibitors have been shown to be effective in treatment of rheumatoid arthritis, psoriasis, and ulcerative colitis (42, 43). The challenge in designing therapies targeting these pathways is understanding how different JAK/STAT members affect signaling and outcomes by specific PRRs and cytokines, how these outcomes vary in different cell types, and at what threshold of signaling various JAKs and STATs decrease or increase specific outcomes (5, 27). Our study identifies roles and mechanisms through which STAT1- and STAT4-immune disease-associated polymorphisms regulate signaling, cytokine secretion, and antimicrobial pathways in myeloid cells, provides further clarification on how these genes might contribute to immune homeostasis and disease pathogenesis, and identifies these pathways as potential beneficial targets in immune-mediated diseases.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants R01DK099097 and R01DK106593 and an ASPIRE Inflammatory Bowel Disease Pfizer Research Award.
Abbreviations used in this article:
- AIEC
adherent-invasive Escherichia coli
- IBD
inflammatory bowel disease
- MDM
monocyte-derived macrophage
- MOI
multiplicity of infection
- poly(I:C)
polyinosinic-polycytidylic acid
- PRR
pattern-recognition receptor
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- SLE
systemic lupus erythematosus
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Abraham C, and Medzhitov R. 2011. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 140: 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. ; Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 110: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. ; International IBD Genetics Consortium (IIBDGC). 2012. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abelson AK, Delgado-Vega AM, Kozyrev SV, Sanchez E, Velazquez-Cruz R, Eriksson N, Wojcik J, Linga Reddy MV, Lima G, D’Alfonso S, et al. ; AADEA group. 2009. STAT4 associates with systemic lupus erythematosus through two independent effects that correlate with gene expression and act additively with IRF5 to increase risk. Ann. Rheum. Dis 68: 1746–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, and Laurence A. 2015. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu. Rev. Med 66: 311–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, and Mansell A. 2014. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol. Cell Biol 92: 761–769. [DOI] [PubMed] [Google Scholar]
- 7.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, et al. 2003. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet 33: 388–391. [DOI] [PubMed] [Google Scholar]
- 8.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, et al. 2011. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med 208: 1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, and Niewold TB. 2009. Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J. Immunol 182: 34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigurdsson S, Nordmark G, Garnier S, Grundberg E, Kwan T, Nilsson O, Eloranta ML, Gunnarsson I, Svenungsson E, Sturfelt G, et al. 2008. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum. Mol. Genet 17: 2868–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hedl M,Lahiri A, Ning K, Cho JH, and Abraham C. 2014. Pattern recognition receptor signaling in human dendritic cells is enhanced by ICOS ligand and modulated by the Crohn’s disease ICOSLG risk allele. Immunity 40: 734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedl M, Yan J, and Abraham C. 2016. IRF5 and IRF5 disease-risk variants increase glycolysis and human M1 macrophage polarization by regulating proximal signaling and Akt2 activation. Cell Rep. 16: 2442–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frank DA, Mahajan S, and Ritz J. 1999. Fludarabine-induced immunosuppression is associated with inhibition of STAT1 signaling. Nat. Med 5: 444–447. [DOI] [PubMed] [Google Scholar]
- 14.Yang Z, Chen M, Fialkow LB, Ellett JD, Wu R, and Nadler JL. 2003. Inhibition of STAT4 activation by lisofylline is associated with the protection of autoimmune diabetes. Ann. N. Y. Acad. Sci 1005: 409–411. [DOI] [PubMed] [Google Scholar]
- 15.Coon ME, Diegel M, Leshinsky N, and Klaus SJ. 1999. Selective pharmacologic inhibition of murine and human IL-12-dependent Th1 differentiation and IL-12 signaling. J. Immunol 163: 6567–6574. [PubMed] [Google Scholar]
- 16.Chen M, Yang Z, Wu R, and Nadler JL. 2002. Lisofylline, a novel anti-inflammatory agent, protects pancreatic beta-cells from proinflammatory cytokine damage by promoting mitochondrial metabolism. Endocrinology 143: 2341–2348. [DOI] [PubMed] [Google Scholar]
- 17.Akira S, and Takeda K. 2004. Toll-like receptor signalling. Nat. Rev. Immunol 4: 499–511. [DOI] [PubMed] [Google Scholar]
- 18.Abraham C, and Cho JH. 2009. Inflammatory bowel disease. N. Engl. J. Med 361: 2066–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedl M, Li J, Cho JH, and Abraham C. 2007. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc. Natl. Acad. Sci. USA 104: 19440–19445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, and Smith PD. 2005. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Invest 115: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, Shaw MH, Kim YG, and Nunez G. 2012. NLRC4-driven production of IL-1b discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol 13: 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hedl M, and Abraham C. 2012. Nod2-induced autocrine interleukin-1 alters signaling by ERK and p38 to differentially regulate secretion of inflammatory cytokines. Gastroenterology 143: 1530–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, and Akira S. 1999. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 10: 39–49. [DOI] [PubMed] [Google Scholar]
- 24.Hedl M, Sun R, Huang C, and Abraham C. 2019. STAT3 and STAT5 signaling thresholds determine distinct regulation for innate receptor-induced inflammatory cytokines, and STAT3/ STAT5 disease variants modulate these outcomes. J. Immunol 203: 3325–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pattison MJ, Mackenzie KF, and Arthur JS. 2012. Inhibition of JAKs in macrophages increases lipopolysaccharide-induced cytokine production by blocking IL-10-mediated feedback. J. Immunol 189: 2784–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Brown J, Gao S, Liang S, Jotwani R, Zhou H, Suttles J, Scott DA, and Lamont RJ. 2013. The role of JAK-3 in regulating TLR-mediated inflammatory cytokine production in innate immune cells. J. Immunol 191: 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hedl M,Proctor DD, and Abraham C. 2016. JAK2 disease-risk variants are gain of function and JAK signaling threshold determines innate receptor-induced proinflammatory cytokine secretion in macrophages. J. Immunol 197: 3695–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burmester GR, Kremer JM, Van den Bosch F, Kivitz A, Bessette L, Li Y, Zhou Y, Othman AA, Pangan AL, and Camp HS. 2018. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 391: 2503–2512. [DOI] [PubMed] [Google Scholar]
- 29.Genovese MC, Fleischmann R, Combe B, Hall S, Rubbert-Roth A, Zhang Y, Zhou Y, Mohamed MF, Meerwein S, and Pangan AL. 2018. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet 391: 2513–2524. [DOI] [PubMed] [Google Scholar]
- 30.Sandborn WJ, Ghosh S, Panes J, Schreiber S, D’Haens G, Tanida S, Siffledeen J, Enejosa J, Zhou W, Othman AA, et al. 2020. Efficacy of upadacitinib in a randomized trial of patients with active ulcerative colitis. Gastroenterology 158: 2139–2149.e14. [DOI] [PubMed] [Google Scholar]
- 31.Sandborn WJ, Feagan BG, Loftus EV Jr., Peyrin-Biroulet L, Van Assche G, D’Haens G, Schreiber S, Colombel JF, Lewis JD, Ghosh S, et al. 2020. Efficacy and safety of upadacitinib in a randomized trial of patients with Crohn’s disease. Gastroenterology 158: 2123–2138.e8. [DOI] [PubMed] [Google Scholar]
- 32.Lahiri A, and Abraham C. 2014. Activation of pattern recognition receptors up-regulates metallothioneins, thereby increasing intracellular accumulation of zinc, autophagy, and bacterial clearance by macrophages. Gastroenterology 147: 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foster SL, Hargreaves DC, and Medzhitov R. 2007. Gene-specific control of inflammation by TLR-induced chromatin modifications. [Published erratum appears in 2008 Nature 451: 102.] Nature 447: 972–978. [DOI] [PubMed] [Google Scholar]
- 34.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, and Ghosh S. 2011. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472: 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, and Colombel JF. 2004. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127: 412–421. [DOI] [PubMed] [Google Scholar]
- 36.Shiloh MU, MacMicking JD, Nicholson S, Brause JE, Potter S, Marino M, Fang F, Dinauer M, and Nathan C. 1999. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 10: 29–38. [DOI] [PubMed] [Google Scholar]
- 37.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, and Green DR. 2007. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257. [DOI] [PubMed] [Google Scholar]
- 38.Mudter J, Weigmann B, Bartsch B, Kiesslich R, Strand D, Galle PR, Lehr HA, Schmidt J, and Neurath MF. 2005. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am. J. Gastroenterol 100: 64–72. [DOI] [PubMed] [Google Scholar]
- 39.Giles EM, Sanders TJ, McCarthy NE, Lung J, Pathak M, MacDonald TT, Lindsay JO, and Stagg AJ. 2017. Regulation of human intestinal T-cell responses by type 1 interferon-STAT1 signaling is disrupted in inflammatory bowel disease. Mucosal Immunol. 10: 184–193. [DOI] [PubMed] [Google Scholar]
- 40.Schreiber S, Rosenstiel P, Hampe J, Nikolaus S, Groessner B, Schottelius A, Kuhbacher T, Hamling J, Folsch UR, and Seegert D. 2002. Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease. Gut 51: 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wirtz S, Finotto S, Kanzler S, Lohse AW, Blessing M, Lehr HA, Galle PR, and Neurath MF. 1999. Cutting edge: chronic intestinal inflammation in STAT-4 transgenic mice: characterization of disease and adoptive transfer by TNF- plus IFN-gamma-producing CD4+ T cells that respond to bacterial antigens. J. Immunol 162: 1884–1888. [PubMed] [Google Scholar]
- 42.Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, and Niezychowski W, Study A3921063 Investigators. 2012. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med 367: 616–624. [DOI] [PubMed] [Google Scholar]
- 43.O’Shea JJ, and Plenge R. 2012. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36: 542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.