

Abstract
Cancer cells express high levels of PD-L1, a ligand of the PD-1 receptor on T cells, allowing tumors to suppress T cell activity. Clinical trials utilizing antibodies that disrupt the PD-1/PD-L1 checkpoint have yielded remarkable results, with anti-PD-1 immunotherapy approved as first-line therapy for lung cancer patients. We used CRISPR-based screening to identify regulators of PD-L1 in human lung cancer cells, revealing potent induction of PD-L1 upon disruption of heme biosynthesis. Impairment of heme production activates the integrated stress response (ISR), allowing bypass of inhibitory upstream open reading frames in the PD-L1 5′ UTR, resulting in enhanced PD-L1 translation and suppression of anti-tumor immunity. We demonstrated that ISR-dependent PD-L1 translation requires the translation initiation factor eIF5B. eIF5B overexpression, which is frequent in lung adenocarcinomas and associated with poor prognosis, is sufficient to induce PD-L1. These findings illuminate mechanisms of immune checkpoint activation and identify targets for therapeutic intervention.
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer-associated deaths worldwide, with limited effective treatments and frequent recurrence 1. Lung tumor cells frequently express high levels of Programmed Death Ligand 1 (PD-L1), a ligand of the PD-1 receptor on T-cells, allowing tumors to directly suppress the host immune response by inhibiting T-cell proliferation and function 2-5. Clinical trials utilizing monoclonal antibodies that disrupt the PD-1/ PD-L1 immune checkpoint have yielded remarkable results, with PD-1 immunotherapy approved as a first-line therapy for human lung cancer patients 6-8. Despite significant progress in targeting this pathway, the mechanisms through which PD-L1 is upregulated in non-small cell lung cancer (NSCLC) and other tumor types is incompletely understood. PD-L1 expression is induced by inflammatory cytokines such as IFN-γ or TNF-α from the tumor microenvironment 4,5 as well as oncogenic driver mutations 9-13. However, mutations in oncogenes including KRAS and ALK do not correlate with tumor PD-L1 expression nor response to immunotherapy 14. Thus, there is a critical need to identify PD-L1 regulators and clinically relevant biomarkers that predict response and resistance to immunotherapy, which may lead to new therapeutic strategies to trigger anti-cancer immune responses and improve clinical outcomes.
In response to diverse cellular stresses, eukaryotic cells activate the integrated stress response (ISR) pathway to re-establish homeostasis 15-17. The critical event that defines ISR activation is phosphorylation of the eukaryotic translation initiation factor 2 alpha (eIF2α) by kinases that sense distinct cellular stresses, leading to reduced global protein synthesis and increased translation of select mRNAs 16,18,19. Recent studies have linked eIF2α phosphorylation to the increased translation of oncogenic transcripts 20 and PD-L1 21 in skin squamous cell carcinoma and liver cancer, respectively. However, the role(s) of the ISR in regulating immune checkpoint proteins and the impact on immune suppression in lung cancer has not been previously investigated.
Here we describe the application of a genome-wide CRISPR screening approach to identify regulators of PD-L1 in human lung cancer cells, revealing potent induction of PD-L1 upon disruption of the heme biosynthesis pathway. Impairment of heme production activated the ISR, allowing bypass of inhibitory open reading frames (uORFS) in the PD-L1 5′ UTR. This culminated in increased PD-L1 translation and suppression of anti-tumor immune responses. Additional stresses that activate the ISR pathway similarly induced PD-L1 protein levels in human lung cancer cells. Moreover, we showed that the alternative translation initiation factor eIF5B is necessary for ISR-dependent PD-L1 translation in human lung cancer cells and syngeneic mouse models. Remarkably, eIF5B overexpression was sufficient to induce PD-L1 even in the absence of ISR activation. Given that eIF5B is commonly upregulated in human lung cancer patients and is associated with poor prognosis, these findings revealed an unanticipated mechanism of immune checkpoint activation in lung cancer with important therapeutic implications.
Results
CRISPR/Cas9 screening identifies the heme synthesis pathway as a regulator of PD-L1
To identify novel regulators of PD-L1 in NSCLC, we performed a genome-wide CRISPR/Cas9 loss of function screen in NCI-H358 (H358) human lung cancer cells (Fig. 1a). These cells express endogenous PD-L1 that could be suppressed by lentiviral expression of Cas9 and a sgRNA targeting PD-L1, or stimulated by IFN-γ treatment (Fig. 1b-c). To screen for new PD-L1 regulators, H358 cells were infected with a previously described genome-wide CRISPR/Cas9 lentiviral library 22. After 2 weeks of growth in puromycin, cells with the highest and lowest surface PD-L1 expression were collected by fluorescence activated cell sorting (FACS). sgRNA representation in the sorted and unsorted populations was assessed by high-throughput sequencing and genes targeted by multiple enriched guides in sorted populations were identified (Fig. 1d-e).
Fig. 1. A genome-wide CRISPR-Cas9 screen identifies regulators of PD-L1 in lung cancer cells.

a, Overview of CRISPR-Cas9 screening approach. b,c, Detection of loss (b) or gain (c) of PD-L1 in H358 cells by flow cytometry in cells expressing a sgRNA targeting PD-L1 (b) or cells treated with 10ng/mL IFN-γ for 24h (c). Data from a single experiment are shown in (b) and (c) and are representative of two independent experiments with 3 independent sgRNA with similar results. RIGER analysis identified positive (d) and negative (e) regulators of PD-L1. CD274 (PD-L1) is highlighted in red. Samples subject to RIGER analysis had cell culture replicates (n=2) as well as library replicates (GeCKO A and B). Genes with bold diamonds were selected for validation. Blue diamond indicates genes involved in mitochondrial function. RIGER analysis is described in Methods. f, Flow cytometry analysis of cell-surface PD-L1 in H1944 cells expressing Cas9 and a control sgRNA or sgRNA targeting MUL1, UQCR10, MRPS12, or UROD.
Among the most significant hits in cells expressing low levels of PD-L1 were PD-L1 (CD274) itself, as well as CKLF-like MARVEL transmembrane domain containing protein 6 (CMTM6), a recently described positive regulator of PD-L1 (Fig. 1d, Extended Data Fig. 1a-b) 23,24, thereby establishing the ability of this approach to identify bona fide PD-L1 regulators. Several additional positive PD-L1 regulators were also validated, including SMAD4, DPAGT1, and DNAJC13 (Fig. 1d, Extended Data Fig. 1a-b).
Prominent among the validated negative regulators of PD-L1 identified in the screen were several genes encoding mitochondrial proteins, including MUL1, MRPS12, and UQCR10 (Fig. 1e-f, Extended Data Fig. 1c). The most significant hit among the putative negative PD-L1 regulators, however, was Uroporphyrinogen Decarboxylase (UROD), a key enzyme in the heme biosynthesis pathway (Fig. 1e-f). Indeed, loss of UROD in multiple NSCLC cell lines robustly induced PD-L1 protein levels (Fig. 1f, Fig. 2a, Extended Data Fig. 2a). Surprisingly, PD-L1 mRNA levels decreased under these conditions (Fig. 2b), suggesting that loss of UROD post-transcriptionally induces PD-L1. Given the potent regulation of PD-L1 by UROD and the previously unknown connection between heme biosynthesis and the PD-1/PD-L1 immune checkpoint, we sought to elucidate the mechanism through which this pathway controls PD-L1 expression.
Fig. 2. Impairment of heme biosynthesis induces PD-L1 in NSCLC cells.

a, Western blot analysis in H1944 cells with control or UROD sgRNA. For this and all subsequent westerns, Tubulin served as a loading control. Data from a single experiment are shown in (a) and are representative of three independent experiments with two independent sgRNAs with similar results. b, Quantitative real-time PCR (qRT-PCR) analysis of PD-L1 mRNA in cells shown in (a). Bar graphs represent PD-L1 mRNA expression normalized to ACTIN and error bars represent SDs from the mean across n=3 independent experiments. A student’s two-tailed t-test was performed to determine statistical significance. ***p =0.0008. c, Schematic of the heme biosynthesis pathway. d, Levels of heme synthesis in control or UROD siRNA-treated H1944 cells 72 hours after transfection. Bar graphs represent heme levels normalized to total protein as pmol/mg and error bars represent SDs from the mean. This experiment was performed one time with n=3 three biological replicates. A student’s two-tailed t-test was performed to determine statistical significance. *** p = 0.004. e, Western blot analysis in H1944 cells after transfection with the indicated siRNAs. f, Western blot analysis in H1944 cells stably expressing Cas9 and the indicated sgRNAs with and without exogenous heme supplementation for 48h. Data from a single experiment are shown in e and f and are representative of two independent experiments with similar results.
UROD catalyzes the step-wise decarboxylation of Uroporphyrinogen III to Coproporphyrinogen III (Fig. 2c), an obligatory step in heme biosynthesis 25. Accordingly, reduction in UROD expression resulted in a 50% decrease in heme synthesis (Fig. 2d). To test if Uroporphyrinogen III accumulation in UROD knockout cells stabilizes PD-L1, we knocked down Uroporphyringen Synthase (URO3S), which encodes the enzyme that functions directly upstream of UROD. PD-L1 was similarly induced upon inhibition of URO3S, UROD, or both together (Fig. 2e), ruling out a role for accumulated Uroporphyrinogen III in PD-L1 upregulation and suggesting that a general impairment of heme synthesis is the trigger for PD-L1 induction. Further supporting this conclusion, chemical inhibition of Aminolevulinic Acid Dehydratase (ALAD) with succinylacetone (SA) or Ferrochelatase (FECH) using N-Methyl Protoporphyrin IX (NMPP) increased PD-L1 protein levels (Extended Data Fig. 2b,d) without increasing PD-L1 transcript levels (Extended Data Fig. 2c,e). Moreover, exogenous hemin suppressed PD-L1 levels in UROD knockout cells (Fig. 2f). Together, these data demonstrate that heme deficiency leads to upregulation of PD-L1 protein in NSCLC cells.
UROD inhibition accelerates tumorigenesis by suppressing anti-tumor immunity
To investigate the effects of UROD inhibition on tumor growth in vivo, we used shRNA to deplete Urod in Lewis Lung Carcinoma (LLC) cells, an established syngeneic tumor model 26. UROD depletion led to a potent increase in PD-L1 protein levels without altering Pd-l1 transcript levels, demonstrating that the pathway is conserved between human and mouse (Fig. 3a, Extended Data Fig. 2f). While Urod-knockdown and control LLC tumors grew at indistinguishable rates in immunodeficient NOD-scid IL2Rgammanull mice (Fig. 3b), Urod knockdown resulted in significantly faster tumor growth in immunocompetent C57BL/6 mice (Fig. 3c, Extended Data Fig. 2g). Moreover, Urod shRNA-expressing tumors exhibited a 50% reduction in CD8+ tumor infiltrating lymphocytes (TILs) compared to control tumors (Fig. 3d, Extended Data Fig. 2h-i) and the growth advantage conferred by Urod knockdown was diminished upon depletion of CD8+ T cells (Fig. 3e). These findings suggest that Urod inhibition promotes tumor growth by suppressing host immune responses mediated by CD8+ T cells.
Fig. 3. Loss of UROD accelerates tumorigenesis in vivo by suppressing CD8+ T-cells.

a, Western blot analysis in Lewis Lung Carcinoma (LLC) cells expressing doxycycline-inducible control shRNA or two independent shRNAs targeting Urod. Data from a single experiment are shown and are representative of two independent experiments with similar results. b,c, Quantification of tumor volumes of LLC cells expressing the indicated shRNAs in immunodeficient NSG mice (n=12 mice per group, p value = 0.3542) (b) or in syngeneic C57BL/6 mice (c) (n=7 mice Scrambled shRNA, n=12 mice Urod shRNA-1 and n=11 mice Urod shRNA-2). p value (Scrambled vs Urod shRNA-1) = 1.016513e-06, p value (Scrambled vs. Urod shRNA-2) = 0.0063. d, Quantification of CD8+ tumor infiltrating lymphocytes (TILs, expressed as a percentage of CD45+ cells). A Two-Sample Fisher-Pitman Permutation Test was used to compare CD8+ TILs between groups. n= 6 Scrambled shRNA and Urod shRNA tumors each, n=4 Scrambled + αPD-1 tumors and n= 5 Urod shRNA+ αPD-1 tumors. p value (Scrambled shRNA vs Urod shRNA) = 0.0038, p value (Urod shRNA vs Urod shRNA+ αPD-1) = 0.0029. e,f, Quantification of tumor volumes in syngeneic C57BL6 mice after treatment with a CD8+ depleting antibody (200ug, IP every 3 days) (n= 8 Scrambled mice, n= 6 Urod shRNA mice, p-value = 0.1491) (e) or αPD-1 antibody (200ug, IP every 3 days) (n = 8 Scrambled mice, n= 7 Scrambled + αPD-1 mice, n= 12 Urod shRNA mice, n=12 Urod shRNA+ + αPD-1 mice). p value (Scrambled shRNA vs Urod shRNA) =1.533e-06, p value (Urod shRNA vs Urod shRNA + αPD-1) = 1.7034e-05. (f). For all tumor growth experiments (b-c and e-f), graphs represent mean tumor volumes and error bars represent SDs from the means. A linear mixed effect model was used to determine statistical significance for all tumor growth assays, as described in Statistics and Reproducibility.
To demonstrate that the immunosuppressive effects of Urod knockdown are dependent upon the PD-1/PD-L1 axis, α-PD1 blocking antibody was administered after establishing LLC tumors. While this treatment had no effect on the growth of control tumors, α-PD1 blocking antibody fully abrogated the increased growth of Urod knockdown tumors (Fig. 3f) and triggered an accumulation of CD8+ TILs (Fig. 3d). Collectively, these data demonstrate that Urod depletion results in activation of a tumor-promoting immune checkpoint through upregulation of PD-L1.
RNA-seq was also performed in LLC cells to assess global gene expression changes in response to Urod depletion. Gene Set Enrichment Analysis (GSEA) identified several upregulated gene sets associated with tumor escape and tumorigenesis, including MYC and HIF1A targets (Extended Data Fig. 3a-b and Supplementary Table 1). Ingenuity Pathway Analysis also identified a significant decrease in expression of genes involved in the recruitment and migration of immune cells (Extended Data Fig. 3c and Supplementary Table 2).
Integrated Stress Response activation induces PD-L1 protein
To probe the mechanisms underlying PD-L1 upregulation in heme-deficient cells, we examined PD-L1 mRNA and protein stability. Consistent with our earlier observation that the steady-state abundance of PD-L1 mRNA is not induced by UROD-depletion, we detected no significant difference in mRNA stability between control and UROD knockout cells after Actinomycin D treatment (Extended Data Fig. 4a). Similarly, cycloheximide treatment revealed that PD-L1 protein degradation occurred at similar rates in control and UROD knockout cells (Extended Data Fig. 4b). Additionally, PD-L1 ubiquitylation was unaffected in UROD-depleted cells (Extended Data Fig. 4c).
Based on these data, we hypothesized that loss of UROD promotes translation of PD-L1 mRNA. Intracellular heme levels regulate mRNA translation through Heme-Regulated Inhibitor (HRI; also known as EIF2AK1), an eIF2α kinase that is inactive when bound by heme 27,28. Under conditions of heme deficiency and other cellular stresses such as arsenite treatment, heat shock, and osmotic stress, HRI is activated and phosphorylates eIF2α at serine 51 (ser-51) 27-29. eIF2α ser-51 phosphorylation by additional kinases is triggered in response to other physiological stresses including viral infection, misfolded protein accumulation, and amino acid deprivation 29,30, leading to inhibition of global translation initiation and preferential translation of selected mRNAs, a phenomenon known as the Integrated Stress Response (ISR) 30-32. Since heme deficiency triggers the ISR through HRI activation, we hypothesized that UROD inhibition promotes translation of PD-L1 mRNA through this pathway. Indeed, phosphorylation of eIF2α in MYC-transformed liver cancer cells was associated with enhanced PD-L1 translation 21. We observed that UROD depletion in lung cancer cells led to potent phosphorylation of eIF2α at ser-51 (Fig. 4a) and a corresponding reduction in global translation (Fig. 4b,c). Treatment with Salubrinal, a selective inhibitor of eIF2α de-phosphorylation 33, was sufficient to induce PD-L1 levels (Fig. 4d). Conversely, treatment with the small molecule ISRIB (Integrated Stress Response Inhibitor), which suppresses the effects of eIF2α phosphorylation by activating eIF2B 34,35, completely reversed PD-L1 upregulation in Salubrinal-treated cells (Fig. 4e). Moreover, ISRIB reduced PD-L1 levels in UROD knockout cells (Extended Data Fig. 5a).
Fig. 4. Integrated Stress Response (ISR) pathway activation induces PD-L1 protein in NSCLC.

a, Western blot analysis of peIF2α and total eIF2α in H1944 cells expressing a control sgRNA or a sgRNA targeting UROD. b, Autoradiography analysis of newly synthesized S-35 labeled proteins in H1944 cells transfected with control or UROD siRNAs, with three biological replicates per sample. Wildtype (WT) cells treated with 20uM cycloheximide (CHX) for 1 hour prior to metabolic labeling served as positive control. c, Quantification of S-35 Met/Cys signal from (b) normalized to control siRNA cells. Error bars represent SDs from the mean, a two-tailed student t test was performed to assess statistical significance. ***p value = 0.0008. d, Western blot analysis in H1944 cells treated with Salubrinal (in μM) for 48h. e, Western blot analysis in H1944 cells treated with 100 μM Salubrinal and/or 200 nM ISRIB for 24 hours. f, Western blot analysis of peIF2α and PD-L1 in cells cultured in normoxic or hypoxic conditions for 24h and 48h. g, Quantitative real-time PCR analysis of PD-L1 mRNA in cells shown in (f). h, Western blot analysis of peIF2α, total eIF2α, and PD-L1 in cells treated with Arsenite (100uM for H1944 cells, 50uM for Calu-6 and 5uM for H358 cells) for 24h. i, Quantitative real-time PCR analysis of PD-L1 mRNA in cells shown in (h). Bar graph represents PD-L1 mRNA expression normalized to ACTIN across three technical replicates, shown as individual data points. j, Western blot analysis in eIF2α wildtype (S/S) or ser51Ala mutant (A/A) cells expressing control or Urod shRNA. Experiments in a-j were performed two independent times with similar results, and data from a representative experiment are shown.
We next investigated the relationship between UROD and IFN-γ, a known regulator of PD-L1 in the tumor microenvironment. UROD levels were unaffected by IFN-γ stimulation (Extended Data Fig. 5b). Treatment with Salubrinal further enhanced PD-L1 levels in IFN-γ treated cells and in cells treated with the JAK inhibitor Ruxolitinib, which inhibits signaling downstream of IFN-γ (Extended Data Fig. 5c). Similarly, UROD knockout increased PD-L1 levels in cells treated with IFN-γ or Ruxolitinib (Extended Data Fig. 5d). These data indicate that the ISR pathway regulates PD-L1 expression independently of IFN-γ signaling.
Stresses known to activate the ISR pathway, including hypoxia and arsenite treatment, induced PD-L1 protein in lung cancer cells, (Fig. 4f-i). Consistent with the effects of UROD inhibition, PD-L1 protein was increased without an associated increase in mRNA levels in cell lines in which these treatments resulted in robust eIF2α phosphorylation. Interestingly, in some cell lines, hypoxia or arsenite induced PD-L1 mRNA, suggesting additional mechanisms of PD-L1 regulation.
To determine whether phosphorylation of eIF2α is necessary for PD-L1 induction, we reduced Urod using shRNA in mouse embryonic fibroblasts (MEFs) expressing either wildtype eIF2α (S/S cells) or mutant eIF2α with serine-51 mutated to alanine (A/A cells). Depletion of Urod led to phosphorylation of eIF2α at ser-51 and a potent induction of PD-L1 protein without increasing Pd-l1 mRNA levels in S/S cells, but not in A/A cells (Fig. 4j, Extended Data Fig. 6a). Taken together, these data demonstrate that UROD depletion activates the ISR pathway and that phosphorylation of eIF2α at ser-51 is necessary for associated PD-L1 protein induction.
ISR activation enhances PD-L1 translation
To test if PD-L1 mRNA translation is selectively enhanced under these conditions, we performed polysome profiling of control and UROD siRNA-treated cells. As expected, both UROD knockdown and salubrinal treatment resulted in an overall decrease in polysomes (Fig. 5a,b). Nevertheless, PD-L1 mRNA redistributed to heavier polysomes under these conditions, indicating increased association with actively translating ribosomes (Fig. 5c,d and Extended Data Fig. 6b-e). Preferential translation of PD-L1 mRNA under these conditions was even more robust than ATF4, a canonical ISR-induced protein (Extended Data Fig. 6f-i) 30,32,36-38.
Figure 5. ISR activation enhances PD-L1 translation.

a,b, Polysome analysis of siRNA-treated (a) or Salubrinal-treated (100 μM, 24 hours) (b) H1944 cells. c, qRT-PCR analysis of PD-L1 mRNA in ribosomal fractions from (a). Experiment was performed with two independent primer pairs with similar results. d, qRT-PCR analysis of PD-L1 mRNA in ribosomal fractions from (b). Fractions associated with <3 ribosomes were grouped together to represent poorly translated mRNAs and fractions with >3 ribosomes were grouped as efficiently translated mRNAs. PD-L1 mRNA expression in each fraction was normalized to Luciferase and PD-L1 mRNA abundance was calculated as the percent of total PD-L1 in all fractions. Luciferase mRNA control was added to each fraction prior to RNA extraction to control for variability in total RNA in fractions during the RNA isolation and reverse transcription reactions. Error bars in c and d represent SDs from the mean from three independent fractions (<3 or >3 ribosomes). A student’s two-tailed t-test was performed to determine statistical significance. *p=0.017; **p= 0.001. Data from individual ribosomal fractions for two independent primer pairs are provided in Extended Data Fig. 6b-e. e, Schematic of the wildtype human PD-L1 5´ UTR with five upstream CTGs cloned upstream of a Firefly luciferase reporter. f, Dual-luciferase assay of MEF cells transfected with the indicated firefly luciferase reporter constructs normalized to co-transfected control Renilla luciferase reporter. Error bars represent SDs from the mean for n=3 biological replicates. A student’s two-tailed t-test was performed to determine statistical significance. **** = p<0.0001 for all constructs except M1 (p=0.14). Data from a single experiment are shown in (f) and are representative of three independent experiments with similar results. g, Dual luciferase reporter analysis of the PD-L1 5′ UTR in H358 cells expressing a control or UROD sgRNA. Error bars represent SDs from the mean relative luciferase activity (Firefly/Renilla) across n=3 biological replicates per group. A student’s two-tailed t-test was performed to assess statistical significance with *p = 0.015. h, Dual luciferase reporter analysis of PD-L1 5’ UTR in MEFs cells with a control or Urod shRNA. Error bars represent SDs from the mean relative luciferase activity (Firefly/Renilla) across n=3 biological replicates per group. A student’s two-tailed t-test was performed to assess statistical significance with **p = 0.0075. Experiments in (g) and (h) were performed two independent times with similar results. Data from a representative experiment are shown.
Several recent studies have demonstrated that activation of the ISR promotes translation of selected mRNAs harboring upstream open reading frames (uORFs) in their 5′ UTRs, such as ATF4, GADD34, and GCN4 20,31,32. Phosphorylation of eIF2α is hypothesized to weaken activity of the eIF2 translation initiation complex, promoting leaky scanning through the 5′ UTR, thereby bypassing inhibitory uORFs and enhancing translation at the canonical translation start site 39,40. The PD-L1 5′ UTR contains several uORFs initiating with the non-canonical start codon CUG (one in-frame and four out-of-frame with the canonical AUG start codon) (Fig. 5e). To test whether these uORFs suppress PD-L1 translation, a series of 5′ UTR reporter constructs were generated with mutations in each CUG, alone and in combination. Consistent with a prior report 21, mutation of the third, fourth, and fifth CUG led to a ~5-8-fold increase in luciferase activity relative to the wild-type (WT) construct (Fig. 5e-f) without affecting luciferase mRNA levels (Extended Data Fig. 6j). Moreover, UROD depletion increased WT reporter activity without increasing reporter mRNA abundance (Fig. 5g-h and Extended Data Fig. 6k,l). Collectively, these results support a model whereby activation of the ISR promotes bypass of inhibitory uORFs in the PD-L1 5′ UTR, thereby enhancing PD-L1 translation and immune checkpoint activation.
ISR-dependent translation requires eIF5B
Alternative translation initiation factors, including eIF2A, eIF2D, and eIF5B, substitute for eIF2α under stress conditions 20,41,42. To determine whether these factors play a role in ISR-dependent PD-L1 translation, each were depleted individually with siRNAs in human UROD knockout cells. EIF2D knockdown had no effect on PD-L1 levels while EIF2A knockdown modestly reduced PD-L1 induction (Fig. 6a,b,d). In contrast, EIF5B depletion strongly downregulated PD-L1 expression in UROD knockout cells (Fig. 6c-d and Extended Data Fig. 7a-c). These data demonstrate that eIF5B is necessary for ISR-dependent PD-L1 translation. Consistent with this finding, eIF5B inhibition in Urod-depleted cells downregulated PD-L1 expression and reduced tumor burden in two independent syngeneic mouse models (Fig. 6e-f and Extended Data Fig. 7d-f). Of note, Eif5b depletion also reduced growth of syngeneic tumors without Urod knockdown, suggesting that translation of additional mRNAs required for tumor growth rely upon eIF5B activity. Accordingly, EIF5B depletion reduced proliferation of multiple human lung cancer cell lines without globally impacting translation rates (Extended Data Fig.8a-b).
Fig. 6. ISR-dependent translation of PD-L1 requires the alternative translation initiation factor eIF5B.

a,b,c, Western blots of H1944 cells expressing the indicated sgRNAs and transfected with siRNAs targeting EIF2D (a), EIF2A (b), or EIF5B (c). d, Quantification of western blots shown in a-c normalized to protein levels in control siRNA-treated cells. e, Western blots of LLC cells expressing the indicated shRNAs targeting Urod, or Eif5b. All experiments (a-c and e) were performed two independent times with similar results, data shown are from a representative experiment. f, Quantification of tumor volumes of LLC cells expressing the indicated shRNAs in syngeneic C57BL/6 mice. Tumor volumes of individual mice are shown (n=12 Scrambled shRNA, n=7 Eif5b shRNA, n=11 Urod shRNA and n=6 Eif5b + Urod shRNA mice). p value (Scrambled shRNA vs Eif5b shRNA = 0.0251, p value (Scrambled shRNA vs Urod shRNA) = 4.075e-10, p value (Urod shRNA vs Urod + Eif5b shRNA) = 8.666e-12. A linear mixed effect model was used to determine statistical significance, as described in Statistics and Reproducibility. g, Western blot analysis in H1944 cells expressing eGFP or EIF5B cDNA. h, qRT-PCR analysis of EIF5B and PD-L1 in H1944 cells shown in g. Bar graph represents mean relative expression of EIF5B and PD-L1 from 3 technical replicates, with individual data points plotted. Experiments in g and h were performed two independent times with similar results, data shown are from a representative experiment. i, Dual luciferase reporter analysis of the PD-L1 5′ UTR in H358 cells with transient overexpression of a control eGFP or EIF5B cDNA. Error bars represent SDs from the mean relative luciferase activity (Firefly/Renilla) across n=3 biological replicates/group. A student’s two-tailed t-test was performed to assess statistical significance with **p = 0.005. j, Dual luciferase reporter analysis of the PD-L1 5′ UTR in MEFs expressing a control eGFP or EIF5B cDNA. Error bars represent SDs from the mean relative luciferase activity (Firefly/Renilla) across n=3 biological replicates/group. A student’s two-tailed t-test was performed to assess statistical significance with ****p = 0.000083. Experiments in i and j were performed two independent times with similar results. Data from representative experiments are shown. k, Model of translational control of PD-L1 in response to ISR activation. Under normal conditions, a functional ternary complex consisting of eIF2α bound to GTP and Met-tRNA initiates translation of PD-L1 at the canonical AUG. Inhibitory upstream open reading frames (uORFs) suppress translation at the canonical AUG, limiting PD-L1 translation. Phosphorylation of eIF2α and ISR activation induce bypass of inhibitory uORFs in the human PD-L1 5′ UTR. In the absence of a functional ternary complex, eIF5B can substitute for eIF2α, culminating in enhanced PD-L1 translation and suppression of anti-tumor immunity.
Examination of TCGA data revealed that EIF5B amplification or mRNA upregulation occurs in 16% of lung adenocarcinomas (Extended Data Fig. 9a), and high expression of EIF5B correlates with poor overall survival of human lung adenocarcinoma patients (Extended Data Fig. 9b). A meta-analysis of the correlation between EIF5B expression and patient survival was also performed across a larger number of lung cancer datasets. This revealed that EIF5B mRNA expression is associated with poor outcome in lung cancer patients (all subtypes) and in lung adenocarcinoma (LUAD) patients, but not in lung squamous cell carcinoma patients (Extended Data Fig. 9c-f). Surprisingly, overexpression of eIF5B in human lung cancer cells was sufficient to induce PD-L1 protein levels without affecting mRNA abundance (Fig. 6g-h) even in the absence of ISR activation. Accordingly, overexpression of eIF5B increased activity of the PD-L1 5′ UTR luciferase reporter without affecting reporter mRNA levels (Fig. 6i-j, Extended Data Fig. 10a-b). Thus, eIF5B gain-of-function represents a previously unrecognized mechanism that is employed by human cancer cells to activate the PD-L1 immune checkpoint.
Discussion
Immune checkpoint blockade has emerged as an indispensable modality in cancer therapy. Understanding tumor intrinsic and extrinsic mechanisms of PD-L1/PD-1 checkpoint regulation will be critical for improving patient outcomes 7,8. Using an unbiased screening approach, we unexpectedly discovered that impairment of the heme biosynthesis pathway potently induces PD-L1 expression in lung cancer cells. Heme deficiency signals HRI to phosphorylate eIF2α and activate the ISR pathway. ISR activation promotes the eIF5B-dependent translation of PD-L1, thereby suppressing CD8+ T cells to sustain tumorigenesis in vivo (Fig. 6k). A recent study demonstrated that pro-tumorigenic Keap1 mutations resulted in an increase in production of Heme Oxygenase (HO) in lung tumors, thereby lowering the amount of active heme 43. While this study did not focus on anti-tumor immunity, the finding that increased HO and reduced heme levels promotes tumorigenesis is intriguing and consistent with our findings. Future studies are warranted to investigate the relationship between heme metabolism and immune checkpoint regulation in human lung cancer.
In agreement with the results reported here, there is a growing appreciation that the ISR pathway contributes to cancer progression. Tumor cells experiencing proteotoxic stress activate the ISR pathway to maintain homeostasis and sustain growth 44. eIF2α phosphorylation triggers an alternative translation program that promotes oncogene expression in a mouse model of skin squamous carcinoma 20. Additionally, transgenic expression of MYC in a mouse model of KrasG12D-induced liver cancer resulted in eIF2α phosphorylation, induction of Pd-l1 translation, and tumor progression 21. Based on these prior findings and the results described here, further investigation of relationship between ISR activation and immune evasion in cancer will be an important priority for future work.
The ISR pathway-targeting drug ISRIB induces cytotoxicity in patient derived xenografts in immunocompromised mice 34,35,44. Our data show that ISRIB also suppresses PD-L1 protein levels in the setting of ISR pathway activation in lung cancer cells, suggesting that ISR pathway inhibition may induce anti-tumor immunity alone or in combination with existing immunotherapies. Perhaps even more importantly, our data pinpoint the alternative initiation factor eIF5B as a key mediator of ISR-dependent PD-L1 translation (Fig. 6k). eIF5B overexpression, a frequent event in lung adenocarcinoma patients that portends poor survival, was sufficient to potently increase PD-L1 levels in lung adenocarcinoma cells. These findings suggest that EIF5B may function as an oncogene in human cancer. Although here we identify eIF5B as an unanticipated regulator of PD-L1 immune checkpoint activation, it is important to note that this is likely one of several mechanisms involved in PD-L1 upregulation in human lung cancers, particularly in contexts where stress response pathways are active. Interestingly, eIF5B was shown to be a key driver of translation under hypoxic conditions or viral challenge, suggesting additional contexts that may engage this mechanism to activate the immune checkpoint in cancer cells 42,45. These findings highlight the importance of future studies to examine the complete translational program orchestrated by eIF5B in cancer and evaluate its potential as a therapeutic target.
METHODS
CRISPR-Cas9 Screening
Lentiviral Library Generation
The human GeCKO v2 library was obtained from Addgene #1000000048 and amplified according to instructions. Plasmids were electroporated and purified from bacterial pellets using the Qiagen plasmid maxi kit. To generate GeCKO v2 lentivirus, 3x106 293T cells were seeded/10 cm dish. Libraries A and B were prepared independently using 20 dishes/library. The next day, each dish was transfected using 10μg of total plasmid (5:3:2 ratio of GeCKO library:psPAX2:pMD2.G), 30 μL of FuGENE HD in 900μL of Opti-MEM. Fresh medium was added the following day. The media containing virus was harvested at 48 and 72h post transfection and filtered through a 0.45 μm Surfactant-free Cellulose Acetate sterile filter (Corning). Library aliquots were snap frozen on dry ice/ethanol and stored at −80°C. Library viral titer was determined as described 22,46.
Lentiviral Infection
The genome-wide CRISPR Cas9 screen was performed in human LUAD H358 cells using both GeCKO v2 libraries A and B, in biological replicates. To achieve 300X or greater coverage, ~50 million cells were transduced/library. For each transduction, ten 12-well plates were seeded with 3.5x105 H358 cells/well. An overnight transduction was performed at a viral MOI of 0.2-0.4 with 8μg/mL polybrene. Cells were trypsinized and pooled, then plated into fresh medium in eight 15cm dishes. 48h later, cells were trypsinized, pooled and plated into fresh medium containing 1ug/mL puromycin. In parallel, a small aliquot of cells was used to confirm that a MOI of 0.2-0.4 was achieved. Cells were passaged in puromycin for 14d before sorting. At every passage, 12×106 cells were seeded/dish into 15cm dishes with medium containing puromycin.
Fluorescence Activated Cell Sorting
Confluent H358 cells were trypsinized and centrifuged at 300g for 5m. Cells were counted (Countess Automated Cell Counter) and washed with cell staining buffer (BioLegend). Cells were incubated in the dark for 20m on ice in cell staining buffer containing APC-PD-L1 antibody (BioLegend) at a concentration of 0.4ug antibody/million cells. Stained cells were centrifuged at 300g for 5m to discard unbound antibody and washed with cell staining buffer 2x. Final cell pellets were resuspended at a cell density of 15 x 106 cells/mL of complete RPMI media. The cells were sorted at the UTSW Flow Cytometry Core Facility (MoFlo cell sorter, Beckman Coulter). The highest and lowest 0.5% cells were gated based on APC fluorescence and collected. 100-200 million cells were sorted to collect 2-4x105 PD-L1 high and low cells. The cells were washed once with PBS and frozen at −80C. Unsorted cells were frozen at −80C.
Genomic DNA Extraction
Genomic DNA was extracted from unsorted cells using the Qiagen DNeasy Blood & Tissue Kit according to manufacturer’s instructions. Extractions were performed on 40-80x106 unsorted cells to ensure 300X coverage or higher of the library. DNA was eluted twice in 125μL nuclease free water and its concentration assessed using the Qubit dsDNA BR assay (Thermo Fisher). To facilitate maximum recovery of gDNA from sorted cells, a previously described method 46 was used with the following modifications: sorted cell pellets were resuspended in 500μL of tissue lysis buffer, consisting of 460μL of STE buffer [1mM EDTA (pH 8.0), 10mM Tris-HCl (pH 8.0), 100mM NaCl] supplemented with 10μL of 0.5 M EDTA, 10μL of proteinase K [10mg/mL in TE buffer containing 10mM Tris-HCl (pH 8.0) and 1 mM EDTA], and 20μL of 10% SDS. Pellets were digested overnight at 55°C while shaking at 600 rpm on a Thermomixer (Eppendorf). The following day, 5μL of 2mg/mL RNase A was added to each tube and incubated at 37°C for 1h while shaking (600 rpm). Extractions were performed with equal volumes of pH 7.9-buffer saturated phenol, followed by phenol:chloroform:isoamyl alcohol (25:24:1) and chloroform. DNA was precipitated in 100% ethanol and glycogen (Roche) at −80°C for 1h followed by centrifugation at 18,000g for 10m at 4°C. Pellets were washed with 75% ethanol, dried, and resuspended in 21μL of water by incubating at 37°C overnight. DNA concentration was determined with the Qubit dsDNA BR assay.
Preparation of Sequencing Libraries
PCR amplicon libraries for deep sequencing were prepared using an adapted protocol 22,46. All primer sequences are provided in Supplementary Table 2. The first round of PCR (PCR I) was performed on DNA from unsorted cells using 6.6μg of gDNA/100μL PCR reaction. To maintain 300X coverage, 20 reactions were assembled/sample. For sorted cells, all extracted gDNA per sample was distributed into two 100μL reactions. 18-20 cycles of amplification were performed using Herculase II Fusion polymerase (Agilent). The reactions from PCR I were pooled together and used as a template for the second round of PCR (PCR II), to add the necessary adapters for Illumina sequencing. To adjust for varying PCR efficiency between samples, the cycle number for PCR II was adjusted such that each library was amplified in a 50μL reaction to generate ~50ng of DNA library. The final DNA for sequencing was purified using AMPure XP beads (Agencourt) according to manufacturer’s instructions with the following modifications: Each 50 uL PCR II reaction was mixed with 25uL of beads and incubated for 5m. Magnetic separation was used to collect the supernatant. The supernatant was mixed with 90μL beads and incubated for 5m. The supernatant was discarded and beads were washed twice with 200μL of 70% ethanol. The beads were dried for 10-15m. Bound DNA was eluted from the beads using 40μL water.
Next Generation Sequencing
Prior to sequencing, all DNA libraries were analyzed using the Bioanalyzer High Sensitivity DNA Analysis Kit (Agilent). Library concentration was determined by qPCR using the KAPA Library Quantification Kit for Illumina platforms. All samples were sequenced on an Illumina HiSeq 2500 or a NextSeq 500 with 75bp single reads. ~15-20 million reads were sequenced/library.
Sequencing Data Analysis
A reference file for all sgRNAs in the library was acquired from Addgene, and identical sgRNAs targeting more than one protein-coding gene were removed. Demultiplexed FASTQ files were mapped to the reference file using Bowtie 2 requiring unique alignments with no mismatches. Normalized read counts were calculated as described previously 22,46. Screen hits were identified using RIGER16 with the following parameters: log fold-change ranking, 1x106 permutations, second best rank (SBR) scoring algorithm.
Ethics statement.
Mice were monitored closely throughout all experimental protocols to minimize discomfort, distress or pain. If any signs of pain and distress were detected (disheveled fur, decreased feeding, significant weight loss (>20% body mass), limited movement, or abnormal gait), the animal was removed from the study and euthanized. All procedures involving mice were performed in accordance with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association and protocols approved by the UTSW Institutional Animal Care and Use Committee. C57BL/6J and NSG mice were obtained from The Jackson Laboratory and the UT Southwestern Breeding core, respectively.
Cell culture.
All human lung cancer cell lines (obtained from Dr. John Minna) were cultured in RPMI 1640 media supplemented with 5% FBS (Sigma) and 1% antibiotic-antimycotic (anti-anti, Invitrogen). All human LUAD cell lines tested (H1944, Calu-6, H2030, H441, H358, and Hop62) harbor a KRAS mutation. Each of these cell lines harbor mutations or deletions in TP53, with the exception of H1944 cells that are TP53 WT. WT and EIF2α mutant Mouse Embryonic Fibroblasts (MEFs), from Dr. Randal Kaufman, were cultured in DMEM supplemented with 10% FBS, 1% anti-anti, 2mM Glutamine, 2% MEM amino acid solutions (Gibco) and 1mM Sodium pyruvate (Invitrogen). Lewis Lung Carcinoma (LLC) (from Dr. Don Gibbons) were cultured in DMEM media supplemented with 10% FBS and 1% penicillin and anti-mycotic. KLN205 cells (from Dr. Rolf Brekken) were cultured in DMEM media supplemented with 10% FBS, 1% penicillin and anti-mycotic, 2mM glutamine and 1% Non-Essential Amino Acids (Sigma). For hemin rescue, control or UROD knockout H1944 cells were treated with 1uM or 10uM Hemin Chloride (Sigma) for 48h. For ISR pathway activation and reversal, H1944 cells were treated with 100uM or 200uM Salubrinal (Tocris) and/or 200nM ISRIB (Sigma) for 24h. For IFN-γ experiments, H1944 cells were treated either with 10ng/mL IFN-γ (R&D systems) or 10uM Ruxolitinib (Sigma) for 24h. For ISR activation, LUAD cells were treated with Arsenite (10-50uM), or grown in a hypoxic chamber for 24 and 48h. All chemicals are listed in Supplementary Table 4. Cell lines have been DNA fingerprinted using the PowerPlex 1.2 kit (Promega) and found to be mycoplasma free using the e-Myco Mycoplasma PCR Detection kit V2.0 (Boca Scientific, catalog #25235) or a direct PCR method with GoTaq Green Master Mix (Promega, M712).
Plasmids.
LentiCRISPR V2, PAX2 and MD2 plasmids were obtained from Addgene (#52961, #12260, #12259). sgRNAs targeting candidates from the screen were selected from human GeCKO libraries (Addgene Library #1000000048, #1000000049) and described in Supplementary Table 3. The pTRIPZ plasmid was obtained from Dharmacon (RHS6371). For Eif5b shRNA experiments, TRIPZ was modified to contain a Blasticidin resistance gene replacing the Puromycin resistance gene using In-fusion cloning (Clontech).
Generation of knockout cell lines using CRISPR-Cas9.
HEK 293T (1x108) cells were co-transfected with lentiCRISPR V2 (10ug) and PAX2 (4ug), MD2 (2.66ug) helper plasmids using Lipofectamine 3000 (Life technologies). Lentiviral supernatant was collected 48h and 72h post transfection and filtered. Recipient cells were infected o/n with viral supernatant containing 8ug/mL polybrene (Sigma) and replenished with fresh media. 48h later, transduced cells were cultured in fresh media containing 1ug/mL puromycin for 10-12 days.
RNA extraction and qRT-PCR analysis.
Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen). For qRT-PCR of mRNA, cDNA synthesis was performed with 1-5μg RNA for reverse transcription using Superscript IV Vilo Master Mix (5X) (Invitrogen). mRNA expression was assessed using quantitative real-time PCR with 2X SYBR Green Master Mix (R&D Systems). mRNA levels were normalized to β-actin or 18S mRNA expression, with gene expression levels measured using a standard curve for each set of primers crossing exon-exon junctions for each gene. All PCR assays were performed in triplicate. PCR primers are provided in Supplementary Table 3. To monitor PD-L1 mRNA decay, cells with control or UROD sgRNA were treated with ActinomycinD (10ug/ml) to halt transcription and RNA was isolated.
Western blotting.
Cells and tissues were lysed in RIPA buffer containing Halt Protease Inhibitor cocktail (Invitrogen) and homogenized using a Bioruptor (Diagenode). Proteins were quantified using the Bicinchoninic Acid (BCA) assay (Thermo Scientific), subject to separation using NuPage Bis-Tris gels (Invitrogen), and transferred to a nitrocellulose membrane. The membranes were blocked for 1h at RT in 5% milk and probed with primary antibodies in 5% milk overnight at 4°C. After incubating the membrane with the appropriate secondary antibody conjugated to horseradish peroxidase, protein levels were detected with SuperSignal Extended Dura substrate (Thermo Scientific). Antibodies are listed in the Reporting Summary. To monitor PD-L1 protein degradation, control or UROD knockout H1944 cells were treated with 20uM Cycloheximide (Sigma) and protein lysates isolated and used to perform westerns to monitor PD-L1 protein stability over time.
Inducible knockdown of Urod and Eif5b in murine lung cancer cells.
HEK 293T cells were co-transfected with pTRIPZ (Dharmacon, RHS5087, Supplementary Table 3) with helper plasmids as described above. LLC or KLN205 cells were infected overnight with scrambled or Urod shRNA (2 independent shRNAs) or Eif5b shRNA (shRNA-1 for LLC cells, shRNA-2 for KLN205 cells) lentiviral supernatant and 8ug/mL polybrene (Sigma). Transduced cells were selected in 2ug/mL puromycin or 4ug/mL blasticidin for 1 week and cultured in 2-3ug/mL doxycycline for 4d. Cells were harvested for RNA/protein to assess knockdown.
Immunoprecipitation assay.
Control or UROD knockdown H1944 cells (90% confluent) were treated with Velcade (10uM) for 24h to inhibit the proteasome. The following day, cells were scraped in cold PBS and lysed in IP lysis buffer (Invitrogen). Lysates were centrifuged at 15,000rpm for 5m and supernatants containing protein quantified using BCA assay. 300mg protein lysate was immunoprecipitated with 5ug PD-L1 antibody using Protein G Dynabeads (Invitrogen). IP samples were subject to western blotting and probed with a Ubiquitin antibody to assess PD-L1 ubiquitylation.
Transient knockdown using siRNA.
H1944 (2 x 106) cells were seeded in 10cm2 dishes. The following day, cells were transfected with siRNA pools (5uM, siGENOME Dharmacon pools targeting UROD, EIF5B or non-targeting control) and Dharmafect solution 4 in Opti-MEM media according to manufacturer’s instructions. Cells were replenished with fresh complete media the next day and harvested 48h later for RNA/western analysis.
Measurement of heme synthesis levels.
Heme synthesis was measured using a published protocol 47. H1944 cells (4 x 105) cells were seeded in triplicate in 6-well plates and transfected with control or UROD siRNAs. 72h post transfection, cells were incubated with 0.3 μCi [14C] 5- alpha aminolevulinic acid (ALA) overnight. The next day, cells were scraped in cold PBS in a radioactive safe area and subject to diethyl ether-HCl phase extractions to extract [14C] labelled heme as previously described 47. Radioactivity was measured in triplicate using a scintillation counter and normalized to total protein to obtain heme synthesis levels.
Metabolic labeling.
H1944 cells (4 x 105) cells were seeded in triplicate in 6-well plates and transfected with control or UROD siRNAs. 72h post transfection, cells (90% confluency) were cultured in methionine/cysteine free RPMI media for 1h and incubated with 75uCi EasyTag Express Protein Labeling Mix [S35] for 15m. Cells were lysed in 1X SDS lysis buffer and proteins subject to separation, then transferred to a nitrocellulose membrane and analyzed for S35 incorporation using autoradiography.
Tumorigenesis assays.
LLC cells (4x105) expressing a scrambled shRNA or Urod shRNA lentiviruses were injected subcutaneously into the right flanks of 6-8-week-old C57BL/6J female mice (Jackson laboratory). Modified KLN205 cells (3x105) were injected into the right flanks of 6-8-week-old DBA/2 female mice (Charles River). For CD8+ T cell depletion, 8-10 mice bearing scrambled shRNA or Urod shRNA tumors were randomized into groups receiving control or CD8+ depleting antibody. Mice were injected with 200ug antibody IP every three days. For PD-1 blockade, on Day 10, 10-15 mice bearing scrambled shRNA or Urod shRNA tumors were randomized into groups receiving control or PD-1 antibody. Mice were injected with 200ug antibody IP every three days. Mice were kept on doxycycline water (2g/L doxycycline, 2% sucrose) for the duration of the experiment. Tumor volume was measured using calipers every 3d until the average tumor mass reached 2cm3. Tumor volume was calculated using the formula (length x width2)/2.
Flow cytometry analysis of tumor infiltrating lymphocytes.
Tumors were excised from euthanized mice and homogenized using a Tissue Chopper. Tumor cells were digested at 37°C at 70 rpm for 1h in digest buffer (RPMI 1640 containing 5% FBS, 0.5mg/mL Hyaluronidase, 0.5mg/mL Collagenase IV (Sigma) and 20ug/mL DNAse I. All subsequent steps were performed on ice. Digested cells were centrifuged at 1500rpm for 5m and the cell pellet resuspended in 5mL Cell Staining Buffer and filtered using a 70uM cell strainer. The cells were centrifuged again and resuspended in 50-100uL blocking-dead mix (CSB containing 5% FBS, 10% mouse serum, 1uL CD16/CD32 Fc blocking antibody and 3uL ef506 viability dye for 30m on ice. The samples were incubated with 50-100uL of primary antibodies (see Reporting Summary) and diluted in CSB for 30m on ice. Cells were washed twice with CSB and filtered through a nylon membrane into flow cytometry tubes for analysis. Samples were run on LSRII (BD Biosciences) and single-color stained C57/BL6 splenocytes were used as compensation controls. FlowJo software was used to calculate the % CD8+ T-cells of CD45+ live cells.
Polysome Profiling.
Sucrose gradients were prepared in advance in Beckman ultracentrifuge tubes as described in 48 and stored at −80°C. One day before the experiment, gradients were allowed to diffuse for 16h at 4°C. The next day, 20-40x106 H1944 cells/sample were trypsinized and washed twice with ice-cold PBS (second wash containing 100ug/mL cycloheximide). After the second wash, PBS was discarded and cell pellets resuspended in 750uL of Polysome Extraction Buffer (20mM Tris-HCl (pH 7.5), 100mM NaCl, 5mM MgCl2, 0.1% NP-40 in distilled water) containing cycloheximide, protease inhibitor cocktail and RNAse inhibitors. Cells were lysed in Eppendorf tubes for 10m on ice and sheared through a 27.5-gauge needle 3-4 times. The lysates were centrifuged at 15,000 rpm for 5m on ice and the supernatant lysate RNA concentration quantified by Nanodrop. Equal amount of lysate (500-600ug RNA) was loaded across all gradients. The gradients were centrifuged at 35,000 rpm for 2h at 4°C and run on a fractionator machine (Biorad) to visualize and collect polysome fractions. Each collected fraction was mixed with 3x volume of 100% ethanol and 20ug glycogen carrier and incubated overnight in −20°C. The next day, fractions were centrifuged at 20,000g for 30m at 4°C to precipitate RNA pellets. Pellets were dried for 20m at RT, resuspended in 100uL Nanopure water and 350uL RNeasy RLT lysis buffer and loaded onto RNeasy columns. The RNeasy kit was used to isolate RNA and cDNA synthesis and real-time PCR performed. 20ng of Luciferase mRNA control (Promega, L4561) was added to each fraction prior to RNA extraction to control for variability in total RNA in fractions during RNA isolation and reverse transcription. Fractions associated with <3 ribosomes were grouped together (poorly translated mRNAs) and fractions with >3 ribosomes were grouped (efficiently translated mRNAs).
Dual luciferase assays.
50 x103 MEF cells were seeded/well in 12-well plates in triplicate and transfected with a Renilla plasmid (20ng), Firefly luciferase pGL3 plasmid expressing PD-L1 wildtype 5’UTR or various mutant constructs (200ng) and a carrier pUC19 plasmid (400ng) per well using Fugene HD (Promega) at a 3:1 Fugene:DNA ratio. For transient EIF5B overexpression, cells were co-transfected with 500 ng pcDNA6.2 GFP or pcDNA6.2 EIF5B/well with luciferase plasmids. Luciferase activity was measured 48h post transfection using a Luminescence plate reader (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity to obtain Relative luciferase levels/sample.
Transient Overexpression of EIF5B.
Human EIF5B cDNA was gateway cloned into pcDNA6.2. On day one, 4x106 H1944 cells were seeded in 10 cm2 dishes. The next day, cells were transfected with 3ug pcDNA6.2 eGFP control or pcDNA6.2 EIF5B plasmid with 16uL enhancer reagent and 60uL Effectene reagent according to manufacturer’s instructions. Media was changed 24h later to remove Effectene-DNA complexes. Cells were harvested for western blot analysis/RNA isolation 48h after transfection.
Cell proliferation assays.
On day 0, 500-1000 human lung cancer cells were seeded in 96-well plates and transfected with control/EIF5B siRNAs. The next day, media was changed and cell proliferation monitored over time using the CellTiter 96 Non-Radioactive cell proliferation assay (G4000, Promega).
RNA sequencing analysis methods.
Raw FASTQ files were analyzed using FastQC v0.11.2 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and FastQ Screen v0.4.4 and reads were quality-trimmed using fastq-mcf (ea-utils/1.1.2-806) 49. The trimmed reads were mapped to the mm10 assembly mouse genome (the University of California, Santa Cruz, version from igenomes) using STAR v2.5.3a 50. Duplicated reads were marked using Picard tools (v1.127; https://broadinstitute.github.io/picard/), RNA counts generated from FeatureCounts 51 were TMM normalized, and differential expression analysis was performed using edgeR 52. For differential expression analysis, statistical cutoffs of FDR ≤ 0.05 and log2CPM ≥ 0 were used to identify statistically significant and biologically relevant differentially regulated genes.
Functional analysis of gene sets.
Pathway and network analysis were conducted using Gene Set Enrichment Analysis (GSEA) 53,54 software and Qiagen’s IPA 55 tool (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis). The GSEAPreranked tool was used for the ranked gene list using rank_score=sign_of_FC * -log (pval) for all expressed genes in the RNASeq dataset with weighted scoring scheme. The mouse RNASeq data was used after projection to the assigned human orthologs using the Jackson Lab report (http://www.informatics.jax.org/downloads/reports/HGNC_homologene.rpt, downloaded in September 2019) using only one to one unique mappings. The functional datasets were from the MSigDB 56 V7 (C2: curated gene sets).
For IPA analysis the cutoffs used were FDR ≤ 0.05, log2CPM ≥ 0 and∣log2 (Fold change)∣ > 1.5. The IPA Core Analysis function was used with default parameters. The p-values were calculated using Fisher’s exact test and IPA z-Score was used as a statistical measure of the comparison between expected direction of change for the uploaded dataset and observed gene expression. Down-regulated pathways have negative z-scores. The pathways with ∣z-score∣ >2 were considered significant.
EIF5B meta-analysis in lung cancer datasets.
The EIF5B meta-analyses were performed using the Lung Cancer Explorer web portal57 (http://lce.biohpc.swmed.edu/lungcancer/). Cox proportional hazard models were fitted to estimate the association between EIF5B mRNA expression and patient survival outcome in each dataset. The hazard ratios (HR)s from individual datasets were then pooled by meta-analysis using a random effects model58,59.
Statistics and reproducibility.
A Student t-test was used for comparisons between two groups with normal data distribution (for real time qPCR, and MTS assays). A Two-Sample Fisher-Pitman Permutation Test was performed for comparing % CD8+ T-cells. For tumor implantation assays, we used a linear mixed effect model (R package nlme version 3.1.140) to model each experimental dataset. The time effect is treated as a random effect for each sample and experimental group treated as a fixed effect. Our model tested the null hypothesis that there are no differences among groups across time points. Reported p-values were adjusted by the Tukey’s method. Representative results from at least two independent repeats are shown, except where specified otherwise in the figure legends.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
CRISPR screening and RNA-sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE129968 and GSE139120, respectively. Kaplan-Meier survival analysis of patients with or without EIF5B alterations are from the TCGA Firehose Legacy lung adenocarcinoma study using cBioPortal. The EIF5B meta-analyses were performed using the Lung Cancer Explorer web portal57 (http://lce.biohpc.swmed.edu/lungcancer/). Source data for Figs.1-6 and Extended Data Figs.1-10 are provided. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Extended Data
Extended Data Fig. 1. Validation of positive and negative regulators of PD-L1 identified in the CRISPR-Cas9 screen.

a, Flow cytometry analysis of cell surface PD-L1 in H358 cells expressing control sgRNA or sgRNAs targeting CMTM6, SMAD4, DPAGT1 or DNAJC13. b, Western blot analysis confirming loss of target protein and a decrease in PD-L1 in cells from (a). Experiments in a and b were repeated two independent times with similar results, data from a representative experiment are shown. Validation was performed with an independent sgRNA for CMTM6 and SMAD4. c, Western blot analysis confirming loss of target protein and an increase in PD-L1 in cells from Fig 1f. Experiments were performed three independent times with similar results, data from a representative experiment are shown.
Extended Data Fig. 2. Effects of heme synthesis inhibition on PD-L1 mRNA and protein, and analysis of Pd-l1 mRNA and tumors from LLC cells.

a, Western blot analysis in Calu-6 and H2030 human lung cancer cell lines expressing the indicated sgRNAs. b, Western blot analysis in cells treated with 10 mM succinyl acetone (ALAD inhibitor) for 48h. c, qRT-PCR analysis of PD-L1 mRNA relative to ACTIN, in cells from (b). Error bars represent SDs from the mean for n=3 independent experiments. A student’s two-tailed t-test was used to assess statistical significance with ***p = 0.0002. d, Western blot analysis of PD-L1 in H1944 cells treated with 50 μM or 100 μM N-Methyl Protoporphyrin IX (FECH inhibitor) for 48h. e, qRT-PCR analysis of PD-L1 mRNA in cells from (d). Data represent mean PD-L1 mRNA expression normalized to ACTIN across n=3 technical replicates, shown as individual data points. All experiments in a-b and d-e were performed two independent times, with similar results. Data from representative experiments are shown. f, qRT-PCR analysis of Pd-l1 mRNA normalized to Actin in LLC cells expressing control or Urod shRNA. Bar graph represents mean normalized Pd-l1 mRNA from n=3 technical replicates, shown as individual data points from a representative experiment. Experiment were performed two independent times with similar results. g, Representative images of LLC tumors expressing control or Urod shRNA. This experiment was repeated twice with similar results. h, Gating strategy for TIL staining of tumors. Cells were gated based on SSC and FSC. Singlet cells were then gated for APC-Cy7-CD45+ and eFLUOR V500+ dead cells were excluded. i, Representative gating for CD4+ and CD8+ cells in one tumor from each experimental group. CD45+ live cells from each group was obtained by gating as in (h) and then gated for FITC-CD8+ and PE-CD4+ cells to obtain % CD8+ (of CD45+ cells).
Extended Data Fig. 3. Urod depletion induces an immune suppressive and pro-tumorigenic transcriptional program in LLC cells.

a-b, Gene Set Enrichment Analysis highlighting expression of top gene sets significantly increased in LLC cells expressing Urod shRNA. c, Ingenuity Pathway Analysis demonstrating a decrease in expression of genes involved in immune cell recruitment and migration. Graph represents biological processes plotted against their bias adjusted z-scores. Transcriptional analysis was performed on n=3 biological (cell culture) replicates per group.
Extended Data Fig. 4. Investigating the mechanism of PD-L1 regulation in UROD depleted cells.

a, qRT-PCR analysis of PD-L1 mRNA decay in H1944 cells expressing control or UROD sgRNA after treatment with 10 μM Actinomycin D in n=3 technical replicates per time point, across 4 time points. b, Quantification of PD-L1 levels, determined by western blot, in H1944 cells expressing control or UROD sgRNA after treatment with 20 μM cycloheximide across 5 time points. c, Immunoprecipitation of endogenous PD-L1 in H1944 cells transfected with control or UROD siRNA treated with 10 μM Velcade for 24 hours. Protein abundance and ubiquitylation monitored by western blot. Experiment in (c) was performed two independent times, with similar results. Data from a representative experiment are shown.
Extended Data Fig. 5. The ISR pathway regulates PD-L1 expression independently of IFN-γ.
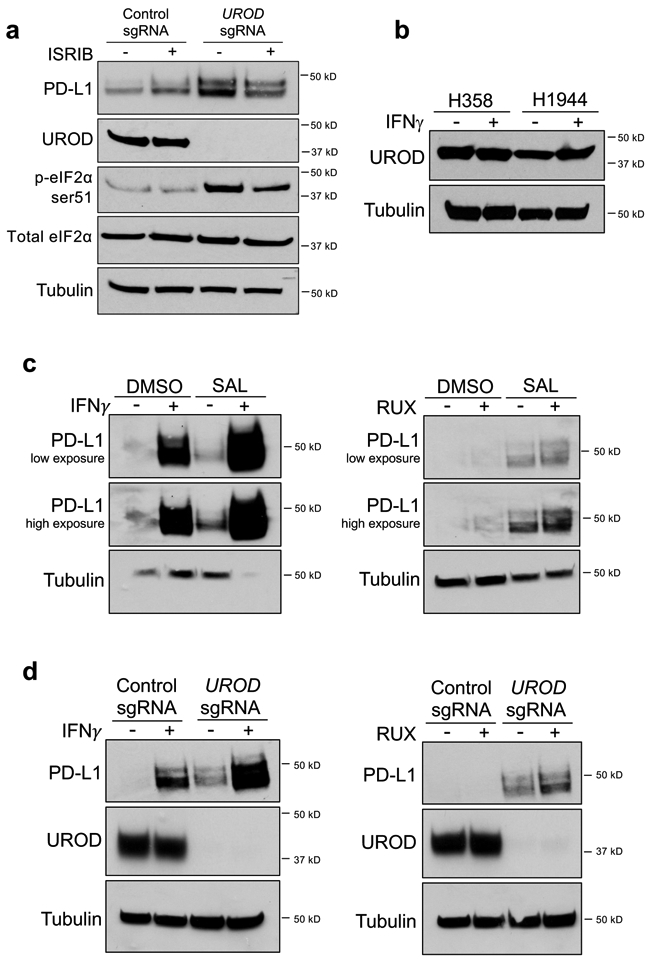
a, Western blot analysis in H1944 cells expressing control or UROD sgRNA after treatment with 200 nM ISRIB for 24 h. b, Western blot analysis of UROD in H358 and H1944 cells treated with 10ng/mL IFN-γ for 24h. c, Western blot analysis of PD-L1 in H1944 cells treated with DMSO or 100uM Salubrinal for 24h +/− 10ng/mL IFN-γ (left) or 10uM Ruxolitinib (right). d, Western blot analysis of PD-L1 in H1944 cells expressing a control or UROD sgRNA +/− 10ng/mL IFN-γ (left) or 10uM Ruxolitinib (right). All experiments were performed three independent times with similar results. Representative data are shown.
Extended Data Fig. 6. ISR pathway activation enhances PD-L1 translation, and regulation of the PD-L1 5′ UTR reporter by UROD depletion.

a, qRT-PCR analysis of mean Pd-l1 mRNA, normalized to Actin, in S/S or A/A MEFs expressing either control or Urod shRNA with 3 technical replicates per sample. Experiment was performed two independent times with similar results. Data from a representative experiment are shown. b,c, qRT-PCR analysis of PD-L1 mRNA in 6 individual ribosomal fractions, with 3 technical replicates per sample for two independent primer pairs. Quantification of combined fractions (<3 and >3) from (b) are shown in main Fig. 5c. d,e, qRT-PCR analysis of PD-L1 mRNA in 6 individual ribosomal fractions, with 3 technical replicates per sample for two independent primer pairs. Quantification of combined fractions (<3 and >3) from (d) are shown in main Fig. 5d. f,g, qRT-PCR analysis of ATF4 mRNA in ribosomal fractions from main Fig. 5a (f) or main Fig. 5b (g). ATF4 mRNA abundance was calculated as described in main Fig. 5c and d. Error bars in f and g represent SDs from the mean from three independent fractions (<3 or >3 ribosomes). A student’s two-tailed t-test was performed to determine statistical significance. ***p=0.00062 (f); ***p=0.0008 (g). h,i, qRT-PCR analysis of ATF4 mRNA in 6 individual ribosomal fractions. j, qRT-PCR analysis of mean Luciferase mRNA, normalized to Actin, in MEF cells transfected with reporter constructs shown in main Fig. 5e, with 3 technical replicates, shown as individual data points. Experiment was performed two independent times, with similar results. Data from a representative experiment are shown. k, qRT-PCR analysis of mean Luciferase mRNA normalized to ACTIN in H358 cells from main Fig. 5g across n=3 technical replicates, shown as individual data points. l, qRT-PCR analysis of mean Luciferase mRNA normalized to Actin in MEF cells from main Fig. 5h across n=3 technical replicates, shown as individual data points. Experiment in (k) was performed in an independent cell line, shown in (l).
Extended Data Fig. 7. eIF5B is necessary for PD-L1 upregulation in UROD depleted human lung cancer cells and the KLN205 syngeneic mouse model.

a, Flow cytometry analysis of cell-surface PD-L1 in independent human lung cancer cells. Bar graph represents Mean Fluorescence Intensity of cell-surface PD-L1 in PD-L1 Low (Calu-6, H2030) and PD-L1 High (H358, H441, Hop62) cell lines. b, Western blot analysis of PD-L1 and eIF5B in Calu-6 and H2030 cells expressing a control or UROD sgRNA transfected with control or EIF5B siRNA. c, Western blot analysis of PD-L1 and eIF5B in H441, H358 and Hop62 cells expressing a control or UROD sgRNA transfected with control or EIF5B siRNA. Experiments in b-c were performed two independent times with similar results. Data from a representative experiment are shown. d, Western blot analysis of PD-L1 in KLN205 cells expressing a Scrambled shRNA or two independent Urod shRNAs. e, Western blot analysis of PD-L1, UROD, and eIF5B in KLN205 cells expressing a control shRNA or Urod shRNA with and without Eif5b shRNA. Experiments in d and e were performed two independent times with similar results. Data from a representative experiment are shown. f, Quantification of tumor volumes of KLN205 cells shown in (e) transplanted to syngeneic DBA/2 mice (n=8 mice for Scrambled shRNA, n=5 for Eif5b shRNA, n=9 for Urod shRNA and n=8 for Urod + Eif5b shRNA). Linear mixed model was used to assess statistical significance, as described in Methods. p value (Scrambled vs Urod shRNA) = 0.0002, p value (Urod shRNA vs Urod + Eif5b shRNA) = 4.9351e-05.
Extended Data Fig. 8. EIF5B depletion reduces cell proliferation without affecting global translation of human lung cancer cells.

a, MTS cell proliferation assay measuring proliferation of H358, H1944, H2030, Calu-6 and H441 cells transfected with control or EIF5B siRNA with n=3 biological replicates. Student’s t-test with Holm-Sidak adjustment was performed per time point to assess statistical significance. ** = p<0.01; *** = p<0.001; **** = p<0.0001. Adjusted p values were as follows for Day 1, 2, 3, 4 and 5 respectively. H358: 0.74, 0.096, 0.064, 0.013 and 0.0001, H1944: 0.495, 0.044, 0.042, 0.0449, 0.018, H2030: 0.801, 0.068, 0.06, 0.039, 0.008, Calu6: 0.57, 0.42, 0.002, 0.016 and 0.008, H441: 0.258, 0.079, 0.01, 0.0002 and 0.012. b, Autoradiography analysis of newly synthesized S-35 labeled proteins in cells from (a). Experiments were performed two independent times for H358 and H1944 cells with similar results. Data from a representative experiment are shown.
Extended Data Fig. 9. Meta-analysis of EIF5B expression correlated with survival across lung cancer datasets.

a, Oncoprint plot depicting mRNA upregulation, amplifications, and mutations in EIF5B in human lung adenocarcinoma samples from the TCGA, Firehose Legacy. b, Kaplan-Meier survival analysis of patients with or without EIF5B alterations shown in (a) from the TCGA Firehose Legacy lung adenocarcinoma study using cBioPortal. n= 230 patients, Log rank test p-value = 0.0238. c, Summary of meta analyses results. d, Forest plot of meta-analysis showing association between EIF5B mRNA expression and patient survival outcome in all lung cancer patients across different studies (26 studies with 4,528 patients in total). e, Forest plot showing association between EIF5B expression and patient outcome in lung adenocarcinoma patients across different studies (20 studies with 2,882 patients in total). f, forest plot showing no association between EIF5B and patient survival in lung squamous cell carcinoma patients (26 studies with 1,277 patients in total). The confidence interval was calculated based on two-sided tests.
Extended Data Fig. 10. Luciferase mRNA levels from the PD-L1 5′ UTR reporter with EIF5B overexpression.

a, qRT-PCR analysis of mean Luciferase mRNA normalized to ACTIN in cells from Fig. 6i across three technical replicates, shown as individual data points. b, qRT-PCR analysis of Luciferase mRNA in cells from Fig. 6j, normalized to Actin across three technical replicates, shown as individual data points.
Supplementary Material
Acknowledgements
We thank the McDermott Center Sequencing Core for sequencing, Dr. Randal Kaufman (SBP Discovery Institute) for sharing eIF2α wild-type (S/S) and mutant (A/A) MEFs, Dr. Don Gibbons (MD Anderson) for sharing LLC cells, Vinnie Tagliabracci and Anju Sreelatha for assistance with heme synthesis experiments, Jenna Jewell and Chase Melick for assistance with S35 labeling, Mike Buszczak and members of his laboratory for assistance with polysome profiling, and Hesham Sadek and members of his lab for assistance with hypoxia treatments. We thank members of the O’Donnell laboratory for critical reading of the manuscript. Funding: K.A.O. was supported by the NCI (R01 CA207763 and P50CA70907), the Sidney Kimmel Foundation (SKF-15-067), the Cancer Prevention Research Institute of Texas (CPRIT, R1101, RP150676), the Welch Foundation (I-1881), the LUNGevity Foundation (2015-03), and the UTSW Friends of the Comprehensive Cancer Center. J.T.M. is an Investigator of the Howard Hughes Medical Institute and was also supported by the NCI (R35CA197311 and P30CA142543, and P50CA196516), CPRIT (RP160249), and The Welch Foundation (I-1961-20180324). S.S. was supported by RP140110 and RP160157 (CPRIT), a NIH T32 grant (1T32GM10977601), and a HHMI Med into Grad Grant (56006776).
Footnotes
Competing interests - The authors declare no competing interests.
REFERENCES
- 1.Bray F et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, doi: 10.3322/caac.21492 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Akbay EA et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer discovery 3, 1355–1363, doi: 10.1158/2159-8290.Cd-13-0310 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong H et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine 8, 793–800, doi: 10.1038/nm730 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Ribas A Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer discovery 5, 915–919, doi: 10.1158/2159-8290.Cd-15-0563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tumeh PC et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571, doi: 10.1038/nature13954 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barber DL et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687, doi: 10.1038/nature04444 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Herbst RS et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (London, England) 387, 1540–1550, doi: 10.1016/s0140-6736(15)01281-7 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Reck M et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. The New England journal of medicine 375, 1823–1833, doi: 10.1056/NEJMoa1606774 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Atefi M et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 3446–3457, doi: 10.1158/1078-0432.Ccr-13-2797 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu-Lieskovan S et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Science translational medicine 7, 279ra241, doi: 10.1126/scitranslmed.aaa4691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casey SC et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (New York, N.Y.) 352, 227–231, doi: 10.1126/science.aac9935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coelho MA et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 47, 1083–1099.e1086, doi: 10.1016/j.immuni.2017.11.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95, doi: 10.1038/nature25015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lan B et al. Association between PD-L1 expression and driver gene status in non-small-cell lung cancer: a meta-analysis. Oncotarget 9, 7684–7699, doi: 10.18632/oncotarget.23969 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dever TE et al. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596 (1992). [DOI] [PubMed] [Google Scholar]
- 16.Pakos-Zebrucka K et al. The integrated stress response. EMBO Rep 17, 1374–1395, doi: 10.15252/embr.201642195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ron D Translational control in the endoplasmic reticulum stress response. J Clin Invest 110, 1383–1388, doi: 10.1172/JCI16784 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donnelly N, Gorman AM, Gupta S & Samali A The eIF2alpha kinases: their structures and functions. Cellular and molecular life sciences : CMLS 70, 3493–3511, doi: 10.1007/s00018-012-1252-6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu PD, Harding HP & Ron D Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167, 27–33, doi: 10.1083/jcb.200408003 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sendoel A et al. Translation from unconventional 5’ start sites drives tumour initiation. Nature 541, 494–499, doi: 10.1038/nature21036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nature medicine 25, 301–311, doi: 10.1038/s41591-018-0321-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science (New York, N.Y.) 343, 84–87, doi: 10.1126/science.1247005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burr ML et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105, doi: 10.1038/nature23643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mezzadra R et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110, doi: 10.1038/nature23669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitby FG, Phillips JD, Kushner JP & Hill CP Crystal structure of human uroporphyrinogen decarboxylase. The EMBO journal 17, 2463–2471, doi: 10.1093/emboj/17.9.2463 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nature communications 5, 5241, doi: 10.1038/ncomms6241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu L, Han AP & Chen JJ Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Molecular and cellular biology 21, 7971–7980, doi: 10.1128/mcb.21.23.7971-7980.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rafie-Kolpin M et al. Two heme-binding domains of heme-regulated eukaryotic initiation factor-2alpha kinase. N terminus and kinase insertion. The Journal of biological chemistry 275, 5171–5178 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Han AP et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. The EMBO journal 20, 6909–6918, doi: 10.1093/emboj/20.23.6909 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harding HP et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell 6, 1099–1108 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Sonenberg N & Hinnebusch AG Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745, doi: 10.1016/j.cell.2009.01.042 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vattem KM & Wek RC Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America 101, 11269–11274, doi: 10.1073/pnas.0400541101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyce M et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science (New York, N.Y.) 307, 935–939, doi: 10.1126/science.1101902 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Sidrauski C, McGeachy AM, Ingolia NT & Walter P The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. eLife 4, doi: 10.7554/eLife.05033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sidrauski C et al. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. eLife 4, e07314, doi: 10.7554/eLife.07314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blais JD et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Molecular and cellular biology 24, 7469–7482, doi: 10.1128/mcb.24.17.7469-7482.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paolini NA et al. Ribosome profiling uncovers selective mRNA translation associated with eIF2 phosphorylation in erythroid progenitors. PloS one 13, e0193790, doi: 10.1371/journal.pone.0193790 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rutkowski DT & Kaufman RJ All roads lead to ATF4. Developmental cell 4, 442–444 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Andreev DE et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife 4, e03971, doi: 10.7554/eLife.03971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hinnebusch AG, Ivanov IP & Sonenberg N Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science (New York, N.Y.) 352, 1413–1416, doi: 10.1126/science.aad9868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kearse MG & Wilusz JE Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev 31, 1717–1731, doi: 10.1101/gad.305250.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White JP, Reineke LC & Lloyd RE Poliovirus switches to an eIF2-independent mode of translation during infection. J Virol 85, 8884–8893, doi: 10.1128/JVI.00792-11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lignitto L et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 178, 316–329 e318, doi: 10.1016/j.cell.2019.06.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen HG et al. Development of a stress response therapy targeting aggressive prostate cancer. Science translational medicine 10, doi: 10.1126/scitranslmed.aar2036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho JJD et al. Oxygen-Sensitive Remodeling of Central Carbon Metabolism by Archaic eIF5B. Cell Rep 22, 17–26, doi: 10.1016/j.celrep.2017.12.031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS ONLY REFERENCES
- 46.Golden RJ et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 542, 197–202, doi: 10.1038/nature21025 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hooda J, Alam M & Zhang L Measurement of Heme Synthesis Levels in Mammalian Cells. Journal of visualized experiments : JoVE, e51579, doi: 10.3791/51579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gandin V et al. Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. Journal of visualized experiments : JoVE, doi: 10.3791/51455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aronesty E Comparison of Sequencing Utility Programs. The Open Bioinformatics Journal 7, 1–8, doi: 10.2174/1875036201307010001 (2013). [DOI] [Google Scholar]
- 50.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, doi: 10.1093/bioinformatics/btt656 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi: 10.1093/bioinformatics/btp616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mootha VK et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273, doi: 10.1038/ng1180 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550, doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kramer A, Green J, Pollard J Jr. & Tugendreich S Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530, doi: 10.1093/bioinformatics/btt703 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740, doi: 10.1093/bioinformatics/btr260 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai L et al. LCE: an open web portal to explore gene expression and clinical associations in lung cancer. Oncogene 38, 2551–2564, doi: 10.1038/s41388-018-0588-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DerSimonian R & Laird N Meta-analysis in clinical trials. Control Clin Trials 7, 177–188, doi: 10.1016/0197-2456(86)90046-2 (1986). [DOI] [PubMed] [Google Scholar]
- 59.Paule RC & Mandel J Consensus values and weighting factors. Journal of Research of the National Bureau of Standards 87, 377–385 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CRISPR screening and RNA-sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE129968 and GSE139120, respectively. Kaplan-Meier survival analysis of patients with or without EIF5B alterations are from the TCGA Firehose Legacy lung adenocarcinoma study using cBioPortal. The EIF5B meta-analyses were performed using the Lung Cancer Explorer web portal57 (http://lce.biohpc.swmed.edu/lungcancer/). Source data for Figs.1-6 and Extended Data Figs.1-10 are provided. All other data supporting the findings of this study are available from the corresponding author on reasonable request.